

Abstract
The response of prostate cells to androgens reflects a combination of androgen receptor (AR) transactivation and transrepression, but how these two processes differ mechanistically and influence prostate cancer risk and disease outcome remain elusive. Given recent interest in targeting AR transrepressive processes, a better understanding of AR/corepressor interaction and responses is warranted. Here, we used transactivation and interaction assays with wild-type and mutant ARs, and deletion AR fragments, to dissect the relationship between AR and the corepressor, silencing mediator for retinoic acid and thyroid hormone receptors (SMRT). We additionally tested how these processes are influenced by AR agonist and antagonist ligands, as well as by variation in the polyglutamine tract in the AR amino terminal domain (NTD), which is encoded by a polymorphic CAG repeat in the gene. SMRT was recruited to the AR ligand binding domain by agonist ligand, and as determined by the effect of strategic mutations in activation function 2 (AF-2), requires a precise conformation of that domain. A distinct region of SMRT also mediated interaction with the AR-NTD via the transactivation unit 5 (TAU5; residues 315–538) region. The degree to which SMRT was able to repress AR increased from 17% to 56% as the AR polyglutamine repeat length was increased from 9 to 42 residues, but critically this effect could be abolished by increasing the SMRT:AR molar ratio. These data suggest that the the extent to which the CAG encoded polyglutamine repeat influences AR activity represents a balance between corepressor and coactivator occupancy of the same ligand-dependent and independent AR interaction surfaces. Changes in the homeostatic relationship of AR to these molecules, including SMRT, may explain the variable penetrance of the CAG repeat and the loss of AR signalling flexibility in prostate cancer progression.
Keywords: androgen receptor, polyglutamine, corepressor, SMRT, NCoR, prostate cancer risk, N/C interaction
1. INTRODUCTION
The AR is a member of the nuclear receptor superfamily of transcription factors sharing a common evolutionary origin, mode of action and structural architecture, with conserved domains for DNA (DBD) and steroid ligand binding (LBD), and an evolutionarily variable amino-terminal domain (NTD) (Fig. 1A). Ultimately, the capacity of nuclear receptors to function in any given cell depends on recruitment of distinct subsets of cofactors, which control conformation, nuclear-cytoplasmic localization, movement, response element recognition, chromatin remodelling, and engagement or disengagement with basal transcription factors (O’Malley et al. 2008; Wolf et al. 2008). At the transcriptional level, coregulators that activate and inhibit these processes are termed coactivators and corepressors respectively. For most nuclear receptors, the binding of agonist ligands induces conformational changes and formation of activation function 2 (AF-2), a conserved protein-protein binding surface (Fig. 1A). The AF-2 pocket binds short LxxLL-like peptide motifs in transcriptional cofactors, such as the p160 family of coactivators comprising NCOA1/SRC-1, NCOA2/GRIP1 and NCOA3/AIB1, and acts in concert with activation functions in the NTD of the receptor to mediate overall transcriptional response (Hur et al. 2004; Ozers et al. 2007).
FIGURE 1. AR and SMRT constructs used in this study.
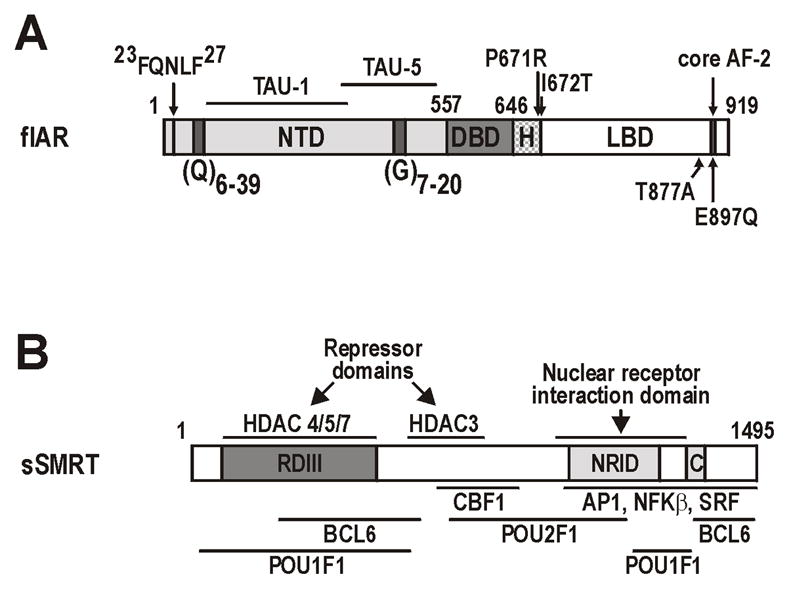
A Schematic representation of the AR showing the N-terminal (NTD), DNA-binding (DBD) and ligand-binding (LBD) domains, and subdomain structures including activation functions (TAU-1, TAU-5, AF-2), homopolymeric repeat sequences in the NTD [polyglutamine, (Q)n; polyglycine, (G)n], and the 23FQNLF27 peptide required for N/C interactions. The location of point mutations in the AR are shown. Numbers represent the amino-acid residues. B. Schematic representation of the smaller sSMRT isoform (Chen and Evans 1995) showing repressor domain III (RDIII), the nuclear receptor interaction domain (NRID), location of the CoRNR box (C), regions with histone deacetylase (HDAC) activity. The interaction surfaces for key SMRT cofactors and substrates are depicted below the schematic. Numbers represent amino-acid residues.
The AR is distinct from other nuclear receptors as the AR AF-2 surface preferentially interacts with the 23FQNLF27 peptide of the AR-NTD in the amino/carboxyl terminal (N/C) interaction, displacing coregulator events to the NTD (He et al. 2006; He et al. 2002a). As a consequence, AR activity is almost exclusively determined by two overlapping NTD transcriptional activation units (TAU), TAU-1 and TAU-5 (Jenster et al. 1995; Need et al. 2009) (Fig. 1A). The AR NTD also contains two polymorphic trinucleotide microsatellite repeats, CAG and GGC, which encode respectively polyglutamine (polyQ) and polyglycine (polyG) tracts with a distribution of 6–39 (mean 21) and 7–20 (mean 16) residues in man (Fig. 1A) (Buchanan et al. 2004b).
While the GGC repeat is less variable and usually categorized as being 16 or non-16 repeats, early studies from our group and others showed that the racial distribution of CAG repeats was inversely related to prostate cancer risk (Coetzee and Ross 1994; Edwards et al. 1999). Subsequent studies confirmed a link between CAG length and risk, age of onset and/or advanced disease at diagnosis (Buchanan et al. 2001a).. While in vitro and animal studies have related CAG length to receptor activity and prostate size (Albertelli et al. 2008; Buchanan et al. 2004b), more extensive clinical analyses have failed to demonstrate a relationship to disease risk, although did identify a correlation between CAG repeat length and serum testosterone levels (Freedman et al. 2005; Price et al. 2010). We cannot easily account for these inconsistent findings, or indeed for the inconclusive role of CAG repeat length in many other diseases (Rajender et al. 2007), perhaps because the precise effect of CAG and GGC repeat sequences on AR function are unknown.
Steroid receptors have classically been distinguished from those for thyroid hormone, retinoic acid and vitamin D by their relationship with DNA (Laudet 1997). While unliganded steroid receptors such as AR are thought to be held in an inactive conformation away from DNA by chaperones, those for thyroid hormone and retinoic acid are constitutively bound to DNA in a multi-subunit corepressor complex that actively represses basal gene transcription (Chen and Evans 1995). In contrast, conventional thought holds that liganded receptors of both classes bind DNA, recruit nuclear receptor coactivators and promote gene transactivation (Biddie et al. 2010; Jepsen et al. 2000; McEwan 2009; McKenna and O’Malley 2000).
