

Abstract
We previously reported the discovery of pyrimido-pyrrolo-quinoxalinedione (PPQ) inhibitors of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel and showed their efficacy in an organ culture model of polycystic kidney disease (PKD) (Tradtrantip et al., J. Med. Chem. 52, 6447–6455, 2009). Here, we report related benzopyrimido-pyrrolo-oxazinedione (BPO) CFTR inhibitors. To establish structure-activity relationships and select lead compound(s) with improved potency, metabolic stability and aqueous solubility compared to the most potent prior compound 8 (PPQ-102, IC50 ~ 90 nM), we synthesized 16 PPQ analogs and 11 BPO analogs. The analogs were efficiently synthesized in 5–6 steps and 11–61 % overall yield. Modification of 8 by bromine substitution at the 5-position of the furan ring, replacement of the secondary amine with an ether bridge, and carboxylation, gave 6-(5-bromofuran-2-yl)-7,9-dimethyl-8,10-dioxo-11-phenyl-7,8,9,10-tetrahydro-6H-benzo[b]pyrimido [4',5':3,4]pyrrolo [1,2-d][1,4]oxazine-2-carboxylic acid 42 (BPO-27), which fully inhibited CFTR with IC50 ~ 8 nM, and, compared to 8, had >10-fold greater metabolic stability and much greater polarity / aqueous solubility. In an embryonic kidney culture model of PKD 42 prevented cyst growth with IC50 ~ 100 nM. Benzopyrimido-pyrrolo-oxazinediones such as 42 are potential development candidates for anti-secretory therapy of PKD.
INTRODUCTION
Polycystic kidney disease (PKD) is one of the most common human genetic diseases and a major cause of chronic renal insufficiency requiring dialysis and kidney transplantation.1 Cyst growth in autosomal dominant polycystic kidney disease (ADPKD) involves progressive fluid accumulation.2, 3 Fluid secretion into the cyst lumen requires chloride secretion by the cystic fibrosis transmembrane conductance regulator (CFTR) protein,4, 5 a cAMP-regulated chloride channel in which loss-of-function mutations cause the genetic disease cystic fibrosis.6 CFTR is expressed strongly in epithelial cells lining cysts in ADPKD.7 Cystic fibrosis (CFTR-deficient) mice are resistant to cyst formation and CFTR inhibitors block cyst formation in cell/organ culture and in vivo models.8, 9 In rare families affected with ADPKD and cystic fibrosis, individuals with both ADPKD and CF have less severe renal disease than those with ADPKD only.10–12 Cyst expansion in ADPKD also requires cyst epithelial cell proliferation involving mTor signaling,13, 14 which is the basis of several `anti-proliferative' therapies under development.15–18 `Anti-secretory' (CFTR inhibition) therapy is predicted to complement antiproliferative therapy or be effective alone in life-long treatment of ADPKD.
We identified several classes of small-molecule CFTR inhibitors by high-throughput screening (reviewed in ref. 19). Thiazolidinone-class CFTR inhibitors such as CFTRinh-172 (Fig. 1) act on the cytoplasmic side of the plasma membrane at a site near the CFTR pore to block CFTR chloride conductance.20–22 CFTRinh-172 has been used widely as a research tool to study CFTR function in cell culture, ex vivo tissues, and animal models. CFTRinh-172 inhibits CFTR with IC50 in the range of 300–3000 nM, depending on cell type and membrane potential, and was shown to have low toxicity and metabolic stability with primarily renal excretion.21, 23 The tetrazolo-substituted thiazolidinone Tetrazolo-172 (Fig. 1) has improved water solubility compared to CFTRinh-172,24 reducing cyst expansion in organ culture and mouse models of PKD.25 A second class of small-molecule CFTR inhibitors, the glycine hydrazides, such as GlyH-101 (Fig. 1), block CFTR at an external site within the CFTR pore.26 Membrane-impermeant, non-absorbable conjugates of glycine hydrazides with polyethylene glycols (Fig. 1)27, 28 and lectins29 inhibit CFTR with IC50 down to 50–100 nM when added at the mucosal cell surface, and were effective in rodent models of secretory diarrheas such as cholera. An absorbable glycine hydrazide, phenyl-GlyH-101 (Fig. 1), reduced cyst growth in PKD.25
Figure 1.
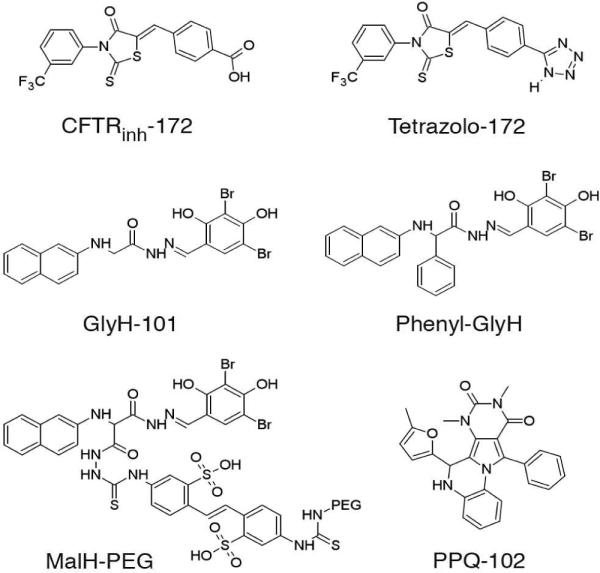
Chemical structures of CFTR inhibitors.
Additional small molecule screening yielded pyrimido-pyrrolo-quinoxalinedione (PPQ) compounds, which were the first uncharged, and thus membrane-potential insensitive, CFTR inhibitors.30 7,9-Dimethyl-11-phenyl-6-(5-methyl furan-2-yl)-5,6-dihydro-pyrimido-[4',5'-3,4]pyrrolo[1,2-a]quinoxaline-8,10-(7H,9H)-dione (PPQ-102, Fig. 1) 8 inhibited CFTR chloride conductance with IC50 ~90 nM by a mechanism involving stabilization of the channel closed-state. 8 prevented cyst expansion and reduced the size of pre-formed cysts in an embryonic kidney organ culture model of PKD. However, as described below, 8 has poor metabolic stability, precluding animal testing, as well as low polarity (clogP 4.92) and hence low aqueous solubility. In order to improve compound stability, water solubility and CFTR inhibition potency, herein we synthesized and characterized a focused library of 27 PPQ analogs. The most potent compound, 42 (BPO-27) had an IC50 ~ 8 nM for CFTR inhibition, more than 10-fold improved metabolic stability and much greater polarity (clogP 1.76) compared to 8, and was effective in preventing renal cyst expansion in a PKD model.
