

Abstract
Methuosis is a novel caspase-independent form of cell death in which massive accumulation of vacuoles derived from macropinosomes ultimately causes cells to detach from the substratum and rupture. We recently described a chalcone-like compound, 3-(2-methyl-1H indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (i.e. MIPP), which can induce methuosis in glioblastoma and other types of cancer cells. Herein we describe the synthesis and structure-activity relationships of a directed library of related compounds, providing insights into the contributions of the two aryl ring systems and highlighting a potent derivative, 3-(5-methoxy, 2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (i.e. MOMIPP) that can induce methuosis at low μM concentrations. We have also generated biologically active azide derivatives that may be useful for future studies aimed at identifying the protein targets of MOMIPP by photoaffinity labeling techniques. The potential significance of these studies is underscored by the finding that MOMIPP effectively reduces the growth and viability of temozolomide-resistant glioblastoma and doxorubicin-resistant breast cancer cells. Thus, it may serve as a prototype for drugs that could be used to trigger death by methuosis in cancers that are resistant to conventional forms of cell death (e.g. apoptosis).
INTRODUCTION
Despite recent advances in developing therapeutic agents that target regulatory pathways unique to certain types of cancer cells, the mainstays for post-surgical adjuvant therapy of many tumors remain radiation and DNA alkylating agents. An example is the highly malignant brain tumor, glioblastoma multiforme (GBM), where the current standard of care involves surgery when possible, followed by adjuvant therapy with radiation and oral temozolomide (TMZ). 1 A limitation of the latter approaches is that they work by damaging DNA, triggering the intrinsic apoptotic pathway. 2 Since GBM cells typically harbor mutations in tumor suppressor genes (e.g., PTEN, p53, pRB), they are relatively insensitive to apoptotic stimuli. 3, 4 Moreover, glioblastoma cells develop resistance to alkylating agents by increasing their capacity to repair DNA lesions. 5 We believe that it may be possible to develop new approaches to treat such drug-resistant cancers through the induction of alternative non-apoptotic forms of cell death, which do not depend on DNA damage as a trigger. Toward this end, we have defined a unique form of cell death termed ‘methuosis’. 6, 7
The hallmark of methuosis is the displacement of much of the cellular cytoplasmic space by vacuoles derived from macropinosomes. 6 The latter are formed when membrane ruffles (lamellipodia) enclose pockets of extracellular fluid and are internalized. 8, 9 In methuosis, impairment of the recycling and lysosome-directed trafficking of macropinocytotic vesicles locks them in an intermediate stage where they fuse to form progressively larger vacuoles. This eventually causes a decrease in metabolic activity and rupture of the cell membrane. 6 Death is considered to be non-apoptotic, as is it is not accompanied by nuclear chromatin condensation, cell blebbing, or nucleosomal DNA fragmentation. Methuosis is also caspase-independent, as it cannot be prevented by broad-spectrum caspase inhibitors such as zVAD-fmk. 6
Methuosis was initially characterized in GBM cells, where this form of cell death was triggered by ectopic expression of activated Ras and Rac GTPases. However, the potential for exploiting this non-conventional cell death pathway to kill cancer cells that are refractory to apoptosis depends on the identification of molecules with drug-like properties that can induce methuosis. We recently described a prototype chalcone-related compound that can induce cell death with the hallmarks of methuosis in both TMZ-resistant and non-resistant GBM cells, as well as other cancer cell lines derived from breast, colon and pancreas. 10 Herein we report synthesis and structure-activity relationship (SAR) studies of a directed library of related compounds leading to: 1) the definition of key features required for methuosis-inducing activity; 2) the identification of a derivative with improved biological activity; and 3) the development of an active azide analog that may be suitable for use as a photoaffinity probe in future target identification efforts.
RESULTS
Structure-Activity Relationships
As we began to seek drug-like small molecules that could trigger methuosis, we noted a report from Kirchhausen and colleagues 11 in which they described a molecule termed vacuolin-1, along with several other triazine-based compounds, that were capable of inhibiting Ca++-dependent lysosomal exocytosis. Although these compounds induced marked cellular vacuolization, they did not cause cell death. However, in the supplementary material included with this report, a structurally distinct vacuole-inducing compound captured our attention because of its resemblance to a class of molecules termed chalcones. The structure of this compound is depicted in Fig. 1 (compound 1). Chalcones consist of a 1,3-diphenyl- 2-propen-1-one framework that serves as a precursor for flavonoid natural products. However, the term chalcones has been applied more broadly to describe a number of synthetic derivatives built on this framework. 12 Several chalcones have been found to have significant anti-cancer activity, but have not been reported to induce vacuolization of cells. 12–14 This prompted us to ask whether compound 1 might represent a novel type of chalcone that can kill cancer cells by inducing methuosis. We found that compound 1 caused extensive vacuolization of glioblastoma cells within a few hours, and substantial loss of cell viability within 48-h. 10 Chemical database searches yielded additional compounds with >75% similarity to 1. Among these, compound 2 (Fig. 1) was selected for further study. It induced vacuoles that were larger and more numerous than those produced by compound 1. Compound 2 was assigned the acronym MIPP: i.e., 3-(2-methyl-1H indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one. Detailed evaluation of the biological effects of MIPP indicated that the form of cell death induced by this compound matched the profile of methuosis. 10 Furthermore, MIPP was effective in reducing the growth and viability of GBM cells that were highly resistant to TMZ. 10
Figure 1.

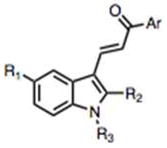
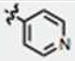
Structures of compounds 1 and 2, initially found to be active inducers of methuosis10 compared with commercially available analogs 3–8, that failed to induce the hallmarks of methuosis in culture U251 GBM cells.
Since concentrations of MIPP ≥10 μM were required to effectively induce methuosis, we began to assemble a directed library of MIPP-related compounds and conducted initial SAR comparisons with the goal of identifying analogs with improved potency. We began by comparing the methuosis-inducing activity of MIPP with several closely related compounds that were commercially available (compounds 3–8 in Fig. 1). The latter did not trigger cellular vacuolization when added to U251 GBM cells at a concentration of 10 μM. Examination of compounds 1–8 (Fig. 1) suggested that the specific relationship between an indole and a pyridinyl ring was likely to be playing a significant role in determining activity. Specifically, comparison of compound 1 (active) with compound 7 (inactive) demonstrated the importance of the pyridinyl ring, because replacement of the latter with a para-methoxy phenyl ring rendered the compound inactive. Therefore, an initial set of analogs was synthesized to investigate the influence of the position of the pyridine nitrogen on biological activity.
Analogs based on the α,β-unsaturated ketone core can be prepared by Claisen-Schmidt condensations between indole-3-carboxaldehydes and aryl ketones 15. Condensation of acetophenone or various acetyl-pyridines with indole-3-carboxaldehyde yielded compounds 9–12 (Scheme 1A). The activities of these compounds were compared to MIPP at a concentration of 10 μM using three criteria: 1) morphological vacuolization of live cells assessed by phase contrast microscopy at 24 and 48 h; 2) cell viability at a 48 h end point assessed by MTT assay, with fresh compound added after the first 24 h; and 3) colony forming assays (two week endpoint) performed on cells exposed to the compounds for 48 h. The results of this analysis (Table 1) indicated that a para-nitrogen orientation of the pyridine ring is a key feature required for activity. The meta and ortho analogs 10 & 11, as well as the acetophenone analog 9 were all relatively ineffective at inducing methuosis compared to MIPP. In contrast, removal of the 2- methyl from the indole ring of MIPP (compound 12) reduced but did not eliminate activity.
Scheme 1.

Synthesis of analogs of MIPP (compound 2). The specific substituents at R1, R2, R3 and Ar for each numbered compound are listed in the chart. For A–D, the specific conditions and reagents were as follows: (A) piperidine, CH3OH, reflux, 19 – 89%; (B) BBr3, CH2Cl2, – 78 °C, 61% and 95% for 15 and 21, respectively; (C) POCl3, DMF, 0 °C, 90%; piperidine, CH3OH, reflux, 89% ; (D) NaH, CH3I, DMF, RT, 82%.
Table 1.
Summary of SAR Studies Performed on MIPP (Compound 2) and Related Compounds Generated in Schemes 1 and 2.
 | |||||||
|---|---|---|---|---|---|---|---|
| cmpd # | R1 | R2 | R3 | Ar | Viability (MTT*) | Colony Formation* | Vacuoles? |
| 2 (MIPP) | H | CH3 | H |
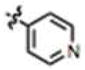
|
24 ± 3 | 8 ± 4 | yes |
| 9 | H | H | H |
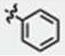
|
109 ± 7 | 76 ± 7 | no |
| 10 | H | H | H |
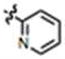
|
96 ± 9 | 53 ± 10 | no |
| 11 | H | H | H |
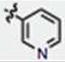
|
72 ± 6 | 71 ± 10 | no |
| 12 | H | H | H |
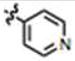
|
58 ± 7 | 22 ± 2 | yes |
| 13 | CH3O | H | H |
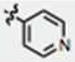
|
58 ± 2 | 2 ± 1 | yes |
| 14 |
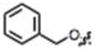
|
H | H |
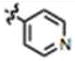
|
56 ± 4 | 8 ± 2 | yes |
| 15 | OH | H | H |
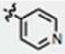
|
107 ± 22 | 72 ± 6 | no |
| 16 | CH3O | H | H |
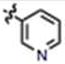
|
90 ± 8 | 99 ± 30 | no |
| 17 | CH3O | H | H |
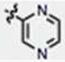
|
88 ± 8 | 96 ± 22 | no |
| 19 (MOMIPP) | CH3O | CH3 | H |
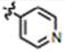
|
12± 2 | 0 | yes |
| 20 | CH3O | H | CH3 |
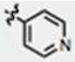
|
73 ± 13 | 52 ± 9 | yes |
| 21 | OH | CH3 | H |
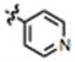
|
101 ± 13 | 43 ± 17 | yes |
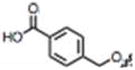
| 26 |
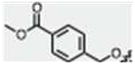
|
CH3 | H |
|
79 ± 2 | 76 ± 8 | no |
| 27 |
|
CH3 | H |
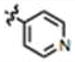
|
79 ± 9 | 71 ± 11 | no |
Results are expressed as percent of controls that received vehicle alone (DMSO). Values are the mean ± SD of quadruplicate (MTT) or triplicate (colony formation) determinations.
We next explored the consequences of functionalizing the 5-position of the indole ring using commercially available 5-methoxy and 5-benzyloxy indole-3-carboxaldehydes to prepare compounds 13 and 14, respectively. The 5-methoxy compound 13 in turn, was demethylated with BBr3 to afford the 5-OH compound 15 (Scheme 1B). As shown in Table 1, the activities of compounds 13 and 14 were similar to MIPP when compared by colony-forming assay and cell morphology, although they were not as cytotoxic in the short-term growth/viability assay. In contrast, the 5-OH substituent, compound 15, exhibited greatly reduced biological activity in all of the assays. To confirm that the location of the pyridine nitrogen was still crucial for activity of the 5-methoxy substituted compounds, analogs 16 and 17 were generated. Loss of activity confirmed the necessity of the para-nitrogen (Table 1).
Since comparisons of compounds 12–15 versus MIPP suggested that modifications at the 5- and 2-positions of the indole ring both affect activity, we generated a 2-methyl-5-methoxy analog starting from commercially available 2-methyl-5-methoxy-indole. Key intermediate 18 was synthesized by a Vilsmeier-Haack formylation of 2-methyl-5-methoxyindole, followed by coupling with 4-acetyl-pyridine to yield 19 (Scheme 1C). The inhibitory activity of this compound exceeded that of MIPP in both the MTT viability assay and the colony formation assay (Table 1). Alternatively, methylation of the indole-nitrogen of compound 13 with NaH/CH3I in DMF (Scheme 1D) to yield 20, produced a compound that induced some cytoplasmic vacuolization but had only modest effects on cell viability (Table 1). Thus, after comparing compounds 13, 19 and 20, it became clear that optimal activity is achieved when the 1- and 2- positions of the indole are occupied by H and methyl, respectively. The 5-OH compound 21, made by BBr3 demethylation of 19 (Scheme 1B), showed a marked reduction in activity compared to the 5-methoxy compound 19 (Table 1), consistent with the detrimental effect of the 5-OH previously observed in compound 15.
One of the limitations for using MIPP under physiological conditions is its sparing solubility in aqueous solutions. Therefore, we explored the effects of modifying the 5-position of the indole with a group that would add polarity at physiological pH. Because our previous SAR studies showed that there was some flexibility at the 5-position of the indole ring (compare compounds 13 and 14), we designed an analog of 14 that added charge and also included a methyl at the indole’s 2- position. We envisaged that the OH analog 21 could be alkylated with methyl 4-(bromomethyl)benzoate, which could then be hydrolyzed to its acid to provide a highly water soluble analog at pH 7.4. However, we were somewhat surprised that no appreciable amount of product could be produced by direct alkylation of 21. Multiple bases were screened with varying pKa’s, from K2CO3 and Cs2CO3, as well as triethylamine, tetramethylguanidine, DBU and NaH. In all reactions, alkylation at the pyridine nitrogen was observed. Stronger bases such as TMG and NaH, even using one equivalent, produced appreciable indole-N-alkylation as well as pyridine and indole-OH alkylation. The yield of singly alkylated product at the 5-indole position was negligible. Thus, a new route was designed by functionalizing the indole before introduction of the pyridine moiety (Scheme 2). Commercially available 2-methyl-5-methoxy-indole was demethylated with BBr3 to provide 22. Alkylation with 4-methyl-(bromomethyl)-benzoate under phase-transfer conditions afforded mono-O-alkylated product 23 16. After formylation with POCl3/DMF, intermediates 24 and 25 were independently prepared. Workup in mild base (NaHCO3) produced the ester 24, while workup in 5 N NaOH gave acid 25. Condensations with 4-acetyl-pyridine provided the corresponding targets 26 and 27. However, testing in glioblastoma cultures revealed that neither 26 nor 27 induced methuosis (Table 1).
Scheme 2.

Analogs with 5′ modifications of the indole ring generated by functionalizing the indole prior to introduction of the pyridine moiety. Conditions and reagents: (compound 22) BBr3, CH2Cl2, – 78 °C, 93%; (compound 23) methyl-4-(bromomethyl)benzoate, NaOH, TBAB, CH2Cl2, RT, 63%; (compound 24) 1. POCl3, DMF, 0 °C; 2. NaHCO3, 90%; (compound 25) 1. POCl3, DMF, 0 °C; 2. 5N NaOH, 99%; (compound 26) 4-acetyl-pyridine, piperidine, MeOH, reflux, 34%; (compound 27) 4-acetyl-pyridine, piperidine, MeOH, reflux, 21%.
