

Abstract
Alkene difunctionalization reactions are important in organic synthesis. We have recently shown that copper(II) complexes can promote and catalyze intramolecular alkene aminooxygenation, carboamination, and diamination reactions. In this contribution, we report a combined experimental and theoretical examination of the mechanism of the copper(II)-promoted olefin aminooxygenation reaction. Kinetics experiments revealed a mechanistic pathway involving an equilibrium reaction between a copper(II) carboxylate complex and the γ-alkenyl sulfonamide substrate and a rate-limiting intramolecular cis-addition of N–Cu across the olefin. Kinetic isotope effect studies support that the cis-aminocupration is the rate-determining step. UV/Vis spectra support a role for the base in the break-up of copper(II) carboxylate dimer to monomeric species. Electron paramagnetic resonance (EPR) spectra provide evidence for a kinetically competent N–Cu intermediate with a CuII oxidation state. Due to the highly similar stereochemical and reactivity trends among the CuII-promoted and catalyzed alkene difunctionalization reactions we have developed, the cis-aminocupration mechanism can reasonably be generalized across the reaction class. The methods and findings disclosed in this report should also prove valuable to the mechanism analysis and optimization of other copper(-II) carboxylate promoted reactions, especially those that take place in aprotic organic solvents.
Keywords: alkenes, aminocupration, copper, EPR spectroscopy, kinetics, reaction mechanisms
Introduction
The discovery and development of copper-catalyzed and -promoted difunctionalizations of unactivated alkenes is a burgeoning area of chemical research.[1–15] The alkene functional group is ubiquitous, the resulting products are useful, and copper is a versatile and relatively inexpensive reagent and catalyst. A number of mechanistic pathways and intermediates are feasible and the precise pathway depends on the copper oxidation state (Cu0, CuI, CuII, or CuIII) and the reaction components.[16] Copper can serve as a Lewis acid, a π-acid, and an oxidant, and organometallic mechanisms as well as redox mechanisms have been implicated in its mode of action.
Copper(II) carboxylate promoted, intramolecular alkene aminofunctionalization reactions, carboamination,[17–20] diamination[21–23] and aminooxygenation [Eq. (1), PMBS = para-methoxybenzenesulfonyl],[24] are a growing class of such reactions that provide useful nitrogen heterocycles functionalized at the 2-position, for example, indolines and pyrrolidines, in good to excellent yield and high diastereoselectivity in many instances.[2] These reactions have been rendered catalytic with the use of ligands and oxidants [MnO2, (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and O2], and when chiral ligands are employed, asymmetric catalytic reactions can result [e.g., Eq. (2)].[23,25–27]
 |
(1) |
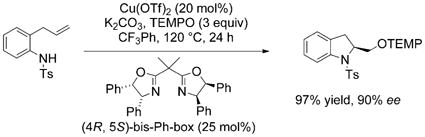 |
(2) |
In our initial mechanistic examination of these three reactions, evidence for both organocopper (C–Cu)[22] and carbon radical (R•)[18,19,22,26,27] intermediates was found. For example, the formation of the terminal C–N bond in the diamination reaction is most consistent with a Kharash–Sosnovsky-type CuIII mechanism,[28–30] which involves addition of a carbon radical to an R2N–CuII complex followed by reductive elimination (Scheme 1). Conversely, formation of the C–C and C–O bonds in the carboamination and aminooxygenation reactions is most consistent with direct addition of a carbon radical to a π-bond or to the stable TEMPO radical, respectively (Scheme 1).[18,19,22,26,27]
Scheme 1.

Proposed mechanisms for C–C, C–N, and C–O bond formations.
We have studied the mechanisms of these reactions using stereochemical and regiochemical trends, isotopic labeling, and radical trapping agents.[18,19,22,26,27] These methods have provided a working mechanistic model that aids in product prediction, but we sought a more rigorous understanding of the nature and structure of the discrete reaction intermediates and the relative energies of the steps on the reaction coordinate. Due to its high efficiency and apparently least complicated reaction sequence, we selected the copper(II) carboxylate promoted, intramolecular aminooxygenation reaction[24] as the first subject for detailed reaction kinetics, isotope effects, and molecular modeling studies. The fact that the soluble TEMPO radical can serve as sole oxidant and oxygen source in the copper-catalyzed aminooxygenation of some substrates[26] also made this a practical choice for comparison with future follow-up studies of the catalytic reaction.
Initial mechanism proposal
A comparison[18] of the high 2,5-cis-pyrrolidine stereoselectivity in the copper(II) carboxylate promoted, alkene aminooxygenation[24] of α-substituted-γ-alkenylsulfonamides [Eq. (1), vide supra] with analogous intramolecular alkene amination reactions that proceed via trans-aminometallation[18,31,32] (outer sphere) and cis-aminometallation[18,33–36] (inner sphere) mechanisms led us to postulate that, stereochemically, the copper-promoted reaction is more similar to reactions that proceed through cis-aminometallation mechanisms than to those that proceed through trans-aminometallations. We proposed that the resulting organocopper(II) intermediate underwent C–Cu homolysis[29,37–39] to generate the carbon radical, and direct trapping of the radical with TEMPO[40,41] provided the observed aminooxygenation product (Scheme 2). We proposed that the cis-pyrrolidine stereochemistry resulted from N–C bond formation through either a chair- or boatlike transition state, both of which place the sterically demanding α- and N-substituents on opposite sides of the ring.[18]
Scheme 2.

Proposed mechanism for the diastereoselective, copper-promoted, aminooxygenation of substituted alkenyl sulfonamides.
An uncoordinated, “naked” nitrogen radical mechanism (the result of N–Cu homolysis) was disfavored, because intramolecular alkene additions that proceed by means of nitrogen radical mechanisms show poor stereoselectivity or selectivity for the 2,5-trans-pyrrolidines.[18,42–44]
Despite this prior analysis, a number of key questions remained. Detailed structures and direct analytical evidence for the individual copper species involved in the reaction were not available from our initial investigations, but might be obtained by UV/Vis and EPR spectroscopy. The ligand environment, geometry, and charge of the copper in the stereochemistry-determining transition state were also poorly understood, but might be analyzed by DFT calculations. In addition, the rate-limiting step for this reaction had yet to be confirmed, but could be probed by kinetics and kinetic isotope effects as well as DFT calculations. Finally, we believed a detailed understanding of the role and localization of the unpaired electron brought to the reaction by CuII and the degree of charge transfer occurring between the nitrogen-containing substrate and the copper(II) promoter might be assessed by EPR spectroscopy and spin analysis of DFT calculated intermediates and transition states.
In this article, we report 1) the kinetics studies conducted to determine the kinetic order in each component of the aminooxygenation reaction, 2) the kinetic isotope effect studies that aid in determining the rate-limiting step, 3) the UV/Vis and EPR spectra of relevant observable [Cu] species, 4) evidence for the direct carbon radical trapping with TEMPO (provided by a deuterium labeling experiment) and 5) DFT calculations of intermediates and transition states along the reaction coordinate.
Results and Discussion
Experimental mechanistic investigations
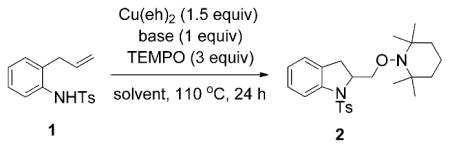
N-Tosyl-o-allylaniline (1) was chosen as the model substrate for kinetics studies due to its high reactivity and efficiency in both the stoichiometric and catalytic aminooxygenation reactions.[18,24,26] Substrate 1 was initially shown to undergo Cu(OAc)2 promoted aminooxygenation in DMF,[18] but subsequent studies with other substrates have shown that the aminooxygenation reaction can be carried out in less polar solvents.[24,26] Thus, several solvents were screened for the copper(II) 2-ethylhexanoate, (Cu(eh)2), promoted aminooxygenation of 1. As shown in Table 1(entries 1–3), toluene, CF3Ph, and xylenes are suitable solvents for the aminooxygenation reaction. As Cu(eh)2 has the highest solubility in toluene and owing to the fact that toluene is the most widely used of these organic solvents, it was selected as the solvent of choice for the kinetics study.
Table 1.
Aminooxygenation reaction of N-tosyl-o-allylaniline using different solvents and bases.
 | |||
|---|---|---|---|
| Entry | Solvent | Base | Yield [%][a] |
| 1 | CF3Ph | Cs2CO3 | 85 |
| 2 | CH3Ph | Cs2CO3 | 87 |
| 3 | xylenes | Cs2CO3 | 90 |
| 4 | CH3Ph | proton sponge | 57 |
| 5 | CH3Ph | NBu4OAc | 96 |
| 6 | CH3Ph | 2,5-di-tBu-4-Me-pyridine | 83 |
| 7 | CH3Ph | – | 70 |
Yield refers to amount of 2 isolated after flash chromatography.
In our previously reported aminooxygenation reactions,[24,26] we used Cs2CO3 or K2CO3 as base. The poor solubility of these bases in toluene prompted us to examine more soluble bases for greater control of concentration in our kinetics experiments. Use of a proton sponge gave poor yield, while the use of NBu4OAc or 2,5-di-tert-butyl-4-methylpyridine gave excellent yields (Table 1, entries 4–6). The reaction was less efficient without use of a base (entry 7, Table 1). Since NBu4OAc gave a higher yield, we used it in our kinetics study. The analogy of this soluble base to the less soluble bases K2CO3 and Cs2CO3 was further explored by UV/Vis spectroscopy (vide infra). From these studies we could conclude that NBu4OAc is a reasonably good model for Cs2CO3, the base of choice for the CuII-promoted reactions.[17–24]
Kinetic order of the reaction
We determined the kinetic order of the aminooxygenation reaction of N-tosyl-o-allylaniline (1) in each of the reaction components. Under pseudo-first-order conditions, we first examined the order in the alkene substrate by combining substrate 1 (50 mM) and 300 mM each of Cu(eh)2, NBu4OAc and TEMPO in toluene at 100°C. The progress of the reaction was monitored up to 80–90% conversion by using high-performance liquid chromatography (see Supporting Information for detailed description).
Initial rate data suggested that the order in concentration of sulfonamide 1 was half-order. The integrated rate law for such a reaction is given in Equation (3).
| (3) |
As shown in Figure 1, a linear plot of 2[1]0.5 versus time was obtained, confirming half-order dependence in the alkene substrate.
Figure 1.