RESULTS
We reported 8 as a small-molecule inhibitor of CFTR chloride conductance with efficacy in preventing and reversing cyst formation in an organ culture model of PKD.30 Preliminary analysis of the metabolic stability and aqueous solubility of 8, done in preparation for in vivo testing in mouse models of PKD, indicated poor metabolic stability and low water solubility (< 2 RM in albumin-free saline). In vitro metabolic stability was determined by compound incubation with hepatic microsomes at 37 °C for specified times in the absence vs. presence of NADPH, following by LC/MS analysis. Fig. 2A shows loss of 8 in hepatic microsomes in the presence of NADPH, with ~60 % disappearance in 30 min. No loss of 8 was seen in the absence of NADPH (data not shown). 8 was undetectable in serum, kidney and urine at 30–60 min after intravenous bolus administration of 300 μg 8 in mice using an LC/MS assay with sensitivity better than 100 nM (data not shown). Though the precise metabolic fate of 8 is not known, structural considerations and the presence of prominent metabolites at +14 and +16 daltons (Fig. 2B) suggested possible oxidation, aromatization, and hydroxylation (Fig. 2C). To improve on the drug-like properties of 8 a series analogs was synthesized and tested.
Figure 2.

Metabolism of compound 8 in hepatic microsomes. A. LC/MS showing disappearance over 30 min during incubation with microsomes in the presence of NADPH. B. Appearance of metabolites at +14 and +16 daltons. C. Schematic of potential sites of metabolism.
PPQ and BPO analog synthesis
Scheme 1 shows the synthesis of dihydroquinoxoline PPQ (Y=N) compounds and Scheme 2 of benzoxazine BPO (Y=O) compounds. Table 1 shows structures and CFTR inhibition data for all synthesized analogs. Our initial efforts focused on improving the synthesis of 8, as the original synthesis had low yield.30 As shown in Scheme 1, 6-methyluracil 1 was exhaustively alkylated using dimethyl sulfate to give 1,3,6-trimethyluracil 2 in 98 % yield. 1,3,6-Trimethyluracil 2 was subject to Friedel-Crafts acylation utilizing benzoyl chloride and anhydrous zinc chloride to give ketone 3 in 66 % yield. Bromination of ketone 3 gave 4 in quantitative yield. At the first point of diversification, 4 was reacted with substituted 1,2-phenylenediamines (2 eq) to give pyrroles 5–7 in 97 % (R2=H), 89 % (R2=NO), and 83 % (R42 =Me) yield. Pyrroles 5–7 were condensed with the appropriately substituted furfural or thiophene carbaldehyde using catalytic acid to give 13–23 and 38 with yields of 57–98 %. 8 was obtained on a gram scale in 83 % yield. Amide analogs were synthesized from 8 and acid halides or anhydrides to give 9–11, in 73 %, 80 % and 79 % yield, respectively. The nitrosamine 12 was synthesized from 8 and t-butyl nitrite in 79 % yield.
Scheme 1.

Synthesis of PPQ analogs
Reagents: (a) NaOH, H2O, dimethylsulfate, r.t. 3 days; (b) benzoyl chloride, ZnCl2, chlorobenzene, reflux; (c) Br2, CH2Cl2, cat. H2O, reflux; (d) 4-R2-1,2-phenylenediamine, EtOH, reflux; (e) R2=H, 4-R5-5-R6-furfural, 1,2-dichloroethane, TsOH, reflux; X=S, 5-methylthiophene carbaldehyde, 1,2-dichloroethane, TsOH, reflux; R2=NO2, 5-bromo-2-furaldehyde CHCl3, TFA reflux; (f) R4= CH3SO2-, methanesulfonyl chloride, DCM, Et3N; R4=ClCH2CO-, Chloroacetyl chloride, DCM, Et3N; R4=CH3CO-, acetic anhydride, DMAP, 100 °C; R4=NO, t-butyl nitrite, DCM; (g) N-methyl-1,2-phenylenediamine, EtOH, reflux; (h) 5-methylfurfural, 1,2-dichloroethane, TsOH, reflux.
Scheme 2.

Synthesis of BPO analogs.
Reagents: (a) R1=H, benzoyl chloride, ZnCl2, chlorobenzene, reflux; R1=Me, m-tolyl chloride, ZnCl2, chlorobenzene, reflux; (b) Br2, CH2Cl2, cat. H2O, reflux; (c) 2-amino-3-R2-4-R3-phenol, EtOH, reflux; (d) 5-R6-furfual, TFA, CHCl3 or 1,2-dichloroethane, 150 °C; (e) KOH, THF, H2O, HCl workup.
Table 1.
CFTR inhibition of PPQ and BPO analogs.
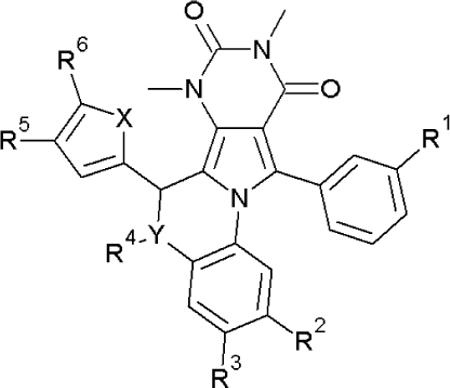
| Compound | X | Y | R1 | R2 | R3 | R4 | R5 | R6 | IC50(μM)A | IC50(μM)B |
|---|---|---|---|---|---|---|---|---|---|---|
| 8 | O | N | H | H | H | H | H | Me | 0.25 | 0.1 |
| 9 | O | N | H | H | H | CH3SO2 | H | Me | inactive | |
| 10 | O | N | H | H | H | ClCH2CO2 | H | Me | inactive | |
| 11 | O | N | H | H | H | CH3CO2 | H | Me | inactive | |
| 12 | O | N | H | H | H | NO | H | Me | 2 | |
| 13 | O | N | H | H | H | Me | H | Me | 1 | |
| 14 | O | N | H | H | H | H | H | Et | 0.3 | |
| 15 | S | N | H | H | H | H | H | Me | 1.7 | |
| 16 | O | N | H | H | H | H | Me | Me | 1.7 | |
| 17 | O | N | H | H | H | H | H | Cl | 0.15 | |
| 18 | O | N | H | H | H | H | H | Br | 0.09 | 0.05 |
| 19 | O | N | H | H | H | H | H | I | 0.17 | 0.1 |
| 20 | O | N | H | H | H | H | H | CF3 | 0.35 | |
| 21 | O | N | H | H | H | H | H | Ph | inactive | |
| 22 | O | N | H | H | H | H | H | CH2OH | inactive | |
| 23 | O | N | H | H | H | H | -(CH)2- | -(CH)2- | inactive | |
| 31 | O | O | H | H | H | H | Br | 0.2 | 0.09 | |
| 32 | O | O | H | H | H | H | I | 0.5 | ||
| 33 | O | O | H | H | H | H | Me | 0.15 | ||
| 34 | O | O | Me | H | H | H | Cl | 0.37 | ||
| 35 | O | O | Me | H | H | H | Me | 0.6 | ||
| 36 | O | O | H | NO2 | H | H | Br | 0.12 | 0.025 | |
| 37 | O | O | H | NO2 | H | H | Me | 0.1 | 0.025 | |
| 38 | O | N | H | NO2 | H | H | H | Br | 0.17 | |
| 39 | O | O | H | Cl | NO2 | H | Me | 0.17 | ||
| 40 | O | O | H | COOEt | H | H | Br | 0.05 | 0.025 | |
| 41 | O | O | H | COOEt | H | H | Me | 0.08 | ||
| 42 | O | O | H | COOH | H | H | Br | 0.04 | 0.008 |
by micro-plate reader assay
by short-circuit current assay
Scheme 2 shows the synthesis of benzoxazine BPO (Y=O) compounds. Ketone 24 (R1=Me) was synthesized from 2 by Friedel-Crafts acylation utilizing m-tolyl chloride in 26 % yield, and subsequently brominated to give 25 in 93 % yield. The condensation of substituted 2-aminophenols with 4 and 25 in ethanol gave pyrroles 26–30 in 96 % (R1=CH3, R2=R3=H), 96 % (R1=R2=R3=H), 94 % (R1=R3=H, R2=NO2), 88 % (R1=H, R2=Cl, R3=NO2), and 98 % (R1=R3=H, R2=COOEt) yield, respectively. Pyrroles 26–30 were then condensed with substituted furfurals using catalytic acid at 150 °C to give 31–37 and 39–41 in yields of 43–84 %. 40 was saponified utilizing KOH in THF and water to give 42 in 83 % yield after acid work-up.