The molecules at the core of the nuclear receptor corepressor complex are the nuclear receptor corepressor (NCOR1) and silencing mediator for retinoic acid and thyroid hormone receptors (SMRT/NCOR2) (Chen and Evans 1995; Cunliffe 2008; Perissi et al. 2010). SMRT and NCOR1 are large scaffold proteins that interact with the hydrophobic AF-2 surface of nuclear receptors using extended LxxLL-like amphipathic alpha helices (LxxI/HIxxxI/L) (the CoRNR boxes) and contain at least three independent repressor domains that alter histone deacetylase (HDAC) activity (Chen and Evans 1995; Perissi et al. 2010; Stanya and Kao 2009). Corepressor recruitment inhibits receptor activation, promotes DNA condensation and attenuates binding of other transcription factors (Cunliffe 2008; Perissi et al. 2010). Thus, receptor occupancy by either repressors or activators is a major determinant of nuclear/steroid receptor transcriptional activity (Glass and Rosenfeld 2000).
In this study, we sought to better define the relationship between the AR and SMRT, and in particular the role played by AR AF-2 and NTD interaction surfaces, canonical and non-canonical ligands, and variation in the length of the CAG encoded polyglutamine repeat in SMRT-mediated repression. We additionally map SMRT interaction surfaces within the AR NTD, and compare the relative specificity and strength of corepressor recruitment to AF-2 with the AR N/C interaction. Our data provide new information on the dynamic role of corepressors in AR signalling, and have implications for understanding AR antagonists, tissue-specific variation in androgen responses, and how the AR CAG repeat might be variably associated with risk and progression of prostate cancer.
2. MATERIALS AND METHODS
2.1 Plasmid vectors
Expression vector for the full-length smaller isoform of SMRT (pCMX-gal4-h-fSMRT; sSMRT) was provided by Dr Ron M. Evans (Howard Hughes Medical Institute, Salk Institute for Biological Studies, La Jolla, California). AR-responsive probasin (ARR3-tk-luc), MMTV (MMTV-luc), PSA540 (pGL3-PSA540), GAL-4 targeted pGK1 luciferase reporter constructs, AR expression vectors encoding different polyQ lengths [pcDNA-AR(CAG)n], the constitutive truncated variant (1–709), mammalian two-hybrid AR-NTD (1–538, 1–555), and mammalian two-hybrid wild-type (618–919, 644–919), E897Q and T877A variant AR-LBD constructs have been previously described (Buchanan et al. 2007; Buchanan et al. 2004b; Need et al. 2009). Mammalian two-hybrid pM-GAL4 and pVP16-AD vectors encoding AR-NTD fragments (encompassing amino acids 1–156, 141–356, 351–426, 427–538, 351–538) were provided by Dr Ryan A. Irvine (NCCC, USC, Los Angeles, CA). pSG5-GRIP1 was provided by Professor Mike Stallcup (University of Southern California, Los Angeles, California). sSMRT fragments for pM and pVP16 vectors were amplified in PCR reactions using Pfu DNA polymerase (Promega, La Jolla, CA) with the following primer pairs:
aa1-1495 (5′-CAGAGAATTCATGGAGGCATGGGACGCC-3′, 5′-GTCATCTAGACTTTAGACAGGCAAGGATGCCG-3′);
aa1-333 (5′-CAGAGAATTCATGGAGGCATGGGACGCC-3′, 5′-GTCATCTAGATGATGGACCCGCGGATGT-3′);
aa298-632 (5′-CAGAGAATTCAGAGCCATCTCCTCAGCCAGCA-3′, 5′-GTCATCTAGAGGTGTCGGGGCAGGTAGTAGGC-3′);
aa598-937 (5′-CAGAGAATTCGGCGTGGACCTGTATCGCAG-3′, 5′-GTCATCTAGAGAAGGCATGGCCGGTGTCT-3′);
aa890-1266 (5′-CAGAGAATTCACATTCCCACCTGCCACCCACT-3′, 5′-GTCATCTAGACTTGCTTCTTGGACTTGACCAT-3′);
a1222-1495 (5′-CAGAGAATTCCTGCTGTACCGGGATGGGGAAC-3′, 5′-GTCATCTAGACTTTAGACAGGCAAGGATGCCG-3′). Products were purified, digested with EcoR1 and XbaI and cloned into the equivalent sites of pM and pVP16 vectors.
2.2 Cell culture, transactivation assays and immunoblot analysis
COS-1 cells (American Type Culture Collection, Rockville, MD) and the PC-3 cell subline PC-3AR+, described previously (Buchanan et al. 2004a) were maintained in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS; SAFC, NSW, Australia). Transactivation and coactivator assays were performed in PC-3AR+ cells (10,000 cells/well in 96-well plates) transfected with 100ng ARR3-tk-Luc and 0.5–2.5ng pCMV-AR3.1 or equivalent molar amount of AR fragments or variant constructs using Lipofectamine 2000™ as previously described (Need et al. 2009). Coactivator assays included 1–50ng pSG5-GRIP1, pCMX-GAL4-h-fSMRT or pCMX-SMRT. Controls always included a molar equivalent amount of the appropriate empty vector, and all comparisons were balanced for total DNA using the prokaryotic pBS-SK plasmid-. Mammalian 2-hybrid N/C interaction assays were performed in COS-1 cells (10,000 cells/well in 96-well plates) transfected similarly with 25ng pGK-1, and 5ng of each of pVP16 and GAL4 expression vectors as previously described (Buchanan et al. 2004b). For both assays, following a 4-hour transfection cells were cultured in RPMI-1640 supplemented with 5% charcoal stripped FBS and 0.01–100nM steroids for 20–24 hours, lysed and analyzed for luciferase activity as previously described (Buchanan et al. 2004b). Data represents six independently transfected and steroid or vehicle treated wells, and is presented as mean (± sem) for each condition. For immunoblot analysis, COS-1 cells (200,000 cells in 6-well dishes) were transfected with 100ng of GAL4-SMRT fragment expression vectors and incubated for 24 hours. Soluble protein (20ug) was resolved by SDS-PAGE, transferred to Hybond C membranes (GE healthcare, NSW, Australia), and immunoblotted using GAL4 antibody (Clontech, Mountain View, CA), detected using HRP conjugated IgG (Dako Australia, VIC, Australia), and visualized using ECL western blotting reagents (GE Healthcare).
2.3 Microarray analysis
Affymetrix U95 microarray data collected as part of our previous studies (Scher et al. 2004) from 23 untreated primary prostate cancers, 17 primary prostate tumors treated with short-term neoadjuvant androgen ablation, and 9 metastatic lesions (manually dissected for prostate cancer epithelial cells) was analyzed for gene expression as previously described (Scher et al. 2004).
3. RESULTS
3.1 SMRT repression of agonist-activated AR
Using a minimal SMRT isoform (sSMRT) (Fig. 1B) (Chen et al. 1996), we examined in detail how the nuclear corepressors inhibit AR transcriptional responses to agonist ligands in cotransfected prostate cancer PC-3 cells (Fig. 2). AR activity stimulated by 0.1–10nM of the potent agonist, 5α-dihydrotestosterone (DHT) was repressed by greater than 50% by sSMRT on both the androgen specific minimal probasin promoter (ARR3) and the more complex mouse mammary tumor virus (MMTV) promoter containing canonical hormone response elements (Fig. 2A–B). To investigate how this repression may be offset by the magnitude of the AR transcriptional response, we utilized receptor variants encoding 9 (Fig. 2C) and 29 (Fig. 2D) glutamine repeats in the NTD; the former having significantly higher maximal activity (Buchanan et al. 2004b). In both cases, repression was achieved at 5ng of sSMRT and was proportionally equivalent across all DHT concentrations (Fig. 2C,D). Increasing the amount of sSMRT to 10ng did not increase repression (Fig. 2C,D). In contrast to p160 and other coactivators (Buchanan et al. 2007; Need et al. 2009; Shen et al. 2005), overexpression of sSMRT did not affect AR sensitivity to ligand (Fig 2C,D).
FIGURE 2. SMRT dampens the transcriptional capacity of the AR but does not alter the specificity or sensitivity of transcriptional responses.