A linear plot of 2[1]0.5 versus time showing half-order dependence in substrate 1.
We next determined the effect of NBu4OAc on the rate of the reaction using 1 (50 mM), 300 mM each of Cu(eh)2 and TEMPO and varying concentrations of NBu4OAc (12 to 500 mM) in toluene at 100°C. The value of the observed rate constant, kobs at each concentration of NBu4OAc was obtained from the slope of the plot of 2[1]0.5 against time (see Supporting Information). The plot of kobs against concentration of NBu4OAc revealed that the observed rate constant increases to a maximum (kobs = (0.62 ± 0.05) mM0.5 min−1) at an OAc− concentration of approximately 150–200 mM, which is about half the concentration of Cu(eh)2, and then decreases at higher concentrations of NBu4OAc (Figure 2). This 2:1 [Cu(eh)2]/[OAc−] ratio appears to be optimal for this reaction. At a NBu4OAc concentration of zero, the extrapolated rate constant is equal to 0.090 mM0.5 min−1, which indicates that the reaction occurs in the absence of NBu4OAc, but at a slower rate. One interpretation of these observations is that the OAc− complexes with the copper(2-ethylhexanoate) dimer to form a more active species. This hypothesis was further examined using UV/Vis spectroscopy (vide infra).
Figure 2.

Plot of kobs against [NBu4OAc] showing the effect of increasing the concentration of NBu4OAc on the rate of the reaction. The rate constant is optimum at 2:1 ratio between [Cu] and [OAc−] (kobs = 0.62 ± 0.05 mM0.5 min−1).
A number of literature reports have demonstrated that copper(II) carboxylates exist in the crystalline phase as carboxylate-bridged dimers.[45–48] Crystallographic evidence exists for the binuclear structure of many of these compounds, and their dimeric structures persist in many nonaqueous solvents.[49,50] Of all the binuclear copper(II) complexes, cupric acetate monohydrate, [Cu2(OCOCH3)4(H2O)2], has been extensively studied.[46] It has a dimeric structure in which the copper ions are bridged by the acetate groups and the axial positions are occupied by water molecules (Figure 3). X-ray crystallographic analyses of various adducts of dimeric copper(II) acetate, [Cu2-(OCOCH3)4(L)2], have also been reported.[46,51,52] Some of the ligands used are methanol, acetic acid, N,N-dimethylformamide, and pyridine. These ligands were shown to occupy the axial position of the dimeric copper acetate complexes.
Figure 3.
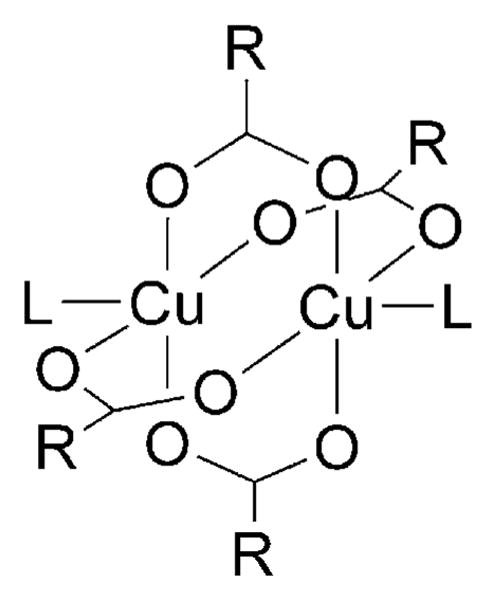
Structure of dimeric copper(II) acetate adducts with R = CH3 and L could be H2O, MeOH, acetic acid, DMF, pyridine.
Based on the NBu4OAc rate information, the order in Cu(eh)2 was determined using a 2:1 ratio of Cu(eh)2 and NBu4OAc. N-Tosyl-2-allylaniline (1) (50 mM), TEMPO (300 mM) and varying concentrations of Cu(eh)2 (300 to 1000 mM) and NBu4OAc (150 to 500 mM), at a constant 2:1 Cu(eh)2:NBu4OAc ratio, were combined in toluene at 100°C. Using an analogous procedure, the disappearance of 1 was monitored and the rate constant at each Cu(eh)2 concentration was determined from the slope of the plot of 2[1]0.5 against time (see Supporting Information). The order in copper was determined by non-linear least squares fit to the equation, kobs = k[Cu(eh)2]n. From the slope of the plot of ln(kobs) versus ln[Cu(eh)2], the order was determined to be (0.52 ± 0.03) (Figure 4). This suggests that the rate law is half-order in total concentration of copper ion, [CuT].
Figure 4.

Plot of ln (kobs) versus ln [Cu(eh)2]. The order in Cu(eh)2 was obtained from the slope of the plot, 0.52 ± 0.03.
Finally, the determination of order in TEMPO was carried out by using sulfonamide 1 (50 mM), 300 mM of Cu(eh)2 and 150 mM of NBu4OAc and varying concentrations of TEMPO (200 to 400 mM) in toluene at 100°C. Rate constants were obtained from the slope of the plot of 2[1]0.5 against time (see Supporting Information). The data from the experiments showed no change in the rate constant upon changing the concentration of TEMPO, which indicates zero-order kinetic dependence in TEMPO. This suggests that TEMPO is not present in the rate-determining step.
The above results are consistent with the rate law given in Equation (4), in which [CuT] is the molar concentration of copper ions, without regard to the nature of the complex(es).
| (4) |
Added acetate affects the value of kobs, increasing it five-fold from 25CuT:1 acetate to a maximum at a 2 CuT:1 acetate ratio and then decreasing. At this 2:1 ratio kobs = (0.036 ± 0.002) min−1 at 100°C.
A mechanistic interpretation of the kinetics study is presented in Scheme 3. An equilibrium between sulfonamide 1 and the complex resulting from the Cu(eh)2 dimer and Bu4NOAc (possibly 3) results in monomers 4 and 5. The monomer–substrate intermediate 5 is the active Cu–N intermediate that undergoes the cis-aminocupration step to form the organocopper intermediate 6 via the transition state A. Carbon radical species 7 is formed by the homolysis of intermediate 6, which is then trapped by TEMPO to give the aminooxygenation product 2. The reaction kinetics indicate that k3, radical trapping with TEMPO, is not rate-limiting. An alternative to 5, 6 and A, in which RCO2H has been dissociated, is also viable (see the section on calculations, for further discussion).
Scheme 3.

Proposed mechanism for the copper(II)-promoted aminooxygenation of alkenes.
Effect of NBu4OAc
From the kinetic plot of kobs versus [NBu4OAc] (Figure 2), we were able to conclude that 1) the OAc− serves to activate Cu(eh)2 rather than as a base to deprotonate the sulfonamide and 2) a 2:1 Cu:OAc− ratio gave the optimum kobs. Kochi and Subramanian reported that the addition of acetate salts to a solution of cupric acetate in acetic acid effects the dissociation of the dimer [Eq. (5)].[53] Tri- (m = 1) and tetracetatocuprate (m = 2) are the anionic species formed; the former is the preferred species and the latter is only available at high concentrations of acetate.[54] The disappearance of the dimer upon the addition of the acetate salts has been qualitatively studied by UV/Vis absorption spectroscopy.[53] Cupric acetate in glacial acetic acid exhibits characteristic absorption bands at 690 and 375 nm. It appears as a green solution due to the broad absorption at 690 nm and the weak absorption band at 375 nm has been attributed to the dimer.[53,55,56] Its color changes to blue when acetate salt is added. At constant copper concentration, the intensity of both bands decreases with increasing amount of acetate salts. This observation indicates the disappearance of the dimer. At 57°C the dimer–monomer equilibrium constant in glacial acetic acid in the absence of acetate is 5010−4; addition of 0.23M sodium acetate increases this to 3.2010−3 and at 1.04M lithium acetate the dimer completely dissociates. Therefore, the acetate adducts of the dimer have greater dissociation equilibrium constants than the adducts of acetic acid.
| (5) |
To support our hypothesis that the OAc− mediates in the dissociation of copper(II) 2-ethylhexanoate, we ran UV/Vis spectra of a series of solutions of Cu(eh)2 in toluene titrated with increasing amount of NBu4OAc (Figure 5). When NBu4OAc was added to a solution of Cu(eh)2, its color turned from green to blue.[53] The spectrum of a solution of Cu(eh)2 in toluene displayed absorption bands at 670 and 380 nm. Changes in the absorption spectra were observed when 6 and 12 mM of NBu4OAc were added to a 12 mM solution of Cu(eh)2. No further decrease in the absorbance was observed upon the addition of more than 12 mM of NBu4OAc. We interpet the spectral changes to indicate formation of first the 2:1 Cu/acetate adduct NBu4[{Cu(eh)2}2-(OAc)] and then the 1:1 Cu/acetate adduct [NBu4]2[{Cu(eh)2}2(OAc)2]. Addition of more acetate has no effect as all available coordination sites are occupied.
Figure 5.