Characterization of PPQ and BPO analogs
Initial compound testing for CFTR inhibition was done using a plate-reader assay in which iodide influx was measured in FRT cells coexpressing human wildtype CFTR and the iodide-sensing yellow fluorescent protein YFP-H148Q/I152L. Fig. 3A (top) shows representative fluorescence data for inhibition of CFTR-mediated iodide influx by one of the synthesized PPQ analogs, showing reduced negative slope after iodide addition with increasing inhibition concentration. Fig. 3A (bottom) shows concentration-inhibition data for selected compounds, with IC50 values for all compounds listed in Table 1. Though relative IC50 values are useful for comparisons, absolute IC50 values from the plate-reader assay are approximate, generally underestimating compound potency because of assay non-linearities, pH-dependent YFP fluorescence, the use of iodide instead of chloride, and compound dilution at the start of the assay.31 Quantitative concentration-inhibition data for the most potent compounds was obtained by analysis of short-circuit current, which represents a definitive electrophysiological measure of compound potency. Current was measured in CFTR-expressing FRT cells in which the basolateral membrane was permeabilized and in the presence of a transepithelial chloride concentration gradient, so that current is a linear measure of CFTR function. CFTR was activated by forskolin, followed by serial additions of increasing concentrations of test compounds. The representative short-circuit current measurement in Fig. 3B shows increased CFTR chloride conductance following addition of the cAMP agonist forskolin, which was reduced in a concentration-dependent manner by a PPQ inhibitor, with complete inhibition at high inhibitor concentration.
Figure 3.

Characterization of PPQ analogs. A. Fluorescence plate-reader assay of CFTR inhibition. FRT cells expressing human wildtype CFTR and iodide-sensing YFP fluorescent dye were incubated with test compound and CFTR agonists, and then subjected to an inwardly directed iodide gradient. (top) Representative data showing kinetics of fluorescence decrease following iodide addition (causing YFP fluorescence quenching) in the absence of cAMP agonists, and in the presence of cAMP agonists and indicated concentrations of 18. (bottom) Summary of concentration-inhibition data for indicated compound (S.E. n=4). See Table 1 for IC50 values. B. Short-circuit current analysis of CFTR inhibition in CFTR-expressing FRT cells in the presence of a transepithelial chloride gradient and basolateral membrane permeabilization. Where indicated, forskolin (20 μM) was added to activate CFTR chloride conductance, following by indicated concentrations of 18. C. (top) LS/MS analysis showing disappearance of 18 and 32 in hepatic microsomes in the presence of NADPH. (bottom) Summary of kinetics of compound disappearance (SEM, n=3). Data for 8 shown for comparison.
Our initial synthesis efforts focused on preventing aromatization, which was initially achieved by derivatizing the secondary amine of 8 to give analogs 9–13. The amides 9–11 were, however, substantially less active than 8. As the loss of the basic amine coincided with a loss in activity we synthesized nitrosamine 12, but our attempts to reduce the nitrosamine to a strongly basic hydrazine only yielded 8. To maintain basicity and prevent aromatization we tried to synthesize the N-methyl analog 13 via reductive alkylation using formaldehyde, but were unsuccessful. Fortunately, the N-methyl precursor 7 was obtained in good yield from N-methyl-1,2-phenylenediamine and 4. Pyrrole 7 was then used to synthesize 13, which was weakly active, and so further derivitization at the secondary amine was abandoned.
Re-examination of SAR data obtained from ~350 commercially available analogs (as reported in ref. 30, and unpublished data) suggested that the 5-position of the furan ring in 8 was privileged. Therefore, compounds 14–23 were synthesized to probe the steric and electronic requirements for activity of the furyl moiety. Substituting bromine at the 5-position of the furyl ring yielded 18, which was substantially more stable than 8 and had similar CFTR inhibition potency (Fig. 3B, 3C). To further probe oxidative aromatization as a metabolic pathway, the secondary amine in 18 was replaced by oxygen forming an ether bridge, which is unable to undergo oxidation. The resulting benzoxazine 31 had similar CFTR inhibition potency (Fig. 3A) and better stability than 8. Synthesis of 32–33 confirmed the CFTR inhibition activity (Table 1) and improved stability (Fig. 3C) imparted by bromine and illustrated the synergy between the 5-bromofuran moiety and the ether bridge.
Previous SAR data30 showed that an m-tolyl moiety (R1=Me) increased CFTR inhibition potency. Unfortunately, synthesis of 34–35 revealed that the m-tolyl moiety reduced CFTR inhibition in the BPO series. To increase the polarity and hence the aqueous solubility of 31, a nitro group was introduced at R2. The resulting compound, 36, had substantially greater CFTR inhibition potency (Fig. 4A) with excellent metabolic stability in hepatic microsomes (Fig. 4B). 37 and 38 showed that the increased CFTR inhibition activity conferred by the nitro substituent at R2 was independent of both the 5-bromo substituent and the ether bridge.
