

Abstract
The subunit composition of multimeric protein complexes is critical in determining their trafficking and functional properties. Despite there being multiple techniques to investigate the trafficking events of individual subunits there are currently limited means to monitor the trafficking properties of heteromeric protein complexes. Here, we combine surface biotinylation with co-immunoprecipitation to monitor the cell surface expression of native, heteromeric AMPA receptor complexes. Using this method, we demonstrate that the surface levels of GluR1/2 and GluR2/3 complexes are reduced following NMDA-evoked long-term depression (NMDA-LTD) in acute hippocampal slices. Finally, we discuss how this method can be adapted to monitor the cell surface expression of other heteromeric protein complexes.
Keywords: AMPA receptor, Heteromeric, Trafficking, Biotinylation, LTD
1. Introduction
The subunit composition of ionotropic receptors dictates their functional and trafficking characteristics (Palmer et al., 2005). α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) are tetrameric, ionotropic glutamate receptors which assemble as a symmetrical dimer of dimers (Mansour et al., 2001). There are four AMPAR subunits (GluR1-4) which display differential expression in different brain regions and exist in multiple splice and edited isoforms (Dingledine et al., 1999). AMPARs are extensively studied in the hippocampus and cerebellum due to their central role in synaptic plasticity (Cull-Candy et al., 2006). In the CA1/CA2 region of rat hippocampus, the majority of AMPARs comprise GluR2 (Ozawa and Iino, 1993) with either GluR1 (GluR1/2) or GluR3 (GluR2/3) (Wenthold et al., 1996). By contrast, at cerebellar stellate cell synapses AMPARs are predominantly GluR2-lacking (Liu and Cull-Candy, 2000).
The subunit composition of AMPARs dramatically affects their function. GluR2 containing AMPARs have low Ca2+ permeability so consequently have outwardly rectifying I–V relations, whereas GluR2-lacking AMPARs have high Ca2+ permeability and are inwardly rectifying (Burnashev et al., 1992; Hollmann et al., 1991). Subunit composition also appears to affect AMPAR trafficking properties. GluR2-lacking receptors are driven into synapses during the early stages of hippocampal LTP, whereas GluR2-containing receptors replace these after 25 min (Plant et al., 2006). Differential movement of GluR2-lacking and GluR2-containing AMPARs has also been reported following ischemic insult. GluR2-lacking AMPARs are directed towards synapses following oxygen-glucose deprivation, whereas GluR2-containing AMPARs are removed from synaptic sites under the same conditions (Liu et al., 2006).
Recently, we have demonstrated that the surface expression of GluR2 and GluR3, but not GluR1, is reduced following NMDA-LTD induction in acute hippocampal slices. From these data we hypothesised that GluR1/2 and GluR2/3 containing AMPARs are differentially trafficked during LTD (Holman et al., 2006). To test this hypothesis further a method was required to monitor changes in the surface levels of heteromeric AMPAR complexes. In this report we develop a method that combines surface biotinylation with co-immunoprecipitation (co-IP) to assess the surface expression of heteromeric AMPARs in dispersed neurons and acute hippocampal slices. Using this method we determine the surface expression of GluR1/2 complexes under basal conditions and detect changes in the surface levels of GluR1/2 and GluR2/3 complexes following NMDA-LTD induction.
2. Materials and methods
2.1. Dispersed neuronal culture and drug treatment
High-density cerebro-cortical cultures were prepared from embryonic day 18 Sprague–Dawley rats. Cortex was dissected in cold Hank’s buffered salt solution (Gibco) and then dissociated using trypsin for 10 min at 37 °C. Neurons were then plated onto poly-l-lysine-coated 6 cm dishes (1 × 106 per 6 cm dish) and maintained at 37 °C, 5% CO2 as described previously (Terashima et al., 2004). After 14 days in vitro (DIV), neurons were treated with 25 μM NMDA for 3 min or left untreated. Cultures were then incubated with normal culture medium for 10 min before being immediately placed on ice to prevent further receptor trafficking.
2.2. Biotinylation and lysis of cultures
Neurons were washed twice with ice-cold PBS and incubated with Sulfo-NHS–SS–biotin (Pierce; 0.25 mg/ml in PBS) for 15 min on ice. Neurons were then washed twice with 50 mM NH4Cl and twice with PBS before being scraped into ice-cold lysis buffer (150 mM NaCl, 20 mM N-2-hydroethylpiperazin-N′-2-ethanesulfonic acid (HEPES), 1% Triton-X-100, 0.1% SDS, 2 mM EDTA, pH 7.4) containing protease inhibitors (protease inhibitor cocktail with EDTA, 1 in 50; Roche). Samples were then sonicated and placed on a head-over-head shaker for 2 h. Samples were then centrifuged at 100,000 × g for 40 min and the pellets were discarded. The protein concentration of the resulting supernatant was determined using a BCA kit (Pierce).