UV/Vis spectra of a series of Cu(eh)2 (A) solution titrated with NBu4OAc (B), K2CO3 and Cs2CO3.
The maximum rate constant, kobs was observed at a Cu:OAc− ratio of 2:1. At OAc− concentration higher than 200 mM, kobs starts to decrease, presumably due to competing reaction (coordination with copper) between OAc− and the sulfonamide substrate 1. It is on the basis of this Cu:OAc− ratio that we propose dimer 3 as a potential structure for the formed complex. We examined the similarity of the 2:1 Cu:OAc− ratio spectra with the spectra obtained from the same ratio with the less soluble bases, K2CO3 and Cs2CO3. As seen in Figure 5, both bases affect the spectra, but the more soluble Cs2CO3 gives a spectrum more similar to that with Bu4NOAc, indicating it is playing a larger role in CuII carboxylate dimer break up. From this data we can conclude that Bu4NOAc is a reasonable soluble base model for Cs2CO3, the base of choice in the majority of our CuII carboxylate promoted reactions [e.g., Eq. (1) vide supra].[2]
Dimeric copper complex 3
Our kinetics results together with literature precedents have led us to postulate that dimer 3 combines with substrate 1 and dissociates to form the active monomeric copper species 5 (Scheme 3). The dimer complex 3 is proposed to contain two copper ions bridged together by 2-ethylhexanoate groups, with one acetate (from NBu4OAc) and an open coordination site on its remaining axial position. This proposed structure is consistent with the ratio that gave the optimum rate constant (2:1 Cu/OAc−). We envisioned that the coordination of substrate 1 with dimer 3 and the breaking down of the resulting intermediate into 4 and 5 is occurring simultaneously. The obtained half-order kinetic dependence in substrate 1 and in copper supports this mechanism (Figures 1 and 4).[57]
Kinetic isotope effects and rate-limiting step analysis
To gain further insight into the nature of the rate-limiting step of this aminooxygenation reaction, we investigated the primary and secondary kinetic isotope effects (kH/kD). The primary kinetic isotope effect was determined by comparing the rate constants for the reaction of 1 and [D]-1a. As shown in Equations (6) and (7), the measured kH/kD was equal to (0.98 ± 0.07) (see Supporting Information for details). This kH/kD is not significant compared to the maximum theoretical KIE of 8.5 (25°C), based on νNH of 3100 cm−1.[58,59] The observed lack of primary isotope effect implies that the amine deprotonation is not the rate-limiting step, thus supporting our depiction of the formation of the N–Cu intermediate 5 as an equilibrium rather than a rate-limiting process (Scheme 3).
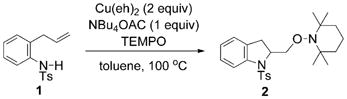 |
(6) |
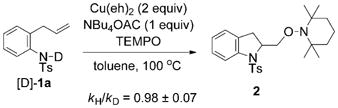 |
(7) |
To determine the secondary isotope effect, a mixture of substrates 1 and [D]-1b was subjected to the aminooxygenation reaction under typical reaction conditions [Eq. (8)]. The isotopic ratio before and after the reaction were determined by mass spectrometry and fractional conversion was determined by HPLC analysis (see Supporting Information). The measured inverse secondary kinetic isotope effect (kH/kD = (0.90 ± 0.03)) supports the addition of N–Cu bond across the olefin (k1, Scheme 3) as the rate-determining step. The inverse nature of the kinetic isotope effect is interpreted as arising from the change in hybridization of the alkene carbon from sp2 to sp3 in the transition state.[60]
 |
(8) |
Rate equation
If the cis-aminocupration is the rate-limiting step (Scheme 3) as suggested by the secondary kinetic isotope effect, the reaction rate for the aminooxygenation can be expressed as Equation (9). After derivation, it can further be expressed as Equation (10), which accounts for the observed rate law.
| (9) |
| (10) |
Competition experiment
The cis-aminocupration step, for example, transition state A (Scheme 3), involves a steric and an electronic component. Sterically, addition to a more substituted alkene should be disfavored. Electronically, addition to a terminally substituted alkene would be more favorable if the transition state held a significant amount of radical character, such that a secondary benzylic carbon radical would be more favored than a primary radical.[61] To ascertain the importance of such electronic stabilization, we ran a competition experiment between terminal alkene 8 and internal alkene 9. Equimolar amounts of 8 and 9 were treated with Cu(eh)2 (1 equiv), NBu4OAc (1 equiv) and TEMPO (3 equiv) in toluene and the solution was heated to 120°C. After 24 h, 70% of the adduct 10 was isolated, while only 14% of 11 was obtained [Eq. (11)]. Thus, the terminal olefin is more reactive than the internal olefin. Therefore, we conclude that the addition of N–Cu to the alkene is more dominated by sterics than electronics. This result disfavors an alternative nitrogen radical mechanism, since it is more favorable for a radical to add to a styrene, to form a benzylic radical, than to add to a terminal alkene to form a primary radical.[61,62] This result is also most supportive of a rate-limiting addition of N–Cu across the alkene versus a rate-limiting C–Cu homolysis, which would be more favorable for formation of a secondary benzylic carbon radical than a primary carbon radical.
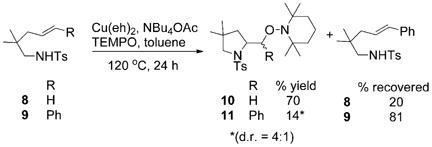 |
(11) |
Mechanistic analysis of C–O bond formation
The existence of a primary carbon radical intermediate in our copper(II)-catalyzed and -promoted reactions has been supported by isotopic labeling studies and regioselectivity trends.[18,22,26,27] In order to differentiate a mechanism involving direct carbon radical trapping with TEMPO versus a potential alkyl copper(II) oxidation with TEMPO to an alkyl copper-(III) complex followed by reductive elimination (Scheme 4), we performed a deuterium-labeling experiment. When the deuterioalkene [D]-12 was treated with the standard amino-oxygenation reaction conditions under partial conversion, 41% of the unscrambled starting material was recovered and 57% of a 1:1 mixture of aminooxygenation product diastereomers was obtained [Eq. (12)].
Scheme 4.

Evidence for direct capture of the carbon radical with TEMPO.
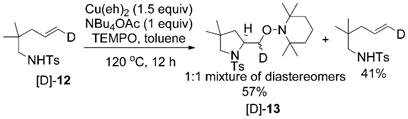 |
(12) |
The observed 1:1 mixture of diastereomers [D]-13 implies that a direct capture of an sp2-hybridized carbon radical intermediate is most likely operative in this reaction (Scheme 4, mechanism 1). This is not surprising as the direct carbon radical trapping with TEMPO is an efficient C–O bond-forming process.[40,41,63,64] If the reaction proceeds through mechanism 2, which is the reductive elimination from an alkyl copper(III) intermediate, then a single (or major) diastereomer would have been observed. This observation is consistent with previous reports in which TEMPO has been used as a catalytic or stoichiometric oxidant in copper-catalyzed cross-coupling and oxidation reactions.[65,66] In each of these reactions, C–O reductive elimination of TEMPO and an alkyl/aryl ligand on copper has never been suggested or observed. Although a mechanism involving an initial C–CuII homolysis and the subsequent recombination of the carbon radical with a TEMPO-coordinated CuII, followed by reductive elimination cannot be entirely ruled out, this mechanism is more complicated and there is no apparent driving force given that the combination of carbon radicals with TEMPO occurs spontaneously in so many instances.[40,41,63,64] Isolation of the starting material γ-alkenyl sulfonamide with complete retention of alkene geometry indicates that the carbon radical intermediate does not revert back to the alkene, implying the N–C bond formation is not reversible after C–Cu homolysis.
This isotope-labeling experiment also rules out a mechanism involving a concerted two-electron electrophilic addition[67,68] of TEMPO (either as a copper-complexed reagent or a oxoammonium ion [O=NR2]+, formed from oxidation of TEMPO by CuII) and the sulfonamide amine to the alkene, as one diastereomer of [D]-13 would also be expected in such a mechanism. The half-order dependence on [Cu] and substrate and, most importantly, zero-order dependence on TEMPO, determined by our reaction kinetics, also rule out such a mechanistic alternative.
Detection of an [N–Cu] intermediate by EPR spectroscopy and kinetics of the intermediate
The proposed mechanism in Scheme 3 postulates the intermediacy of a mononuclear copper–amido complex. To test the viability of such an intermediate we sought to independently synthesize a complex analogous to the proposed intermediate and to determine its ability to undergo aminooxygenation. Electron paramagnetic resonance (EPR) spectroscopy was used to detect an [N–Cu] intermediate. The EPR spectra were collected at the X-band at 100 K (Figure 6).
Figure 6.