Figure 4.

BPO CFTR inhibitors with high potency, metabolic stability and water solubility. A. Short-circuit current analysis showing CFTR inhibition by 36 and 42. B. Compound stability in hepatic microsomes in the presence of NADPH.
To further increase compound polarity, we replaced the nitro functionality at R2 in 36 with a carboxyl moiety to give 42. Compound 42 was found the most potent CFTR inhibitor to date with IC50 ~ 8 nM (Fig. 4A), and had excellent stability in hepatic microsomes with <5 % compound loss in 30 min (Fig. 4B). At physiological pH 42 is deprotonated and thus substantially more polar than 8 (clogP 1.76 vs. 4.92), with solubility ~17 μM in a pH 7.4 aqueous phosphate buffer. 42 was synthesized by hydrolysis of the ethyl ester 40, which also strongly inhibited CFTR, and could potentially serve as a pro-drug of 42.
Inhibition of cyst growth in a kidney organ culture model of PKD
An established embryonic ex vivo kidney organ culture model of PKD was used to test the efficacy of 42 in reducing cAMP agonist-induced renal cystogenesis. Fig. 5A shows progressive renal cyst formation and growth in 8-Br-cAMP agonist-treated cultures, as seen by transmitted light microscopy. Kidney growth without cyst formation was seen in the absence of 8-Br-cAMP. Cyst growth in 8-Br-cAMP-treated kidneys was remarkably reduced by inclusion of 42 in the culture medium. As quantified by percentage area occupied by cysts, 42 inhibited cyst growth with IC50 ~ 100 nM (Fig. 5B), much better than that of > 500 nM measured for 8.30
Figure 5.

Compound 42 reduces renal cytogenesis. A. Transmission light micrographs of E13.5 embryonic kidneys cultured for indicated days without or in the presence of 100 μM 8-Br-cAMP, and with 0, 0.1 or 1 μM 42. B. Summary of percentage cyst areas at 5 days in cultures (SEM, n=4–6, * P < 0.001
DISCUSSION AND CONCLUSIONS
The best compound emerging from this study, 42, had substantially improved metabolic stability (by > 10-fold) over 8, as well as greater polarity and potency for inhibition of CFTR chloride conductance in vitro (~10-fold) and renal cystogenesis ex vivo (> 5-fold). SAR analysis suggested that the greater stability of 42 over 8 is the consequence of the 5-Br substituted furan and the ether bridge, which largely prevent hydroxylation and aromatization modifications. The greatly improved water solubility of 42 over 8 is a consequence of the carboxylic acid substituent, which is charged at physiological pH. The carboxylic acid addition also, unexpectedly, improved CFTR inhibition potency. The high CFTR inhibition potency of the esterified form of 42, 40, suggests its possible use as a pro-drug for efficient intestinal absorption and cell accumulation following de-esterification by ubiquitous intracellular esterases.
Several noteworthy observations were made in synthesizing the PPQ analogs. In an experiment intended to improve yield, bromination of ketone 3 was conducted under strict anhydrous conditions. Ketone 3 was refluxed in CH2Cl2 and bromine (1 eq) but after several hours TLC showed little product. On a whim, several drops of wet CH2Cl2 were added to the reaction, which resulted in remarkably rapid discharge of the bromine color and evolution of fuming HBr gas. After several minutes TLC indicated a near quantitative yield of 4, which spontaneously crystallized when dried in vacuo. When 4 was condensed with N-methyl-1,2-phenylenediamine, the more reactive secondary amine can undergo alkylation by displacement of the readily accessible bromide in 4. The high regioselectivity evident in the 83% yield of 5 suggests that the formation of the pyrrole ring in the initial reaction in is one of imine formation rather than alkylation, despite the crowded reaction center.
ADPKD is a major health care burden with a prevalence of 1 in 500 to 1000 individuals.1 There is no approved drug at present to slow the progression of renal disease in PKD. As mentioned in the Introduction, there is compelling rationale and experiment proof-of-concept for CFTR inhibitor anti-secretory therapy for PKD. An alternative anti-secretory therapy, vasopressin V2 antagonism, is in clinical trials for PKD and is based on the idea that cysts in PKD often express V2 receptors, which, when stimulated by antidiuretic hormone, elevate cytoplasmic cAMP and activate both CFTR chloride conductance and mTOR signaling.15, 32 Our lab introduced an alternative, V2 receptor-independent strategy to reduce cAMP involving small-molecule phosphodiesterase activators, which were shown to resist growth in an in vitro PKD model.33 Pure anti-proliferative therapies, most based on the central role of mTOR signaling in proliferation of cyst-lining epithelial cells, are also in clinical trials, as well as renin-angiotensin inhibitors and statins.16–18 Whether anti-proliferative or anti-secretory therapy alone, or in combination, will slow the progression of kidney destruction in PKD awaits human clinical trials.
Compound 42, with IC50 ~ 8 nM, is substantially more potent than existing CFTR inhibitors. In addition to their potential utility for anti-secretory therapy of PKD, potent small-molecule CFTR inhibitors have potential efficacy in the therapy of secretory diarrheas, and as research tools in defining CFTR-dependent cellular and physiological processes. Secretory diarrheas caused by enterotoxins, such as cholera and Travellers' diarrhea (enteropathogenic E. coli), require functional CFTR for primary chloride secretion into the intestinal lumen, which secondarily drives sodium and water secretion.34, 35 Testing of 42 and related PPQ analogs is needed to establish their efficacy in animal models of secretory diarrheas, as has been done for thiazolidinone36 and glycine hydrazide28, 29 CFTR inhibitors. CFTR inhibitors are also useful to create the CF phenotype in ex vivo human and animal tissues, as has been done, for example, in studies of the role of CFTR in airway submucosal gland fluid secretion.37 Finally, though mouse, pig and ferret models of CFTR gene deletion exist, pharmacological creation of the CFTR phenotype in animals by CFTR inhibitors might provide complementary data on CFTR function in the absence of compensatory phenomena that can occur in transgenic animal models.
In conclusion, the synthesis and evaluation of a rationally designed series of PPQ analogs produced compounds with much improved metabolic stability, CFTR inhibitory potency, and aqueous solubility compared with reference compound 8. Compound 42 and related analogs are thus suitable for in vivo testing of efficacy and represent potential development candidates for antisecretory therapy of PKD.