PC-3 cells (15,000 per well in 96 well plates) were transiently transfected with AR, either sSMRT or molar equivalent of empty vector, and one of probasin (ARR3), PSA (PSA540) or MMTV luciferase reporter constructs, and treated with vehicle (ethanol) or different concentrations of agonists DHT or medroxyprogesterone acetate (MPA) as indicated. Data represent the mean (±SEM) of 5–8 independently transfected wells. A–B. Effect of sSMRT on DHT-mediated AR transactivaiton on ARR3 and MMTV promoters shown as relative light units (RLU). C–D. Ability of sSMRT to repress transactivation capacity of AR with short (Q9) or long (Q29) polyQ repeat length. Data are presented as percent luciferase activity for AR without sSMRT at 10−9M (1nM) DHT. E. sSMRT repression of AR activity is similar for both natural (i.e. DHT) and non-classical (i.e. MPA) receptor agonists on the ARR3 promoter. Data are presented as percent AR activity (±SEM) with control. F. Increasing the comparative amount of transfected full-length (fl) AR partially rescues AR activity from sSMRT repression. Data are presented as percent AR activity with control in the presence of DHT for each transfection condition.
To investigate agonist specificity of SMRT responses, we compared DHT with medroxyprogesterone acetate (MPA), an AR agonist that in contrast to DHT does not induce a direct N-terminal/C-terminal (N/C) interaction (Birrell et al. 2007). The repression of AR responses to DHT and MPA by sSMRT were similar on the ARR3 promoter (Fig. 2E) and a similar response was observed for the tandem KLK3 enhancer and promoter construct, PSA540 (data not shown). Increasing the amount of AR in the experiment was able to partially rescue AR activity from sSMRT inhibition (Fig. 2F). These data imply that the molar ratio of AR and SMRT is a more important determinant of the degree of repression than the AR agonist, magnitude of the transcriptional response or origin of the reporter. Moreover, they indicate that SMRT dampens the transcriptional response of the receptor rather than altering the overall dynamics of AR action.
3.2 SMRT repression is modulated by length of the AR CAG repeat/AR polyQ tract in a process dependent on the AR:SMRT ratio
The inverse relationship between increasing polyQ length (encoded by the CAG repeat) and AR activity is dependent on cell and promoter context, and modulated by p160 and other NTD coactivators (Beilin et al. 2000; Buchanan et al. 2004b; Hsiao et al. 1999; Irvine et al. 2000). In contrast, polyQ length does not affect ligand binding characteristics, steady-state protein levels of the receptor, or activity of the isolated AF-1 region (Buchanan et al. 2004b). We demonstrate here that the degree to which sSMRT represses activity of full-length AR is directly proportional to polyQ/CAG length, increasing stepwise (normalized for each variant in the absence of sSMRT) from 17(±7)%, 31(±4)% and 56(±3)% for glutamine repeat lengths of 9, 21 and 42 respectively (Fig. 3A). Critically however, repression of AR became largely independent of polyQ length as the amount of transfected sSMRT was increased. At the highest amount of SMRT tested, repression of AR variants with between 9 and 42 glutamine residues showed only minimal variation (67(±3) to 71(±2)% repression; Fig. 3B). These results suggest that the effect of the polyQ/CAG repeat on AR function is critically dependent on the ratio between AR and repressors, and can be eliminated when SMRT is in excess.
FIGURE 3. The degree to which SMRT repression is modulated by AR CAG repeat length is dependent on the AR:SMRT ratio.

Experiments were conducted in PC-3 cells as detailed in Fig. 2, but using AR vectors encoding polyQ repeat lengths of 9–42 residues as indicated. A. Repression of AR by sSMRT is proportional to polyQ repeat length. Data are presented as percent maximal activity of each AR alone. B. The polyQ effect on sSMRT repression is diminished/abolished by increasing transfected amounts of the corepressor. Data is presented as the mean percent repression by sSMRT of each AR in the presence of 1nM DHT. Lines of best fit across polyQ length for each amount of sSMRT are shown.
3.3 SMRT repression of AR is dependent on the AR/corepressor ratio
Although it is well established that the response of AR to cofactors in vitro is critically dependent on the relative levels of each protein (Irvine et al. 2000; Ma et al. 1999), this concept is almost universally overlooked in clinical analyses. Indeed, we have previously shown that the mean AR:cofactor ratio in a cohort of prostate cancer samples is more informative of progression than the average of each protein alone (Buchanan et al. 2007). When we interrogated our published prostate cancer microarray data (Holzbeierlein et al. 2004; Scher et al. 2004), the ratio of AR expression to both SMRT (NCOR2) and NCOR1 was found to be more variable in metastatic compared with primary samples, primarily due to changes in AR expression (Fig. 4). Given our findings above, this implies that the relative contribution of the AR CAG repeat to androgen signalling may actually change during disease progression.
FIGURE 4. Change in the relative level of AR and corepressors in prostate cancer progression.
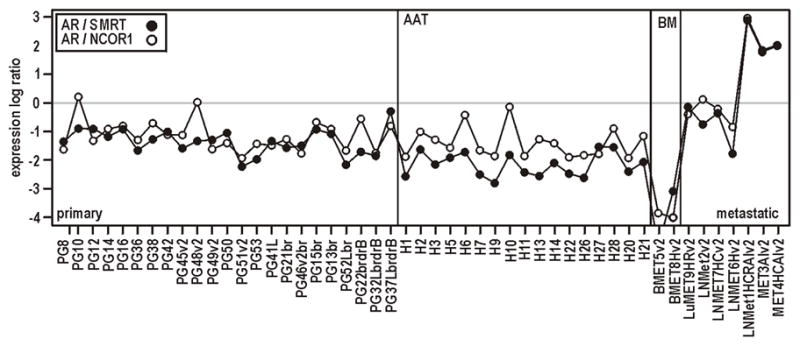
Expression of AR and the corepressors NCOR1 and NCOR2/SMRT derived from published Affymetrix array profiling experiments in clinical prostate cancer samples (Holzbeierlein et al. 2004; Scher et al. 2004). Relative log2 expression of AR/SMRT or AR/NCOR1. primary, primary prostate cancer; AAT, primary cancer following short-term androgen deprivation therapy; BM, bone metastases; metastatic, metastatic prostate cancer.
3.4 SMRT can repress transactivation driven by both AF-1 and AF-2
It was previously reported that SMRT and/or NCOR1 are recruited to both the NTD and LBD of steroid receptors (Dotzlaw et al. 2002; Varlakhanova et al. 2010). To assess in detail the consequences of these interactions for AR activity derived from either AF-1 or AF-2, we created AR constructs depleted of these two activation functions but retaining the DBD (i.e. AF-1, amino acids 1–648; AF-2, amino acids 538–919; Fig. 5A). In PC3 cells, sSMRT repressed both the weak agonist activity derived from isolated AF-2 (Fig. 5B) and the strong ligand-independent activity derived from isolated AF-1 (Fig. 5C). Conversely, coexpression of the p160 protein GRIP1 amplified activity derived from both domains (Fig. 5B–C), consistent with the common but opposing modes of action for these two cofactor classes. The inhibition of intrinsic AF-1 activity was dependent on the amount of sSMRT added (Fig. 5D), but not on promoter context (Fig. 5E–F). Previous studies from our group identified a protein-protein interaction surface in the LBD (Buchanan et al. 2001b), now termed BF-3, that is mutated in prostate cancer and coordinates the response of cofactors at AF-2 (Buchanan et al. 2001b; Estebanez-Perpina et al. 2007). sSMRT was able to mediate a similar degree of repression of both loss (P671A) and gain (I672T) of function AR BF-3 variants compared to full-length AR (Fig. 5G).
FIGURE 5. SMRT is capable of repressing the AR-LBD in a ligand dependent and the AR-NTD in a ligand independent manner.