Biological Activity of Compound 19 (MOMIPP) versus Compound 2 (MIPP)
The foregoing studies identified compound 19 as the most potent for inducing methuosis. Hereafter we refer to this compound by the acronym ‘MOMIPP’ for 3-(5-methoxy, 2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one. Superior activity of MOMIPP versus compound 2 (MIPP) was confirmed in studies with U251 glioblastoma cells, using MTT viability assays, cell growth assays, morphological assessment, and colony forming assays to compare MOMIPP and MIPP. Figure 2A shows the dose-response curves for the effects of the drugs on cell viability. Each compound was added at the indicated concentration for two days, with medium and compounds replenished after the first day. The relative IC50 for MOMIPP was 1.9 μM, versus 4.8 μM for MIPP. To obtain a measure of the comparative duration of activity for each compound, their effects on cell growth and survival were assessed by counting the number of cells in parallel cultures treated for three consecutive days with 2.5 μM, 5 μM, or 10 μM compound (Fig. 2B). Unlike the viability studies, in these experiments the compounds were added at the beginning of the study and were not replenished for the duration. Under these conditions, MOMIPP was clearly more effective than MIPP in reducing cell growth and viability. The reduction of cell number in the cultures treated with MOMIPP coincided with massive early vacuolization of the cells and loss of nonviable cells from the substratum (Fig. 3A & B). In contrast, the cells treated with MIPP initially underwent vacuolization on days 1 and 2, but tended to recover, especially at the 2.5 μM concentration (Fig. 3A). These studies demonstrate that a single application of MOMIPP has a much more sustained effect than MIPP on cell morphology and cell viability. The difference between the MOMIPP and MIPP was underscored when colony-forming assays were used to evaluate proliferative capacity and long-term cell viability (Fig. 4). MOMIPP was clearly more effective than MIPP in reducing colony formation when cells were treated for 2 days (Fig. 4A). If treatment was shortened to just 4 h, MOMIPP was still more effective than MIPP, but higher concentrations of both compounds were required to reduce colony formation (Fig.4B).
Figure 2.

Comparison of the effects of MOMIPP (compound 19) and MIPP (compound 2) on growth and viability of U251 GBM cells. (A). One day after plating the cells in 96 well dishes MOMIPP (●) or MIPP (■) was added at the indicated concentrations. Controls consisted of cells in parallel wells treated with an equivalent volume of vehicle (DMSO). Medium containing fresh compound was added after 24 h and MTT assays were performed after a total 48 h treatment. Each point represents the mean (± S.D.) from four separate wells, with the results expressed as percent of the mean of the parallel control wells. (B) U251 cells seeded in parallel 35 mm dishes were treated with MOMIPP (●), MIPP (■) or an equivalent volume of DMSO (▲) and cells were harvested for counting on each of three consecutive days. Each point is a mean ± S.D. from three separate cultures.
Figure 3.

Comparison of the abilities of MOMIPP and MIPP to induce the morphological hallmarks of methuosis. One day after plating, U251 GBM cells were treated with MOMIPP or MIPP at final concentrations of 2.5 μM (A) or 10 μM (B). Controls received an equivalent volume of vehicle (DMSO). Cells were observed by phase contrast microscopy on three sequential days after addition of the compounds, without changing the medium or replenishing the compounds. Methuosis is characterized by extensive accumulation of phase-lucent cytoplasmic vacuoles, with eventual cell rounding and detachment from the substratum as viability is compromised. 6,10
Figure 4.

Comparison of the abilities of MOMIPP and MIPP to inhibit survival of U251 GBM cells in colony-forming assays. (A) Cells were plated for colony-forming assays as described in the Experimental Section. One day after plating the cells, MOMIPP (●) or MIPP (■) was added to the medium at the indicated concentrations and cells were maintained in the presence of the compounds for 48 h. Thereafter the compounds were removed and colonies >50 cells were counted after two weeks. Each point represents the mean (± S.D.) from three separate dishes, with the results expressed as percent of the mean of the parallel control dishes containing DMSO. (B) The effects MOMIPP (●) and MIPP (■) on colony formation were compared as in panel A, except that cells were exposed to the compounds for only 4 h instead of 48 h.
To determine if the methuosis-inducing compounds might be effective for targeting TMZ-resistant glioblastoma, we used a previously generated TMZ-resistant U251 cell line that was essentially unaffected by concentrations of TMZ as high as 100 μM 10 (Fig. 5A). As shown in Fig. 5B, both MIPP and MOMIPP were able to induce methuosis and reduce cell viability in the TMZ-resistant cells, but the efficacy of MOMIPP was clearly superior to MIPP. The ability of MOMIPP to kill drug-resistant tumor cells by methuosis extends beyond glioblastoma. For example, we observed similar MOMIPP-induced vacuolization and loss of colony forming ability in both parental and doxorubicin-resistant MCF-7 breast cancer cells (Fig. 5C & D). To determine if the concentration of MOMIPP that is maximally toxic in cancer cells might also be cytotoxic to normal cells, we treated human mammary epithelial cells (HMEC) with 10 μM MOMIPP. As shown in Fig. 5E, HMECs underwent extensive cytoplasmic vacuolization within 24 h after addition of MOMIPP. By 48h, cell viability was reduced approximately 40%, compared with a 70% reduction seen in MCF7 breast carcinoma cells treated for the same period at a similar cell density (Fig. 5F). To further assess the effects of MOMIPP on normal cells, we applied the compound to human skin fibroblasts. As in all the other cell lines tested, 10 μM MOMIPP induced obvious vacuolization in the fibroblasts by 24h (Fig. 5G). Similar to the HMECs, normal fibroblasts plated at sub-confluent density to maintain exponential growth during exposure to MOMIPP exhibited a 40% decline in viability by 48h. However, when the fibroblasts were plated at a high initial density, so that they would be in stationary phase at the time that MOMIPP was added, cell viability was essentially unaffected (Fig. 5H), even though the cells underwent vacuolization (not shown). These observations suggest that although MOMIPP can be cytotoxic to normal cells, the latter are somewhat less sensitive to the compound than the cancer cell lines, U251 and MCF7. A possible explanation for this is suggested by the differential effects of MOMIPP on sub-confluent versus confluent fibroblasts, which imply that the disruptions of endosomal trafficking typified by vacuolization may be relatively well tolerated in cells that are not dividing, compared with cells that are actively proliferating.
Figure 5.

MOMIPP effectively inhibits the viability of drug-resistant GBM and breast cancer cells. (A) TMZ-resistant U251 glioblastoma cells (U251-TR) were derived as described previously.10 The graph, reprinted from ref. 10 (with permission), shows that in contrast to the parental U251 cells, the viability of U251-TR cells is not reduced by treatment with TMZ. (B) The U251-TR cells were treated with the indicated concentrations of MIPP or MOMIPP for 48 h and then subjected to colony-forming assays as described in the Experimental Section. Each point is based on colony counts from three dishes (mean ±S.D.), with the results expressed as percent of the mean from parallel control dishes treated with an equivalent volume of vehicle alone (DMSO). (C) Parental and doxorubicin-resistant (DoxR) MCF-7 breast cancer cells56 were treated with 10 μM MOMIPP for 24 h and then examined by phase-contrast microscopy. (D) Long-term viability of parental or DoxR MCF-7 cells was assessed by colony-forming assay after a 48 h treatment with the indicated concentrations of MOMIPP. The results are the mean ± S.D. of determinations performed on three parallel dishes. (E) Normal HMECs were treated with 10 μM MOMIPP or an equivalent volume of DMSO (control) and examined by phase contrast microscopy after 24 h. (F) MCF-7 cells or HMECs were plated in 96-well plates. After 48h, while the cells were still sub-confluent, fresh medium containing 10 μM MOMIPP or an equal volume of vehicle (DMSO) was added. MTT viability assays were performed at a 48h end-point. Values are the means (+ S.D.) from four separate wells. (G) Normal human skin fibroblasts were treated with 10 μM MOMIPP or an equivalent volume of DMSO (control) and examined by phase contrast microscopy after 24 h. (H) Normal human fibroblasts were seeded in 96-well plates at sub-confluent density (1,000 cells/well) or confluent density (10,000 cells/well). After 24h, fresh medium containing 10 μM MOMIPP or an equal volume of vehicle (DMSO) was added. MTT viability assays were performed at a 48h end-point. Values are the means (+ S.D.) from four separate wells.
Synthesis and Biological Activity of Potential Photoaffinity Labels
Our SAR studies showed that very subtle changes to the structures of MIPP or MOMIPP (e.g., modifications of the pyridine ring) can abolish their capacity to induce methuosis. Although chalcones are widely recognized as electrophiles, the level of structural specificity required for induction of methuosis suggests that the immediate effects of MIPP and MOMIPP are most likely due to their interactions with one or more distinct molecular targets. Several examples of protein-specific modifications by electrophilic compounds have been reported (see Discussion). Therefore, an important goal for the future is to identify the targets of methuosis-inducing chalcones. A common approach for identifying drug targets is affinity chromatography. 17 However, at this point our SAR studies have not revealed any non-essential sites that could be linked to a bulky affinity tag (e.g. biotin) or tethered to a solid matrix without loss of activity. Therefore, we focused on generating minimally modified biologically active analogs with reactive groups that might be useful for photoaffinity-based drug target discovery efforts. 18
Initially, we designed a pseudo-benzophenone analog of MIPP as a potential photoaffinity probe. Benzophenones are commonly incorporated into active molecules for target identification studies. 18–21 Based on the structure of compound 14, we postulated that a 5-benzoyl analog might retain activity and function as a photoaffinity probe. The benzophenone analog (compound 30, benzoyl-MIPP) was prepared in two steps, starting with acylation of the Vilsmeier-Haack intermediate of 2-methylindole (Scheme 3A). With unmethylated indole, this system has been shown to direct acylation to the 5- and 6- positions. 22 In our hands, acylation of the Vilsmeier-Haack intermediate of 2-methylindole gave a 1:3 mixture of 5- and 6-benzoyl-2-methylindole-3-carboxaldehyde respectively (28 and 29). This regioselectivity was characterized by 1-D nuclear Overhauser effect (nOe) as described in the experimental section. In the second step of the scheme, the final compounds 30 and 31 were made by coupling the aldehydes with 4-acetyl pyridine in the presence of piperidine. Subsequent morphology and viability studies revealed that unlike the benzyloxy derivative (compound 14), neither of the benzophenone analogs (30 and 31) induced methuosis when tested in U251 cells (data not shown).
Scheme 3.

Synthesis of 5′ and 6′ azido derivatives of MIPP. (A) Conditions and reagents: (compounds 28 & 29) 1. COCl2, DMF, 1,2-DCE, 0 °C; 2. AlCl3, benzoyl chloride, 0 °C RT, 13% and 41% for 28 and 29, respectively; (compound 30) 4-acetyl-pyridine, piperidine, MeOH, reflux, 13%; (compound 31) 4-acetyl-pyridine, piperidine, MeOH, reflux, 74%; (B) Conditions and reagents: (compound 32) NaNO3, H2SO4, 0 °C, 95%; (compound 33) H2, 10% Pd/C, EtOH, RT, 97%; (compound 34) 1. NaNO2; 2. NaN3, 75% AcOH, 0 °C, 66%; (compound 35) POCl3, DMF, 0 °C, 88%; (compound 36) 4-acetyl-pyridine, piperidine, MeOH, reflux, 39%; (compound 37) NaNO3, H2SO4, 0 °C, 62%; (compound 38) H2, 10% Pd/C, EtOH, RT, 99%; (compound 39) 1. NaNO2; 2. NaN3, 75% AcOH, 0 °C, 53% (compound 40) POCl3, DMF, 0 °C, 69%; (compound 41) 4-acetyl-pyridine, piperidine, MeOH, reflux, 39%.
Aromatic azides represent another photoreactive moiety commonly used in photoaffinity labeling. 18, 23–25 Thus, we next explored the possibility that an azide could be added to either the 5 or 6 position of the indole ring without loss of methuosis-inducing activity, as the azide represents less of a structure change from MOMIPP than the benzoyl group of compound 30. The synthesis of compound 36 (5-azido MIPP) was based on the directed nitration of 2-alkylindoles toward the 5 position26, with further conversion to an azide by diazotization (see Scheme 3B). 2-Methylindole was selectively nitrated in concentrated sulfuric acid to yield 32 (see experimental details for validation of regiospecificity), which was then hydrogenated to amine 33. Under mild diazotization conditions, the aminoindole was converted to the aromatic azide 34. 27 The azide was formylated to obtain 35 and condensed with 4-acetyl-pyridine, providing 36, 5-azido-MIPP. Separately, the same reaction scheme was performed on 2-methyl-5-methoxyindole, wherein nitration led predominately to the 6-nitrated product 37. The same sequence of reaction steps described above were then completed to obtain 41, 6-azido-MOMIPP.
Evaluation of the biological activities of 36 (5-azido MIPP) and 41 (6-azido MOMIPP) indicated that both compounds retained methuosis-inducing activity as judged by both cellular vacuolization (Fig. 6A) and inhibition of colony formation (Fig. 6B&C) in U251 cells. Although not as potent as MOMIPP, the 5-azido compound was clearly superior to the 6-azido compound in terms of its biological activity.
Figure 6.
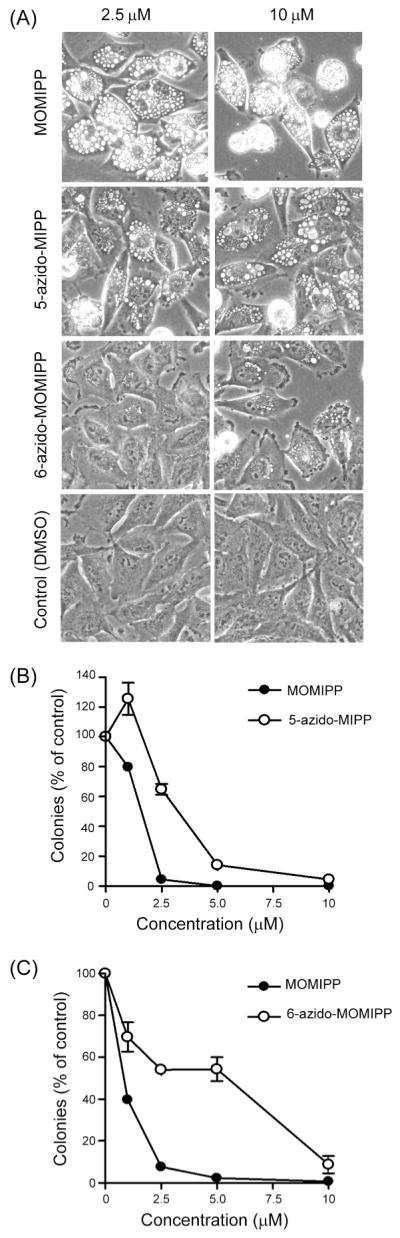
Comparison of the effects of MOMIPP (compound 19), 5-azido-MIPP (compound 36) and 6-azido-MOMIPP (compound 41) on morphology and viability of U251 glioblastoma cells. (A) The indicated compounds were added to the cells at concentrations of 2.5 μM or 10 μM and the cells were observed by phase-contrast microscopy after 48 h. (B & C) U251 cells were treated with varying concentrations of the indicated compounds for 48 h and long term cell viability was assessed by colony-forming assays. Each represents the counts from three dishes (mean ±S.D.), with the results expressed as percent of the mean from parallel control dishes treated with an equivalent volume of vehicle alone (DMSO).