EXPERIMENTAL SECTION
Cell culture and platereader assay of CFTR inhibition
Fischer rat thyroid (FRT) cells coexpressing human wild type CFTR and the halide indicator YFP-H148Q were cultured in 96-well black-walled microplates (Corning Costar) at a density of 20,000 cells per well in Coon's modified F12 medium containing 10% fetal bovine serum, 2 mM L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. CFTR chloride conductance was assayed at 48 h after plating on a FluoStar fluorescence platereader (BMG Lab Technologies) as described.21 Each well was washed 3 times with PBS, leaving 60 μL PBS. Test compounds were added and incubated with the cells for 45 min. Then, 5 μL of a CFTR-activating cocktail (10 μM forskolin, 100 μM IBMX, 20 μM apigenin in PBS) was added. After 15 min, each well was assayed for iodide influx by recording fluorescence continuously (200 ms per point) for 2 s (baseline) and then for 10 s after rapid addition of 160 μL of isosmolar PBS in which 137 mM chloride was replaced by iodide. The initial rate of iodide influx was computed from fluorescence data by non-linear regression.
Short-circuit current
Snapwell inserts containing CFTR-expressing FRT cells were mounted in an Ussing chamber. The hemichambers contained 5 mL of buffer containing 75 mM NaCl and 75 mM Na gluconate (apical) and 150 mM NaCl (basolateral) (pH 7.3), and the basolateral membrane was permeabilized with 250 μg/mL amphotericin B, as described.27 Short-circuit current was recorded continuously using a DVC-1000 voltage clamp (World Precision Instruments) using Ag/AgCl electrodes and 3 M KCl agar bridges.
Liquid chromatography / mass spectrometry
Compounds (each 5 μM) were incubated for specified times at 37 °C with rat liver microsomes (1 mg protein/ml; Sigma-Aldrich, St. Louis, MO) in potassium phosphate buffer (100 mM) containing NADPH (0 or 1 mM). The mixture was then chilled on ice, and 0.5 ml of ice-cold ethyl acetate was added. Samples were centrifuged for 15 min at 3,000 rpm, and the supernatant was evaporated to dryness under nitrogen. The residue was dissolved in 150 μl mobile phase (acetonitrile:water 3:1, containing 0.1% formic acid) for LC/MS analysis. Reverse-phase HPLC separations were carried out using a Waters C18 column (2.1 × 100 mm, 3.5 mm particle size) equipped with a solvent delivery system (Waters model 2690, Milford, MA). The solvent system consisted of a linear gradient from 5 to 95% acetonitrile run over 16 min (0.2 mL/min flow rate). Mass spectra were acquired on an Alliance HT 2790 + ZQ mass spectrometer using negative ion detection, scanning from 150 to 1500 Da. The electrospray ion source parameters were: capillary voltage 3.5 kV (positive ion mode), cone voltage 37 V, source temperature 120 °C, desolvation temperature 250 °C, cone gas flow 25 L/h, and dessolvation gas flow 350 L/h.
Embryonic organ culture model of PKD
Mouse embryos were obtained at embryonic day 13.5 (E13.5). Metanephroi were dissected and placed on transparent Falcon 0.4-mm diameter porous cell culture inserts as described.30 To the culture inserts was added DMEM/Ham's F-12 nutrient medium supplemented with 2 mM L-glutamine, 10 mM HEPES, 5 μg/mL insulin, 5 μg/mL transferrin, 2.8 nM selenium, 25 ng/ml prostaglandin E, 32 pg/ml T3, 250 U/ml penicillin and 250 μg/ml streptomycin. Kidneys were maintained in a 37 °C humidified CO2 incubator for up to 8 days. Culture medium containing 100 μM 8-Br-cAMP, with or without test compound, was replaced (in the lower chamber) every 12 h. Kidneys were photographed using a Nikon inverted microscope (Nikon TE 2000-S) equipped with 2× objective lens, 520 nm bandpass filter, and high-resolution CCD camera. Percentage cyst area was calculated as total cyst area divided by total kidney area.
Synthesis Procedures
NMR spectra (1H at 600 MHz; 13C at 150 MHz) were obtained in methylene chloride (CD2Cl2), chloroform (CDCl3), acetonitrile (CD3CN) or dimethyl sulfoxide (DMSO-d6) using a 600-MHz Varian Spectrometer. Chemical shifts are expressed in parts per million relative to the solvent. NMR spectra for PPQ and BPO compounds were acquired at −20 °C due to excessive broadening of the 11-phenyl protons at ambient temperature. NMR spectra for the intermediates were obtained at ambient temperature. Mass spectrometry was done using a Waters LC/MS instrument with MS: electrospray (+) ionization, mass ranging from 100 to 900 Da, 20-V cone voltage; LC: Xterra MS C18 column (2.1 mm × 50 mm × 3.5 μm), 0.2 mL/min water/acetonitrile (containing 0.1% TFA). Purity was judged by the peak area percentage of the UV absorbance signal. Compound purities by RP-HPLC were >98%. Flash chromatography was done using EM silica gel (230–400 mesh), and thin-layer chromatography was done on EMD silica gel 60 F254 plates (Darmstadt, Germany). Melting points are uncorrected. To follow is the synthesis procedure for 42. See Supporting Information for synthesis of all PPQ and BPO compounds, and analytical data.
1,3,6-Trimethyl-1H,3H-pyrimidine-2,4-dione (2)38
In a 250 mL round bottom flask, 6-methyluracil (1; 15.0 g, 119 mmol) and NaOH (9.55 g, 239 mmol) were dissolved in water (150 mL) at low heat. The solution was brought to 25 °C in an ice bath and maintained at 25°C while dimethyl sulfate (23 mL, 30.59 g, 243 mmol) was added dropwise over 20 min with vigorous stirring. After 22 h, the reaction mixture contained a white precipitate and had pH 9. NaOH (5.0 g, 125 mmol) was added to make the solution homogenous, and an ice bath was used to maintain a temperature of 25 °C while dimethyl sulfate (12 ml, 15.96 g, 127 mmol) was added dropwise over 10 min. The reaction was stirred for 72 h, then NaOH (2 g, 50 mmol) was added and the reaction extracted with CHCl3 (5×50 mL). The chloroform extracts were pooled, dried over Na2SO4, and concentrated in vacuo to yield 2 (18 g, 98% yield); mp 114–115 °C. 1H NMR (600 MHz, CDCl3) δ 5.67 (s, 1H), 3.45 (s, 3H), 3.35 (s, 3H), 2.29 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 162.6, 152.5, 151.8, 101.2, 31.9, 28.1, 20.6. MS (ES+) (m/z): [M+1]+ calculated for C7H11N2O2, 155.17, found 155.14.