Experiments for B–G were conducted and presented as detailed in Fig. 2, although where indicated substituting the p160 coactivator, GRIP1 for sSMRT. A. Schematic representation of the AR as in Fig. 1, showing the location of point mutations introduced into the AR, as well as fragments created as isolated expression vectors. Numbers represent the amino-acid residues. B. The small transactivation capacity directed by the isolated AR-LBD [i.e. AR(538–919)] is amplified by GRIP1 and inhibited by SMRT in a ligand dependent manner. Data are expressed as percent activity of full length (flAR) in the presence of 1nM DHT. C. The constitutive transactivation capacity mediated by TAU-1/5 of AR(1–648) is decreased by sSMRT and amplified by GRIP1 independent of ligand. Data are presented as percent constitutive activity for AR alone in the absence of ligand. D. The transactivation capacity of the constitutive AR(1–709) variant is decreased by sSMRT in a dose-dependent manner. E–F. Both ligand dependent and independent AR transactivation capacity is repressed to a similar extent by SMRT on either ARR3 or MMTV promoters. Data are presented as in A. G. sSMRT can repress DHT-mediated transactivation of somatic mutations that confer either increased or decreased receptor function. Data are presented as arbitrary light units (ALU).
3.5 SMRT interaction with the AR-LBD is driven by agonist ligands and dependent on structural integrity of the AF-2 surface
To determine how SMRT might be affecting the response of the AR to different agonists, we first investigated protein-protein interaction between full-length sSMRT and the complete AR-LBD (i.e. 644–919) (Fig. 6A). We observed a strong DHT dose-dependent interaction between these two proteins that was up to 79-fold higher than control (Fig. 6B–C). These results imply that sSMRT interacts via a ligand-dependent interaction surface on the AR, of which AF-2 is most likely (Hodgson et al. 2008; Liao et al. 2003). We took advantage of two known characteristics of AF-2 to investigate this further: (i) the relationship between AF-2 formation and agonist ligands, and (ii) mutations in the core of the AF-2 that alter the structure of the surface upon ligand binding (Birrell et al. 2007; He et al. 2006). First, recruitment of sSMRT to the wild-type AR-LBD segregated with ligands known to elicit an androgenic response [i.e. DHT, MPA, androstenedione (ASD)], whereas no increased signal was seen with AR antagonists [hydroxyflutamide (OHF), bicalutamide (BIC)] or the non-androgenic ligand progesterone (PROG; Fig. 6D). Introducing the T877A substitution from the LNCaP human prostate cancer cell line, which broadens AR agonist-like responses to include adrenal androgens, progesterone and hydroxyflutamide (Veldscholte et al. 1992; Veldscholte et al. 1990), clearly demonstrates the relationship between agonist action and sSMRT recruitment to the AF-2 surface (Fig. 6D). Consistent with this interpretation, the non-steroidal anti-androgen BIC is known to retain antagonist activity on the LNCaP AR (Veldscholte et al. 1992) and fails to induce SMRT recruitment (Fig. 6D). Conversely, introducing the E897Q substitution that disrupts the charge clamp responsible for holding LxxLL-like helices within the AF-2 pocket, but not agonist responses, eliminated ligand-dependent sSMRT recruitment for all ligands tested (Fig. 6D). Together, the above observations suggest that a precise agonist-induced conformation of AF-2 is required for sSMRT recruitment.
FIGURE 6. SMRT interaction with the AR-LBD is driven by agonist ligands and is dependent on the structural integrity of the AF-2 surface.

For B–D, COS-1 cells (20,000 per well in 96 well plates) were transfected with equivalent molar amounts of VP16-AD and GAL-DBD fusion constructs, and with the GAL4-responsive pGK1 reporter. AR and SMRT negative interaction controls were SV40-T antigen and p53. Transfected cells were treated with vehicle or DHT, progesterone (PROG), medroxyprogesterone acetate (MPA), bicalutamide (BIC), hydroxyflutamide (OHF) or androstenedione (ASD) as indicated. Data is in relative light units (RLU), and represents the mean (±SEM) of 4–6 independently transfected wells. A. Schematic representation of the AR as in Fig. 1, showing the location of point mutations introduced into the AR, as well as fragments created as GAL4 and/or VP16 fusion constructs. Numbers represent the amino-acid residues. B–C. Analysis of DHT-dependent recruitment of sSMRT to the AR-LBD. D. Analysis of the relationship between sSMRT recruitment to the AR-LBD, different ligands, and by mutations analysis, the conformation of AF-2. Tested LBDs were wild-type (wt) AR, T877A containing the promiscuous AR variant from the LNCaP cell line, and E897Q that disrupts the charge clamp of the AF-2 pocket but not transactivation (Veldscholte et al. 1990).
3.6 Role of the AF-2 surface in SMRT recruitment and N/C interaction
Analogous to the AR 23FQNLF27 region mediating the N/C interaction, peptide library screens have consistently identified FxxLF sequences as the highest affinity peptides for the agonist-induced AF-2 surface (Chang and McDonnell 2002; Hur et al. 2004). Our aims here were to (i) compare the sensitivity of the AF-2 surface for recruitment of sSMRT and the NTD (i.e. N/C), and (ii) determine whether either interaction is affected by other regions of the AR, such as the hinge region (H; Fig. 7A) which is known to attenuate AF-2 activity (Moilanen et al. 1997). In the mammalian two-hybrid assay, extending the AR-LBD construct to include amino acids 618–643 of the hinge decreased both sSMRT recruitment and the N/C interaction to a similar extent, but for both AR constructs N/C occurred at DHT concentrations 10-fold lower than sSMRT recruitment (Fig. 7B-C). Compared to N/C however, sSMRT recruitment was relatively more sensitive to MPA and ASD (Fig. 7D–E). Both DHT-induced interactions could be antagonized to a similar extent by increasing amounts of the non-permissive ligands, OHF and PROG (Fig. 7F–G). These results confirm the requirement for AF-2 in both interactions and an inhibitory effect of the hinge region, but imply that N/C and SMRT may require a different conformation of the AF-2 surface.
FIGURE 7. Agonist recruitment of SMRT to the AR-LBD parallels the N/C interaction.

Experiments in B–G were conducted and presented as described in Fig. 6, except for D–E where data is shown as percent of the interaction mediated by DHT. A. Schematic representation of the AR as in Fig. 1, showing fragments created as GAL4 and/or VP16 fusion constructs. Numbers represent the amino-acid residues. B–C. Effect of including residues encompassing the C-terminal extension of the DBD (i.e. amino acids 618–643) on recruitment of sSMRT and the AR-NTD by the AR-LBD. D–E. The relative ability of non-canonical ligands to induce recruitment of sSMRT and the AR-NTD to the AR-LBD. Data in C is from Fig. 6C. F–G. DHT induced recruitment of both sSMRT and the AR-NTD to the AF-2 surface can be potently inhibited by non-agonist ligands OHF and PROG. Inhibition of N/C may be slightly less sensitive to disruption.
3.7 SMRT recruitment to the AR-NTD maps to a region containing the polyglycine repeat
The effect of polyQ length on SMRT repression presented above (Fig. 3) and on p160 responses in other studies (Buchanan et al. 2004b; Irvine et al. 2000), is consistent with interaction of both cofactor classes with the AR-NTD (Ma et al. 1999; Need et al. 2009). Nevertheless, unlike the full-length receptor above (Fig. 3), the relative repression of the AR-NTD by SMRT was independent of CAG repeat length (data not shown). Using a series of sSMRT constructs tethered to the GAL4-DBD (Fig. 8A), we mapped using the mammalian two-hybrid assay a significant increase in response to VP16-AR NTD (amino acids 1–538) with sSMRT residues 1–333, but not the full-length repressor (Fig. 8B). The latter cannot be explained by variation in expression of the different sSMRT fragments (Fig. 8C), but instead may be due to steric effects associated with binding full-length sSMRT directly to DNA and/or recruitment of the complete cellular repressor machinery (reviewed in Glass and Rosenfeld 2000; Perissi et al. 2004). Mapping in the reverse orientation using sSMRT fragments tethered to VP16, captured an increased signal for full-length sSMRT and the AR-NTD(1–557) compared with controls (Fig. 8D), but only limited apparent recruitment of smaller sSMRT fragments (Fig. 8E). In this orientation, the direct tethering of repressor functions to the VP16 activation domain may be producing false negatives. Irrespective, using sSMRT(1–333) and a series of AR-NTD fusion fragments (Fig. 8D), we were able to map this interaction to a region of the AR-NTD encompassing TAU-5 residues 315–538, which is centred on the polyglycine repeat (Fig. 8D,F).