EPR spectra of solutions of a) Cu(eh)2 and Bu4NOAc, b) Cu(eh)2, Bu4NOAc and sulfonamide 1 (heated at 50 °C for 15 min) and c) Cu(eh)2 and deprotonated sulfonamide 1 in toluene at 100 K.
The EPR spectrum of the [Cu2(eh)4] dimer (frozen glass in toluene, 100 K) is very weak and broad (EPR silent), indicating a strong degree of CuII–CuII spin coupling due to the dimer structure. In contrast, Figure 6a shows the spectrum of Cu(eh)2 and Bu4NOAc (2:1 ratio) in frozen toluene glass and has the parameters g|| = 2.33, g⊥ = 2.07, A||Cu = 157 MHz and A ⊥Cu = 19 MHz. The strong signal indicates the unpaired electron of CuII is being observed. No change in the EPR spectrum was observed when sulfonamide 1 was added to the Cu(eh)2/Bu4NOAc mixture (Figure 6b; 1/Cu(eh)2/BuNOAc = 2:2:1) which indicates that no [N–Cu] intermediate is detectable as a component of the equilibrium reaction under these conditions (Scheme 3). A change in the EPR spectrum was observed (Figure 6c), however, when the Li amide of 1, formed from the deprotonation of 1 with nBuLi, was added to a solution of Cu(eh)2 in toluene (R2NLi/Cu(eh)2 = 1:1). The EPR spectrum of the R2NLi/Cu(eh)2 product (Figure 6c) exhibits the typical four-line pattern expected for a single CuII center with N–Cu super-hyperfine splitting (ACu = 35 MHz and AN = 4 MHz). This spectrum (Figure 6c) is clearly different from the others (Figure 6a and b).[69] The broadness of some peaks in spectrum (Figure 6c) may be due to the presence of more than one species (for example, R2NCu(eh)2Li and R2NCu(eh)). The small N–Cu superhyperfine splitting indicates a low spin density located on the nitrogen (DFT calculated spin densities of the proposed observed intermediate are in agreement, see Supporting Information for a discussion and calculations). The spectra resemble EPR spectra recorded for other N–CuII species[70,71] and do not indicate a significant degree of aminal-radical–CuI character (due to internal redox).[72] The EPR spectrum (Figure 6c) indicates the [R2N-CuII] species under observation is more isotropic than would be expected of a square-planar CuII complex (lack of significant g|| signals).
We demonstrated that the [R2N–Cu] intermediate observed in Figure 6c was kinetically competent. Upon the addition of TEMPO and subsequent heating of the solution to 110°C the aminooxygenation product was produced in 89% yield [Eq. (13)].
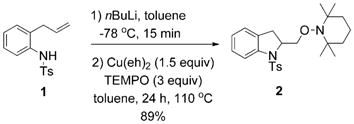 |
(13) |
The kinetics of the transformation of this [R2N–Cu] complex to aminooxygenation product 2 were determined at 100°C. A linear plot of ln[1] against time indicates that the reaction is first-order in the [R2N–Cu] complex (Figure 7). Neither excess of Cu(eh)2 nor TEMPO had an effect on the observed rate constant (kobs = (1.6 ± 0.1)010−2 min−1).
Figure 7.

Kinetic plot of the conversion of [N–Cu] complex to 2.
The rate of the reaction of the [R2N–Cu] complex at an initial concentration of 50 mM (0.75 mMmin−1) is about half the rate of the reaction of 1 promoted by the 2:1 [Cu(e-h)2]/[OAc−] ratio at 50 mM concentrations of 1 and [CuT] (1.8 mMmin−1 based on the experimentally determined rate law and rate constant). Therefore, the intermediate in the [Cu(eh)2]/[OAc−]-promoted reaction (which is present in non-detectable equilibrium concentrations) must be more reactive than the deprotonated [R2N–Cu] complex. Nonetheless, the EPR spectrum and the kinetic behavior of the [R2N–Cu] intermediate support the cis-aminocupration mechanism and suggest it is the rate-determining step. Calculated relative aminocupration activation energies of the respective [R2N–Cu] intermediates involved in these reactions (Scheme 6, vide infra) are consistent with the finding that the intermediate formed from R2NLi and Cu(OR)2 is less reactive (vide infra).
Scheme 6.

Cation effects on relative energies of aminocupration activation energy. Relative energies only are being compared since different atoms are involved in each path. GTS(C–A) = 2.8 kcalmol−1.
Theoretical investigation of the aminooxygenation reaction mechanism
Calculations were carried out at the Kohn–Sham hybrid-DFT B3LYP[73,74] level of theory using the Gaussian 09[75] and GaussView v5.0.8 programs. The GenECP method was employed with a 6-31G(d) basis set applied to all atoms (i.e., H, C, N, O, S) except copper, which was computed using the Los Alamos LAN2DZ[76–79] basis set. This combination was used because the sole use of the 6-31G(d) basis set resulted in exaggerated basis set superposition error (BSSE), while sole use of the LanL2DZ basis set did not properly account for the hypervalent nature of sulfur. For ease of calculations, Cu(OAc)2 was used instead of Cu(eh)2. The calculations (in vacuo) were carried out at the temperatures most relevant to the reaction being studied. These calculations were performed in vacuo owing to lower computational time required compared to use of an implicit solvation model. This is largely a reasonable simplification due to the low dielectric constant of toluene (ε = 2.38) and our use of relative energies for comparison of mechanistic scenarios. Introduction of error from this simplification would have most impact on species with a large charge separation. See the Supporting Information for a brief comparison of calculated relative energies using in vacuo versus implicit solvation models.
Calculations of aminooxygenation reaction mechanism possibilities
Detailed mechanistic alternatives for the Cu(OAc)2/Bu4NOAc-promoted aminooxygenation reaction of N-tosyl-2-allylaniline (1) were calculated (110°C) and the optimized transition states were confirmed to be first-order saddle points by the appearance of a single imaginary frequency (i). Furthermore, the respective product complexes immediately following from the transition states were located using the intrinsic reaction coordinate (IRC)[80–82] method and confirmed to be minima by the appearance of only real vibrational modes. Thereafter, each intermediate shown was located by optimization to the lowest energy minima of the corresponding geometry.
Two mechanistic scenarios (pathways A and B) were envisioned for this aminooxygenation reaction (Scheme 5). The two mechanisms differ in their possible starting [N–Cu] complexes, in particular, the number of coordinated acetate ligands. Path A starts from [(R1R2N)Cu(OAc)(HOAc)] (I-5; R1 = tosyl (Ts), R2 = 2-allylphenyl) and is the two-acetate, neutral path. Path B starts from [(R1R2N)CuOAc] (I-1B) + HOAc and is the one-acetate, neutral path. We found that one-acetate-ligand mechanism, path B, has a lower aminocupration activation barrier than the two-acetate neutral mechanism, path A, by about 1.2 kcalmol−1 (Figure 8 and Scheme 5). (The early steps of a two-acetate anionic pathway were considered briefly but deemed less likely due to a much higher relative aminocupration activation barrier, see Supporting Information for further discussion and calculations.)
Scheme 5.

Mechanistic alternative for copper(II)-promoted alkene aminoxygenation.
Figure 8.