5-Benzoyl-1,3,6-trimethylpyrimidine-2,4(1H,3H)-dione (3)39
In a 100 mL round bottom flask equipped with a condenser and an air lock was added 2 (5.00 g, 32.4 mmol), anhydrous zinc chloride40 (freshly dried, 4.45 g, 32.6 mmol), dry chlorobenzene (20 mL), and benzoyl chloride (freshly distilled, 4 ml, 4.84 g, 34.4 mmol). The reaction was refluxed in an oil bath equipped with an air lock and vigorously stirred for 3 h. The reaction was allowed to cool and the condenser arranged for distillation. Water (40 mL) was added dropwise at first and then with increasing speed. Chlorobenzene (30 mL) and water were distilled off and the mixture was cooled on ice. Diethyl ether (30 ml) was added while stirring causing a precipitate to form. The precipitate was collected by filtration and recrystallized from 2-propanol (50 mL) to yield 3a (5.53 g, 66%); mp 143.2–143.7 °C. 1H NMR (600 MHz, CD3CN) δ 8.74 (d, J = 7.1 Hz, 2H), 8.46 (t, J = 6.0 Hz, 1H), 8.37 – 8.29 (m, 2H), 4.25 (s, 3H), 4.07 (s, 3H), 2.99 (s, 3H). 13C NMR (151 MHz, CD3CN) δ 195.6, 162.2, 153.7, 153.2, 139.2, 134.9, 130.5, 130.0, 113.4, 32.8, 28.6, 18.3. MS (ES+) (m/z): [M+1]+ calculated for C14H15N2O3, 259.28, found 259.11.
5-Benzoyl-6-(bromomethyl)-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (4)39
In a 50 mL round bottom flask 3 (1.00 g, 38.7 mmol) was dissolved in CCl4 (5 mL) and CH2Cl2 (4 mL) at 50 °C. Bromine (200 μL, 0.621 g, 38.8 mmol) was mixed with CCl4 (5 mL) and CH2Cl2 (5 mL) in an addition funnel and added dropwise to the solution of 3 such that the brown color disappeared between drops. The last few drops caused the reaction to remain brown. The reaction was then brought to reflux for 10 min before the brown color was discharged by the addition of a few drops of acetone. The reaction was refluxed for 30 min to remove HBr. The reaction was quantitative by TLC and the product crystallized when concentrated in vacuo to yield 4 (1.305 g, 100%). The product was recrystallized from 2-propanol as colorless needles; mp 171.0–171.7 °C. 1H NMR (600 MHz, CD2Cl2) δ 7.88 – 7.80 (m, 2H), 7.65 – 7.60 (m, 1H), 7.52 – 7.45 (m, 2H), 4.24 (s, 2H), 3.59 (s, 3H), 3.31 (s, 3H). 13C NMR (151 MHz, CD2Cl2) δ 192.9, 160.9, 152.0, 149.6, 137.8, 134.5, 129.8, 129.2, 114.8, 32.0, 28.6, 23.7. MS (ES+) (m/z): [M+1]+ calculated for C14H14BrN2O3, 338.18, found 338.81.
Ethyl 3-(1,3-dimethyl-2,4-dioxo-5-phenyl-3,4-dihydro-1H-pyrrolo[3,4-d]pyrimidin-6(2H)-yl)-4-hydroxybenzoate (30)
In a 100 mL round bottom flask ethyl 3-amino-4-hydroxybezoate (1.10 g, 6.07 mmol) and 4 (1.00 g, 2.98 mmol) were refluxed in ethanol (50 mL). After 2 h, the condenser was rearranged for distillation and ethanol (25 mL) was distilled off. The resulting solution was slowly poured into a vigorously stirred solution of ice cold water (200 mL) and citric acid (50 mg) giving a pink solid precipitate. The mixture was stirred for 10 min and then the solid was collected by filtration and rinsed with cold water to give 30 (1.23 g, 98.5%) after drying; m.p. 129.2–130 °C. 1H NMR (600 MHz, CD2Cl2) δ 8.58 (s, 1H), 7.89 (dd, J = 2.1 Hz, 8.7, 1H), 7.67 (d, J = 2.1 Hz, 1H), 7.36 – 7.29 (m, 2H), 7.28 – 7.18 (m, 3H), 6.97 (d, J = 8.7 Hz, 1H), 4.25 (q, J = 7.1 Hz, 2H), 3.27 (s, 3H), 3.17 (s, 3H), 1.30 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CD2Cl2) δ 165.4, 159.7, 156.8, 151.9, 136.1, 132.1, 130.9, 130.7, 129.5, 129.0, 128.8, 127.8, 125.7, 122.9, 117.7, 104.7, 103.1, 61.2, 32.0, 28.1, 14.2. MS (ES+) (m/z): [M+1]+ calculated for C23H22N3O5, 420.16, found 420.13.
Ethyl 6-(5-Bromofuran-2-yl)-7,9-dimethyl-8,10-dioxo-11-phenyl-7,8,9,10-tetrahydro-6H-benzo[b]pyrimido [4',5':3,4]pyrrolo[1,2-d][1,4]oxazine-2-carboxylate (40)
Pyrrole 30 (500 mg, 1.19 mmol), 5-bromofurfural (240 mg, 1.37 mmol), chloroform (7 mL), TFA (10 RL, 14.8 mg, 130 Rmol), and 3 Å molecular sieves (2.0 g, 8–12 mesh beads) were sealed in an Emrys 10–20 mL process vial and submerged to the level of solvent in an oil bath at 150 °C. The reaction was stirred for 24 min then removed from the oil bath. Once the internal pressure dropped the reaction vial was rapidly cooled in water. After cooling, the reaction was filtered through celite into a 50 mL recovery flask and the dried in vacuo. The residue was dissolved in a minimum amount of CH2Cl2 and quickly diluted with warm ethanol (25 mL). Fine crystals began to form immediately. The mixture was then placed on a rotary evaporator and the CH2Cl2 was removed to increase the quantity of crystals. The mixture was then chilled, filtered, and the crystals rinsed with cold ethanol to give 40 (0.500 g, 76.4%) as fine white needle like crystals. No m.p. (slow decomposition). 1H NMR (600 MHz, CD2Cl2) δ 7.81 (d, J = 7.7 Hz, 1H), 7.68 (dd, J = 1.9 Hz, 8.4, 1H), 7.63 (t, J = 7.5 Hz, 1H), 7.52 (t, J = 7.5 Hz, 1H), 7.34 (t, J = 7.5 Hz, 1H), 7.23 (d, J = 1.8 Hz, 1H), 7.09 (d, J = 8.4 Hz, 1H), 7.06 (d, J = 7.7 Hz, 1H), 6.86 (s, 1H), 6.14 (d, J = 3.4 Hz, 1H), 5.98 (d, J = 2.9 Hz, 1H), 4.11 (dq, J = 7.2 Hz, 10.7, 1H), 4.00 (dq, J = 7.1 Hz, 10.7, 1H), 3.48 (s, 3H), 3.26 (s, 3H), 1.14 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, CD2Cl2) δ 165.0, 159.3, 151.9, 151.8, 149.1, 131.8, 131.3, 130.0, 129.8, 129.7, 128.8, 128.8, 128.3, 125.4, 124.7, 124.5, 124.4, 121.6, 119.6, 114.9, 112.4, 105.9, 105.7, 68.4, 61.3, 32.4, 28.0, 14.2. MS (ES+) (m/z): [M+1]+ calculated for C28H23BrN3O6, 576.08, found 576.04.