DISCUSSION
Methuosis is a newly discovered form of non-apoptotic cell death that is triggered by alterations of macropinocytotic vesicular trafficking, resulting in massive cellular vacuolization and loss of cellular metabolic integrity. 6 We recently described MIPP as a chalcone-like small molecule that is capable of inducing methuosis in GBM and other cancer cell lines.10 Here we present SAR studies that have led to the identification of a 5-methoxy analog termed MOMIPP, which demonstrates improved potency and stability in cell culture systems. We have also generated active azido compounds that may be useful for future studies aimed at identifying the protein targets of MOMIPP.
In the course of generating a directed library of compounds for SAR studies, we carried out Claisen-Schmidt condensation reactions between various indole-3-carboxaldehydes and aromatic ketones catalyzed by piperidine, which efficiently provided indole-chalcones. As piperidine acts as a catalyst, we initially carried out the aldol condensations in the presence of catalytic amounts of piperidine. However, we generally observed higher yields when piperidine was used in excess. In almost all instances, the products precipitated from solution, and were thus easily purified from excess catalyst (or starting material) by simple rinsing.
Our SAR studies provided several useful insights into the molecular features required for the methuosis-inducing activity of this class of compounds. First, regarding the indole ring, methylation of the nitrogen (compound 20) or removal of the 2- methyl group (compound 13) reduced but did not eliminate activity. At the 5-position, different modifications had opposite consequences. The activity of the 5-methoxy compound (19, MOMIPP) was substantially improved and the 5-benzyloxy (compound 14) was similar to unsubstituted MIPP. In contrast, modification of the 5- position with OH (compound 21), p-methyl ester benzyloxyl (compound 26), and p-COOH benzyloxyl (compound 27) all caused a major loss of activity. Together these findings demonstrate that there may be some flexibility at these positions on the indole ring for future attempts to further improve potency, create more water-soluble derivatives, or tether the compound to an affinity matrix. Regarding the second aryl system of the chalcone framework, we learned that the para-nitrogen orientation of the pyridine ring is critical for activity. Analogs with a 2-pyridine (compound 10), 3-pyridine (compound 11 and 16), pyrazine (compound 17) or phenyl (compound 9) were inactive.
In considering the potential mechanisms through which MOMIPP induces cell death by methuosis, one possibility is that it could be acting as an electrophile (i.e., Michael acceptor). Compounds possessing electrophilic moieties that render them potential substrates to cellular nucleophiles are not often used in drug design because they can arbitrarily modify many biomolecules. This can lead to off-target effects, including the formation of immunoreactive haptens. 28 The SAR studies summarized in Table 1 show that while many of the compounds in our series possess the α,β-unsaturated ketone scaffold and could act as putative Michael acceptors, only MOMIPP and a few other compounds were effective inducers of methuosis at micromolar concentrations. Thus, it seems unlikely that MOMIPP is inducing cell death via general electrophilic modification of proteins. It remains possible that MOMIPP may be functioning as a target-specific Michael acceptor, with certain features of the molecule mediating association with a specific protein, where proximity of the α,β-unsaturated ketone to nucleophilic residues (e.g., cysteine) may then promote covalent modification. Examples of such protein-specific modifications have been reported for compounds that bind to the receptor protein ErbB2, 29 the TRPA1 ion channel, 30 the nuclear protein KSRP/FUBP2, 31 and sortase enzymes in Gram-positive bacteria. 32 These types of observations have spurred a general resurgence of interest in drugs that work by covalent modification of their specific targets.33
Compounds broadly classified as chalcones have been shown to exhibit anticancer activity through multiple mechanisms, including disruption of p53 interactions with MDM2 or HDM2, 13, 34 inhibition of p-glycoprotein, 35–37 and disruption of microtubule polymerization. 38–40 Many of the latter compounds were designed as analogs of colchicine and combretastatins, natural products known to bind β-tubulin. A number of publications have since established that chalcones and related molecules can act as antimitotic agents, and substantial progress has been made in understanding their SAR.41, 42 While our active methuosis-inducing compounds (e.g., MOMIPP) can be classified as chalcones, their specific features are quite distinct from most of the anti-mitotic chalcones previously described. The stringent structural specificity for induction of methuosis, with dependence on the specific substitution patterns of both the indolyl and pyridinyl moieties, appears to differentiate MOMIPP from chalcones previously reported as antimitotic agents. We have not observed mitotic arrest prior to loss of viability in cells treated with these compounds (unpublished observation). Conversely, massive endosomal vacuolization akin to what we have observed with MOMIPP has not been reported with the anti-mitotic chalcones. Of all the cytotoxic chalcones described in the literature, the one most related to MOMIPP contains an unsubstituted indole ring linked to a 3,4,5-trimethoxy phenyl moiety.14 However, we found that replacement of the 4-pyridine of MOMIPP with a 3,4,5-trimethoxy phenyl group yielded a compound that did not induce appreciable cellular vacuolization or death when applied to U251 cells at 10 μM for 48h (not shown). This is consistent with the lack of tolerance for modifications to the 4-pyridinyl moiety, and suggests that the cytotoxicity previously reported for the trimethoxyphenyl indolyl chalcone 14 probably was not due to methuosis.
In general, cell death induced by anti-mitotic chalcones is thought to occur by classical apoptosis, not methuosis. A possible exception was noted in a recent report where a chalcone-derivative termed ‘C2’ may have induced death in glioblastoma cells by a non-apoptotic mechanism involving accumulation of autophagic vacuoles 43. However, as we have previously reported, the vacuoles induced during methuosis arise from macropinosomes and endosomes, which are distinct from autophagosomes 6, 10. It would be premature to rule out the possibility that MOMIPP might bind tubulin in a manner similar to colchicine and related chalcones, but so far the preponderance of evidence suggests that the compounds described in this study act by a different mechanism to trigger abnormal macropinocytosis, swelling of endosomal compartments, and non-apoptotic cell death.10 Ultimately, clarification of this mechanism will depend on identification of the specific molecular target(s) of MOMIPP and related compounds.
The future identification of the specific target(s) of MOMIPP will be important for several reasons: 1) The expression level or activity of the identified target(s) might have predictive value for determining which types of tumors would be most susceptible to the compound; 2) understanding the function(s) of the proteins targeted by MOMIPP could be helpful for assessing the potential toxicity to normal cells; and 3) knowledge about the target protein(s) will facilitate analysis of the drug binding site that could suggest modifications to increase potency or specificity. In this respect, our finding that incorporation of a photoreactive azide at the 5-position of the indole ring of MIPP yields a derivative that retains good methuosis-inducing activity (Fig. 6) offers several avenues for protein target identification using established strategies. Aside from the photoreactive azide 36, MOMIPP’s core structure contains two other features that may render it suitable for target identification studies, potentially bypassing the need for incorporation of a photoreactive azide. First, MOMIPP itself may be photoreactive and could behave similarly to an excited benzophenone. The core scaffold of MOMIPP is a pseudo-diarylketone (as one side contains a double vinylogous amide through the indole, while the other contains a 4-pyridine), and could potentially exhibit photoexcitation chemistry similar to that of a benzophenone. We are currently pursuing experiments to characterize MOMIPP’s inherent photoreactivity and its ability to insert into other molecules. The second characteristic of MOMIPP’s core structure that may render it suitable for target identification is its electrophilic α,β-unsaturated ketone moiety, which may be responsible for a covalent modification of a nucleophilic target residue (e.g. cysteine). Thus, if MOMIPP is covalently binding to its target protein(s), there may be no need for photo-initiated reactive intermediate formation. This possibility is also undergoing further study.
The key observation that MOMIPP effectively induced methuosis in TMZ-resistant GBM cells, as well as doxorubicin-resistant breast cancer cells, raises the possibility that further development of this compound could lead to useful therapeutic agents for treating cancers that are resistant to drugs that commonly work by inducing apoptosis. Ultimately, deployment of MOMIPP or related compounds as anti-cancer agents will need to address some challenges. Preliminary studies indicate that MOMIPP’s ability to induce vacuolization is not restricted to cancer cells (Fig. 5E–H). However, the studies with normal HMECs and skin fibroblasts suggest that the consequences of vacuolization for cell viability are more severe for rapidly dividing cancer cells than normal cells, particularly when the normal cells enter stationary phase at high cell density (Fig. 5H). This raises a possibility that a therapeutic window might be identified for selective effects on cancer cells. A second challenge relates to the poor aqueous solubility of MOMIPP and most of its active analogs. However, similar solubility issues have been encountered with other hydrophobic anti-cancer drugs (e.g. taxol, camptothecin) and have been circumvented through the use of appropriate excipients 44 or novel nanoparticle 45, 46 or liposome 47, 48 based delivery strategies. The latter strategies may offer the additional benefit of allowing selective targeting of non-specific agents by surface modification of the delivery vehicle with tumor-specific peptides or antibodies. 45, 49, 50 Thus, given the observed cytotoxic affects of MOMIPP on drug-resistant cancer cells, and the range of available options for its delivery in vivo, we believe that MOMIPP can serve as a valuable prototype for further preclinical testing.
EXPERIMENTAL SECTION
Chemistry
General methods
Reagents and starting materials were obtained from commercial suppliers without further purification. Thin layer chromatography (TLC) was done on 250 μm fluorescent silica gel 1B–F plates and visualized with UV light. Flash column chromatography was performed using silica gel 230–400 μm mesh size. Melting points (MP) are uncorrected. Nuclear magnetic resonance (NMR) spectra were recorded on either a 600, 400 or 200 MHz instrument. Peak locations were referenced using the residual solvent peak (7.26 and 77.16 for CDCl3 1H and 13C, respectively, and 2.50 and 39.51 for DMSO 1H and 13C). Proton coupling constants (J values) and signals are expressed in hertz using the following designations: s (singlet), d (doublet), br s (broad singlet), m (multiplet), t (triplet), dd (doublet of doublets) and qd (quintet of doublets). NMR spectra are available in the supplementary information. All synthesized compounds were at least 95% pure as determined by elemental analysis (Atlantic Microlabs, Norcross, GA).
2-Methylindole-3-carboxaldehyde (2a)51
To a dried 300 mL two-neck round bottom flask under argon at 0 °C, N,N-dimethylformamide (3 mL) was added, followed by POCl3 (1.05 mL, 11.3 mmol). After stirring for ten minutes, 2-methylindole (1.23 g, 9.37 mmol) dissolved in DMF (6 mL) was added dropwise via an addition funnel under argon. After two hours, 1 N NaOH (70 mL) was added slowly, upon which a white precipitate formed. The solid was filtered and dried under vacuum, yielding 1.32 g (89%) of white solid. 1H NMR (600 MHz, d6-DMSO): δ 11.998 (s, 1H, N-H), 10.050 (s, 1H, CHO), 8.044-8.032 (d, J = 7.2, 1H, indole-4H), 7.388-7.375 (d, J = 7.8, 1H, indole-7H), 7.183-7.135 (m, 2H, indole-5,6H), 2.679 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 184.3, 148.6, 135.4, 125.6, 122.7, 121.9, 120.0, 113.7, 111.4, 11.5. Melting Point: 195 – 200 °C (published51: 197–200 °C). TLC (ethyl acetate:hexanes 4:1) Rf = 0.39. Elemental analysis calculated for C10H9NO: C, 75.45; H, 5.70; N, 8.80; found: C, 75.23; H, 5.70; N, 8.93.
trans-3-(2-Methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (2)52
To a dried 500 mL round bottom flask under argon, 2-methylindole-3-carboxaldehyde (400 mg, 2.51 mmol) was dissolved in anhydrous MeOH (10 mL). 4-Acetyl-pyridine (305 μL, 2.76 mmol, 1.1 equiv.) and piperidine (82 μL, 0.83 mmol) were added and the reaction was stirred under reflux. A red-orange precipitate gradually formed, and after twelve hours this solid was isolated by filtration, rinsed with chilled methanol and dried under vacuum, producing 458 mg (69%) of yellow solid. NMR: 1H NMR (600 MHz, d6-DMSO): δ 12.017 (s, 1H, N-H), 8.816-8.806 (dd, J1 = 4.5, J2 = 2.1, 2H, pyr-2,6H), 8.121-8.095 (d, J = 15.6, 1H, C=CH), 8.077-8.063 (m, 1H, indole-4H), 7.975-7.965 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-3,5H), 7.489-7.464 (d, J = 15.6, 1H, C=CH), 7.415-7.400 (m, 1H, indole-7H), 7.220-7.200 (m, 2H, indole-5,6H), 2.596 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.0, 150.7, 145.6, 144.9, 139.5, 136.3, 125.8, 122.4, 121.50, 121.46, 120.4, 113.1, 111.7, 109.4, 11.9. Melting Point: 268 – 272 °C. TLC: (ethyl acetate:hexanes 4:1) Rf = 0.18. Elemental analysis calculated for C17H14N2O: C, 77.84; H, 5.38; N, 10.68; found: C, 77.96; H, 5.28; N, 10.59.
trans-3-(1H-Indol-3-yl)-1-phenyl-2-propen-1-one (9)15
In a dried, 25 mL round bottom flask under argon, indole-3-carboxaldehyde (300 mg, 2.07 mmol) was dissolved in anhydrous methanol (8 mL). Acetophenone (240 μL, 2.07 mmol) and piperidine (100 μL, 1.00 mmol) was added. The reaction was stirred under reflux for 18 hours. 10% Acetic acid was added (10 mL), precipitating 248 mg of a crude yellow solid. This was recrystallized in 100% EtOH, filtered, and dried under vacuum, yielding a pure, yellow solid (198 mg, 39%, 247.29 MW). 1H NMR (600 MHz, d6-DMSO): δ 8.133-8.114 (m, 3H, phenyl-2,6H, indole-2H), 8.095-8.081 (m, 1H, indole-4H), 8.075-8.049 (d, J = 15.6, 1H, C=CH), 7.668-7.642 (d, J = 15.6, 1H, C=CH), 7.655-7.629 (m, 1H, phenyl-4H), 7.581-7.556 (t, J = 1.5, 2H, phenyl-3,5H), 7.504-7.490 (m, 1H, indole-7H), 7.254-7.231 (m, 2H, indole-5,6H). 13C NMR (150 MHz, d6-DMSO): δ 188.8, 139.1, 138.5, 137.5, 133.4, 132.4, 128.7, 128.2, 125.1, 122.8, 121.2, 120.4, 115.3, 112.8, 112.5. Melting point: 167 – 170 °C (published15: 166–167 °C). TLC (in 2:1 ethyl acetate:hexanes) Rf = 0.48. Elemental analysis calculated for C17H13NO: C, 82.57; H, 5.30; N, 5.66; found: C, 82.19; H, 5.35; N, 5.63.