Potential energy landscapes for the two-acetate-ligand neutral path A (blue diamond) and the one-acetate-ligand path B (red square) at 110°C. Energy scale in kcalmol−1. GTSA = 17.2 kcalmol−1, GTSB = 16.0 kcal mol−1.
We also calculated the activation barrier for the aminocupration starting from the [(R1R2N)Cu(OAc)2Li] intermediate (most relevant to Figure 7, vide supra) and found it was 2.8 kcalmol−1 higher than the activation barrier for transition state A, path A at 100°C, the temperature the kinetics experiments were run at (compare aminocupration paths A and C, Scheme 6). This is consistent with experimental findings that the reaction involving 1 deprotonated with nBuLi is half the rate of the reaction involving 1 and Bu4NOAc (vide supra). The fact that there is a difference in rates between the two series indicates that the copper centers in each intermediate are not likely to lose an acetate ligand to form intermediate I-1B (path B, Scheme 5), since then the rates would be expected to be the same.
Upon further investigation of reaction coordinates A and B (Scheme 5 and Figure 8), a dramatic ligand effect is observed. The most stable aminocopper intermediate to emerge from TS-B is I-2B, which still maintains N–Cu coordination. Loss of this coordination and homolysis to carbon radical I-7 and CuOAc resulted in a 26.5 kcalmol−1 higher energy state. The majority of the energy cost appears to be loss of the C–Cu bond (17.5 kcalmol−1), but loss of the N–Cu bond is also endothermic (9.0 kcalmol−1). The organocopper intermediate emerging from TS-A is I-2A, which also maintains N–Cu coordination. Loss of N–Cu coordination generates a re-oriented minimized C–Cu intermediate, I-6, which has an acetate and HOAc ligand bound to it. In the path A series, the re-oriented intermediate I-6 is actually lower in energy than I-2A by 2.0 kcalmol−1. The different relative change in energy between the two C–Cu intermediates in the two series might be explained by the formal charge on Cu in the two series. In series A, I-2A has a Cu with a formal charge of −2, so when it releases the N ligand, it becomes Cu with a formal charge of −1, and this change in formal charge on Cu is apparently favorable even though the Cu becomes tricoordinate in going to I-6. In series B, I-2B has a formal charge of −1, so when it releases the N ligand to yield I-3B, it becomes Cu with formal charge of 0. This is apparently not as beneficial a change compared to becoming the tricoordinate C–Cu intermediate I-3B (the acetate is bidentate), so I-2B to I-3B is an endothermic process. Homolysis of I-6 to give the carbon radical I-7 and Cu(OAc) (HOAc) is exothermic, but by only 1.0 kcal mol−1 (it is possible this step is actually slightly endothermic if solvation is taken into account, see supporting information for further discussion). Trapping of the carbon radicals with TEMPO was found to be highly exothermic, resulting in the optimized aminooxygenation product 2 and the respective CuI salt (Figure 8 and Scheme 5).
The large difference in product energies between paths A and B is the result of the copper(I) species, Cu(OAc) + HOAc in the one-acetate-ligand path B, and Cu(OAc)- (HOAc) for the two-acetate-ligand path A. Thus, it appears the ligation of the second acetate greatly stabilizes the CuI homolysis product, thereby impacting the overall reaction profile. It is thus proposed that the two-acetate-ligand (neutral) mechanism (path A, blue diamond, Figure 8) is overall lower in energy and thus a reasonable mechanism. Another mechanistic possibility, however, would be a “cross-over” mechanism in which aminocupration proceeds through the one-acetate-ligand pathway (path B, red square, Figure 8) resulting in the [C–Cu] complex I-2B, which then picks up an acetate (as HOAc to maintain overall neutrality) to generate intermediate I-6. At this point, the two-ligand, neutral mechanism (path A) is embarked upon whereby C–Cu homolysis is followed by capture of the resulting carbon radical I-7 by TEMPO to yield the exothermic aminooxygeantion product 2 and the respective CuI(OAc) (HOAc) salt. Bond dissociation energies were used in this analysis because the C–Cu homolysis transition states were difficult and in most cases impossible to locate.[83,84]
The calculated aminocupration transition states TS-A (path A) and TS-B (path B) have distorted square-planar copper geometries (Figure 9). Spin density analysis of transition states TS-A and TS-B indicate the majority of spin density resides on Cu and little increase in nitrogen spin from ground state to transition state is observed. The spin on nitrogen never exceeds 15% and thus the nitrogen does not possess significant radical character. Wiberg bond indices[85–89] were analyzed to quantify the extent to which the N–C and Cu–C bonds are formed at the transition states. Accordingly, the Cu–C bond is 73% formed in TS-A, while the N–C bond is only 49% formed. Similarly, the Cu–C bond is 71% formed at TS-B, while the N–C bond is only 49% formed. This indicates the alkene amincupration is being led by Cu π-bond activation. In the most stable C–Cu intermediates, I-6 and I-2B, significant spin density, 40 and 36%, respectively, resides on the terminal carbon, which leads to the prediction that this carbon will behave like a radical, which it subsequently does.
Figure 9.
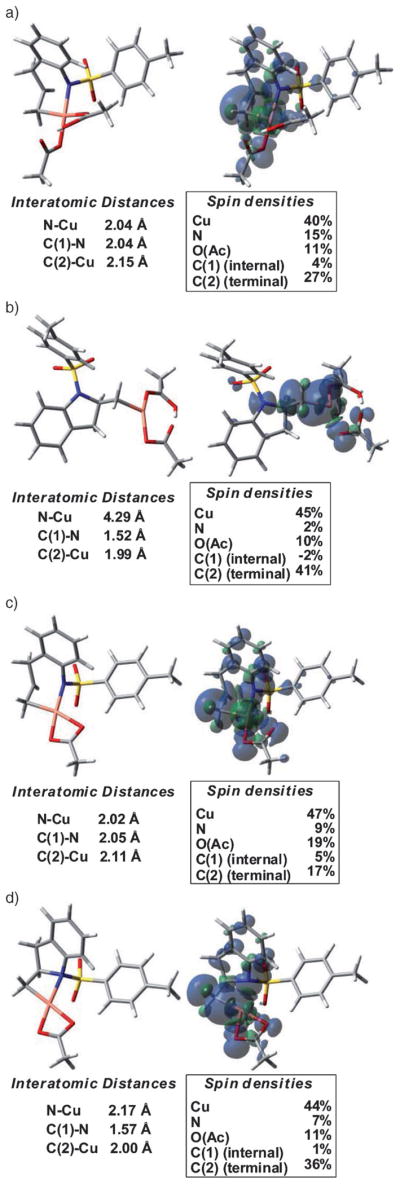
Optimized structures of aminocupration transition states, the resulting C–Cu intermediates, and spin density analysis thereof. C(1) = internal (alkene) carbon, C(2) = terminal (alkene) carbon. a) Transition state TS-A, b) intermediate I-6, c) transition state TS-B, d) intermediate I-2B.
An alternative aminooxygenation mechanism involving a nitrogen radical pathway, by means of homolysis of the N–Cu bond of I-5, was also considered. This pathway was calculated to be significantly higher in energy compared to pathway A, and thus was not considered competitive. See Supporting Information for a discussion and the calculation data.
Theoretical treatment of observed reaction diastereoselectivity
The Cu(eh)2 promoted aminooxygenation reaction of α-methyl-γ-alkenyl sulfonamide 14 occurred in about 83% yield and >20:1 cis/trans diastereoselectivity irrespective of whether Cs2CO3 or Bu4NOAc was used [Eq. (14)]. This 2,5-cis selectivity is consistent for a number of substrates.[18,24] We calculated eight possible aminocupration transition states for this reaction, four that lead to the observed 2,5-cis-pyrrolidine 15 and four that lead to the 2,5-trans-pyrrolidine 16.
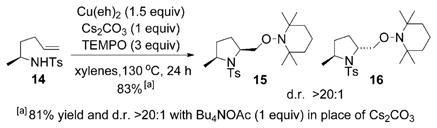 |
(14) |
The four potential aminocupration transition states for the two-ligand, neutral path D are shown in Figure 10a–d. Transition states cis-TS-Dch and cis-TS-Dbt (ch = chair, bt = boat) are favored (lower in energy by at least 2.5 kcalmol−1) in which the steric interaction is minimized between the methyl substituent and the tosyl group, leading to the 2,5-cis-product. The relative energies are shown in Figure 10(see Supporting Information for a potential energy landscape for the four possible path D transition states). The lowest energy transition state, cis-TS-Dch, most closely resembles a chairlike geometry, but it is within 0.2 kcalmol−1 of the cis-TS-Dbt transition state, which has a boatlike geometry.
Figure 10.
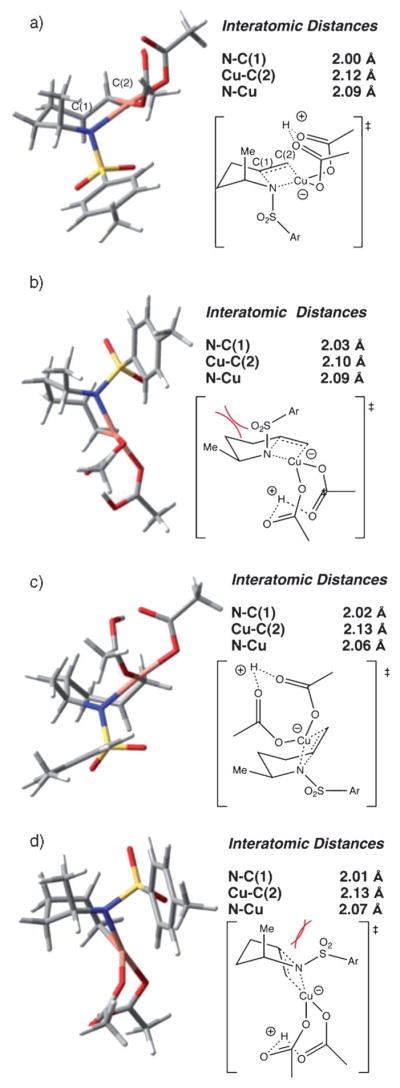
Path D transition states: a) cis-chair TS, relative energy = 0 kcalmol−1; b) trans-chair TS, relative energy = 3.5 kcalmol−1; c) cis-boat TS, relative energy = 0.2 kcalmol−1; d) trans-boat TS, relative energy = 2.7 kcalmol−1. C(1) = internal carbon, C(2) = terminal carbon.
The aminocupration transition states for the one ligand pathway E are shown in Figure 11a–d. The cis-product is favoured due to the lower energy (lower by at least 3.1 kcal mol−1) of transition states of cis-TS-Ech and cis-TS-Ebt compared with trans-TS-Ech and trans-TS-Ebt. The relative energies are shown in Figure 11 (see Supporting Information for a potential energy landscape for the four possible path E transition states). The lowest energy transition state is the cis-TS-Ebt, which is boatlike, and lower in energy than the chairlike cis-TS- Dch transition state by 0.2 kcalmol−1.
Figure 11.

Path E transition states: a) cis-chair TS, relative energy = 0.2 kcalmol−1; b) trans-chair TS, relative energy = 3.3 kcalmol−1; c) cis-boat TS, relative energy = 0 kcalmol−1; d) trans-boat TS, relative energy = 4.9 kcalmol−1. C(1) = internal carbon, C(2) = terminal carbon.
The finding that both the boat and chair transition states that lead to the 2,5-cis-pyrrolidines are within 0.2 kcalmol−1 is not consequential to the diastereoselectivity of the reaction since both lead to the same product. However, if this trend is similar with CuII catalysts coordinated by chiral bis(oxazoline) ligands, it could translate to low levels of enantioselectivity in some reactions (e.g., substrates with α-substituents). In point of fact, this has actually been observed in a recently performed attempted desymmetrization reaction.[90]
A comparison between the two mechanistic paths, D and E, is depicted in Figure 12 and Scheme 7, in which the lowest energy aminocupration transition states are supplemented by three additional minima on the energy landscape. The one-ligand mechanism (path E) is calculated to have a lower activation energy for aminocupration by 3.1 kcal mol−1. The one-ligand mechanism, however, is disfavoured to undergo C–Cu homolysis due to the higher energy state of the CuIOAc species. Again, a crossover mechanism may be active wherein aminocupration occurs with one ligand bound to copper, followed by coordination of a free HOAc ligand to facilitate homolysis.
Figure 12.

Potential energy landscape for the two possible pathways for the aminooxygenation of -methyl–al-kenylsulfonamide 14 at 130°C (energy scale in kcalmol−1).
Scheme 7.