2.3. Acute hippocampal slice preparation
Hippocampal slices (400 μm) were prepared from P21 to P23 male Wistar rats and immediately placed in ice-cold artificial cerebrospinal fluid (ACSF; composition in mM: 124NaCl; 3KCl; 26NaHCO3; 1.25NaH2PO4; 2CaCl2; 1MgSO4; 10D-glucose; saturated with 95% O2 and 5% CO2). After removing the CA3 region, slices were transferred to a submersion storage chamber where they were maintained in ACSF for 1 h at room temperature.
2.4. Slice treatment for biochemical analysis
To compare multiple NMDA- and ACSF (control)-treated slices simultaneously we used a two-chamber perfusion system. Before treating the slices the cortex was removed leaving the CA1/CA2 region. Alternate slices were then placed into one of two groups, those that would be treated with NMDA and those that would be treated with ACSF. These slices (four to seven slices per group) were then equilibrated at room temperature for 1 h before being placed into the chambers of the two-chamber perfusion system (maintained at 28 °C). Following ACSF perfusion for 40 min, one chamber was perfused with ACSF plus 20 μM NMDA for 5 min, while the other chamber was perfused with ACSF alone. Both sets of slices were then perfused with ACSF for a further 90 min prior to biochemical analysis.
2.5. Slice biotinylation, homogenisation and lysis
Previously it has been shown that acute hippocampal slices can be effectively biotinylated using Sulfo-NHS–SS–biotin. Moreover, it has been demonstrated that biotin can reach all layers of a 400 μm thick slice without labelling intracellular proteins (Holman et al., 2006; Thomas-Crusells et al., 2003). Given these details we were confident that the surface co-IP assay would be suitable for monitoring the surface expression of heteromeric protein complexes in acute hippocampal slices.
Slices were washed once with ice-cold ACSF (5 min) and then incubated with Sulfo-NHS–SS–biotin (Pierce; 0.5 mg/ml in ACSF) for 30 min on ice. Excess biotin was removed by two brief washes with 50 mM NH4Cl (in ACSF) and two ACSF washes. Slices were then homogenised in 1 ml of homogenisation buffer (320 mM Sucrose; 10 mM Tris; pH 7.4) and centrifuged at 1000 × g for 5 min to remove nuclear material and cell debris. Post-nuclear supernatants were centrifuged at 100,000 × g for 1 h and supernatants were discarded. The hippocampal cell membranes were resuspended in 1 ml of lysis buffer, sonicated and placed on a head-over-head shaker for 2 h. Samples were then centrifuged at 100,000 × g for 40 min and the pellets were discarded. The protein concentration of the resulting supernatant was then determined as described above.
2.6. Streptavidin pull down
Streptavidin beads (40 μl; Sigma) were washed three times with lysis buffer. Lysed biotinylated samples were added to the beads (50 μg total protein), volumes were made up to 200 μl with lysis buffer and mixed on a head-over-head shaker for 4 h. Beads were then centrifuged at 800 × g. Loading buffer (4×; containing β-mercaptoethanol) was added to the supernatant to make a final volume of 266 μl. The beads were washed three times with lysis buffer, residual lysis buffer was removed and 133 μl of 2 × loading buffer was added. Samples were then heated at 90 °C for 5 min and beads were centrifuged at 800 × g. The bead supernatant was then diluted with 133 μl of lysis buffer and stored at −20 °C until further use.
2.7. Immunoprecipitation
Protein G beads (20 μl; Sigma) were washed three times with lysis buffer and centrifuged at 800 × g. Lysis buffer (1 ml) containing 2 μg of polyclonal anti-GluR2 (Chemicon) or polyclonal anti-Myc (Upstate) was added to the beads and incubated for 2 h on a head-over-head shaker. Excess antibody was removed by two lysis buffer washing steps. Lysed sample (50–200 μg) was added to the beads and incubated for 6 hr on a head-over-head shaker. Beads were centrifuged at 800 × g and the supernatant was used for a second round of GluR2 immunoprecipitation. This additional step was performed to maximise the IP efficiency. After the second round, beads were centrifuged and loading buffer (containing β-mercaptoethanol) was added to the beads and supernatant as described for the streptavidin pull down.