FIGURE 8. Amino acids 1–333 in RDIII of SMRT mediate interaction with the TAU-5 region of the AR-NTD encompassing the polyglycine repeat.

Experiments for B–C and E–F were conducted and presented as described in Fig. 6. A. Schematic representation of the sSMRT isoform as in Fig. 1B showing fragments generated as fusions in mammalian two-hybrid GAL4 DNA binding domain and VP16 activation domain vectors. Numbers represent amino-acid residues. B. GAL-4 sSMRT fusion constructs used to map the interaction with the AR-NTD implicates sSMRT amino acids 1–333 of the RDIII region. The AR N/C interaction is presented as a positive control. C. Immunoblot analysis showing the expected molecular weight of each sSMRT fragment. D. Schematic representation of the AR as in Fig. 1, showing fragments created as GAL4 and/or VP16 fusion constructs. Numbers represent the amino-acid residues. E. VP16 sSMRT fusion constructs used to map the interaction with the AR-NTD (the converse experiment to B) identifies full-length sSMRT but not individual fragments. F. AR-NTD fragments used to map interaction with the 1–333 RDIII region of sSMRT identifies the AR TAU-5 region of the receptor encompassing the polyglycine repeat
4. DISCUSSION
The dynamic nature of steroid responses in different tissues necessitates a flexible system of transcriptional regulation. A component of this process is the relationship of the steroid receptors with their coregulators, which act to amplify (coactivators) or inhibit (corepressors) transcriptional activity in a locus and lineage specific manner (Carroll et al. 2005; Heemers and Tindall 2007). This dynamic relationship implies that transrepression is just as important as transactivation for overall cellular response (reviewed in Perissi et al. 2010). It is now emerging that this mechanism breaks down in the evolution of cancer, with malignant cells adopting a more rigid steroidogenic response (reviewed in Battaglia et al. 2010). In prostate cancer, the AR evolves the capacity to target genes involved in proliferation at the expense of those controlling cell cycle (Hendriksen et al. 2006; Wang et al. 2009), which may be mediated by changes in the relationship of AR with key coregulators. Although numerous studies have reported changes in the expression of AR and its coregulators during disease progression, including SMRT (reviewed in Battaglia et al. 2010; Chmelar et al. 2007), we nonetheless know little of how they alter the AR-driven transcriptional network.
Several studies have shown that antagonists of androgen and estrogen (ER) receptors might be conducive to recruitment of SMRT and NCOR over p160 coactivators, thereby repressing partial-agonist responses (Hodgson et al. 2005; Hodgson et al. 2007; Hodgson et al. 2008; Peterson et al. 2007). In this manner, corepressors would play a key role in conferring agonist or antagonist capacity. The data in this study supports the recognition that SMRT and NCOR1 can bind AR in the presence of agonist ligands such as DHT (Hodgson et al. 2005; Yoon and Wong 2006), thereby dampening the receptor’s ability to activate gene transcription and thereby promote prostate cancer cell growth (Eisold et al. 2009). Indeed, numerous microarray and functional studies have demonstrated that agonist-activated steroid receptors are as likely to repress transcription as activate it (Cunliffe 2008; Perissi et al. 2010; Yoon and Wong 2006). Similarly, inhibitors of histone deacetylase activity increase AR levels, promote corepressor/AR interaction on DNA, and augment AR activity and chromatin remodelling of integrated reporters in a receptor- and DHT-dependent manner (List et al. 1999; Trtkova et al. 2010).
In this study, we have shown that SMRT is able to reduce agonist-activated AR responses on multiple responsive sites. . Interaction of SMRT with the AR-LBD requires agonist binding, is weaker than the N/C interaction at the same site, and is antagonized by non-canonical ligands. Nevertheless, AF-2 core mutations effect parallel changes on SMRT recruitment, N/C interaction, ligand agonist capacity and, as previously shown, p160 coactivator recruitment (Berrevoets et al. 1998; He et al. 2000; He et al. 2002a; He et al. 2002b). As such, these interactions appear to require a similar folded agonist-induced AF-2 surface, while the variable sensitivity of these interactions may reflect the nature of the interacting peptide (Chang and McDonnell 2002; Hur et al. 2004), or a more complex interaction surface. These findings are consistent with our data showing that SMRT interaction with AF-2 is mediated by a region distinct from the well-characterised CoRNR boxes and is considerably weaker than interaction of the full sSMRT molecule. Indeed, distinct but overlapping surfaces in AF-2 are responsible for coactivator and corepressor binding to other nuclear receptors (Glass and Rosenfeld 2000; Perissi et al. 2004).
Key to understanding corepressor effects on the AR is the recognition that agonist binding has greater consequences on the structure of receptor LBDs than the repositioning of helix 12 (Pissios et al. 2000), and translates stabilizing conformational changes to the NTD that promote interaction with coregulator molecules including SMRT (Fischer et al. 2010; McEwan 2004; McEwan et al. 2007). Indeed, the high affinity N/C interaction that occurs rapidly in the cytoplasm upon agonist binding (van Royen et al. 2007), likely induces structural order within the NTD and primes the DNA-bound receptor for cofactor binding. Like the p160 coactivators (Need et al. 2009; Shen et al. 2005), our data suggests that SMRT may utilize different surfaces to form a bridge between TAU-5 and AF-2 regions of the receptor. In this model, the response of AR to agonists thus reflects the degree of induced NTD structural order and competitive ability of coactivators and/or corepressors to bind to and bridge, the N and C termini of the receptor. In support of this model, the greater relative ability of MPA and ASD to recruit SMRT to the LBD rather than the N/C interaction parallels the reduced relative agonist activity of those ligands compared with DHT (Buchanan et al. 2005). This model also explains why SMRT is more effective at repressing the intrinsic activity of isolated NTD and LBD fragments than that of the full-length receptor. For receptor antagonists, the inability to form a precise AF-2 surface would limit N/C-induced conformational changes and be permissive for cofactor binding only in the NTD. Indeed, binding of SMRT and/or NCOR1 to full length AR, as well as for ERα, PGR and glucocorticoid receptor (GR), reportedly occurs only via the NTD (Jackson et al. 1997; Smith et al. 1997; Smith and McLaughlin 1997; Zhang et al. 1998). That antagonists mostly inhibit transcriptional responses raises the possibility that unstructured receptor NTDs favour interaction with corepressors over coactivators.
There has been considerable interest over the last two decades about the length of the AR CAG repeat and its association with the risk of developing prostate cancer and other diseases (Coetzee and Ross 1994; Freedman et al. 2005; Giovannucci et al. 1997; Price et al. 2010). The belief that the relationship between CAG length and disease relates to a gradient of AR transcriptional competence is supported by a recently described mouse model comparing different AR CAG repeat lengths (Albertelli et al. 2008; Robins et al. 2008), which demonstrated that shorter AR CAG repeat length led to increased AR transcriptional activity and earlier onset of tumorigenesis. This polyQ effect on AR activity may be partly mediated by altered cofactor responses. For example ras-related nuclear protein (RAN) binds to the polyQ region and enhances AR activity in a polyQ size-dependent manner (Hsiao et al. 1999). Despite interacting with a distinct NTD region, the same as identified in this study for the AR NTD-SMRT interaction, all three p160 coactivators exaggerate the polyQ effect on AR transactivation (Irvine et al. 2000). Additionally, we found that the AR:SMRT ratio has a marked effect on the extent to which activity of the full-length receptor, but not of the isolated NTD, is influenced by CAG repeat/polyQ length. These results can be explained in terms of the structural model presented above. The polyQ acts as a flexible spacer that separates regions of the NTD involved in N/C interaction and cofactor driven transactivation, and influences activity of the full-length AR but not the isolated NTD (Buchanan et al. 2004b). The longer the polyQ tract the greater the intrinsic disorder in the NTD, which by our structural model will favour SMRT binding over coactivator recruitment. Increasing the availability of SMRT will push the AR towards a corepressor-bound state, squelching the polyQ effect. This has important implications in understanding the contribution of the AR CAG repeat in prostate cancer (Buchanan et al. 2001a). At a basic level, we know that an increase in AR is perhaps the only common defining factor in prostate cancer progression (Chen et al. 2004; Scher and Sawyers 2005). For a given amount of SMRT, this would not only increase overall transcriptional output from androgen signaling but enhance the influence of the CAG repeat on AR transcriptional responses. This could explain why the polyQ size effect varies in different cell lines (Beilin et al. 2000), and why some studies have failed to detect a relationship between CAG repeat length and prostate cancer risk/progression (Freedman et al. 2005; Price et al. 2010). The phenotypic effects of the AR-CAG polymorphism in prostate cancer and other diseases should therefore be considered in a tissue-specific manner in the context of the level of AR and availability of key cofactors such as SMRT.