Potential reaction coordinates for the cis-pyrrolidine selective aminooxygenation reaction. Path D involves a two-acetate neutral copper(II) complex and Path E involves a one-acetate neutral copper(II) complex.
Taken together, it is important to note that despite having a higher activation energy for aminocupration, overall, the two-ligand, neutral mechanism (pathway D) is favored over the one-acetate-ligand mechanism (pathway E) due to the relative energetics of the C–Cu homolysis step.
The two-acetate, neutral pathway D for the α-methyl-γ-alkenylsulfonamide 14 (Scheme 7 and Figure 12) closely resembles the analogous path A for the 2-allylaniline sulfonamide 1 (Scheme 5 and Figure 8). The C–Cu homolysis in each of these reactions is exothermic by about 1 kcalmol−1. The homolysis mechanism for the one-acetate neutral pathway E for the α-methyl-γ-alkenylsulfonamide 14 (Scheme 7 and Figure 12) is also quite similar to the analogous path B for the 2-allylaniline sulfonamide 1 (Scheme 5 and Figure 8). Breaking the N–Cu interaction in the initial aminocupration product to give the re-oriented intermediate, for example, I-2E to I-3E in the α-methyl series is endothermic by 7.0 kcal mol−1. Likewise, in the aniline series, the conversion of I-2B to I-3B is endothermic by 9.0 kcalmol−1. Similarly, C–Cu homolysis is endothermic in both series (I-3E to I-4D/E, +16.7 kcalmol−1; I-3B to I-7, +17.5 kcalmol−1). The energy differences I-2B to I-7 [+Cu(OAc) +HOAc)] and I-2E to ID/E [+Cu(OAc) +HOAc] are similar (26.5 and 23.7 kcal mol−1); C–Cu homolysis through pathways B and E are energetically unfavorable and thus not reasonable mechanisms.
In practice, the N-tosyl-2-allylaniline substrate 1 is more reactive than the α-methyl-γ-alkenyl sulfonamide 14 (1 reacts at 110°C, while 14 requires heating to 130°C). This trend in reactivity is better reflected by the two-acetate neutral pathways (paths A and D, ΔGTS = 17.2 and 17.9 kcal mol−1, respectively) than the one-acetate pathways (paths B and E, ΔGTS = 16.0 and 14.8 kcalmol−1, respectively). Thus, it is possible that paths A and D more closely resemble the actual reaction mechanism, even in the aminocupration step. Additionally, since the diastereoselectivity is determined in the aminocupration step and the major diastereomer of the aminooxygenation, diamination and carboamination reactions [Eqs. (1) and (14), Scheme 1] are the same irrespective of base and ligands or conditions (catalytic versus stoichiometric copper catalyst),[24,90] we can infer that the cis-aminocupration mechanism is relevant to all reactions in the class.
Conclusion
In this study we report mechanistically rigorous evidence for a rate-limiting cis-aminocupration step in the CuII-promoted alkene aminooxygenation/cyclization. Although we had previously proposed this mechanism based on empirical observations, this study, involving reaction kinetics, EPR spectroscopy, and DFT calculations, has given much stronger experimental evidence for the proposed mechanism. A detailed understanding of alkene aminometallation is required to further develop stereoselective metal-promoted alkene addition reactions. While significant work has been conducted to study the mechanism of alkene aminopalladation[91–93] and aminoauration,[94,95] few rigorous mechanistic studies have been conducted with respect to the alkene aminocupration.[18] Our reaction kinetics and observed secondary kinetic isotope effect support a rate-limiting, cis-aminocupration step. The cis-aminocupration mechanism is also supported by the kinetic competency of a preformed [N–Cu] intermediate with a first-order kinetic profile. Competition experiments further support aminocupration as the alkene addition mode and help rule out an aminyl radical mechanism. The EPR experiments and DFT calculations directly addressed the possible contribution of nitrogen radical chemistry in the mechanism and we were able to conclusively eliminate the involvement of a nitrogen radical in the productive steps of the reactions mechanism. Isotopic-labeling studies support C–O bond formation through carbon radical trapping with TEMPO and rule out more concerted reaction processes.
In this study we have also observed an interesting and unexpected role of acetate ion in the break-up of copper(II) carboxylate dimer in a pre-equilibrium step. The prominence of this pre-equilibrium is established by the observed half-order kinetics in [CuT] and in [substrate]. This under-recognized role of CuII carboxylates as acceptors of acetate ions may explain their ability to facilitate a variety of reactions that require CuII additives.[96–98] To the best of our knowledge, this is also the first study that documents kinetic order to illustrate the importance of copper(II) carboxylate dimer breakup on the rate of a copper(II) carboxylate promoted reaction. Although half-order [CuT] kinetics would be expected when starting from dimeric copper(II) carboxylates, the half-order substrate kinetics, which indicates the substrate is involved in the break-up of the CuII dimer, is more unusual. There is some evidence that a dimer break-up step may be important in other copper(II) carboxylate promoted reactions, for example, the known Chan–Lam Cu(OAc)2-promoted cross-coupling reaction of amines and organoboronic acids.[99,100]
We further document for the first time a copper(II) carboxylate promoted reaction in which the rate of the reaction can be dependent upon the ratio of copper(II) carboxylate to added base or nucleophile, in this case, acetate ion. This finding may also be relevant to other copper(II) carboxylate promoted reactions. For example, Lam has indicated in a recent review of the Chan–Lam coupling reaction, that while solubility and color changes are observed upon addition of nucleophiles to Cu(OAc)2 solutions, further insight into the influence of nucleophiles and ligands on the identity of the copper(II) catalyst is needed.[99] Reaction kinetics have recently been investigated in the Chan–Lam coupling.[16, 101] In these studies, the reaction conditions for the kinetic study were catalytic in CuII and performed in alcohol solvent.[16, 101] The majority of Chan–Lam couplings, however, are performed by using a stoichiometric amount of Cu(OAc)2 in aprotic, organic solvents, much like our reactions.[98,99,102] Thus, the methods of reaction study disclosed herein may be useful in the kinetic study of other CuII carboxylate promoted reactions that take place in aprotic, organic solvents. Furthermore, although numerous copper(II) carboxylate promoted reactions have been developed, the role of the dimeric nature of the copper(II) carboxylate salt is rarely considered in mechanism discussions.[98,99,102–104] Our studies indicate that consideration of the dimeric state of copper(II) carboxylates could lead to a more rational reaction optimization approach in some cases. Reaction kinetics of copper(II) carboxylate promoted reactions that take place in aprotic organic solvent are rare, perhaps due to the difficulties associated with Cu(OAc)2 solubility. We propose that Cu(2-ethylhexanoate)2 can be used in the kinetic analysis of any reaction previously run with the less soluble Cu(OAc)2.
The molecular modeling studies are largely consistent with the experimental results and also indicate a further role for the acetate ligands throughout the reaction coordinate. In our calculations we have found a strong ligand effect on the energies of ligand dissociation and C–CuII homolysis. There are few theoretical studies of C–CuII homolysis[84] and a ligand effect on bond dissociation energy has not previously been noted, although calculated ligand effects on single electron transfer involving copper complexes have been recently reported.[105]
The mechanistic work described in this paper sets the stage for a subsequent detailed mechanistic analysis of our recently reported enantioselective Cu-catalyzed aminooxygenation of alkenes, which can open the door to a more detailed understanding of catalytic, enantioselective, Cu-catalyzed, alkene amination reactions.[26] While we do not expect the reaction kinetics to be identical, since we start from monomeric rather than dimeric copper complexes in the enantioselective reaction,[26] the stereochemical trends (diastereoselectivity) of the reactions[24,90] are similar enough for us to anticipate similar kinetic isotope effects and rate-limiting step. Although CuII-promoted and -catalyzed alkene amination reactions are relatively novel, the attractive qualities CuII salts have to offer with respect to low expense, high versatility, and stereoselectivity ensure this reaction class will grow, thus a detailed mechanistic understanding as described in this paper should aid in the rational development of this class of reactions.