trans-3-(1H-Indol-3-yl)-1-(2-pyridinyl)- 2-propen-1-one (10)52
Indole-3-carboxaldehyde (200 mg, 1.38 mmol) was added to a dried 100 mL round bottom flask under argon and anhydrous methanol (8 mL) was added. 2-Acetyl-pyridine (232 μL, 2.07 mmol) and piperidine (69 μL, 0.7 mmol) were added and the reaction was stirred under reflux for 24 hours, after which still no precipitate had formed (AcOH did not lead to precipitation). The crude reaction mixture was concentrated and directly applied to a silica column for chromatography (ethyl acetate:hexanes 1:1). The product was partially separated from the aldehyde starting material (some product coeluted with aldehyde and was discarded), and 80 mg (23%) of purified product was isolated. NMR: 1H NMR (600 MHz, d6-DMSO): δ 11.989 (s, 1H, N-H), 8.831-8.819 (m, pyr-H2), 8.220-8.194 (d, J = 15.6, 1H, C=CH), 8.154-8.105 (m, 3H, C=CH, indole-H2, pyr-H3), 8.052-8.027 (m, 1H, pyr-H4), 7.976-7.962 (m, 1H, indole-H4), 7.679-7.656 (m, 1H, pyr-H5), 7.522-7.508 (m, 1H, indole-H7), 7.285-7.262 (m, 2H, indole-H5,6). 13C NMR (150 MHz, d6-DMSO): δ 188.2, 154.3, 149.1, 139.3, 137.7, 137.6, 134.2, 127.1, 125.1, 122.3, 122.2, 121.4, 120.1, 114.3, 113.1, 112.7. Melting Point: 141 – 145 °C. TLC: (ethyl acetate:hexanes 4:1) Rf = 0.40. Elemental analysis calculated for C16H12N2O • 0.1 C4H8O2: C, 76.62; H, 5.02; N, 10.90; found: C, 76.41; H, 5.03; N, 10.80.
trans-3-(1H-Indol-3-yl)-1-(3-pyridinyl)- 2-propen-1-one (11)53
Indole-3-carboxaldehyde (100 mg, 0.69 mmol) was added to a dried 100 mL round bottom flask and dissolved in anhydrous methanol (5 mL). 3-Acetyl-pyridine (113 μL, 1.03 mmol, 1.5 equiv.) and piperidine (69 μL, 0.7 mmol) were added and the reaction stirred under reflux. After twelve hours, the reaction was cooled to room temperature, upon which a precipitate formed. The solid was filtered, yielding 54 mg. However, there was significant aldehyde present on a crude NMR. This mixture was dry-loaded onto silica and purified by column chromatography (methylene chloride:methanol 9:1), producing 33 mg pure, yellow solid (19%). NMR: 1H NMR (600 MHz, d6-DMSO): δ 12.004 (s, 1H, N-H), 9.292-9.289 (d, J = 1.8, 1H, pyr-H2), 8.808-8.797 (dd, J1 = 4.8, J2 = 1.8, 1H, pyr-H6), 8.467-8.447 (dt, J1 = 7.8, J2 = 2.1, 1H, pyr-H4), 8.174 (s, 1H, indole-H2), 8.154-8.140 (dd, J1 = 6.6, J2 = 1.8, 1H, indole-H4), 8.120-8.095 (d, J = 15.0, 1H, C=CH), 7.665-7.639 (d, J = 15.6, 1H, C=CH), 7.607-7.585 (m, 1H, pyr-H5), 7.506-7.492 (dd, J1 = 6.9, J2 = 1.5, 1H, indole-H7), 7.268-7.223 (qd, J1 = 6.6, J2 = 1.5, 1H, indole-H5,6). 13C NMR (150 MHz, d6-DMSO): δ 187.9, 152.7, 149.3, 139.9, 137.6, 135.7, 134.1, 133.7, 125.1, 123.9, 122.9, 121.3, 120.7, 115.0, 112.9, 112.5. Melting Point: 192 – 194 °C (published53: 191 °C). TLC (methylene chloride:methanol 9:1) Rf = 0.26. Elemental analysis calculated for C16H12N2O: C, 77.40; H, 4.87; N, 11.28; found: C, 77.00; H, 4.80; N, 11.12.
trans-3-(1H-Indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (12)53
In a dried, 25 mL round bottom flask under argon, indole-3-carboxaldehyde (300 mg, 2.07 mmol) was dissolved in anhydrous methanol (8 mL). 4-Acetyl-pyridine (229 μL, 2.07 mmol) and piperidine (100 μL, 1.00 mmol) were added. The reaction was stirred under reflux; gradually, yellow product precipitated during the reaction. After 14 hours, the reaction was cooled to room temperature, filtered and washed with chilled methanol and hexanes. Drying under vacuum for two hours yielded a pure, yellow solid (343 mg, 67%). 1H NMR (600 MHz, d6-DMSO): δ 8.826-8.816 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-H2,6), 8.186-8.182 (d, J = 2.4, 1H, indole-H2), 8.128-8.117 (m, 1H, indole-H4), 8.121-8.095 (d, J = 15.6, 1H, C=CH), 7.978-7.968 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-H3,5), 7.592-7.566 (d, J = 15.6, 1H, C=CH), 7.512-7.498 (m, 1H, indole-H7), 7.267-7.243 (m, 2H, indole H-5,6). 13C NMR (150 MHz, d6-DMSO): δ 188.4, 150.7, 144.7, 140.9, 137.6, 134.6, 125.0, 123.0, 121.5, 121.4, 120.6, 114.6, 112.9, 112.6. Melting point: 266 – 268 °C (published53: 257–258 °C). TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.23. Elemental analysis calculated for C16H12N2O: C, 77.40; H, 4.87; N, 11.28; found: C, 77.35; H, 4.84; N, 11.23.
trans-3-(5-Methoxy-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (13)
In a dried, 25 mL round bottom flask under argon, 5-methoxyindole-3-carboxaldehyde (100 mg, 0.57 mmol) was dissolved in anhydrous methanol (3 mL). 4-Acetyl-pyridine (63 μL, 0.57 mmol) and piperidine (30 μL, 0.3 mmol) were added. The reaction was stirred under reflux, during which a crude yellow solid precipitated. After 15 hours, 10% acetic acid (10 mL) was added to further the precipitation. The solid was filtered and dried under vacuum, yielding a pure, yellow solid (121 mg, 76%). 1H NMR (600 MHz, d6-DMSO): δ 8.821-8.811 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-H2,6), 8.163-8.159 (d, J = 2.4, 1H, indole-H2), 8.117-8.091 (d, J = 15.6, 1H, C=CH), 7.953-7.943 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-H3,5), 7.532-7.506 (d, J = 15.6, 1H, C=CH), 7.489-7.485 (d, J = 2.4, 1H, indole-H4), 7.405-7.391 (d, J = 8.4, 1H, indole-H7), 6.902-6.883 (dd, J1 = 9.0, J2 = 2.4, 1H, indole-H6), 3.866 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.4, 155.2, 150.7, 144.9, 140.9, 134.2, 132.4, 126.0, 121.5, 114.3, 113.3, 112.7, 112.4, 102.4, 55.6. Melting point: 235 – 237 °C. TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.20. Elemental analysis calculated for C17H14N2O2: C, 73.37; H, 5.07; N, 10.07; found: C, 73.55; H, 5.00; N, 10.04.
trans-3-(5-Phenylmethoxy-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (14)
In a dried, 25 mL round bottom flask under argon, 5-benzyloxyindole-3-carboxaldehyde (100 mg, 0.40 mmol) was dissolved in anhydrous methanol (3 mL). 4-Acetyl-pyridine (75 μL, .68 mmol) and piperidine (20 μL, 0.2 mmol) were added. The reaction was stirred under reflux, during which a crude yellow solid precipitated. The solid was filtered, rinsed with cold methanol and dried under vacuum. This crude product (107 mg) was purified from residual aldehyde by column chromatography in ethyl acetate:hexanes (1:1 → 3:1 gradient), yielding pure, yellow solid (70 mg, 49%). 1H NMR (600 MHz, d6-DMSO): δ 8.837-8.827 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-2,6H), 8.148-8.143 (d, J = 3.0, 1H, indole-2H), 8.095-8.069 (d, J = 15.6, 1H, C=CH), 7.932-7.922 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-3,5H), 7.556-7.553 (d, J = 1.8, 1H, indole-4H), 7.523-7.511 (d, J = 7.2, 2H, phenyl-2,6H), 7.460-7.434 (d, J = 15.6, 1H, C=CH), 7.411-7.370 (m, 3H, phenyl-3,5H, indole-7H), 7.330-7.306 (t, J = 7.2, 1H, phenyl-4H), 6.979-6.961 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-6H), 5.252 (s, 2H, methylene). 13C NMR (150 MHz, d6-DMSO): 188.4, 154.1, 150.7, 144.9, 140.9, 137.7, 134.6, 132.5, 128.4, 127.7, 127.6, 125.8, 121.5, 114.2, 113.3, 113.1, 112.7, 104.1, 69.8. Melting point: 218 – 221 °C. TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.26. Elemental analysis calculated (for C23H19N2O2 • 0.2 C6H14 • 0.05 H2O): C, 78.02; H, 5.93; N, 7.52; found: C, 77.69; H 5.62; N 7.24.
trans-3-(5-Hydroxy-1H-Indol-3-yl)-1-(4-pyridinyl)- 2-propen-1-one (15)
To a dried two-neck 250 mL round bottom flask under argon at − 40 °C, 13 (352 mg, 0.99 mmol) was partially dissolved in CH2Cl2 (30 mL). BBr3 (10 mL, 1.0 M in CH2Cl2, 10 mmol) was added dropwise via an addition funnel under argon. After four hours the reaction was poured onto ice and treated with 5 N NaOH until pH ≈ 12. The aqueous solution was isolated and treated with 5 N HCl until pH ≈ 7, forming a brown precipitate which was extracted with ethyl acetate (3 × 40 mL). Extracts were combined, dried with Na2SO4, filtered, concentrated and dried under vacuum to yield 158 mg of an orange solid (61%). NMR: 1H NMR (600 MHz, d6-DMSO): δ 11.840 (s, 1H, N-H), 9.109 (s, 1H, O-H), 8.826-8.816 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-2,6H), 8.063-8.058 (d, J = 3.0, 1H, indole-2H), 8.048-8.022 (d, J = 15.6, 1H, C=CH), 7.902-7.892 (dd, J1 = 4.5, J2 = 1.5, 2H, pyr-3,5H), 7.349-7.346 (d, J = 1.8, 1H, indole-4H), 7.307-7.293 (d, J = 8.4, 1H, indole-H7), 6.762-6.744 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-H6). 13C NMR (150 MHz, d6-DMSO): δ 188.2, 152.9, 150.6, 145.0, 141.4, 134.8, 131.7, 126.0, 121.3, 113.6, 113.1, 112.7, 112.3, 104.9. Melting Point: 262 – 268 °C. TLC (in ethyl acetate:hexane 4:1) Rf = 0.35. Elemental analysis calculated for C16H12N2O2 • 0.1 H2O: C, 72.22; H, 4.62; N, 10.53; found: C, 72.21; H, 4.42; N, 10.26.
trans-3-(5-Methoxy-1H-indol-3-yl)-1-(3-pyridinyl)-2-propen-1-one (16)
In a dried, 50 mL round bottom flask under argon, 5-methoxyindole-3-carboxaldehyde (100 mg, 0.57 mmol) was dissolved in anhydrous methanol (4 mL). 3-Acetyl-pyridine 63 μL, 0.57 mmol) and piperidine (30 μL, 0.30 mmol) were added. The reaction was stirred under reflux, during which a crude orange solid precipitated. After twenty hours, the solid was isolated by vacuum filtration, rinsed with chilled methanol and dried under vacuum, yielding a pure, orange solid (98 mg, 60%). 1H NMR (600 MHz, d6-DMSO): δ 11.891 (s, 1H, N-H), 9.276-9.273 (d, J = 1.8, 1H, pyr-2H), 8.798-8.790 (m, 1H, pyr-6H), 8.440-8.421 (m, 1H, pyr-4H), 8.155-8.150 (d, J = 3.0, 1H, indole-2H), 8.113-8.088 (d, J = 15.0, 1H, C=CH), 7.607-7.582 (d, J = 15.0, 1H, C=CH), 7.598-7.584 (m, 1H, pyr-5H), 7.500-7.497 (d, J = 1.8, 1H, indole-4H), 7.397-7.383 (d, J = 8.4, 1H, indole-7H), 6.892-6.874 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-6H), 3.867 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 187.9, 155.1, 152.6, 149.3, 139.9, 135.7, 133.8, 133.7, 132.3, 126.1, 123.9, 114.7, 113.2, 112.7, 112.4, 102.6, 55.6. Melting point: 169 – 173 °C. TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.17. Elemental analysis calculated C17H14N2O2: C, 73.37; H, 5.07; N, 10.07; found: C, 73.09; H, 5.10; N, 10.01.
trans-3-(5-Methoxy-1H-indol-3-yl)-1-(pyrazine)-2-propen-1-one (17)
In a dried, 25 mL round bottom flask under argon, 5-methoxyindole-3-carboxaldehyde (75 mg, 0.43 mmol) was dissolved in anhydrous methanol (4 mL). Acetyl-pyrazine (52 mg, .43 mmol) and piperidine (23 μL, 0.23 mmol) were added. The reaction was stirred under reflux, during which a crude, yellow solid precipitated. After three hours, the solid was isolated by vacuum filtration, rinsed with chilled methanol and dried under vacuum. This crude product was recrystallized in EtOH (8 mL) to remove residual aldehyde, yielding a pure, yellow solid (28 mg, 24%). 1H NMR (600 MHz, d6-DMSO): δ 11.960 (s, 1H, N-H), 9.244 (s, 1H, pyr-2H), 8.905-8.873 (m, 2H, pyr-4,5H), 8.204-8.178 (d, J = 15.6, 1H, C=CH), 8.155 (s, 1H, indole-2H), 7.987-7.961 (d, J = 15.6, 1H, C=CH), 7.427-7.412 (m, 2H, indole-4,7H), 6.934-6.915 (dd, J1 = 9.0, J2 = 2.4, 1H, indole-6H), 3.853 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 187.3, 155.1, 148.8, 147.6, 144.0, 143.7, 140.2, 134.6, 132.5, 126.0, 113.4, 113.2, 112.8, 111.9, 102.9, 55.6. Melting point: 176 – 180 °C. TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.30. Elemental analysis calculated C16H13N3O2: C, 68.81; H, 4.69; N, 15.05; found: 68.76; H, 4.64; N, 14.99.