Highlights.
Repression by SMRT increases with the length of the polymorphic AR CAG repeat
SMRT represses AR agonist activity by binding to both activation functions 1 and 2
Ligands discriminate recruitment of SMRT and the receptors NTD (N/C interaction)
An induced structural model of corepressor recruitment and AR responses
Acknowledgments
Funding: This work was funded by the Prostate Cancer Foundation of Australia (GB ID#YI02), the Australian Research Council (GB&WDT; DP110101101), the National Health and Medical Research Council of Australia (WDT 627185), the NIH/NCI (GAC, R01CA109147 and R01CA136924), The Prostate Cancer Foundation (GAC), and the Department of Defense Prostate Cancer Research Program (WDT PC060443). VCT holds an EJ Whitten Foundation Fellowship. TBM holds a Cancer Council of South Australia W Bruce Hall Fellowship. LMB holds a senior research fellowship from the Cancer Council of South Australia. EFN holds a Freemasons Foundation Postdoctoral Research Fellowship. GB holds an NHMRC CDA level 1 Fellowship (ID#627018).
The authors would like to thank Cindy G.M. Koh and Albert Cheong for technical assistance, Dr Howard C. Shen and Professor Michael R. Stallcup for invaluable discussion, and Professors Howard I. Scher, and William L. Gerald for microarray data. Dr Ryan A. Irvine kindly provided AR expression vectors for variable length polyQ repeats and some AR-NTD fragments. Professor Ron M. Evans kindly provided pCMX-SMRT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albertelli MA, O’Mahony OA, Brogley M, Tosoian J, Steinkamp M, Daignault S, Wojno K, Robins DM. Glutamine tract length of human androgen receptors affects hormone-dependent and -independent prostate cancer in mice. Hum Mol Genet. 2008;17:98–110. doi: 10.1093/hmg/ddm287. [DOI] [PubMed] [Google Scholar]
- Alen P, Claessens F, Verhoeven G, Rombauts W, Peeters B. The androgen receptor amino-terminal domain plays a key role in p160 coactivator-stimulated gene transcription. Mol Cell Biol. 1999;19:6085–6097. doi: 10.1128/mcb.19.9.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia S, Maguire O, Campbell MJ. Transcription factor co-repressors in cancer biology: roles and targeting. Int J Cancer. 2010;126:2511–2519. doi: 10.1002/ijc.25181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilin J, Ball E, Favaloro J, Zajac J. Effect of the androgen receptor CAG repeat polymorphism on transcriptional activity: specificity in prostate and non-prostate cell lines. J Mol Endocrinol. 2000;25:85–96. doi: 10.1677/jme.0.0250085. [DOI] [PubMed] [Google Scholar]
- Berrevoets CA, Doesburg P, Steketee K, Trapman J, Brinkmann AO. Functional interactions of the AF-2 activation domain core region of the human androgen receptor with the amino-terminal domain and with the transcriptional coactivator TIF2 (transcriptional intermediary factor2) Mol Endocrinol. 1998;12:1172–1183. doi: 10.1210/mend.12.8.0153. [DOI] [PubMed] [Google Scholar]
- Biddie SC, John S, Hager GL. Genome-wide mechanisms of nuclear receptor action. Trends Endocrinol Metab. 2010;21:3–9. doi: 10.1016/j.tem.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell SN, Butler LM, Harris JM, Buchanan G, Tilley WD. Disruption of androgen receptor signaling by synthetic progestins may increase risk of developing breast cancer. Faseb J. 2007;21:2285–2293. doi: 10.1096/fj.06-7518com. [DOI] [PubMed] [Google Scholar]
- Bolton EC, So AY, Chaivorapol C, Haqq CM, Li H, Yamamoto KR. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21:2005–2017. doi: 10.1101/gad.1564207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan G, Birrell SN, Peters AA, Bianco-Miotto T, Ramsay K, Cops EJ, Yang M, Harris JM, Simila HA, Moore NL, Bentel JM, Ricciardelli C, Horsfall DJ, Butler LM, Tilley WD. Decreased androgen receptor levels and receptor function in breast cancer contribute to the failure of response to medroxyprogesterone acetate. Cancer Res. 2005;65:8487–8496. doi: 10.1158/0008-5472.CAN-04-3077. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Craft PS, Yang M, Cheong A, Prescott J, Jia L, Coetzee GA, Tilley WD. PC-3 cells with enhanced androgen receptor signaling: A model for clonal selection in prostate cancer. Prostate. 2004a;60:352–366. doi: 10.1002/pros.20079. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Irvine RA, Coetzee GA, Tilley WD. Contribution of the androgen receptor to prostate cancer predisposition and progression. Cancer Metastasis Rev. 2001a;20:207–223. doi: 10.1023/a:1015531326689. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Ricciardelli C, Harris JM, Prescott J, Yu ZC, Jia L, Butler LM, Marshall VR, Scher HI, Gerald WL, Coetzee GA, Tilley WD. Control of androgen receptor signaling in prostate cancer by the cochaperone small glutamine rich tetratricopeptide repeat containing protein alpha. Cancer Res. 2007;67:10087–10096. doi: 10.1158/0008-5472.CAN-07-1646. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Yang M, Cheong A, Harris JM, Irvine RA, Lambert PF, Moore NL, Raynor M, Neufing PJ, Coetzee GA, Tilley WD. Structural and functional consequences of glutamine tract variation in the androgen receptor. Hum Mol Genet. 2004b;13:1677–1692. doi: 10.1093/hmg/ddh181. [DOI] [PubMed] [Google Scholar]
- Buchanan G, Yang M, Harris JM, Nahm HS, Han G, Moore N, Bentel JM, Matusik RJ, Horsfall DJ, Marshall VR, Greenberg NM, Tilley WD. Mutations at the boundary of the hinge and ligand binding domain of the androgen receptor confer increased transactivation function. Mol Endocrinol. 2001b;15:46–56. doi: 10.1210/mend.15.1.0581. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Chang CY, McDonnell DP. Evaluation of ligand-dependent changes in AR structure using peptide probes. Mol Endocrinol. 2002;16:647–660. doi: 10.1210/mend.16.4.0818. [DOI] [PubMed] [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- Chen JD, Umesono K, Evans RM. SMRT isoforms mediate repression and anti-repression of nuclear receptor heterodimers. Proc Natl Acad Sci U S A. 1996;93:7567–7571. doi: 10.1073/pnas.93.15.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmelar R, Buchanan G, Need EF, Tilley W, Greenberg NM. Androgen receptor coregulators and their involvement in the development and progression of prostate cancer. Int J Cancer. 2007;120:719–733. doi: 10.1002/ijc.22365. [DOI] [PubMed] [Google Scholar]
- Coetzee GA, Ross RK. Re: Prostate cancer and the androgen receptor. J Natl Cancer Inst. 1994;86:872–873. doi: 10.1093/jnci/86.11.872. [DOI] [PubMed] [Google Scholar]
- Cunliffe VT. Eloquent silence: developmental functions of Class I histone deacetylases. Curr Opin Genet Dev. 2008;18:404–410. doi: 10.1016/j.gde.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotzlaw H, Moehren U, Mink S, Cato ACB, Iniguez Lluhi JA, Baniahmad A. The Amino Terminus of the Human AR Is Target for Corepressor Action and Antihormone Agonism. Mol Endocrinol. 2002;16:661–673. doi: 10.1210/mend.16.4.0798. [DOI] [PubMed] [Google Scholar]