2.8. Surface co-immunoprecipitation assay
GluR2-containing AMPARs were immunoprecipitated from biotinylated neuronal samples as described above (see Section 2.7). After washing the beads three times with lysis buffer, residual buffer was removed and bead–IP–AMPAR interactions were disrupted by incubating beads with 50 μl of 1% SDS for 80 min at 37 °C. Beads were centrifuged at 800 g and the supernatant was mixed with 150 μl of lysis buffer excluding SDS (the final concentration of SDS was 0.25%). Samples were then used for streptavidin pull downs as described above.
2.9. Quantitative immunoblotting
Proteins were resolved by SDS-PAGE and immunoblotting was performed using a rabbit polyclonal antibody to GluR1 (Upstate; 0.6 μg/ml) and mouse monoclonal antibodies to PSD-95 (Upstate; 0.5 μg/ml), GluR2 (Chemicon; 1 μg/ml), GluR3 (Chemicon; 1 μg/ml) and β-tubulin (Sigma; 0.5 μg/ml). Quantitative densitometric analysis was performed using NIH Image J.
2.10. Calculation of AMPAR subunit surface expression
Increasing amounts (10, 25 and 50 μg) of total protein were resolved alongside the streptavidin bead bound and unbound fractions. Following Western blot analysis, optical density values were obtained (within a linear range) for each of the bands representing the input and bound fractions. By plotting these values the percentage surface expression of GluR1 and GluR2 was determined by normalising the bound optical band density value to the input band density values.
2.11. Calculation of AMPAR complex surface expression
Since the surface co-IP assay has multiple steps we reasoned that it would be more appropriate to calculate the surface expression of GluR1/2 complexes by using the bound and the unbound fractions rather than the input and bound fractions. Varying amounts of the bound (100 and 50 μl) and unbound (100, 50 and 25 μl) fractions were resolved and optical density values were obtained for each of the bands. The surface expression of each AMPAR complex was determined by normalising the surface fraction band density values to the unbound fraction band density values.
3. Results and discussion
By combining surface biotinylation with co-immunoprecipitation we devised a method to monitor the cell surface expression of heteromeric protein complexes. We optimised this assay to monitor the surface expression of GluR1/2 or GluR2/3 containing AMPAR complexes. The method involved: (1) biotinylation of surface complexes; (2) neuronal lysis and isolation of GluR2 containing complexes by immunoprecipitation; (3) disruption and release of bead-antibody-subunit complexes; (4) recovery of biotin labelled GluR1 or GluR3 by streptavidin pull down (see Fig. 1 for schematic of assay).
Fig. 1.
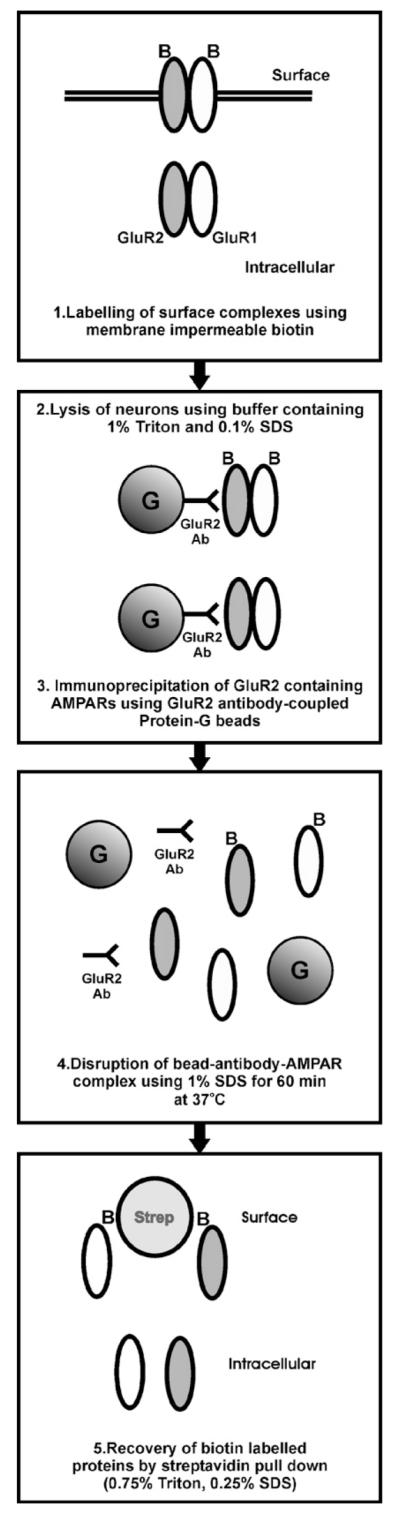
Schematic of surface co-immunoprecipitation protocol. Neuronal cultures or acute hippocampal slices were biotinylated (panel 1). After cell lysis, GluR2 containing complexes were isolated by GluR2 immunoprecipitation using GluR2 antibody-coupled protein-G beads (panel 2). Bead–antibody–AMPAR interactions were disrupted by 1% SDS (panel 3) and biotinylated subunits were recovered by streptavidin pull down (panel 4).