6-(5-Bromofuran-2-yl)-7,9-dimethyl-8,10-dioxo-11-phenyl-7,8,9,10-tetrahydro-6H-benzo[b]pyrimido [4',5':3,4]pyrrolo [1,2-d][1,4]oxazine-2-carboxylic acid (42)
In a 500 mL round bottom flask equipped with stir bar, 40 (1.00 g, 1.73 mmol) was dissolved in THF (100 mL) with gentle heat and then allowed to cool. A mixture of water (70 mL) and KOH (768 mg, 13.7 mmol) was quickly added to the vigorously stirred solution which formed a white suspension. After 3 days the mixture was a homogenous yellow solution free of 40 as determined by LC/MS. The THF was removed using a rotary evaporator and warm water bath leaving behind a viscous aqueous solution. The solution was made strongly acidic to litmus using 1% aq. HCl while stirring vigorously with a glass rod. The resulting gel was shaken with EtOAc (125 mL) and then quickly poured into a 1 liter separatory funnel where a precipitate formed in the organic layer. The 500 mL flask was rinsed with additional EtOAc (125 mL). Additional EtOAc (400 mL) was added until all solids dissolved. After settling, the lower yellow aqueous layer was discarded. The EtOAc layer was washed with brine, dried over Na2SO4 and dried on a rotary evaporator. The resulting slightly yellow amorphous powder was loosened by swirling with CH2Cl2 (15 mL) and then diluted with diethyl ether (15 mL). The solids were collected by filtration and rinsed with CH2Cl2:Et2O (1:1) to give 42 (791 mg, 83.2%) as a white solid. No m.p. (slow decomposition). 1H NMR (600 MHz, 91% CD2Cl2, 9% DMSO-d6) δ 12.30 (s, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.63 (dd, J = 1.9 Hz, 8.4, 1H), 7.58 (t, J = 7.6 Hz, 1H), 7.46 (t, J = 7.5 Hz, 1H), 7.29 (t, J = 7.5 Hz, 1H), 7.17 (d, J = 1.7 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 7.7 Hz, 1H), 6.89 (s, 1H), 6.12 (d, J = 3.4 Hz, 1H), 5.96 (d, J = 3.3 Hz, 1H), 3.44 (s, 3H), 3.21 (s, 3H). 13C NMR (151 MHz, 91% CD2Cl2, 9% DMSO-d6) δ 166.7, 159.1, 151.9, 151.6, 149.0, 131.8, 131.0, 129.7, 129.7, 129.5, 128.6, 128.5, 128.5, 125.3, 125.2, 124.4, 124.2, 121.9, 119.4, 114.8, 112.3, 106.0, 105.7, 68.2, 32.3, 27.8. MS (ES+) (m/z): [M+1]+ calculated for C26H19BrN3O6, 548.05, found 548.00.
Supplementary Material
ACKNOWLEDGMENTS
Supported by NIH grants DK86125, DK72517, HL73856, EB00415, DK35124 and EY13574, and Research Development Program and Drug Discovery grants from the Cystic Fibrosis Foundation.
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- BPO
benzopyrimido. pyrrolo.oxazinedione
- CFTR
cystic fibrosis transmembrane conductance regulator
- CPT.cAMP
chlorophenylthio.cAMP
- IBMX
isobutylmethylxanthine
- PKD
polycystic kidney disease
- PPQ
pyrimido.pyrrolo.quinoxalinedione
- YFP
yellow fluorescent protein
Footnotes
SUPPORTING INFORMATION Detailed synthesis procedures and NMR spectra for all compounds. This material is available free of charge via the internet http://pubs.acs.org.
REFERENCES
- 1.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 2.Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1:148–157. doi: 10.2215/CJN.00330705. [DOI] [PubMed] [Google Scholar]
- 3.Ye M, Grantham JJ. The secretion of fluid by renal cysts from patients with autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:310–313. doi: 10.1056/NEJM199307293290503. [DOI] [PubMed] [Google Scholar]
- 4.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. doi: 10.1681/ASN.V1171179. [DOI] [PubMed] [Google Scholar]
- 5.Magenheimer BS, St John PL, Isom KS, Abrahamson DR, De Lisle RC, Wallace DP, Maser RL, Grantham JJ, Calvet JP. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na+K+2Cl− cotransporter-dependent cystic dilation. J Am Soc Nephrol. 2006;17:3424–3437. doi: 10.1681/ASN.2006030295. [DOI] [PubMed] [Google Scholar]
- 6.Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 7.Brill SR, Ross KE, Davidow CJ, Ye M, Grantham JJ, Caplan MJ. Immunolocalization of ion transport proteins in human autosomal dominant polycystic kidney epithelial cells. Proc Natl Acad Sci U S A. 1996;93:10206–10211. doi: 10.1073/pnas.93.19.10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davidow CJ, Maser RL, Rome LA, Calvet JP, Grantham JJ. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996;50:208–218. doi: 10.1038/ki.1996.304. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Findlay IA, Sheppard DN. The relationship between cell proliferation, Cl- secretion, and renal cyst growth: a study using CFTR inhibitors. Kidney Int. 2004;66:1926–1938. doi: 10.1111/j.1523-1755.2004.00967.x. [DOI] [PubMed] [Google Scholar]
- 10.Cotton CU, Avner ED. PKD and CF: an interesting family provides insight into the molecular pathophysiology of polycystic kidney disease. Am J Kidney Dis. 1998;32:1081–1083. doi: 10.1016/s0272-6386(98)70088-5. [DOI] [PubMed] [Google Scholar]
- 11.O'Sullivan DA, Torres VE, Gabow PA, Thibodeau SN, King BF, Bergstralh EJ. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am J Kidney Dis. 1998;32:976–983. doi: 10.1016/s0272-6386(98)70072-1. [DOI] [PubMed] [Google Scholar]
- 12.Xu N, Glockner JF, Rossetti S, Babovich-Vuksanovic D, Harris PC, Torres VE. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J Nephrol. 2006;19:529–534. [PubMed] [Google Scholar]
- 13.Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20:2493–2502. doi: 10.1681/ASN.2008111186. [DOI] [PubMed] [Google Scholar]
- 14.Torres VE, Boletta A, Chapman A, Gattone V, Pei Y, Qian Q, Wallace DP, Weimbs T, Wuthrich RP. Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin J Am Soc Nephrol. 2010;5:1312–1329. doi: 10.2215/CJN.01360210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Belibi FA, Edelstein CL. Novel targets for the treatment of autosomal dominant polycystic kidney disease. Expert Opin Investig Drugs. 2010;19:315–328. doi: 10.1517/13543781003588491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Masoumi A, Reed-Gitomer B, Kelleher C, Schrier RW. Potential pharmacological interventions in polycystic kidney disease. Drugs. 2007;67:2495–2510. doi: 10.2165/00003495-200767170-00004. [DOI] [PubMed] [Google Scholar]