5-Methoxy-2-methyl-1H-indole-3-carboxaldehyde (18)54
To a dried two-neck 25 mL round bottom flask at 0 °C, POCl3 (1.00 mL, 10.8 mmol) was added to N,N-dimethylformamide (2.5 mL). After ten minutes of stirring, 2-methyl-5-methoxyindole (1.45 mg, 9.00 mmol) dissolved in DMF (5 mL) was added dropwise. After 45 minutes, 1N NaOH (50 mL) was slowly added, forming a white precipitate. The solid was isolated by filtration, washed with cold H2O and dried under vacuum, yielding 1.52 g of white solid (90 %). 1H NMR (600 MHz, d6-DMSO): δ 11.875 (s, 1H, N-H), 10.009 (s, 1H, CHO), 7.570-7.566 (d, J = 2.4, 1H, indole-4H), 7.282-7.268 (d, J = 8.4, 1H, indole-7H), 6.797-6.778 (dd, J1 = 9.0, J2 = 2.4, 1H, indole-6H), 3.764 (s, 3H, o-methyl), 2.646 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 184.1, 155.5, 148.6, 130.0, 126.4, 113.7, 112.1, 111.8, 102.3, 55.2, 11.5. Melting Point: 188 – 192 °C (published54: 191–194 °C). TLC (ethyl acetate:hexane 4:1) Rf = 0.37. Elemental analysis calculated for C11H11NO2: C, 69.83; H, 5.86; N, 7.40; found: C, 69.65; H, 5.95; N, 7.25.
trans-3-(5-Methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (19)
To a dried 250 mL two-neck round bottom flask under argon, 2-methyl-5-methoxy-1H-indole-3-carboxaldehyde (1.51 g, 7.98 mmol) was dissolved in anhydrous MeOH (30 mL). 4-Acetyl-pyridine (1.32 mL, 11.97 mmol, 1.5 equiv.) and piperidine (0.788 mL, 7.98 mmol) were added and the reaction was stirred under reflux. An orange solid gradually precipitated, and this was isolated by filtration, rinsed with chilled MeOH and dried under vacuum, yielding 2.08 g of orange solid (89%). 1H NMR (600 MHz, d6-DMSO): δ 11.909 (s, 1H, N-H), 8.812-8.802 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-2,6H), 8.097-8.072 (d, J = 15.0, 1H, C=CH), 7.947-7.937(dd, J1 = 4.2, J2 = 1.8, 2H, pyr-3,5H), 7.434-7.430 (d, J = 2.4, 1H, indole-4H), 7.374-7.349 (d, J = 15.0, 1H, C=CH), 7.315-7.301 (d, J = 8.4, 1H, indole-7H), 6.851-6.833 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-6H), 3.860 (s, 3H, o-methyl), 2.572 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.1, 155.2, 150.6, 145.8, 145.1, 136.6, 131.0, 126.6, 121.5, 112.8, 112.3, 110.9, 109.3, 103.5, 55.6, 12.2. Melting Point: 252 – 256 °C. TLC (ethyl acetate:hexane 4:1) Rf = 0.16. Elemental analysis calculated for C18H16N2O2: C, 73.95; H, 5.52; N, 9.58; found: C, 73.76; H, 5.46; N, 9.47.
trans-3-(5-Methoxy-1-methyl-Indol-3-yl)-1-(4-pyridinyl)- 2-propen-1-one (20)
N,N-dimethylformamide (3 mL) was added to a dried 100 mL two-neck round bottom flask under argon containing NaH (21 mg, 0.52 mmol, 60% in mineral oil, 1.2 equiv., unwashed). After stirring for five minutes, the starting material (120 mg, 0.43 mmol) dissolved in DMF (1 mL) was added slowly and stirred for five minutes until a homogenous red-solution was formed. Methyl iodide (40 μL, 0.65 mmol, 1.5 equiv.) was added slowly. After two hours, sat. NH4Cl (20 mL) and H2O (20 mL) was added; this was extracted with ethyl acetate (3 × 30 mL), dried with Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (methylene chloride:methanol 95:5), providing 103 mg (82%) of solid. NMR: 1H NMR (400 MHz, d6-DMSO): δ 8.821-8.806 (dd, J1 = 4.4, J2 = 1.6, 2H, pyr-2,6H), 8.143 (s, 1H, indole-2H), 8.081-8.042 (d, J = 15.6, 1H, C=CH), 7.950-7.935 (dd, J1 = 4.4, J2 = 1.6, 2H, pyr-3,5H), 7.517-7.473 (m, 3H, C=CH, indole-4,7H), 6.971-6.942 (dd, J1 = 9.2, J2 = 2.4, 1H, indole-6H), 3.875 (s, 3H, o-methyl), 3.838 (s, 3H, n-methyl). 13C NMR (100 MHz, d6-DMSO): δ 188.2, 155.5, 150.6, 144.9, 140.2, 137.3, 133.1, 126.5, 121.5, 114.2, 112.3, 111.9, 111.5, 102.9, 55.7, 33.4. Melting Point: 178 – 182 °C. TLC (in methylene chloride:methanol 95:5) Rf = 0.35. Elemental analysis calculated for C18H16N2O2 • 0.1 H2O: C, 73.50; H, 5.55; N, 9.52; found: C, 73.34; H, 5.77; N, 9.25.
trans-3-(5-Hydroxy-1H-Indol-3-yl)-1-(4-pyridinyl)- 2-propen-1-one (21)
In a dried 250 mL two-neck round bottom flask under argon, 19 (500 mg, 1.71 mmol) was partially dissolved in anhydrous CH2Cl2 (35 mL) and placed at − 78 °C. BBr3 (17 mL, 1.0 M in CH2Cl2, 17 mmol) was added dropwise via an addition funnel under argon. After addition, the reaction was warmed to room temperature and stirring continued for one hour. Ice-water (50 mL) was added, followed by 1N NaOH until the pH ≈ 12 (~ 60 mL). The CH2Cl2 was further extracted with 1N NaOH (3 × 20 mL). The basic extracts were neutralized with 8N HCl to a neutral pH (~ 8 mL), upon which the product precipitated. The precipitate was filtered and dried under vacuum, providing 454 mg (95%) of a yellow solid. NMR: 1H NMR (600 MHz, d6-DMSO): δ 11.833 (s, 1H, N-H), 9.072 (s, 1H, O-H), 8.818 (s, 2H, pyr-2,6H), 8.064-8.039 (d, J = 15.0, 1H, C=CH), 7.897 (s, 2H, pyr-3,5H), 7.342 (s, 1H, indole-4H), 7.282-7.257 (d, J = 15.0, 1H, C=CH), 7.209-7.195 (d, J = 8.4, 1H, indole-7H), 6.687-6.674 (d, J = 7.8, 1H, indole-6H), 2.542 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 187.7, 153.0, 150.7, 145.9, 145.4, 140.0, 130.2, 126.8, 121.3, 112.2, 112.0, 111.6, 109.1, 105.2, 12.0. Melting Point: 296 – 299 °C. TLC (in ethyl acetate:hexane 4:1) Rf = 0.19. Elemental analysis calculated for C17H14N2O2 • 0.1 CH2Cl2: C, 71.55; H, 4.99; N, 9.79; found: C, 71.89; H, 5.02; N, 9.67.
2-Methyl-1H-indol-5-ol (22)55
A dried 250 mL two-neck round bottom flask was charged with 2-methyl-5-methoxyindole (1.00 g, 6.20 mmol), purged with argon, and CH2Cl2 (30 mL) was added and stirred vigorously until the indole was dissolved. After placing the reaction at −78 °C, BBr3 (37.2 mL, 1.0 M in CH2Cl2, 37.2 mmol, 6 equiv.) was added dropwise via an addition funnel under argon. After addition, the reaction was allowed to slowly warm to room temperature. Thirty minutes after removing the cooling bath, the reaction was poured into ice-water (~ 50 mL) and sat. NaHCO3 (50 mL), to a neutral pH. This was extracted with CH2Cl2 (3 × 50 mL); the aqueous phase retained a yellow color and was acidified to pH ~3 (with 5N HCl) and extracted with ethyl acetate (2 × 50 mL). The combined organic extracts were washed with brine, dried with Na2SO4, filtered and concentrated to a brown oil. After drying under vacuum for 6 hours, 845 mg of a pure brown solid was isolated (93%, 147.17 MW). 1H NMR (600 MHz, d6-DMSO): δ 10.538 (s, 1H, N-H), 8.476 (s, 1H, O-H), 7.028-7.014 (d, J = 8.4, 1H, indole-7H), 6.701-6.698 (d, J = 1.8, 1H, indole-4H), 6.487-6.468 (dd, J1 = 9.0, J2 = 2.4, 1H, indole-6H), 5.906 (s, 1H, indole-3H), 2.305 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 150.4, 135.8, 130.6, 129.4, 110.6, 109.8, 103.3, 98.5, 13.5. Melting point: 131 – 134 °C (published55: 134 °C). TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.28. Elemental analysis calculated for C9H8NO: C, 73.45; H, 6.16; N, 9.52; found: C, 73.09; H, 6.29; N, 9.28.
5-(4-Methylbenzoate)methoxy-2-methyl-1H-indole (23)
In a 250 mL round bottom flask, 2-methyl-1H-indole-5-ol (670 mg, 4.55 mmol) was partially dissolved in CH2Cl2 (50 mL). Tetra-n-butylammonium bromide (808 mg, 2.5 mmol) was added, followed by NaOH (50 mL of a 5 N solution, 250 mmol), and methyl 4-(bromomethyl)benzoate (1.15 g, 5.01 mmol, 1.1 equiv.). After 8 hours, the organic layer was removed, and the aqueous phase was extracted with CH2Cl2 (1 × 30 mL). The combined extracts were washed with brine, dried with Na2SO4, filtered and concentrated to an oil. Purification by column chromatography (1:3 ethyl acetate:hexane) provided 839 mg of pure product (63%, 295.33 MW) [followed by the 3,5-doubly alkylated product (115 mg, 5%)]. 1H NMR (600 MHz, d6-DMSO): δ 10.755 (s, 1H, N-H), 7.980-7.966 (d, J = 8.4, 2H, phenyl-3,5H), 7.602-7.588 (d, J = 8.4, 2H, phenyl-2,6H), 7.150-7.136 (d, J = 8.4, 1H, indole-7H), 6.982-6.978 (d, J = 2.4, 1H, indole-4H) 6.717-6.699 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-6H), 6.005 (s, 1H, indole-3H), 5.157 (s, 2H, methylene), 3.849 (s, 3H, o-methyl), 2.331 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 166.1, 151.9, 143.6, 136.3, 131.4, 129.3, 129.0, 128.7, 127.4, 111.0, 110.1, 102.8, 99.0, 69.0, 52.1, 13.4. Melting point: 151 – 154 °C. TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.49 (doubly alkylated product Rf = 0.42). Elemental analysis calculated for C19H17NO3: C, 73.20; H, 5.80; N, 4.74; found: C, 73.02; H, 5.82; N, 4.55.
5-(4-Methylbenzoate)methoxy-2-methyl-1H-indole-3-carboxaldehyde (24)
To a dried two-neck 250 mL round bottom flask under argon, POCl3 (325 μL, 3.5 mmol) was added to N.N-dimethylformamide (8 mL) at 0 °C. After stirring for five minutes, 23 (320 mg, 1.08 mmol) dissolved in DMF (4 mL) was added dropwise. The yellow solution was slowly warmed to room temperature and after one hour sat. NaHCO3 was added (50 mL), producing a white precipitate, followed by 1N NaOH (20 mL) to complete precipitation (direct workup with NaOH resulted in roughly 1:1 mixture of ester product to ester-hydrolyzed analog). The solid was filtered, rinsed with cold H2O and dried under vacuum, yielding 315 mg of white solid (90%). 1H NMR (600 MHz, d6-DMSO): δ 11.902 (s, 1H, N-H), 10.000 (s, 1H, CHO), 7.991-7.977 (d, J = 8.4, 2H, phenyl-3,5H), 7.664-7.660 (d, J = 2.4, 1H, indole-4H), 7.633-7.619 (d, J = 8.4, 2H, phenyl-2,6H), 7.304-7.290 (d, J = 8.4, 1H, indole-7H), 6.906-6.887 (dd, J1 = 9.0, J2 = 2.4, 1H, indole-6H), 5.211 (s, 2H, methylene), 3.854 (s, 3H, o-methyl), 2.646 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 184.0, 166.1, 154.3, 148.7, 143.2, 130.4, 129.3, 128.8, 127.4, 126.4, 113.6, 112.3, 112.2, 104.0, 69.0, 52.1, 11.5. Melting point: 224 – 227 °C. TLC: (in ethyl acetate:hexanes 4:1) Rf = 0.35. Elemental analysis calculated for C19H17NO4: C, 70.58; H, 5.30; N, 4.33; found: C, 70.45; H, 5.41; N, 4.51.
5-(4-Benzoate)methoxy-2-methyl-1H-indole-3-carboxaldehyde (25)
To a dried two-neck 250 mL round bottom flask under argon, POCl3 (565 μL, 6.1 mmol) was added to N.N-dimethylformamide (12 mL) at 0 °C. After stirring for five minutes, 23 (600 mg, 2.03 mmol) dissolved in DMF (9 mL) was added dropwise. The yellow solution was slowly warmed to room temperature. After one hour, the reaction was cooled to 0 °C and 5N NaOH (90 mL) was added. After stirring for 30 minutes, 5N HCl (95 mL) was added to precipitate the product, which was filtered and dried overnight under vacuum, yielding 626 mg (99%) of white solid. 1H NMR (600 MHz, d6-DMSO): δ 11.944 (s, 1H, N-H), 10.001 (s, 1H, CHO), 7.961-7.947 (d, J = 8.4, 2H, phenyl-3,5H), 7.664-7.660 (d, J = 2.4, 1H, indole-4H), 7.590-7.576 (d, J = 8.4, 2H, phenyl-2,6H), 7.304-7.290 (d, J = 8.4, 1H, indole-7H), 6.902-6.884 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-6H), 5.194 (s, 2H, methylene), 2.647 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 184.0, 167.3, 154.4, 148.8, 142.5, 130.4, 129.4, 127.3, 127.2, 126.4, 113.6, 112.3, 112.2, 104.0, 69.1, 11.5. Melting point: 267 – 270 °C. TLC: (in ethyl acetate:hexanes 4:1) Rf = 0.25.
trans-3-[5-((4-Methylbenzoate)methoxy)-1H-Indol-3-yl)]-1-(4-pyridinyl)-2-propen-1-one (26)
In a dried 100 mL two-neck round bottom flask under argon, 24 (50 mg, 0.15 mmol) was partially dissolved in anhydrous methanol (2 mL). 4-Acetyl-pyridine (26 μL, 0.23 mmol) and piperidine (7 μL, 0.075 mmol) were added and the reaction was refluxed. A yellow precipitate gradually formed, and after 24 hours the reaction was brought to room temperature and the solid was isolated by filtration, rinsed with cold methanol and dried under vacuum, yielding 36 mg. However, crude NMR showed 1:0.15 product:aldehyde. This mixture was dry loaded onto silica and purified by column chromatography (ethyl acetate:hexanes 2:1), providing 22 mg pure product (34 %). 1H NMR (600 MHz, d6-DMSO): δ 11.937 (s, 1H, N-H), 8.829-8.819 (d, J = 6.0, 2H, pyr-2,6H), 8.069-8.044 (d, J = 15.0, 1H, C=CH), 7.971-7.957 (d, J = 8.4, 2H, phenyl-3,5H), 7.905-7.896 (d, J = 5.4, 2H, pyr-3,5H), 7.662-7.649 (d, J = 7.8, 2H, phenyl-2,6H), 7.479-7.475 (d, J = 2.4, 1H, indole-4H), 7.326-7.312 (d, J = 8.4, 1H, indole-7H), 7.284-7.259 (d, J = 15.0, 1H, C=CH), 6.938-6.920 (dd, J1 = 8.7, J2 = 2.1, 1H, indole-6H), 5.358 (s, 2H, methylene), 3.838 (s, 3H, o-methyl), 2.561 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.1, 166.0, 153.9, 150.6, 146.0, 145.1, 143.5, 139.5, 131.3, 129.3, 128.8, 127.4, 126.4, 121.4, 112.8, 112.4, 111.9, 109.3, 104.8, 69.2, 52.2, 12.1. Melting Point: 236 – 239 °C. TLC (ethyl acetate:hexanes 4:1) Rf = 0.26. Elemental analysis calculated for C26H22N2O4 • 0.25 C4H8O2: C, 72.31; H, 5.39; N, 6.25; found: C, 72.62; H, 5.64; N, 5.95.
trans-3-[5-((4-Carboxyphenyl)-methoxy)-1H-Indol-3-yl)]-1-(4-pyridinyl)-2-propen-1-one (27)
In a dried 100 mL round bottom flask under argon, 25 (200 mg, 0.65 mmol) was partially dissolved in anhydrous methanol (15 mL). 4-Acetyl-pyridine (107 μL, 0.97 mmol, 1.5 equiv.) and piperidine (256 μL, 0.65 mmol) were added and the reaction was refluxed. After 24 hours, the solid precipitate was isolated by filtration, yielding 75 mg of product contaminated with aldehyde. Due to both solubility and poor filtration, plenty of product remained in the filtrate; thus, the filtrate was dry loaded onto silica and purified by column chromatography (methylene chloride:methanol 9:1), yielding 85 mg of purified acid product (21%). 1H NMR (600 MHz, d6-DMSO): δ 11.947 (s, 1H, N-H), 8.831-8.821 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-2,6H), 8.073-8.047 (d, J = 15.6, 1H, C=CH), 7.954-7.941 (d, J = 7.8, 2H, phenyl-3,5H), 7.904-7.894 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-3,5H), 7.630-7.616 (d, J = 8.4, 2H, phenyl-2,6H), 7.479-7.476 (d, J = 1.8, 1H, indole-4H), 7.326-7.312 (d, J = 8.4, 1H, indole-7H), 7.289-7.264 (d, J = 15.0, 1H, C=CH), 6.939-6.920 (dd, J1 = 9.0, J2 = 2.4, 1H, indole-6H), 5.349 (s, 2H, methylene), 2.562 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.1, 167.3, 154.0, 150.6, 146.0, 145.1, 142.8, 139.6, 131.2, 130.3, 129.5, 127.2, 126.4, 121.4, 112.8, 112.4, 111.9, 109.3, 104.8, 69.3, 12.1. Melting point: 269 – 273 °C. TLC (methylene chloride:methanol 9:1) Rf = 0.41. Elemental analysis calculated for C25H20N2O4 • 0.25 CH2Cl2 • 0.25 CH3OH: C, 69.34; H, 4.91; N, 6.34; found: 69.28; H, 5.22; N, 6.72.