Experimental Section
N-(2-Allyl-2-[D1]-phenyl)-4-methylbenzenesulfonamide ([D]-1b)
A solution of LiAlD4 (3.6 g, 85.6 mmol, 1.6 equiv) in dry Et2O (285 mL) was cooled to −10°C and a solution of propargyl alcohol (3.0 g, 53.5 mmol, 1 equiv) in Et2O (33 mL) was added through an addition funnel over 30 min. The resulting solution was warmed to room temperature and stirred for 14 h. The mixture was cooled to 0°C and was quenched slowly with H2O (4.0 mL). The solution was stirred for another 15 min and then 15% aqueous NaOH solution (4.0 mL) and H2O (4.0 mL) were added. The white slurry was filtered through a short pad of Celite and was washed with Et2O (300 mL). The filtrate was concentrated in vacuo to give the crude allyl-2-d1 alcohol4 as a yellow oil (3.0 g). 1H NMR (500 MHz, CDCl3): δ = 5.22 (s, 1H), 5.09 (s, 1H), 4.08 (s, 2H), 3.0 ppm (brs, 1H).
The crude allyl-2-[D1] alcohol (3.0 g, 50.8 mmol, 1 equiv) was added to a stirring solution of PBr3 (2.4 mL, 25.5 mmol, 0.5 equiv) in Et2O (21 mL) dropwise at 0°C. The resulting solution was stirred at 0°C for 1 h and then carefully quenched by the addition of brine (12 mL). The layers were separated and the combined organic extracts were washed with a saturated solution of NaHCO3, brine and dried over Na2SO4. Excess solvent was removed via careful distillation (45–50°C). The crude allyl-2-[D1] bromide was obtained as colorless liquid (2.1 g, 32% yield over 2 steps).[5] 1H NMR (500 MHz, CDCl3): δ = 5.31 (s, 1H), 5.14 (s, 1H), 3.94 ppm (s, 2H).
Into an oven-dried round bottom flask were added the crude allyl-2-[D1] bromide (1.5 mL, 12.5 mmol, 1.0 equiv), aniline (4.5 mL, 37.0 mmol, 3.0 equiv), K2CO3 (5.0 g, 37.0 mmol, 3.0 equiv) and DMF (20 mL).[6] The flask was equipped with a stopper and the reaction mixture was heated to 70°C overnight. The mixture was allowed to cool to room temperature and was washed with water (20 mL). The aqueous phase was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine, dried with Na2SO4 and concentrated in vacuo. Purification by flash chromatography on SiO2 (10% EtOAc in hexanes) gave compound N-allyl-2-[D1] aniline (0.7 g, 45% yield). Data: 1H NMR (500 MHz, CDCl3): δ = 7.18 (t, J = 7.5 Hz, 2H), 6.72 (t, J = 7.5 Hz, 1H), 6.64 (d, J = 8.0 Hz, 2H), 5.29 (s, 1H), 5.17 (s, 1H), 3.78 ppm (s, 2H); 13C NMR (75 Hz, CDCl3): δ = 148.0, 135.1 (t, J = 23.0 Hz), 129.2, 117.5, 116.1, 112.9, 46.4 ppm; HRMS (ESI): m/z calcd for [M]+ C9H11DN1: 135.1027, found: 135.1022.
To an oven-dried pressure tube were added N-allyl-2-[D1] aniline (0.7 g, 5.2 mmol, 1 equiv) and xylenes (13 mL). The solution was cooled to −78°C and BF3·Et2O (1.3 mL, 10.4 mmol, 2.0 equiv) was added dropwise. The resulting solution was warmed to room temperature and was heated to 160°C for 4 h.[7] The reaction mixture was then cooled to room temperature and was placed in an ice–water bath and was quenched with 2M NaOH (6 mL). The organic layer was separated and aqueous layer was extracted with Et2O (3 × 10 mL). The organics were combined, dried with Na2SO4, filtered and concentrated in vacuo. Purification by flash chromatography on SiO2 (10% EtOAc in hexanes) gave o-allyl-2-[D1] aniline (0.5 g, 70% yield). 1H NMR (500 MHz, CDCl3): δ = 7.05 (t, J = 8.0 Hz, 2H), 6.75 (t, J = 7.5 Hz, 1H), 6.64 (d, J = 8.0 Hz, 1H), 5.12 (s, 1H), 5.10 s, 1H), 3.66 (brs, 2H), 3.30 ppm (s, 2H); HRMS (ESI): m/z calcd for [M+ H]+ C9H11DN1: 135.1012, found: 135.1009.
The o-allyl-2-[D1] aniline (0.5 g, 3.7 mmol, 1 equiv) was dissolved in dry CH2Cl2 (20 mL) and the solution was treated with pyridine (1.18 mL, 14.9 mmol, 4 equiv) followed by p-toluenesulfonyl chloride (0.85 g, 4.5 mmol, 1.2 equiv). The mixture was stirred at room temperature for 24 h, washed with 1N HCl (15 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo. Flash chromatography of the resulting crude compound on SiO2 (5–10% EtOAc in hexanes) afforded compound [D]-1b as white solid (1.0 g, 96% yield). M.p. 62–65°C; 1H NMR (500 MHz, CDCl3): δ = 7.61 (d, J = 8.5 Hz, 2H), 7.41 (d, J = 8.5 Hz, 1H), 7.23 (d, J = 8.5 Hz, 2H), 7.19 (m, 1H), 7.13–7.05 (m, 2H), 6.55 (b. s., 1H), 5.11 (s, 1H), 4.93 (s, 1H), 3.00 (s, 2H), 2.39 ppm (s, 3H); 13C NMR (75 Hz, CDCl3): δ = 143.7, 136.7, 135.2 (t, J = 24 Hz), 134.9, 131.9, 130.4, 129.5, 127.6, 127.0, 126.2, 124.4, 116.9, 36.0, 21.5 ppm; IR (neat): ν̃ = 3266, 3076, 3030, 2967, 2913, 2849, 2243, 1922, 1623, 1596, 1492, 1451, 1401, 1333, 1161, 1089, 1039, 1016, 921, 817, 754, 663 cm−1; HRMS (ESI): m/z calcd for [M]+ C16H16DO2N1S1: 288.1037, found: 288.1036.
Supplementary Material
Acknowledgments
We acknowledge the National Institutes of Health, NIGMS (GM-078383) for funding this research. We are grateful to Dr. Alice Bergmann and Ying Long for assistance with mass spectrometry. We thank Prof. Dan Kosman (UB Biochemistry) for use of his EPR instrument and invaluable assistance in interpretation of the EPR spectra.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201101703.
References
- 1.Chemler SR, Fuller PH. Chem Soc Rev. 2007;36:1153–1160. doi: 10.1039/b607819m. [DOI] [PubMed] [Google Scholar]
- 2.Chemler SR. J Organomet Chem. 2011;696:150–158. doi: 10.1016/j.jorganchem.2010.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley LM, Sibi MP. Chem Rev. 2008;108:2887–2902. doi: 10.1021/cr078371m. [DOI] [PubMed] [Google Scholar]
- 4.Pellissier H. Tetrahedron. 2010;66:1509–1555. [Google Scholar]
- 5.Noack M, Gottlich R. Chem Commun. 2002:536–537. doi: 10.1039/b111656h. [DOI] [PubMed] [Google Scholar]
- 6.Manzoni MR, Zabawa TP, Kasi D, Chemler SR. Organometallics. 2004;23:5618–5621. [Google Scholar]
- 7.Michaelis DJ, Shaffer CJ, Yoon TP. J Am Chem Soc. 2007;129:1866–1867. doi: 10.1021/ja067894t. [DOI] [PubMed] [Google Scholar]
- 8.Michaelis DJ, Williamson KS, Yoon TP. Tetrahedron. 2009;65:5118–5124. doi: 10.1016/j.tet.2009.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao B, Yuan W, Du H, Shi Y. Org Lett. 2007;9:4943–4945. doi: 10.1021/ol702061s. [DOI] [PubMed] [Google Scholar]
- 10.Du H, Zhao B, Yuan W, Shi Y. Org Lett. 2008;10:4231–4234. doi: 10.1021/ol801605w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao B, Peng X, Cui S, Shi Y. J Am Chem Soc. 2010;132:11009–11011. doi: 10.1021/ja103838d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornwall RG, Zhao B, Shi YA. Org Lett. 2011;13:434–437. doi: 10.1021/ol102767j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Du H. J Org Chem. 2010;75:3503–3506. doi: 10.1021/jo100413p. [DOI] [PubMed] [Google Scholar]
- 14.Huang L, Jiang H, Qi C, Liu X. J Am Chem Soc. 2010;132:17652–17654. doi: 10.1021/ja108073k. [DOI] [PubMed] [Google Scholar]
- 15.Mancheno DE, Thornton AR, Stoll AH, Kong A, Blakey SB. Org Lett. 2010;12:4110–4113. doi: 10.1021/ol101702w. [DOI] [PubMed] [Google Scholar]
- 16.King AE, Huffman LM, Casitas A, Costas M, Ribas X, Stahl SS. J Am Chem Soc. 2010;132:12068–12073. doi: 10.1021/ja1045378. [DOI] [PubMed] [Google Scholar]
- 17.Sherman ES, Chemler SR, Tan TB, Gerlits O. Org Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+. [DOI] [PubMed] [Google Scholar]
- 18.Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuller PH, Chemler SR. Org Lett. 2007;9:5477–5480. doi: 10.1021/ol702401w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majumdar KC, Taher A, Nandi RK. Synlett. 2010;9:1389–1393. [Google Scholar]
- 21.Zabawa TP, Kasi D, Chemler SR. J Am Chem Soc. 2005;127:11250–11251. doi: 10.1021/ja053335v. [DOI] [PubMed] [Google Scholar]
- 22.Zabawa TP, Chemler SR. Org Lett. 2007;9:2035–2038. doi: 10.1021/ol0706713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sequeira FC, Turnpenny BW, Chemler SR. Angew Chem. 2010;122:6509–6512. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:6365–6368. doi: 10.1002/anie.201003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paderes MC, Chemler SR. Org Lett. 2009;11:1915–1918. doi: 10.1021/ol9003492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao L, Haque I, Manzoni MR, Tham WS, Chemler SR. Org Lett. 2010;12:4739–4741. doi: 10.1021/ol102233g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andrus MB, Lashley JC. Tetrahedron. 2002;58:845–866. [Google Scholar]
- 29.Mansano-Weiss C, Epstein DM, Cohen H, Masarwa A, Meyerstein D. Inorg Chim Acta. 2002;339:283–291. [Google Scholar]
- 30.Huffman LM, Stahl SS. J Am Chem Soc. 2008;130:9196–9197. doi: 10.1021/ja802123p. [DOI] [PubMed] [Google Scholar]
- 31.Harding KE, Burks SR. J Org Chem. 1981;46:3920–3922. [Google Scholar]