- 17.Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens. 2009;18:99–106. doi: 10.1097/MNH.0b013e3283262ab0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torres VE. Treatment strategies and clinical trial design in ADPKD. Adv Chronic Kidney Dis. 2010;17:190–204. doi: 10.1053/j.ackd.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verkman AS, Galietta LJ. Chloride channels as drug targets. Nat Rev Drug Discov. 2009;8:153–171. doi: 10.1038/nrd2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caci E, Caputo A, Hinzpeter A, Arous N, Fanen P, Sonawane N, Verkman AS, Ravazzolo R, Zegarra-Moran O, Galietta LJ. Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem J. 2008;413:135–142. doi: 10.1042/BJ20080029. [DOI] [PubMed] [Google Scholar]
- 21.Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taddei A, Folli C, Zegarra-Moran O, Fanen P, Verkman AS, Galietta LJ. Altered channel gating mechanism for CFTR inhibition by a high-affinity thiazolidinone blocker. FEBS Lett. 2004;558:52–56. doi: 10.1016/S0014-5793(04)00011-0. [DOI] [PubMed] [Google Scholar]
- 23.Sonawane ND, Muanprasat C, Nagatani R, Jr., Song Y, Verkman AS. In vivo pharmacology and antidiarrheal efficacy of a thiazolidinone CFTR inhibitor in rodents. J Pharm Sci. 2005;94:134–143. doi: 10.1002/jps.20228. [DOI] [PubMed] [Google Scholar]
- 24.Sonawane ND, Verkman AS. Thiazolidinone CFTR inhibitors with improved water solubility identified by structure-activity analysis. Bioorg Med Chem. 2008;16:8187–8195. doi: 10.1016/j.bmc.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang B, Sonawane ND, Zhao D, Somlo S, Verkman AS. Small-molecule CFTR inhibitors slow cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19:1300–1310. doi: 10.1681/ASN.2007070828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muanprasat C, Sonawane ND, Salinas D, Taddei A, Galietta LJ, Verkman AS. Discovery of glycine hydrazide pore-occluding CFTR inhibitors: mechanism, structure-activity analysis, and in vivo efficacy. J Gen Physiol. 2004;124:125–137. doi: 10.1085/jgp.200409059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sonawane ND, Hu J, Muanprasat C, Verkman AS. Luminally active, nonabsorbable CFTR inhibitors as potential therapy to reduce intestinal fluid loss in cholera. FASEB J. 2006;20:130–132. doi: 10.1096/fj.05-4818fje. [DOI] [PubMed] [Google Scholar]
- 28.Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Nanomolar CFTR inhibition by pore-occluding divalent polyethylene glycol-malonic acid hydrazides. Chem Biol. 2008;15:718–728. doi: 10.1016/j.chembiol.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sonawane ND, Zhao D, Zegarra-Moran O, Galietta LJ, Verkman AS. Lectin conjugates as potent, nonabsorbable CFTR inhibitors for reducing intestinal fluid secretion in cholera. Gastroenterology. 2007;132:1234–1244. doi: 10.1053/j.gastro.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 30.Tradtrantip L, Sonawane ND, Namkung W, Verkman AS. Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J Med Chem. 2009;52:6447–6455. doi: 10.1021/jm9009873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Galietta LV, Jayaraman S, Verkman AS. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am J Physiol Cell Physiol. 2001;281:C1734–C1742. doi: 10.1152/ajpcell.2001.281.5.C1734. [DOI] [PubMed] [Google Scholar]
- 32.Torres VE. Role of vasopressin antagonists. Clin J Am Soc Nephrol. 2008;3:1212–1218. doi: 10.2215/CJN.05281107. [DOI] [PubMed] [Google Scholar]
- 33.Tradtrantip L, Yangthara B, Padmawar P, Morrison C, Verkman AS. Thiophenecarboxylate suppressor of cyclic nucleotides discovered in a small-molecule screen blocks toxin-induced intestinal fluid secretion. Mol Pharmacol. 2009;75:134–142. doi: 10.1124/mol.108.050567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kunzelmann K, Mall M. Electrolyte transport in the mammalian colon: mechanisms and implications for disease. Physiol Rev. 2002;82:245–289. doi: 10.1152/physrev.00026.2001. [DOI] [PubMed] [Google Scholar]
- 35.Thiagarajah JR, Verkman AS. CFTR pharmacology and its role in intestinal fluid secretion. Curr Opin Pharmacol. 2003;3:594–599. doi: 10.1016/j.coph.2003.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Thiagarajah JR, Broadbent T, Hsieh E, Verkman AS. Prevention of toxin-induced intestinal ion and fluid secretion by a small-molecule CFTR inhibitor. Gastroenterology. 2004;126:511–519. doi: 10.1053/j.gastro.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Thiagarajah JR, Song Y, Haggie PM, Verkman AS. A small molecule CFTR inhibitor produces cystic fibrosis-like submucosal gland fluid secretions in normal airways. FASEB J. 2004;18:875–877. doi: 10.1096/fj.03-1248fje. [DOI] [PubMed] [Google Scholar]
- 38.Azas N, Rathelot P, Djekou S, Delmas F, Gellis A, Di Giorgio C, Vanelle P, Timon-David P. Antiparasitic activity of highly conjugated pyrimidine-2,4-dione derivatives. Farmaco. 2003;58:1263–1270. doi: 10.1016/j.farmac.2003.07.009. [DOI] [PubMed] [Google Scholar]
- 39.Tsupak EB. Pyrrolopyrimidines. 5. Reaction of 6-amino-1,3-dimethylpyrrolo[3,4-d]pyrimidine-2,4(1H,3H)-diones with 1,3-diketones. Chem Heterocycl Comp. 2003;39:953–959. [Google Scholar]
- 40.Pray AP. Inorganic Syntheses. Vol. XXVIII. J. Wiley & Sons; New York: 1990. pp. 321–322. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.