3.1. Validation of immunoprecipitation step
First, we assessed the effectiveness of our solubilisation protocol. The majority of GluR1, GluR2, GluR3 and PSD-95 were present in the soluble fraction (See Fig. 2A) indicating that both synaptic and nonsynaptic proteins were effectively solubilised. Next, we validated the GluR2 immunoprecipitation protocol by performing IPs using a polyclonal GluR2 antibody or a control myc antibody. As expected, GluR2 was detected in the bound fraction of the GluR2 IP but not the myc IP (Fig. 2B). Next, we identified an input-antibody ratio (50 μg of neuronal sample for 2 μg of antibody) where the GluR2 IP isolated ~95% of the GluR2 whole-cell pool. Using this ratio, we determined that ~79% of the GluR1 whole-cell pool was isolated by the GluR2 IP (Fig. 2C). This finding suggests that the majority of GluR1 is assembled with GluR2 in dispersed cerebro-cortical cultures, which is entirely consistent with previous estimates using tissue from the CA1/CA2 region of adult rat hippocampus (Wenthold et al., 1996).
Fig. 2.

Validation of neuronal lysis and GluR2 co-Immunoprecipitation protocol. DIV 14–18 cerebro-cortical cultures were lysed using a buffer containing 1% Triton and 0.1% SDS. The majority of GluR1, GluR2, GluR3 and PSD-95 were present in the soluble fraction (A). GluR2 containing AMPARs were immunoprecipitated with a rabbit polyclonal GluR2 antibody (B). A rabbit polyclonal myc antibody was unable to IP GluR2 under the same conditions (B). The GluR2 IP was ~95% efficient when using a 2:50 μg antibody-input ratio. Under these conditions, the GluR2 IP isolated ~79% of GluR1 (C). GluR2 containing AMPAR were eluted from the beads using 1% SDS as well as the heavy chain of the IP IgG (D). No AMPARs were eluted from the beads under lysis buffer control conditions (D).
Having established that the GluR2 IP reliably pulls down GluR2 containing AMPARs, a method was required to elute these complexes from the protein-G beads. We investigated multiple IP disruption conditions (pH 1.8 and 2.5, 0.1% SDS, 1% SDS and various temperatures) and identified that the most effective was 1% SDS for 60 min at 37 °C (data not shown). Under these conditions the majority of GluR2 was eluted from the beads as well as the heavy chain of the GluR2 antibody (Fig. 2D).
3.2. The surface expression of GluR1/2 complexes under basal conditions
Before calculating the surface expression of GluR1/2 complexes we determined the surface expression of the individual subunits. Consistent with previous reports (Greger et al., 2002; Hall and Soderling, 1997), the surface expression of GluR1 was higher that that of GluR2 in our cerebro-cortical cultures (Fig. 3A and B; GluR1 59 ± 9%; GluR2 34 ± 4%). Importantly, the intracellular protein β-tubulin was exclusively in the unbound fraction (Fig. 3A) indicating that only surface proteins were labelled.
Fig. 3.

Surface expression of GluR1/2 complexes in neuronal cultures. DIV 14–18 cerebro-cortical cultures were biotinylated and lysed. The surface expression of GluR1 and GluR2 was determined by quantitative immunoblotting (see Section 2). (A) Representative blots for GluR1, GluR2 and β-tubulin. (B) Proportion of AMPARs surface expressed (mean ± S.E.M. of three independent experiments). The surface expression of GluR1/2 complexes was determined by surface co-IP (see Section 2). (C) Representative blots for GluR1 and GluR2. (D) Proportion of GluR1/2 complexes surface expressed. The surface expression of GluR2 which was immunoprecipitated by the GluR2 IP was also determined (mean ± S.E.M. of three independent experiments).