- Edwards SM, Badzioch MD, Minter R, Hamoudi R, Collins N, Ardern-Jones A, Dowe A, Osborne S, Kelly J, Shearer R, Easton DF, Saunders GF, Dearnaley DP, Eeles RA. Androgen receptor polymorphisms: association with prostate cancer risk, relapse and overall survival. Int J Cancer. 1999;84:458–465. doi: 10.1002/(sici)1097-0215(19991022)84:5<458::aid-ijc2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Eisold M, Asim M, Eskelinen H, Linke T, Baniahmad A. Inhibition of MAPK-signaling pathway promotes the interaction of the corepressor SMRT with the human androgen receptor and mediates repression of prostate cancer cell growth in the presence of antiandrogens. J Mol Endocrinol. 2009;42:429–435. doi: 10.1677/JME-08-0084. [DOI] [PubMed] [Google Scholar]
- Estebanez-Perpina E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer K, Kelly SM, Watt K, Price NC, McEwan IJ. Conformation of the mineralocorticoid receptor N-terminal domain: evidence for induced and stable structure. Mol Endocrinol. 2010;24:1935–1948. doi: 10.1210/me.2010-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman ML, Pearce CL, Penney KL, Hirschhorn JN, Kolonel LN, Henderson BE, Altshuler D. Systematic evaluation of genetic variation at the androgen receptor locus and risk of prostate cancer in a multiethnic cohort study. Am J Hum Genet. 2005;76:82–90. doi: 10.1086/427224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannucci E, Stampfer MJ, Krithivas K, Brown M, Brufsky A, Talcott J, Hennekens CH, Kantoff PW. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc Natl Acad Sci U S A. 1997;94:3320–3323. doi: 10.1073/pnas.94.7.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Rosenfeld MG. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 2000;14:121–141. [PubMed] [Google Scholar]
- He B, Gampe RT, Jr, Hnat AT, Faggart JL, Minges JT, French FS, Wilson EM. Probing the Functional Link between Androgen Receptor Coactivator and Ligand-binding Sites in Prostate Cancer and Androgen Insensitivity. J Biol Chem. 2006;281:6648–6663. doi: 10.1074/jbc.M511738200. [DOI] [PubMed] [Google Scholar]
- He B, Kemppainen JA, Wilson EM. FXXLF and WXXLF sequences mediate the NH2-terminal interaction with the ligand binding domain of the androgen receptor. J Biol Chem. 2000;275:22986–22994. doi: 10.1074/jbc.M002807200. [DOI] [PubMed] [Google Scholar]
- He B, Lee LW, Minges JT, Wilson EM. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J Biol Chem. 2002a;277:25631–25639. doi: 10.1074/jbc.M202809200. [DOI] [PubMed] [Google Scholar]
- He B, Minges JT, Lee LW, Wilson EM. The FXXLF motif mediates androgen receptor-specific interactions with coregulators. J Biol Chem. 2002b;277:10226–10235. doi: 10.1074/jbc.M111975200. [DOI] [PubMed] [Google Scholar]
- Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- Hendriksen PJ, Dits NF, Kokame K, Veldhoven A, van Weerden WM, Bangma CH, Trapman J, Jenster G. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006;66:5012–5020. doi: 10.1158/0008-5472.CAN-05-3082. [DOI] [PubMed] [Google Scholar]
- Hodgson MC, Astapova I, Cheng S, Lee LJ, Verhoeven MC, Choi E, Balk SP, Hollenberg AN. The Androgen Receptor Recruits Nuclear Receptor CoRepressor (N-CoR) in the Presence of Mifepristone via Its N and C Termini Revealing a Novel Molecular Mechanism for Androgen Receptor Antagonists. J Biol Chem. 2005;280:6511–6519. doi: 10.1074/jbc.M408972200. [DOI] [PubMed] [Google Scholar]
- Hodgson MC, Astapova I, Hollenberg AN, Balk SP. Activity of Androgen Receptor Antagonist Bicalutamide in Prostate Cancer Cells Is Independent of NCoR and SMRT Corepressors. Cancer Res. 2007;67:8388–8395. doi: 10.1158/0008-5472.CAN-07-0617. [DOI] [PubMed] [Google Scholar]
- Hodgson MC, Shen HC, Hollenberg AN, Balk SP. Structural basis for nuclear receptor corepressor recruitment by antagonist-liganded androgen receptor. Mol Cancer Ther. 2008;7:3187–3194. doi: 10.1158/1535-7163.MCT-08-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, Ryan C, Smith S, Scher H, Scardino P, Reuter V, Gerald WL. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao PW, Lin DL, Nakao R, Chang C. The linkage of Kennedy’s neuron disease to ARA24, the first identified androgen receptor polyglutamine region-associated coactivator. J Biol Chem. 1999;274:20229–20234. doi: 10.1074/jbc.274.29.20229. [DOI] [PubMed] [Google Scholar]
- Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. Recognition and Accommodation at the Androgen Receptor Coactivator Binding Interface. PLoS Biol. 2004;2:E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RA, Ma H, Yu MC, Ross RK, Stallcup MR, Coetzee GA. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet. 2000;9:267–274. doi: 10.1093/hmg/9.2.267. [DOI] [PubMed] [Google Scholar]
- Jackson TA, Richer JK, Bain DL, Takimoto GS, Tung L, Horwitz KB. The partial agonist activity of antagonist-occupied steroid receptors is controlled by a novel hinge domain-binding coactivator L7/SPA and the corepressors N-CoR or SMRT. Mol Endocrinol. 1997;11:693–705. doi: 10.1210/mend.11.6.0004. [DOI] [PubMed] [Google Scholar]
- Jenster G, van der Korput HA, Trapman J, Brinkmann AO. Identification of two transcription activation units in the N-terminal domain of the human androgen receptor. J Biol Chem. 1995;270:7341–7346. doi: 10.1074/jbc.270.13.7341. [DOI] [PubMed] [Google Scholar]
- Jenster G, van der Korput JA, Trapman J, Brinkmann AO. Functional domains of the human androgen receptor. J Steroid Biochem Mol Biol. 1992;41:671–675. doi: 10.1016/0960-0760(92)90402-5. [DOI] [PubMed] [Google Scholar]
- Jepsen K, Hermanson O, Onami TM, Gleiberman AS, Lunyak V, McEvilly RJ, Kurokawa R, Kumar V, Liu F, Seto E, Hedrick SM, Mandel G, Glass CK, Rose DW, Rosenfeld MG. Combinatorial roles of the nuclear receptor corepressor in transcription and development. Cell. 2000;102:753–763. doi: 10.1016/s0092-8674(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Jia L, Berman BP, Jariwala U, Yan X, Cogan JP, Walters A, Chen T, Buchanan G, Frenkel B, Coetzee GA. Genomic androgen receptor-occupied regions with different functions, defined by histone acetylation, coregulators and transcriptional capacity. PLoS ONE. 2008;3:e3645. doi: 10.1371/journal.pone.0003645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laudet V. Evolution of the nuclear receptor superfamily: early diversification from an ancestral orphan receptor. J Mol Endocrinol. 1997;19:207–226. doi: 10.1677/jme.0.0190207. [DOI] [PubMed] [Google Scholar]
- Liao G, Chen L-Y, Zhang A, Godavarthy A, Xia F, Ghosh JC, Li H, Chen JD. Regulation of Androgen Receptor Activity by the Nuclear Receptor Corepressor SMRT. J Biol Chem. 2003;278:5052–5061. doi: 10.1074/jbc.M206374200. [DOI] [PubMed] [Google Scholar]
- List HJ, Smith CL, Rodriguez O, Danielsen M, Riegel AT. Inhibition of histone deacetylation augments dihydrotestosterone induction of androgen receptor levels: an explanation for trichostatin A effects on androgen-induced chromatin remodeling and transcription of the mouse mammary tumor virus promoter. Exp Cell Res. 1999;252:471–478. doi: 10.1006/excr.1999.4638. [DOI] [PubMed] [Google Scholar]