2-Methyl-5-benzoyl-indole-3-carboxaldehyde and 2-Methyl-6-benzoyl-indole-3-carboxaldehyde (28 and 29)
In a dried two-neck 250 mL round bottom flask under argon, N,N-dimethylformamide (339 μL, 4.4 mmol) was added to 1,2-dichloroethane (8 mL). The reaction was cooled to 0 °C and oxalyl chloride (377 μL, 4.4 mmol) dissolved in 1,2-DCE (8 mL) was slowly added, forming a white heterogeneous mixture. The mixture was allowed to warm to room temperature while stirring. After fifteen minutes, the reaction was cooled to 0 °C and 2-methylindole (577 mg, 4.0 mmol) dissolved in 1,2-DCE (8 mL) was slowly added, forming a dark red solution. After one hour, AlCl3 (1.96 g, 14.7 mmol) was added and stirred vigorously. Benzoyl chloride (510 μL, 4.4 mmol) dissolved in 1,2-DCE (4 mL) was slowly added and the reaction was warmed to room temperature and stirred overnight. After 24 hours, cold H2O (50 mL) was added, followed by 5N NaOH (10 mL), and the mixture was stirred. After one hour, 5N HCl (18 mL) was added and this was extracted with methylene chloride (3 × 50 mL). The combined 1,2-DCE and methylene chloride extracts were combined, dried with Na2SO4, filtered and concentrated. The crude product mixture was purified by column chromatography (ethyl acetate: hexanes 1:1 → 4:1), yielding 5-benzoyl product 28 (142 mg, 13%) and 6-benzoyl product 29 (429 mg, 41%) (1:3 regioselectivity for 5 vs. 6 benzoylation).
2-Methyl-5-benzoyl-indole-3-carboxaldehyde (28)
1H NMR (600 MHz, d6-DMSO): δ 12.371 (s, 1H, N-H), 10.073 (s, 1H), CHO, 8.462 (s, 1H, indole-4H), 7.723-7.711 (m, 2H, phenyl-2,6H), 7.673-7.656 (m, 2H, indole-6H, phenyl-4H), 7.587-7.561 (t, J = 7.8, 2H, phenyl-3,5H), 7.553-7.539 (d, J = 8.4, 1H, indole-7H), 2.728 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 196.1, 184.8, 150.8, 138.3, 138.0, 132.1, 130.9, 129.5, 128.5, 124.9, 124.7, 123.4, 114.4, 111.6, 11.6. 1-D nOe: irradiation of peak at δ 12.371 (N-H): nOe signal enhancement seen at δ 7.549 (C-H) and 2.728 (CH3). The H peak at 7.549 has ortho coupling (J = 8.4), proving the benzoyl group inserted at the 5 position, not 6. Separately, irradiation of peak at δ 7.540 (C-H with ortho coupling): nOe signal enhancement seen at δ 12.370 (N-H), again proving the proximity of N-H to an ortho-coupled C7-H; signal enhancement also seen at δ 7.66 for C6-H (the benzoyl 2H triplet peak at δ 7.574 was also irradiated by proximity, thus signal enhancement was seen for the other benzoyl C-H peaks at δ 7.72 and δ 7.67). Melting point: 227 – 230 °C. TLC (ethyl acetate:hexanes 4:1) Rf = 0.32. Elemental analysis calculated for C17H13NO2 • 0.2 C4H8O2 (trace ethyl acetate): C, 76.11: H, 5.24; N, 4.99; found: C, 75.74; H, 4.94; N, 5.06.
2-Methyl-6-benzoyl-indole-3-carboxaldehyde (29)
1H NMR (600 MHz, d6-DMSO): δ 12.323 (s, 1H, N-H), 10.116 (s, 1H, CHO), 8.173-8.159 (d, J = 8.4, 1H, indole-4H), 7.776-7.774 (d, J = 1.2, 1H, indole-7H), 7.741-7.729 (m, 2H, phenyl-2,6H), 7.683-7.659 (t, J = 7.2, 1H, phenyl-4H), 7.627-7.611 (dd, J1 = 7.8, J2 = 1.8, 1H, indole-5H), 7.583-7.558 (t, J = 7.5, 2H, phenyl-3,5H), 2.744 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 196.3, 185.4, 152.2, 138.7, 135.3, 132.8, 131.8, 130.1, 129.8, 129.1, 124.4, 120.3, 114.7, 114.4, 12.5. 1-D nOe: irradiation of peak at δ 12.323 (N-H): nOe signal enhancement seen at δ 7.775 (C-H with meta coupling, thus proving benzoyl addition to position 6 of indole) and δ 2.744 (CH3). Separately, irradiation of the peak at δ 7.775 (C-H with meta coupling: J = 1.2) led to signal enhancement at δ 12.323 (N-H peak), thus proving proximity of meta-coupled C-H to N-H (the benzoyl 2,6 2H peak at 7.73 was also irradiated by proximity, leading to signal enhancement of benzoyl 3,5 2H peak at δ 7.57). Melting point: 192 – 196 °C. TLC (ethyl acetate:hexanes 4:1) Rf = 0.40. Elemental analysis calculated for C17H13NO2: C, 77.55; H, 4.98; N, 5.32; found: C, 77.28; H, 4.97; N, 5.15.
trans-3-(5-Benzoyl-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (30)
In a dried 100 mL round bottom flask under argon, 28 (50 mg, 0.19 mmol) was dissolved in anhydrous methanol (3 mL). 4-Acetyl-pyridine (21 μL, 0.19 mmol) and piperidine (4 μL, 0.04 mmol) were added and the reaction was refluxed. After twelve hours, 0.3 equivalents of 4-acetyl-pyridine (6.3 μL,.06 mmol) was added. After a total of 24 hours reaction time, the reaction was cooled and the precipitate was isolated by filtration and rinsed with cold methanol, providing 37 mg of yellow solid. A crude 1H NMR showed a 1:3.5 ratio of product to aldehyde. The filtrate was concentrated and added to the product/aldehyde mixture, and 0.8 equivalents of both 4-acetyl-pyridine (17 μL, 0.15 mmol) and piperidine (15 μL, 0.15 mmol) were added to the reaction. This was refluxed for another 24 hours, after which the reaction was cooled, filtered and rinsed with cold methanol, providing 9 mg (13%) pure, yellow product. 1H NMR (600 MHz, d6-DMSO): δ 12.397 (s, 1H, N-H), 8.815-8.805 (m, 2H, pyr-2.6H), 8.321 (s, 1H, indole-4H), 8.081-8.056 (d, J = 15.0, 1H, C=CH), 7.820-7.808 (d, J = 7.2, 2H, phenyl-2,6H), 7.761-7.728 (m, 4H, phenyl-4H, indole-6H, pyr-3,5H), 7.644-7.619 (t, J = 7.5, 2H, phenyl-3,5H), 7.587-7.573 (d, J = 8.4, 1H, indole-7H), 7.347-7.321 (d, J = 15.6, 1H, C=CH), 2.639 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 195.7, 187.9, 150.6, 146.9, 144.7, 138.8, 138.5, 138.4, 131.9, 130.1, 129.4, 128.4, 125.2, 123.9, 123.6, 121.1, 114.6, 111.9, 110.1, 12.1. Melting point: 258 – 262 °C. TLC (ethyl acetate:hexanes 4:1) Rf = 0.24. Elemental analysis calculated for C24H18N2O2 • 0.65 H2O: C, 76.24; H, 5.14; N, 7.41; C, 75.96; H, 4.75; N, 7.16.
trans-3-(6-Benzoyl-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (31)
In a dried 100 mL round bottom flask under argon, 29 (50 mg, 0.19 mmol) was dissolved in anhydrous methanol (3 mL). 4-Acetyl-pyridine (21 μL, 0.19 mmol) and piperidine (19 μL, 0.19 mmol) were added and the reaction was refluxed. After 24 hours, the reaction was cooled, filtered and rinsed with cold hexanes and dried under vacuum, yielding 52 mg (74 %) of pure yellow solid. 1H NMR (600 MHz, d6-DMSO): δ 12.350 (s, 1H, N-H), 8.832-8.222 (d, J = 6.0, 2H, pyr-2,6H), 8.255-8.241 (d, J = 8.4, 1H, indole-4H), 8.125-8.099 (d, J = 15.6, 1H, C=CH), 8.008-7.999 (d, J = 5.4, 2H, pyr-3,5H), 7.801 (s, 1H, indole-7H), 7.766-7.754 (d, J = 7.2, 2 H, phenyl-2,6H), 7.683-7.642 (m, 2H, phenyl-4H, indole-5H), 7.585-7.563 (m, 3H, C=CH, phenyl-3,5H), 2.659 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 195.5, 188.2, 150.6, 148.5, 144.6, 138.7, 138.2, 135.4, 132.0, 130.6, 129.4, 129.2, 128.4, 123.1, 121.5, 119.9, 114.5, 114.1, 109.6, 12.3. Melting point: 267 – 270 °C. TLC (ethyl acetate:hexanes 4:1) Rf = 0.29. Elemental analysis calculated for C24H18N2O2 • 0.05 H2O: C, 78.48; H, 4.97; N, 7.63; found: C, 78.08; H, 5.01; N, 7.50.
2-Methyl-5-nitro-1H-indole (32)26
In a 250 mL round bottom flask, 2.62 grams of 2-methyl-indole (20 mmol) was dissolved in 20 mL of H2SO4 after vigorous stirring. In a separate flask, 1.87 grams of NaNO3 (1.1x, 22 mmol, 84.99 MW) was dissolved in 20 mL of H2SO4, also after vigorous stirring, and added dropwise via addition funnel to the indole. After addition, the reaction was stirred for another 10 minutes, and then poured into 400 mL of ice-water, precipitating a yellow product. The product was isolated via filtration and washed with cold water. After 12 hours of drying under vacuum, 3.35 grams of yellow product was isolated (95%). 1H NMR (600 MHz, d6-DMSO): δ 11.703 (s, 1H, N-H), 8.4003-8.400 (d, J = 1.8, 1H, indole-4H), 7.916-7.898 (dd, J1 = 9.0, J2 = 1.8, 1H, indole-6H), 7.422-7.407 (d, 1H, J = 9.0, 1H, indole-7H), 6.408 (s, 1H, indole-3H), 2.419 (s, 1H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 140.5, 140.0, 139.4, 128.0, 115.9, 115.7, 110.7, 101.6, 13.4. 1-D nOe: irradiation of peak at δ 7.422-7.407 (C7-H): nOe signal enhancement seen at δ 11.690 (N-H) and 7.916-7.897 (C6-H). Irradiation of peak at δ 11.690 (N-H): nOe signal enhancement seen at δ 7.422-7.407 (C7-H) and 2.419 (C2-CH3) (nOe observed between the C-H proton with ortho coupling and N-H proton, thus proving NO2 inserted at indole C-5 vs. C-6). Melting point: 166 – 169 °C (published26: 176–176.5 °C). TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.38. Elemental analysis calculated for C9H8N2O2: C, 61.36; H, 4.58; N, 15.90; found: C, 61.54; H, 4.63; N, 15.89.
2-Methyl-5-amino-1H-indole (33)26
32 (1.00 grams, 5.68 mmol) was dissolved in 75 mL ethanol. 10% Pd/C was added (200 mg) and the mixture was subjected to H2 (38 psi) using a Parr hydrogenator for 3.5 hours. The mixture was filtered over celite, which was washed with methanol. After concentration and drying under vacuum, 801 mg of a brown powder (97%) was isolated. 1H NMR (600 MHz, d6-DMSO): δ 10.370 (s, 1H, N-H), 6.943-6.929 (d, J = 8.4, 1H, indole-7H), 6.552-6.549 (d, J = 1.8, 1H, indole-4H), 6.373-6.355 (dd, J1 = 8.4, J2 = 2.4, 1H), 5.815 (s, 1H, indole-6H), 4.436 (bs, 2H, NH2), 2.288 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 140.8, 135.0, 129.9, 129.6, 110.5, 110.3, 102.9, 98.0, 13.5. Melting point: 147 – 150 °C (published26: 157–159 °C). TLC (in 3:1 ethyl acetate:hexanes) Rf = 0.30; (in 1:1 ethyl acetate:hexanes) Rf = 0.16. Elemental analysis calculated for C9H10N2 • 0.05 C2H6O: C, 73.61; H, 6.99; N, 18.87; found: C, 73.48; H, 6.77; N, 18.81.
2-Methyl-5-azido-1H-indole (34)
In an oven dried, 250 mL round bottom flask flushed with argon, 33 (400 mg, 2.74 mmol, 146.19 MW) was dissolved in 90 % AcOH (20 mL). After complete solvation, the reaction was placed at 0 °C and protected from light with foil. NaNO2 (1.1x, 3.01 mmol, 208 mg, 69.00 MW) dissolved in cold H2O (2 mL) was added dropwise and stirred for ten minutes. NaN3 (1.1x, 3.01 mmol, 196 mg, 65.01 MW) dissolved in cold H2O (2 mL ) was added dropwise. After one hour the reaction was poured into water (40 mL), which was extracted 3 times with CH2Cl2 (40 mL each). The extracts were washed with sodium bicarbonate and brine (100 mL each), dried with sodium sulfate, filtered and concentrated. Purification by silica flash chromatography (100% CH2Cl2) yielded the azide as a light brown solid (310 mg, 66%). 1H NMR (600 MHz, d6-DMSO): δ 11.025 (s, 1H, N-H), 7.295-7.281 (d, J = 8.4, 1H, indole-7H), 7.131-7.127 (d, J = 2.4, 1H, indole-4H), 6.737-6.719 (dd, J1 = 8.4, J2 = 2.4, 1H, indole-6H), 6.108-6.106 (d, J = 1.2, 1H, indole-3H), 2.369 (s, 1H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 137.5, 133.9, 130.0, 129.5, 111.7, 111.6, 108.5, 99.0, 13.4. Melting point: 57 – 60 °C. TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.56. IR (film): 3409 cm.−1, 2111 (aryl-azide). Elemental analysis calculated for C9H8N4: C, 62.78; H, 4.68; N, 32.54; found: C, 62.95; H, 4.68; N, 32.53.