Similar to that for GluR1 and GluR2 alone, the surface expression of GluR1/2 complexes was higher than the surface levels of GluR2 isolated by the GluR2 IP (Fig. 3D; GluR1/2 54 ± 2 %; GluR2 isolated by the GluR2 IP 38 ± 5%). This is consistent with a previous proposal that GluR1 promotes the exit of GluR2 from the endoplasmic reticulum (Greger et al., 2003).
3.3. Application of surface co-IP assay: NMDA-LTD induction causes surface downregulation of GluR1/2 and GluR2/3 complexes
Before monitoring the cell surface expression of GluR1/2 and GluR2/3 complexes following NMDA-LTD induction in acute hippocampal slices we assessed the surface expression of GluR1/2 complexes following transient NMDA application in cerebro-cortical cultures. Consistent with a previous report, showing reduced surface levels of GluR1 and GluR2 following NMDA application (Hanley and Henley, 2005), the surface expression of GluR1/2 complexes was markedly lower in the NMDA treated cultures (3 min, 25 μM NMDA) compared to control cultures (Fig. 4a and b). This was an important result as it confirmed that the surface co-IP assay could be used to monitor activity-induced changes in heteromeric AMPAR cell surface expression.
Fig. 4.

Down-regulation of surface GluR1/2 and GluR2/3 complexes 90 min following NMDA-LTD induction. DIV 14–18 cerebro-cortical cultures (A and B) or acute hippocampal slices from P21 to 23 rats (C and D) were treated with NMDA, biotinylated and lysed (see Section 2). GluR2 IPs were performed using protein matched samples from NMDA and control treated cultures/slices. GluR2 complexes were eluted from the beads using 1% SDS and the surface receptors were isolated using streptavidin pull downs. The bound fraction of each streptavidin pull down was then resolved by SDS-PAGE and blots were probed with antibodies specific for GluR1 or GluR3. (A and C) Representative blots for GluR1 or GluR3 (IP: antibody used for immunoprecipitation; IB: antibody used for immunoblotting). (B and D) Surface expression of receptor complexes (data represent the mean ± S.E.M. of three experiments (*P < 0.05)).
Previously, we have described that the surface levels of GluR2 and GluR3 are reduced, while that of GluR1 is unaltered, following NMDA-LTD induction (Holman et al., 2006). We were therefore interested to use the surface co-IP assay to monitor any changes in GluR1/2 and GluR2/3 surface expression using the same cell lysates as those prepared for the previous study. As expected, the surface expression of GluR2/3 complexes was markedly lower in the NMDA-treated slices compared to control (Fig. 4). This finding suggests that GluR2/3 complexes are redistributed from surface to intracellular locations during LTD. This is consistent with the observation that exogenous GluR2 homomers, which are thought to have trafficking properties similar to that of endogenous GluR2/3 heteromers, are removed from synapses following low frequency stimulation (LFS)-induced LTD in organotypic culture (Seidenman et al., 2003). Taken together, these data suggest that prolonged loss of surface GluR2/3 complexes may contribute to LTD expression in acute hippocampal slices.
Interestingly, the surface expression of GluR1/2 complexes was also significantly lower in the NMDA treated slices compared to control (Fig. 4c and d). This was an intriguing result given our previous observation that GluR1 surface levels are unchanged 90 min following NMDA-LTD induction (Holman et al., 2006). As ~80% of GluR1 is in a complex with GluR2/3 in the CA1/CA2 region of rat hippocampus (Wenthold et al., 1996), we propose that GluR1 homomers or GluR2-lacking GluR1 heteromers (GluR1/3 and GluR1/4) are inserted to maintain surface GluR1 levels 90 min following NMDA-LTD induction. Another, less energetically favourable, possibility is that surface GluR1/2 complexes disassemble following NMDAR activation allowing GluR2 subunits to be selectively removed from the surface membrane leaving GluR1 surface levels unaffected.
3.4. Other applications of surface co-IP assay
In this study we developed a surface co-IP assay to monitor NMDA-induced changes in GluR1/2 and GluR2/3 surface expression. Other potential applications of this assay include monitoring the cell surface expression of GluR2-lacking AMPAR complexes (GluR1/3, GluR1/4 and GluR3/4) and assessing the cell surface expression of all heteromeric AMPAR complexes following LTP induction (Kopec et al., 2006) and ischemic insult (Liu et al., 2006).