- Ma H, Hong H, Huang SM, Irvine RA, Webb P, Kushner PJ, Coetzee GA, Stallcup MR. Multiple signal input and output domains of the 160-kilodalton nuclear receptor coactivator proteins. Mol Cell Biol. 1999;19:6164–6173. doi: 10.1128/mcb.19.9.6164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan I. Molecular mechanisms of androgen receptor-mediated gene regulation: structure-function analysis of the AF-1 domain. Endocr Relat Cancer. 2004;11:281–293. doi: 10.1677/erc.0.0110281. [DOI] [PubMed] [Google Scholar]
- McEwan IJ. Nuclear receptors: one big family. Methods Mol Biol. 2009;505:3–18. doi: 10.1007/978-1-60327-575-0_1. [DOI] [PubMed] [Google Scholar]
- McEwan IJ, Lavery D, Fischer K, Watt K. Natural disordered sequences in the amino terminal domain of nuclear receptors: lessons from the androgen and glucocorticoid receptors. Nucl Recept Signal. 2007;5:e001. doi: 10.1621/nrs.05001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna NJ, O’Malley BW. From ligand to response: generating diversity in nuclear receptor coregulator function. J Steroid Biochem Mol Biol. 2000;74:351–356. doi: 10.1016/s0960-0760(00)00112-6. [DOI] [PubMed] [Google Scholar]
- Moilanen A, Rouleau N, Ikonen T, Palvimo JJ, Janne OA. The presence of a transcription activation function in the hormone-binding domain of androgen receptor is revealed by studies in yeast cells. FEBS Lett. 1997;412:355–358. doi: 10.1016/s0014-5793(97)00791-6. [DOI] [PubMed] [Google Scholar]
- Need EF, Scher HI, Peters AA, Moore NL, Cheong A, Ryan CJ, Wittert GA, Marshall VR, Tilley WD, Buchanan G. A novel androgen receptor amino terminal region reveals two classes of amino/carboxyl interaction-deficient variants with divergent capacity to activate responsive sites in chromatin. Endocrinology. 2009;150:2674–2682. doi: 10.1210/en.2008-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley BW, Qin J, Lanz RB. Cracking the coregulator codes. Curr Opin Cell Biol. 2008;20:310–315. doi: 10.1016/j.ceb.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozers MS, Marks BD, Gowda K, Kupcho KR, Ervin KM, Rosier TD, Qadir N, Eliason HC, Riddle SM, Shekhani MS. The Androgen Receptor T877A Mutant Recruits LXXLL and FXXLF Peptides Differently than Wild-Type Androgen Receptor in a Time-Resolved Fluorescence Resonance Energy Transfer Assay. Biochemistry. 2007;46:683–695. doi: 10.1021/bi061321b. [DOI] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–123. doi: 10.1038/nrg2736. [DOI] [PubMed] [Google Scholar]
- Peterson TJ, Karmakar S, Pace MC, Gao T, Smith CL. The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor alpha transcriptional activity. Mol Cell Biol. 2007;27:5933–5948. doi: 10.1128/MCB.00237-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissios P, Tzameli I, Kushner P, Moore DD. Dynamic stabilization of nuclear receptor ligand binding domains by hormone or corepressor binding. Mol Cell. 2000;6:245–253. doi: 10.1016/s1097-2765(00)00026-5. [DOI] [PubMed] [Google Scholar]
- Price DK, Chau CH, Till C, Goodman PJ, Baum CE, Ockers SB, English BC, Minasian L, Parnes HL, Hsing AW, Reichardt JK, Hoque A, Tangen CM, Kristal AR, Thompson IM, Figg WD. Androgen receptor CAG repeat length and association with prostate cancer risk: results from the prostate cancer prevention trial. J Urol. 2010;184:2297–2302. doi: 10.1016/j.juro.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajender S, Singh L, Thangaraj K. Phenotypic heterogeneity of mutations in androgen receptor gene. Asian J Androl. 2007;9:147–179. doi: 10.1111/j.1745-7262.2007.00250.x. [DOI] [PubMed] [Google Scholar]
- Robins DM, Albertelli MA, O’Mahony OA. Androgen receptor variants and prostate cancer in humanized AR mice. J Steroid Biochem Mol Biol. 2008;108:230–236. doi: 10.1016/j.jsbmb.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronacher K, Hadley K, Avenant C, Stubsrud E, Simons SS, Jr, Louw A, Hapgood JP. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299:219–231. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. 2004;11:459–476. doi: 10.1677/erc.1.00525. [DOI] [PubMed] [Google Scholar]
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Shen HC, Buchanan G, Butler LM, Prescott J, Henderson M, Tilley WD, Coetzee GA. GRIP1 mediates the interaction between the amino- and carboxyl-termini of the androgen receptor. Biol Chem. 2005;386:69–74. doi: 10.1515/BC.2005.009. [DOI] [PubMed] [Google Scholar]
- Smith CL, Nawaz Z, O’Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- Smith SA, McLaughlin LW. Probing contacts to the DNA backbone in the trp repressor-operator sequence-specific protein-nucleic acid complex using diastereomeric methylphosphonate analogues. Biochemistry. 1997;36:6046–6058. doi: 10.1021/bi9700781. [DOI] [PubMed] [Google Scholar]
- Stanya KJ, Kao HY. New insights into the functions and regulation of the transcriptional corepressors SMRT and N-CoR. Cell Div. 2009;4:7. doi: 10.1186/1747-1028-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trtkova K, Paskova L, Matijescukova N, Kolar Z. Formation of AR-SMRT binding in prostate cancer cells treated with natural histone deacetylase inhibitor. Cancer Biomark. 2010;7:79–90. doi: 10.3233/CBM-2010-0150. [DOI] [PubMed] [Google Scholar]
- van Royen ME, Cunha SM, Brink MC, Mattern KA, Nigg AL, Dubbink HJ, Verschure PJ, Trapman J, Houtsmuller AB. Compartmentalization of androgen receptor protein-protein interactions in living cells. J Cell Biol. 2007;177:63–72. doi: 10.1083/jcb.200609178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varlakhanova N, Snyder C, Jose S, Hahm JB, Privalsky ML. Estrogen receptors recruit SMRT and N-CoR corepressors through newly recognized contacts between the corepressor N terminus and the receptor DNA binding domain. Mol Cell Biol. 2010;30:1434–1445. doi: 10.1128/MCB.01002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, Trapman J, Brinkmann AO, Mulder E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J Steroid Biochem Mol Biol. 1992;41:665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- Veldscholte J, Voorhorst-Ogink MM, Bolt-de Vries J, van Rooij HC, Trapman J, Mulder E. Unusual specificity of the androgen receptor in the human prostate tumor cell line LNCaP: high affinity for progestagenic and estrogenic steroids. Biochim Biophys Acta. 1990;1052:187–194. doi: 10.1016/0167-4889(90)90075-o. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, Chen Z, Beroukhim R, Wang H, Lupien M, Wu T, Regan MM, Meyer CA, Carroll JS, Manrai AK, Janne OA, Balk SP, Mehra R, Han B, Chinnaiyan AM, Rubin MA, True L, Fiorentino M, Fiore C, Loda M, Kantoff PW, Liu XS, Brown M. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf IM, Heitzer MD, Grubisha M, DeFranco DB. Coactivators and nuclear receptor transactivation. J Cell Biochem. 2008;104:1580–1586. doi: 10.1002/jcb.21755. [DOI] [PubMed] [Google Scholar]
- Yoon H-G, Wong J. The Corepressors Silencing Mediator of Retinoid and Thyroid Hormone Receptor and Nuclear Receptor Corepressor Are Involved in Agonist- and Antagonist-Regulated Transcription by Androgen Receptor. Mol Endocrinol. 2006;20:1048–1060. doi: 10.1210/me.2005-0324. [DOI] [PubMed] [Google Scholar]
- Zhang X, Jeyakumar M, Petukhov S, Bagchi MK. A nuclear receptor corepressor modulates transcriptional activity of antagonist-occupied steroid hormone receptor. Mol Endocrinol. 1998;12:513–524. doi: 10.1210/mend.12.4.0089. [DOI] [PubMed] [Google Scholar]