2-Methyl-3-carboxaldehyde-5-azido-1H-indole (35)
In an oven dried, 100 mL round bottom flask purged with argon, POCl3 (1.5x, 3.14 mmol, 291 μL) was added to anhydrous DMF (2.0 mL) at 0 °C. 34 (dissolved in DMF, 2 mL) was added dropwise. The stirred reaction was slowly warmed to RT. 1N NaOH (25 mL) and water (25 mL) was added, forming a precipitate which was filtered and rinsed with cold water. The resulting white solid was dried under vacuum, affording 369 mg (88%). 1H NMR (600 MHz, d6-DMSO): δ 12.078 (s, 1H, N-H), 10.031 (s, 1H, CHO), 7.754-7.751 (d, J = 1.8, 1H, indole-4H), 7.423-7.409 (d, J = 8.4, 1H, indole-7H), 6.913-6.896 (dd, J1 = 8.4, J2 = 1.8, 1H, indole-6H), 2.676 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 184.3, 149.7, 133.4, 133.0, 126.6, 114.4, 113.4, 112.8, 109.4, 11.5. Melting point: degradation ca. 150 °C. TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.19. Elemental analysis calculated for C10H8N4O: C, 59.99; H, 4.03; N, 27.99; found: C, 60.03; H, 4.07; N, 27.96.
trans-3-(5-Azido-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (36)
In a dried, 100 mL round bottom flask under argon and protected from light, 35 (80 mg, 0.40 mmol) was partially dissolved in anhydrous methanol (5 mL). 4-Acetyl-pyridine (1.5 equiv., 66 μL, 0.60 mmol) and piperidine (40 μL, 0.40 mmol) were added and the reaction was stirred at 40 °C. After 24 hours, the reaction was cooled to RT and the yellow precipitate was isolated via filtration and rinsed with cold methanol. A crude 1H NMR of this product showed a ca. 1:3 ratio of product to the indole starting material. This crude product was redissolved in anhydrous methanol (5 mL) along with the concentrated filtrate, and another equivalent of 4-acetyl-pyridine (44 μL, 2.5 equiv. total) and five equivalents of piperidine (200 μL, 6 equiv. total) were added. The stirred reaction was heated at 40 °C. After 12 hours, a yellow precipitate was filtered and rinsed with cold methanol and dried under vacuum, affording 47 mg (39%) of pure product. 1H NMR (600 MHz, d6-DMSO): δ 12.105 (s, 1H, N-H), 8.816-8.806 (dd, J1 = 4.8, J2 = 1.8, 2H, pyr-2,6H), 8.066-8.041 (d, J = 15, 1H, C=CH), 7.956-7.945 (dd, J1 = 4.8, J2 = 1.8, 2H, pyr-3,5H), 7.661-7.657 (d, J = 2.4, 1H, indole-4H), 7.455-7.441 (d, J = 8.4, 1H, indole-7H), 7.401-7.375 (d, J = 15.6, 1H, C=CH), 7.001-6.984 (dd, J1 = 8.4, J2 = 1.8, indole-6H), 2.587 (s, 3H, methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.2, 150.6, 146.5, 144.9, 139.0, 133.9, 132.9, 126.8, 121.5, 113.9, 113.8, 113.0, 110.3, 109.2, 12.2. Melting point: decomposes ca. 190 °C. TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.21. Elemental analysis calculated for C17H13N5O: C, 67.32; H, 4.32; N, 23.09; found: C, 67.37; H, 4.33; N, 23.12.
2-Methyl-5-methoxy-6-nitro-1H-indole (37)
In a 100 mL round bottom flask, 2-methyl-5-methoxyindole (370 mg, 2.30 mmol) was dissolved in H2SO4 (4 mL) after vigorous stirring, then placed at 0 °C. In a separate flask, 1.87 grams of NaNO3 (214 mg, 2.52 mmol) was dissolved in H2SO4, (4 mL) also after vigorous stirring, chilled to 0 °C, and added dropwise to the indole. After addition, the reaction was stirred for another 10 minutes, and then poured into ice-water (200 mL), precipitating a yellow product, which was extracted with ethyl acetate (3 × 100 mL). Extracts were washed with sat. NaHCO3 and brine, dried with Na2SO4, filtered and concentrated. Purification by column chromatography (ethyl acetate:hexane 2:3 → 3:2) yielded 296 mg of pure product (62%), (followed by 73 mg of 4-nitrated product, 15%; 4:1 regioselectivity). 1H NMR (600 MHz, CDCl3): δ 8.350 (s, 1H, N-H), 7.983 (s, 1H, indole-7H), 7.066 (s, 1H, indole-4H), 6.211 (s, 1H, indole-3H), 3.958 (s, 3H, o-methyl), 2.477 (s, 3H, c-methyl). 13C NMR (150 MHz, CDCl3): δ 148.7, 142.8, 134.7, 134.2, 129.1, 109.2, 102.7, 101.3, 57.1, 14.2. Melting point: 134 – 138 °C. TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.37 (4-nitro product Rf = 0.25). Elemental analysis calculated for C10H10N2O3 • 0.1 C6H14: C, 59.27; H, 5.35; N, 13.04; found: C, 59.33; H, 5.10; N, 12.98.
2-Methyl-5-methoxy-6-amino-1H-indole (38)
In a hydrogenation flask, 37 (270 mg, 1.31 mmol) was dissolved in 100% EtOH (35 mL) and 10 % Pd/C (40 mg) was added. The mixture was hydrogenated on a Parr hydrogenator at 40 p.s.i. for 45 minutes. The red-pink solution was filtered over celite and rinsed with MeOH, concentrated and dried under vacuum, yielding 230 mg of pure product (99%). 1H NMR (400 MHz, CDCl3): δ 7.516 (s, 1H, N-H), 6.907 (s, 1H), 6.607 (s, 1H), 6.052 (s, 1H, indole-3H), 3.873 (s, 3H, o-methyl), 3.733 (bs, 2H, NH2), 2.365 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 142.8, 132.9, 131.5, 131.4, 119.5, 100.5, 98.7, 95.7, 55.6, 13.5 Melting point: 144 – 148 °C. TLC (in 3:1 ethyl acetate:hexanes) Rf = 0.33. Elemental analysis calculated for C10H10N2O3 • 0.1 C6H14: C, 59.27; H, 5.35; N, 13.04; found: C, 59.33; H, 5.10; N, 12.98.
2-Methyl-5-methoxy-6-azido-1H-indole (39)
In a dried, 100 mL round bottom flask under argon, 38 (227 mg, 1.29 mmol) was dissolved in 90% AcOH (10 mL) and placed at 0 °C. The flask was covered with foil and the reaction was conducted in low light. NaNO2 (98 mg, 1.43 mmol, 1.1 equiv) dissolved in H2O (1 mL) was added dropwise, and the mixture stirred for 10 minutes. NaN3 (92 mg, 1.43 mmol, 1.1 equiv.) dissolved in H2O (1 mL) was added dropwise. After 45 minutes, the mixture was slowly poured into H2O (25 mL) and sat. K2CO3 (19 mL) to form a neutral pH. This was extracted with ethyl acetate (4 × 50 mL), washed with brine, dried with Na2SO4, filtered and concentrated. The crude product was purified by column chromatography (ethyl acetate:hexanes 1:4), yielding 139 mg of pure oil (53 %). 1H NMR (600 MHz, CDCl3): δ 7.782 (s, 1H, N-H), 7.007 (s, 1H), 6.883 (s, 1H), 6.145 (s, 1H, indole-3H), 3.890 (s, 3H, o-methyl), 2.388 (s, 3H, c-methyl). 13C NMR (150 MHz, CDCl3): δ 147.0, 136.1, 130.7, 126.5, 123.1, 102.4, 102.0, 100.4, 56.5, 13.7. TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.60.
2-Methyl-3-carboxaldehyde-5-methoxy-6-azido-1H-indole (40)
In an oven dried, 100 mL round bottom flask, purged with argon and protected from light with foil, POCl3 (84 μL, 0.90 mmol, 1.5 equiv.) was added to anhydrous DMF (1.0 mL) at 0 °C and stirred for 5 minutes. Next, 39 (dissolved in DMF, 1 mL) was added dropwise. The stirred reaction was slowly warmed to RT. 1N NaOH (10 mL) and water (10 mL) were added, and this was extracted with CH2Cl2 (3 × 20 mL). Extracts were washed with brine, dried with Na2SO4, filtered, concentrated and dried overnight under vacuum, producing 95 mg pure product (69%). 1H NMR (600 MHz, d6-DMSO): δ 11.865 (s, 1H, N-H), 10.004 (s, 1H, CHO), 7.645 (s, 1H, indole-4H), 7.032 (s, 1H, indole-7H), 3.857 (s, 3H, o-methyl), 2.645 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 184.8, 149.5, 149.4, 130.3, 124.1, 124.0, 114.4, 104.2, 103.5, 56.9, 12.2. TLC (in 1:1 ethyl acetate:hexanes) Rf = 0.12.
trans-3-(6-Azido-5-methoxy-2-methyl-1H-indol-3-yl)-1-(4-pyridinyl)-2-propen-1-one (41)
In a dried, 100 mL round bottom flask under argon and protected from light, 40 (70 mg, 0.30 mmol) was partially dissolved in anhydrous methanol (5 mL). 4-Acetyl-pyridine (50 μL, 0.45 mmol, 1.5 equiv.) and piperidine (148 μL, 1.5 mmol, 5 equiv.) were added and the reaction was stirred at 40 °C. After 7 hours, the reaction was cooled to RT and the precipitate was isolated via filtration and rinsed with cold methanol. A crude 1H NMR of this product showed a mixture of aldehyde with trace product. This mixture was redissolved in the filtrate and more 4-acetyl-pyridine (165 μL, 5 equiv.) and piperidine (148 μL, 5 equiv.) was added and the mixture was set to react at 40 °C. After 48 hours, although starting materials are still seen on TLC, the crude reaction mixture was dry loaded onto silica and purified by column chromatography (ethyl acetate:hexane 3:1), eluting first the aldehyde, followed by the ketone and finally the product as a yellow solid (39 mg, 39%). 1H NMR (600 MHz, d6-DMSO): δ 11.784 (s, 1H, N-H), 8.815-8.805 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-2,6H), 8.073-8.047 (d, J = 15.6, 1H, C=CH), 7.957-7.947 (dd, J1 = 4.2, J2 = 1.8, 2H, pyr-3,5H), 7.538 (s, 1H, indole-4H), 7.410-7.384 (d, J = 15.6, 1H, indole C=CH), 7.051 (s, 1H, indole-7H), 3.982 (s, 3H, o-methyl), 2.569 (s, 3H, c-methyl). 13C NMR (150 MHz, d6-DMSO): δ 188.2, 150.6, 148.5, 145.6, 145.0, 139.1, 130.6, 123.6, 123.3, 121.5, 113.5, 109.5, 103.8, 103.7, 56.9, 12.3. Melting point: degrades to black substance ca. 160 °C. TLC (in 4:1 ethyl acetate:hexanes) Rf = 0.15. Elemental analysis calculated for C18H15N5O2 • C4H8O2 (trace ethyl acetate): C, 64.72; H, 4.60; N, 20.74; found: C, 65.11; H, 4.71; N, 20.39.
Library Compounds
Compounds 1–8 used for initial screening of methuosis-inducing activity (Fig. 1) were obtained from Hit2Lead.com, a division of Chembridge Corp. The identification numbers of the compounds were: 1, 5224450; 2, 5224466; 3, 5312531; 4, 7995005; 5, 7916760; 6, 6161388; 7, 5267766; 8, 6155359. All compounds are certified by the vendor to be at least 90% pure with NMR confirmation of structure.
Cell Culture. Cell Culture
U251 human glioblastoma cells were purchased from the DCT Tumor Repository (National Cancer Institute, Frederick, MD). MCF-7 mammary carcinoma cells were obtained from The American Type Culture Collection, Rockville, MD. Temozolomide-resistant U251 cells (U251-TR) were derived in our laboratory as described previously. 10 MCF-7 cells selected for resistance to doxorubicin56 were provided by Amadeo Parissenti, Northeastern Ontario Regional Cancer Centre. Normal human skin fibroblasts were derived from a skin biopsy as described previously. 57 Unless stated otherwise, cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% (v/v) fetal bovine serum (FBS) (JR Scientific, Woodland, CA) at 37°C in an atmosphere of 5% CO2/95% air. Normal pre-stasis human mammary epithelial cells (specimen 184) were provided by Martha Stampfer, Lawrence Berkeley Lab, Berkeley, CA. The HMECs were maintained in M87A medium supplemented with cholera toxin and oxytocin, essentially as described. 58
Cell Proliferation and Morphology
To generate cell growth curves, U251 cells were plated in 35 mm dishes (100,000 cells/dish, Fig. 4B) and allowed to attach for 24 h. Thereafter, cells were treated with the indicated compounds dissolved in DMSO or with vehicle alone. At daily intervals, three parallel cultures were harvested from each group by trypsinization and aliquots of cell suspension were counted in a Coulter Z1 particle counter. Phase-contrast images of live cells were obtained using an Olympus IX70 inverted microscope equipped with a digital camera and SPOT imaging software (Diagnostic Instruments, Inc., Sterling Heights, MI).
Cell Viability
Cells were seeded in 96-well plates, with four replicate wells for each culture condition. After addition of the indicated concentrations of compounds, cell viability was determined at a 48 h end-point using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)-based assay as described. 10 Absorbance at 570 nm was quantified on a SpectraMax Plus 384 plate reader (Molecular Devices, Sunnyvale, CA).
Colony Formation
Cells were plated in 100 mm dishes at 2,500 (U251 and U251-TR) or 1,500 (MCF-7 and MCF-7 DoxR) cells per dish. Beginning on the day after plating, the cells were exposed to the indicated compounds for the periods of time noted in the figure legends. The medium was then replaced (without compounds) and cells were incubated for 10 days, with fresh medium added every 2 d. Colonies were visualized by washing with phosphate-buffered normal saline, fixing for 10 min with ice-cold 100% methanol, and staining with 1% (w/v) crystal violet (Acros Organics, Fisher Scientific, Pittsburgh, PA) in 35% methanol. After 2–3 washes with water, colonies containing at least 50 cells were counted using a dissecting microscope or a Protocol 2 colony counter (Synbiosis, Frederick MD).
Supplementary Material
Acknowledgments
The work was supported by grant R01 CA115495 (W. Maltese) from the National Institutes of Health.