The surface co-IP assay could also be adapted to study the surface expression of other heteromeric protein complexes. To achieve this, an excellent subunit-specific antibody is required for the immunoprecipitation procedure. Once this is identified the next step is to determine an input:antibody ratio where the IP is at least 90% efficient. Using this approach, it should then be possible to assess the surface expression of your heteromeric protein complex, under basal and stimulated conditions, by quantitative immunoblotting. It is noteworthy, however, that the heavy chain of the IP antibody will be present in the eluate after the 1% SDS elution step. This may be an issue for those groups wishing to monitor the intracellular levels of small heteromeric protein complexes (~50 kDa), since proteins of this size will resolve at the same level as the heavy chain of the IP antibody. This problem can be easily overcome by covalently cross-linking the IP antibody to the protein-G beads. The absence of the heavy chain in the eluate should then enable easy detection of intracellular proteins of 50 kDa by Western blotting.
Finally, a potential refinement of this approach could be used to study the surface-synaptic localisation of these receptors. This could be achieved by isolating post-synaptic density (PSD) fractions (Phillips et al., 2001) from drug-treated, biotinylated slices (>10 slices would be required per group). These samples could then be used to perform surface co-immunoprecipitation experiments from which useful information about synaptic complex composition could be acquired.
In conclusion, we have developed an assay to monitor the cell surface expression of native heteromeric protein complexes that has potential applications for a wide range of studies. Using this assay we determined the surface expression of GluR1/2 complexes under basal conditions and detected reductions in the surface levels of GluR1/2 and GluR2/3 complexes following NMDA-LTD induction in acute hippocampal slices.
Acknowledgements
DH is a post-doctoral fellow supported by the EU (GRIP-PANT, PL 005320). We also thank the MRC, the Wellcome Trust and the EU (ENINET LSHM-CT-2005-019063) for financial assistance.
References
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–98. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca(2+)-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–97. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Greger IH, Khatri L, Ziff EB. RNA editing at arg607 controls AMPA receptor exit from the endoplasmic reticulum. Neuron. 2002;34:759–72. doi: 10.1016/s0896-6273(02)00693-1. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40:763–74. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- Hall RA, Soderling TR. Quantitation of AMPA receptor surface expression in cultured hippocampal neurons. Neuroscience. 1997;78:361–71. doi: 10.1016/s0306-4522(96)00525-8. [DOI] [PubMed] [Google Scholar]
- Hanley JG, Henley JM. PICK1 is a calcium-sensor for NMDA-induced AMPA receptor trafficking. EMBO J. 2005;24:3266–78. doi: 10.1038/sj.emboj.7600801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA— gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–3. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Holman D, Feligioni M, Henley JM. Differential redistribution of native AMPA receptor complexes following LTD induction in acute hippocampal slices. Neuropharmacology. 2006 doi: 10.1016/j.neuropharm.2006.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–9. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, et al. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–19. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–8. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Mansour M, Nagarajan N, Nehring RB, Clements JD, Rosenmund C. Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron. 2001;32:841–53. doi: 10.1016/s0896-6273(01)00520-7. [DOI] [PubMed] [Google Scholar]
- Ozawa S, Iino M. Two distinct types of AMPA responses in cultured rat hipocampal neurons. Neurosci Lett. 1993;155:187–90. doi: 10.1016/0304-3940(93)90704-o. [DOI] [PubMed] [Google Scholar]
- Palmer CL, Cotton L, Henley JM. The molecular pharmacology and cell biology of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Pharmacol Rev. 2005;57:253–77. doi: 10.1124/pr.57.2.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, et al. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, et al. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–4. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Seidenman KJ, Steinberg JP, Huganir R, Malinow R. Glutamate receptor subunit 2 Serine 880 phosphorylation modulates synaptic transmission and mediates plasticity in CA1 pyramidal cells. J Neurosci. 2003;23:9220–8. doi: 10.1523/JNEUROSCI.23-27-09220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima A, Cotton L, Dev KK, Meyer G, Zaman S, Duprat F, et al. Regulation of synaptic strength and AMPA receptor subunit composition by PICK1. J Neurosci. 2004;24:5381–90. doi: 10.1523/JNEUROSCI.4378-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Crusells J, Vieira A, Saarma M, Rivera C. A novel method for monitoring surface membrane trafficking on hippocampal acute slice preparation. J Neurosci Meth. 2003;125:159–66. doi: 10.1016/s0165-0270(03)00050-5. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Petralia RS, Blahos J, II, Niedzielski AS. Evidence for multiple AMPA receptor complexes in hippocampal CA1/CA2 neurons. J Neurosci. 1996;16:1982–9. doi: 10.1523/JNEUROSCI.16-06-01982.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]