- 32.Harding KE, Marman TH. J Org Chem. 1984;49:2838–2840. [Google Scholar]
- 33.Ney JE, Wolfe JP. Angew Chem. 2004;116:3689–3692. [Google Scholar]; Angew Chem Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]
- 34.Neukom JD, Perch NS, Wolfe JP. J Am Chem Soc. 2010;132:6276–6277. doi: 10.1021/ja9102259. [DOI] [PubMed] [Google Scholar]
- 35.Gribkov DV, Hultzsch KC. Angew Chem. 2004;116:5659–5663. [Google Scholar]; Angew Chem Int Ed. 2004;43:5542–5546. doi: 10.1002/anie.200460880. [DOI] [PubMed] [Google Scholar]
- 36.Fujita H, Tokuda M, Nitta M, Suginome H. Tetrahedron Lett. 1992;33:6359–6362. [Google Scholar]
- 37.Navon N, Golub G, Cohen H, Meyerstein D. Organometallics. 1995;14:5670–5676. [Google Scholar]
- 38.Goldstein S, Czapski G, Cohen H, Meyerstein D. Inorg Chem. 1992;31:2439–2444. [Google Scholar]
- 39.Kochi JK. Acc Chem Res. 1974;7:351–360. [Google Scholar]
- 40.Root KS, Hill CL, Lawrence LM, Whitesides GM. J Am Chem Soc. 1989;111:5405–5412. [Google Scholar]
- 41.Hayes P, Suthers BD, Kitching W. Tetrahedron Lett. 2000;41:6175–6179. [Google Scholar]
- 42.Clark AJ, Deeth RJ, Samuel CJ, Wongtap H. Synlett. 1999;4:444–446. [Google Scholar]
- 43.Senboku H, Kajizuka Y, Hasegawa H, Fujita H, Suginome H, Orito K, Tokuda M. Tetrahedron. 1999;55:6465–6474. [Google Scholar]
- 44.Hasegawa H, Senbuku H, Kajizuka Y, Orito K, Tokuda M. Tetrahedron. 2003;59:827–832. [Google Scholar]
- 45.Turpeinen U, Hamalainen R, Mutikainen I. Acta Crystallogr. 1995;C51:2544–2546. [Google Scholar]
- 46.Rao VM, Sathyanarayana DN, Manohar H. J Chem Soc Dalton Trans. 1983:2167–2173. [Google Scholar]
- 47.Jotham RW, Kettle SFA, Marks JA. J Chem Soc Dalton Trans. 1972:428–438. [Google Scholar]
- 48.Kato M, Jonassen HB, Fanning JC. Chem Rev. 1964;64:99–128. [Google Scholar]
- 49.Tsuchida R, Yamada S, Nakamura H. Nature. 1956;178:1192–1193. [Google Scholar]
- 50.Martin RL, Whitley A. J Chem Soc. 1958:1394–1402. [Google Scholar]
- 51.Barclay GA, Kennard CHL. J Chem Soc. 1961:5244–5251. [Google Scholar]
- 52.Hanic F, Stempelova D, Hanicova K. Acta Crystallogr. 1964;17:633–639. [Google Scholar]
- 53.Kochi JK, Subramanian RV. Inorg Chem. 1965;4:1527–1533. [Google Scholar]
- 54.Lundqvist I. Acta Chem Scand. 1964;18:858–870. [Google Scholar]
- 55.Reimann CW, Kokoszka GF, Gordon G. Inorg Chem. 1965;4:1082–1084. [Google Scholar]
- 56.Graddon DP. J Inorg Nucl Chem. 1960;14:161–168. [Google Scholar]
- 57.Collum D, McNeil A, Ramirez A. Angew Chem. 2007;119:3060–3077. doi: 10.1002/anie.200603038. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2007;46:3002–3017. doi: 10.1002/anie.200603038. [DOI] [PubMed] [Google Scholar]
- 58.Bell RP. The Proton in Chemistry. 2. Cornell University Press; Ithaca, New York: 1973. [Google Scholar]
- 59.Melander L, Saunders WH., Jr . Reaction Rates of Isotopic Molecules. John Wiley and Sons; New York: 1980. [Google Scholar]
- 60.Carey FA, Sundberg RJ. Advanced Organic Chemistry, Part A: Structure and Mechanisms. 4. Springer; New York: 2000. [Google Scholar]
- 61.Fischer H, Random L. Angew Chem. 2001;113:1380–1414. [Google Scholar]; Angew Chem Int Ed. 2001;40:1340–1371. doi: 10.1002/1521-3773(20010417)40:8<1340::aid-anie1340>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 62.Martinez E, Newcomb M. J Org Chem. 2006;71:557–561. doi: 10.1021/jo051953o. [DOI] [PubMed] [Google Scholar]
- 63.Gilbert BC, Kalz W, Lindsay CI, McGrail PT, Parsons AF, Whittaker DTE. Tetrahedron Lett. 1999;40:6095–6098. [Google Scholar]
- 64.Blank O, Wetzel A, Ullrich D, Heinrich MR. Eur J Org Chem. 2008:3179–3189. [Google Scholar]
- 65.Sheldon RA, Arends IWCE, ten Brink G-J, Dijksman A. Acc Chem Res. 2002;35:774–781. doi: 10.1021/ar010075n. [DOI] [PubMed] [Google Scholar]
- 66.Lam PYS, Vincent G, Clark CG, Deudon S, Jadhav PK. Tetrahedron Lett. 2001;42:3415–3418. [Google Scholar]
- 67.Van Humbeck JF, Simonovich SP, Knowles RR, Mac-Millan DWC. J Am Chem Soc. 2010;132:10012–10014. doi: 10.1021/ja1043006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Michel C, Belanzoni P, Gamez P, Reedijk J, Baerends EJ. Inorg Chem. 2009;48:11909–11920. doi: 10.1021/ic902155m. [DOI] [PubMed] [Google Scholar]
- 69.Peisach J, Blumberg WE. Arch Biochem Biophys. 1974;165:691–708. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 70.Macías B, Villa MV, Gomez B, Borras J, Alzuet G, Gonzalez-Alvarez M, Castineiras A. J Inorg Biochem. 2007;101:444–451. doi: 10.1016/j.jinorgbio.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 71.Ghattas W, Giorgi M, Mekmouche Y, Tanaka T, Rockenbauer A, Reglier M, Hitomi Y, Simaan AJ. Inorg Chem. 2008;47:4627–4638. doi: 10.1021/ic702303g. [DOI] [PubMed] [Google Scholar]
- 72.Mankad NP, Antholine WE, Szilagyi RK, Peters JC. J Am Chem Soc. 2009;131:3878–3880. doi: 10.1021/ja809834k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 74.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 75.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision C.02. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 76.Dunning THJ, Hay PJ. In: Methods of Electronic Structure Theory (Modern Theoretical Chemistry) Schaefer HF III, editor. Vol. 3. Plenum; New York: 1977. pp. 1–28. [Google Scholar]
- 77.Hay PJ, Wadt WR. J Chem Phys. 1985;82:270–283. [Google Scholar]
- 78.Hay PJ, Wadt WR. J Chem Phys. 1985;82:299–310. [Google Scholar]
- 79.Wadt WR, Hay PJ. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 80.Hratchian HP, Schlegel HB. In: Theory and Applications of Computational Chemistry: The First Forty Years. Dykstra CE, Frenking G, Kim KS, Scuseria GE, editors. Elsevier; Amsterdam: 2005. pp. 195–249. [Google Scholar]
- 81.Hratchian HP, Schlegel HB. J Chem Theory Comput. 2005;1:61–69. doi: 10.1021/ct0499783. [DOI] [PubMed] [Google Scholar]
- 82.Hratchian HP, Schlegel HB. J Chem Phys. 2004;120:9918–9924. doi: 10.1063/1.1724823. [DOI] [PubMed] [Google Scholar]
- 83.Lorant F, Behar F, Goddard WA, Tang Y. J Phys Chem A. 2001;105:7896–7904. [Google Scholar]
- 84.Goj LA, Blue ED, Delp SA, Gunnoe TB, Cundari TR, Petersen JL. Organometallics. 2006;25:4097–4104. [Google Scholar]
- 85.Foster JP, Weinhold F. J Am Chem Soc. 1980;102:7211–7218. [Google Scholar]
- 86.Reed AE, Weinhold F. J Chem Phys. 1983;78:4066–4073. [Google Scholar]
- 87.Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 88.Reed AE, Weinhold F. J Chem Phys. 1985;83:1736–1740. [Google Scholar]
- 89.Reed AE, Curtiss LA, Weinhold F. Chem Rev. 1988;88:899–926. [Google Scholar]
- 90.Paderes MC, Chemler SR. Eur J Org Chem. 2011:3679–3684. doi: 10.1002/ejoc.201100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu G, Stahl SS. J Am Chem Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u. [DOI] [PubMed] [Google Scholar]
- 92.He W, Yip K-T, Zhu N-Y, Yang D. Org Lett. 2009;11:5626–5628. doi: 10.1021/ol902348t. [DOI] [PubMed] [Google Scholar]
- 93.Neukom JD, Perch NS, Wolfe JP. Organometallics. 2011;30:1269–1277. [Google Scholar]
- 94.LaLonde RL, Brenzovich WE, Benitez D, Tkatchouk E, Kelley K, Goddard WA, Toste FD. Chem Sci. 2010;1:226–233. doi: 10.1039/C0SC00255K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.de Haro T, Nevado C. Angew Chem. 2011;123:936–940. [Google Scholar]; Angew Chem Int Ed. 2011;50:906–910. doi: 10.1002/anie.201005763. [DOI] [PubMed] [Google Scholar]
- 96.Ye Y, Ball ND, Kampf JW, Sanford MS. J Am Chem Soc. 2010;132:14682–14687. doi: 10.1021/ja107780w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang X, Truesdale L, Yu J-Q. J Am Chem Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s. [DOI] [PubMed] [Google Scholar]
- 98.Jensen KH, Webb JD, Sigman MS. J Am Chem Soc. 2010;132:17471–17482. doi: 10.1021/ja108106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Qiao JX, Lam PYS. Synthesis. 2011:829–856. [Google Scholar]
- 100.Gonza’lez I, Mosquera J, Guerrero C, Rodri’guez R, Cruces J. Org Lett. 2009;11:1677–1680. doi: 10.1021/ol802882k. [DOI] [PubMed] [Google Scholar]
- 101.King AE, Brunold TC, Stahl SS. J Am Chem Soc. 2009;131:5044–5045. doi: 10.1021/ja9006657. [DOI] [PubMed] [Google Scholar]
- 102.Shade RE, Hyde AM, Olsen J-C, Merlic CA. J Am Chem Soc. 2010;132:1202–1203. doi: 10.1021/ja907982w. [DOI] [PubMed] [Google Scholar]
- 103.Wang Q, Schreiber SL. Org Lett. 2009;11:5178–5180. doi: 10.1021/ol902079g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DeMartino MP, Chen K, Baran PS. J Am Chem Soc. 2008;130:11546–11560. doi: 10.1021/ja804159y. and references therein. [DOI] [PubMed] [Google Scholar]
- 105.Jones GO, Liu P, Houk KN, Buchwald SL. J Am Chem Soc. 2010;132:6205–6213. doi: 10.1021/ja100739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.