ABBREVIATIONS USED
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- GBM
glioblastoma multiforme
- HMEC
human mammary epithelial cells
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- RT
room temperature
- TMZ
temozolomide
Footnotes
Spectral data for all synthesized compounds are available free of charge via the internet.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Norbury CJ, Zhivotovsky B. DNA damage-induced apoptosis. Oncogene. 2004;23:2797–2808. doi: 10.1038/sj.onc.1207532. [DOI] [PubMed] [Google Scholar]
- 3.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequnt co-alterations of TP53, p16/CDKN2A, p14arf, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 5.Bocangel DB, Finkelstein S, Schold SC, Bhakat KK, Mitra S, Kokkinakis DM. Multifaceted resistance of gliomas to temozolomide. Clin Cancer Res. 2002;8:2725–2734. [PubMed] [Google Scholar]
- 6.Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res. 2008;6:965–977. doi: 10.1158/1541-7786.MCR-07-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhanot H, Young AM, Overmeyer JH, Maltese WA. Induction of non-apoptotic cell death by activated Ras requires inverse regulation of Rac1 and Arf6. Mol Cancer Res. 2010;8:1358–1374. doi: 10.1158/1541-7786.MCR-10-0090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swanson JA, Watts C. Macropinocytosis. Trends Cell Biol. 1995;5:424–428. doi: 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 9.Donaldson JG, Porat-Shliom N, Cohen LA. Clathrin-independent endocytosis: A unique platform for cell signaling and PM remodeling. Cell Signal. 2009;21:1–6. doi: 10.1016/j.cellsig.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Overmeyer JH, Young AM, Bhanot H, Maltese WA. A Chalcone-Related Small Molecule That Induces Methuosis, a Novel Form of Non-Apoptotic Cell Death, in Glioblastoma Cells. Mol Cancer. 2011;10:69. doi: 10.1186/1476-4598-10-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cerny J, Feng Y, Yu A, Miyake K, Borgonovo B, Klumperman J, Meldolesi J, McNeil PL, Kirchhausen T. The small chemical vacuolin-1 inhibits Ca(2+)-dependent lysosomal exocytosis but not cell resealing. EMBO Rep. 2004;5:883–888. doi: 10.1038/sj.embor.7400243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Go ML, Wu X, Liu XL. Chalcones: an update on cytotoxic and chemoprotective properties. Curr Med Chem. 2005;12:481–499. doi: 10.2174/0929867053363153. [DOI] [PubMed] [Google Scholar]
- 13.Kamal A, Ramakrishna G, Raju P, Viswanath A, Ramaiah MJ, Balakishan G, Pal-Bhadra M. Synthesis and anti-cancer activity of chalcone linked imidazolones. Bioorg Med Chem Lett. 2010;20:4865–4869. doi: 10.1016/j.bmcl.2010.06.097. [DOI] [PubMed] [Google Scholar]
- 14.Kumar D, Kumar NM, Akamatsu K, Kusaka E, Harada H, Ito T. Synthesis and biological evaluation of indolyl chalcones as antitumor agents. Bioorg Med Chem Lett. 2010;20:3916–3919. doi: 10.1016/j.bmcl.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 15.Lindwall HG, Order RBV. 3-Indole aldehyde and certain of its condensation products. J Org Chem. 1945;10:128–133. [Google Scholar]
- 16.Pasquini S, Mugnaini C, Brizzi A, Ligresti A, Di MV, Ghiron C, Corelli F. Rapid combinatorial access to a library of 1,5-disubstituted-3-indole-N-alkylacetamides as CB2 receptor ligands. J Comb Chem. 2009;11:795–798. doi: 10.1021/cc9000955. [DOI] [PubMed] [Google Scholar]
- 17.Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem Biol. 2011;6:34–46. doi: 10.1021/cb100294v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hatanaka Y, Sadakane Y. Photoaffinity labeling in drug discovery and developments: chemical gateway for entering proteomic frontier. Curr Top Med Chem. 2002;2:271–288. doi: 10.2174/1568026023394182. [DOI] [PubMed] [Google Scholar]
- 19.Galardy RE, Craig LC, Printz MP. Benzophenone triplet: a new photochemical probe of biological ligand-receptor interactions. Nat New Biol. 1973;242:127–128. doi: 10.1038/newbio242127a0. [DOI] [PubMed] [Google Scholar]
- 20.Liu D, O’Leary B, Iruthayanathan M, Love-Homan L, Perez-Hernandez N, Olivo HF, Dillon JS. Evaluation of a novel photoactive and biotinylated dehydroepiandrosterone analog. Mol Cell Endocrinol. 2010;328:56–62. doi: 10.1016/j.mce.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Hasegawa M, Miura T, Kuzuya K, Inoue A, Won KS, Horinouchi S, Yoshida T, Kunoh T, Koseki K, Mino K, Sasaki R, Yoshida M, Mizukami T. Identification of SAP155 as the target of GEX1A (Herboxidiene), an antitumor natural product. ACS Chem Biol. 2011;6:229–233. doi: 10.1021/cb100248e. [DOI] [PubMed] [Google Scholar]
- 22.Demopoulos VJ, Nicolaou I. Electrophilic substitution of indole on the benzene moiety: A synthesis of 5-acyl- and 5-aroylindoles. Synthesis. 1998;1998:1519–1522. [Google Scholar]
- 23.Miller DK, Gillard JW, Vickers PJ, Sadowski S, Leveille C, Mancini JA, Charleson P, Dixon RA, Ford-Hutchinson AW, Fortin R. Identification and isolation of a membrane protein necessary for leukotriene production. Nature. 1990;343:278–281. doi: 10.1038/343278a0. [DOI] [PubMed] [Google Scholar]
- 24.Palnitkar SS, Bin B, Jimenez LS, Morimoto H, Williams PG, Paul-Pletzer K, Parness J. [3H]Azidodantrolene: synthesis and use in identification of a putative skeletal muscle dantrolene binding site in sarcoplasmic reticulum. J Med Chem. 1999;42:1872–1880. doi: 10.1021/jm9805079. [DOI] [PubMed] [Google Scholar]
- 25.He G, Luo W, Li P, Remmers C, Netzer WJ, Hendrick J, Bettayeb K, Flajolet M, Gorelick F, Wennogle LP, Greengard P. Gamma-secretase activating protein is a therapeutic target for Alzheimer’s disease. Nature. 2010;467:95–98. doi: 10.1038/nature09325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noland WE, Smith LR, Johnson DC. Nitration of indoles: II. The mononitration of methylindoles. J Org Chem. 1963;28:2262–2266. [Google Scholar]
- 27.Melhado LL, Leonard NJ. An efficient synthesis of azidoindoles and azidotryptophans. J Org Chem. 1983;48:5130–5133. [Google Scholar]
- 28.Uetrecht J. Idiosyncratic drug reactions: current understanding. Annu Rev Pharmacol Toxicol. 2007;47:513–539. doi: 10.1146/annurev.pharmtox.47.120505.105150. [DOI] [PubMed] [Google Scholar]
- 29.Raja SM, Clubb RJ, Ortega-Cava C, Williams SH, Bailey TA, Duan L, Zhao X, Reddi AL, Nyong AM, Natarajan A, Band V, Band H. Anticancer activity of Celastrol in combination with ErbB2-targeted therapeutics for treatment of ErbB2-overexpressing breast cancers. Cancer Biol Ther. 2011;11:263–276. doi: 10.4161/cbt.11.2.13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF, Patapoutian A. Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature. 2007;445:541–545. doi: 10.1038/nature05544. [DOI] [PubMed] [Google Scholar]
- 31.Yamakoshi H, Kanoh N, Kudo C, Sato A, Ueda K, Muroi M, Kon S, Satake M, Ohori H, Ishioka C, Oshima Y, Osada H, Chiba N, Shibata H, Iwabuchi Y. KSRP/FUBP2 is a binding protein of GO-YO86, a cytotoxic curcumin analogue. ACS Med Chem Lett. 2010;1:273–276. doi: 10.1021/ml1000454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maresso AW, Wu R, Kern JW, Zhang R, Janik D, Missiakas DM, Duban ME, Joachimiak A, Schneewind O. Activation of inhibitors by sortase triggers irreversible modification of the active site. J Biol Chem. 2007;282:23129–23139. doi: 10.1074/jbc.M701857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 34.Stoll R, Renner C, Hansen S, Palme S, Klein C, Belling A, Zeslawski W, Kamionka M, Rehm T, Muhlhahn P, Schumacher R, Hesse F, Kaluza B, Voelter W, Engh RA, Holak TA. Chalcone derivatives antagonize interactions between the human oncoprotein MDM2 and p53. Biochemistry. 2001;40:336–344. doi: 10.1021/bi000930v. [DOI] [PubMed] [Google Scholar]
- 35.Bois F, Boumendjel A, Mariotte AM, Conseil G, Di Petro A. Synthesis and biological activity of 4-alkoxy chalcones: potential hydrophobic modulators of P-glycoprotein-mediated multidrug resistance. Bioorg Med Chem. 1999;7:2691–2695. doi: 10.1016/s0968-0896(99)00218-7. [DOI] [PubMed] [Google Scholar]
- 36.Hadjeri M, Barbier M, Ronot X, Mariotte AM, Boumendjel A, Boutonnat J. Modulation of P-glycoprotein-mediated multidrug resistance by flavonoid derivatives and analogues. J Med Chem. 2003;46:2125–2131. doi: 10.1021/jm021099i. [DOI] [PubMed] [Google Scholar]
- 37.Liu XL, Tee HW, Go ML. Functionalized chalcones as selective inhibitors of P-glycoprotein and breast cancer resistance protein. Bioorg Med Chem. 2008;16:171–180. doi: 10.1016/j.bmc.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Edwards ML, Stemerick DM, Sunkara PS. Chalcones: a new class of antimitotic agents. J Med Chem. 1990;33:1948–1954. doi: 10.1021/jm00169a021. [DOI] [PubMed] [Google Scholar]
- 39.Dupeyre G, Chabot GG, Thoret S, Cachet X, Seguin J, Guenard D, Tillequin F, Scherman D, Koch M, Michel S. Synthesis and biological evaluation of (3,4,5-trimethoxyphenyl)indol-3-ylmethane derivatives as potential antivascular agents. Bioorg Med Chem. 2006;14:4410–4426. doi: 10.1016/j.bmc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 40.Boumendjel A, McLeer-Florin A, Champelovier P, Allegro D, Muhammad D, Souard F, Derouazi M, Peyrot V, Toussaint B, Boutonnat J. A novel chalcone derivative which acts as a microtubule depolymerising agent and an inhibitor of P-gp and BCRP in in-vitro and in-vivo glioblastoma models. BMC Cancer. 2009;9:242. doi: 10.1186/1471-2407-9-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boumendjel A, Ronot X, Boutonnat J. Chalcones derivatives acting as cell cycle blockers: potential anti cancer drugs? Curr Drug Targets. 2009;10:363–371. doi: 10.2174/138945009787846416. [DOI] [PubMed] [Google Scholar]
- 42.Ducki S. Antimitotic chalcones and related compounds as inhibitors of tubulin assembly. Anticancer Agents Med Chem. 2009;9:336–347. doi: 10.2174/1871520610909030336. [DOI] [PubMed] [Google Scholar]
- 43.Champelovier P, Mininno M, Duchamp E, Nicolle E, Curri V, Boumendjel A, Boutonnat J. Cytotoxicity of chalcone derivatives towards glioblastoma. Anticancer Res. 2011;31:3213–3218. [PubMed] [Google Scholar]
- 44.Chao TC, Chu Z, Tseng LM, Chiou TJ, Hsieh RK, Wang WS, Yen CC, Yang MH, Hsiao LT, Liu JH, Chen PM. Paclitaxel in a novel formulation containing less Cremophor EL as first-line therapy for advanced breast cancer: a phase II trial. Invest New Drugs. 2005;23:171–177. doi: 10.1007/s10637-005-5863-8. [DOI] [PubMed] [Google Scholar]
- 45.McCarron PA, Marouf WM, Quinn DJ, Fay F, Burden RE, Olwill SA, Scott CJ. Antibody targeting of camptothecin-loaded PLGA nanoparticles to tumor cells. Bioconjug Chem. 2008;19:1561–1569. doi: 10.1021/bc800057g. [DOI] [PubMed] [Google Scholar]
- 46.Danhier F, Lecouturier N, Vroman B, Jerome C, Marchand-Brynaert J, Feron O, Preat V. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133:11–17. doi: 10.1016/j.jconrel.2008.09.086. [DOI] [PubMed] [Google Scholar]
- 47.Emerson DL. Liposomal delivery of camptothecins. Pharm Sci Technolo Today. 2000;3:205–209. doi: 10.1016/s1461-5347(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 48.Yang T, Cui FD, Choi MK, Lin H, Chung SJ, Shim CK, Kim DD. Liposome formulation of paclitaxel with enhanced solubility and stability. Drug Deliv. 2007;14:301–308. doi: 10.1080/10717540601098799. [DOI] [PubMed] [Google Scholar]
- 49.Park YS. Tumor-directed targeting of liposomes. Biosci Rep. 2002;22:267–281. doi: 10.1023/a:1020190606757. [DOI] [PubMed] [Google Scholar]
- 50.Ruoslahti E, Bhatia SN, Sailor MJ. Targeting of drugs and nanoparticles to tumors. J Cell Biol. 2010;188:759–768. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolby LJ, Rodia RM. The periodate oxidation of heterocycles: II. 2-Methylindole and 2,3-diphenylindole. J Org Chem. 1970;35:1493–1496. [Google Scholar]
- 52.Note: Although no published analytical data were found, the following compounds have CAS numbers: compound 2, 373618-91-8; compound 10, 1237055–51–4. 2012.
- 53.Tsukerman SV, Nikitchenko VM, Bugai AI, Lavrishin VF. Synthesis of analogs of chalcones and derivatives of delta 2-pyrazoline based on 3-formylindole. Khimiia Geterotsiklicheskikh Soedinenii. 1969;2:268–272. [Google Scholar]
- 54.Gillard JW, Belanger P. Metabolic synthesis of arylacetic acid antiinflammatory drugs from arylhexenoic acids. 2. Indomethacin. J Med Chem. 1987;30:2051–2058. doi: 10.1021/jm00394a020. [DOI] [PubMed] [Google Scholar]
- 55.Beer RJS, Clarke K, Davenport HF, Robertson A. The chemistry of the melanins. III/. The synthesis of hydroxyindoles from p-benzoquinones. J Chem Soc. 1951:2029–2032. [Google Scholar]
- 56.Guo B, Hembruff SL, Villeneuve DJ, Kirwan AF, Parissenti AM. Potent killing of paclitaxel- and doxorubicin-resistant breast cancer cells by calphostin C accompanied by cytoplasmic vacuolization. Breast Cancer Res Treat. 2003;82:125–141. doi: 10.1023/B:BREA.0000003969.21267.81. [DOI] [PubMed] [Google Scholar]
- 57.Maltese WA, De Vivo DC. Cholesterol and phospholipids in cultured skin fibroblasts from patients with dystonia. Ann Neurol. 1984;16:250–252. doi: 10.1002/ana.410160215. [DOI] [PubMed] [Google Scholar]
- 58.Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler HS, Wyrobek AJ, Stampfer MR. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009;69:7557–7568. doi: 10.1158/0008-5472.CAN-09-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.