

Abstract
Angiogenic factors, such as vascular endothelial-derived growth factor (VEGF) and IGF-I, play pivotal roles in endothelial proliferation and migration. IGF binding protein-3 (IGFBP-3) is emerging as a key regulator of cell growth and apoptosis, both as an IGF antagonist and as an independent molecule. We investigated the role of IGFBP-3 in VEGF-mediated survival of human macrovascular umbilical vein endothelial cells (HUVEC). Specific commercial ELISAs quantified cell proliferation and apoptosis, and Akt phosphorylation was assessed by immunoblots and confocal microscopy. IGF-I and VEGF significantly stimulated HUVEC proliferation and survival. Addition of IGFBP-3 reversed both IGF- and VEGF-induced proliferation and prevented the survival induced by these factors. The antiproliferative and proapoptotic effects of exogenous IGFBP-3 upon VEGF-induced HUVEC survival were not inhibited by blockade of the type 1 IGF receptor with αIR-3 immunoglobulin, which fully prevented IGF actions. An IGFBP-3 mutant, which binds IGFs normally but has a substituted mid-region domain, lost the ability to inhibit VEGF actions. VEGF-induced phosphorylation of Akt, as evident by both specific immunoblots and confocal microscopy, was significantly and rapidly reduced in the presence of IGFBP-3, as well as wortmannin.
Endothelial cells comprise the critical border between the arterial wall and the blood and, by virtue of their unique location, play several key roles in angiogenesis. Through elaboration of several growth factors, the endothelium regulates metabolism, migration, proliferation, and apoptosis of underlying smooth muscle cells as well as production of the extracellular matrix (1). Disorders of endothelial function have been closely associated with vascular diseases (2), the leading causes of morbidity and mortality for diabetic patients in the United States (3). Endothelial-derived growth factors—such as vascular endothelial derived growth factor (VEGF), TGF-β, angiotensin II, IGF-I, and insulin—have been implicated in the development of diabetic angiopathy (4, 5). The phosphatidylinositol 3′-kinase (PI3-kinase)/Akt pathway transmits a survival signal common to these and other essential growth factors (6-8).
Ligand binding activates the VEGF receptor, kinase insert domain-containing region (KDR), resulting in tyrosine ring autophosphorylation, which allows the membrane binding of PI3-kinase, and its subsequent activation and phosphorylation (9-12). The phosphorylation provides docking sites for Akt and phosphatidylinositol-dependent kinase-1 (13). Phosphatidylinositol-dependent kinase-1 catalyzes the phosphorylation of Akt at position Thr 308. Phosphorylated Akt is thought to be responsible for enhancing cell survival though the inhibition of apoptosis via downstream intracellular pathways (14, 15).
It has been noted that IGF-I up-regulates VEGF expression supporting its role as an angiogenic factor (16). VEGF, in turn, regulates the expression of IGF binding proteins (IGFBPs) -3, -4, and -5 (17, 18). Among the six high-affinity IGFBPs (IGFBP-1 to -6), IGFBP-3 is the most abundant in human serum, and circulating IGFBP-3 is primarily under the regulation of GH (19). IGFBP-3 is produced locally in several tissues and exerts its major paracrine and autocrine actions related to cellular growth via IGF-dependent and IGF-independent mechanisms (20, 21). Transfection of murine fibroblasts lacking the type 1 IGF receptor (IGF1R) with IGFBP-3 resulted in growth inhibition that correlated directly with the level of IGFBP-3 expression (22). IGFBP-3 has been observed in the nucleus of several cell types (23). Our group has recently reported direct interactions between IGFBP-3 and nuclear retinoic X receptor-α, which result in apoptosis (24). In addition, IGFBP-3 has been shown to interact with a number of other proteins, some well defined (25) and others still being molecularly characterized (26). VEGF has recently been shown to down-regulate IGFBP-3 in cultured bovine aortic endothelial cells (17). Considering the IGF-independent actions of IGFBP-3 on apoptosis and other cellular events, we hypothesized that IGFBP-3 may modulate VEGF-mediated HUVEC survival.
Materials and Methods
VEGF, wortmannin, BSA, dextrose, glycine, bromophenol blue, methanol, and 4′,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma (St. Louis, MO). Pharmacia Corp. (Peapack, NJ) generously supplied recombinant human IGF-I. Rabbit, antihuman Akt, and α-IR3 IgG antibodies were purchased from Cell Signaling, Inc. (Beverly, MA). Goat antirabbit IgG was obtained from Chemicon (Temecula, CA). Fluorescein isothiocyanate (FITC)-conjugated, goat antirabbit IgG was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Tween, FluoroGuard, skim milk, and sodium dodecyl sulfate (SDS)-PAGE reagents were purchased from Bio-Rad Laboratories, Inc. (Hercules, CA). Acrylamide (30%) was purchased from Roche (Indianapolis, IN). N-N-N′-N′-tetramethylenediamine was provided by Life Technologies, Inc. (Grand Island, NY). Amersham Pharmacia Biotechnology, Inc. (Piscataway, NJ) supplied enhanced chemiluminescence solutions. Desmond Mascarenhas of Protigen (Mountain View, CA) generously provided nonglycosylated recombinant human IGFBP-3. The IGFBP-3 mutant, manipulated to replace the heparin-binding domain (HBD) domain with the corresponding region from IGFBP-1, was generously donated by David Powell (Baylor University, Houston, TX). Human umbilical vein endothelial cells (HUVEC) and Microvascular Endothelial Cell Growth Medium Bullet Kit-2 (EGM-2-MV Bullet Kit) were purchased from Clonetics (San Diego, CA). Culture plates were purchased from Nalge Nunc (Rochester, NY).
Cell culture
HUVEC were grown in a modified MCDB 131 formulation (27), EGM2-MV from Clonetics. Cell monolayers were cultured in uncoated 75-cm2 flasks at 37 C in a humidified atmosphere containing 5% carbon dioxide. At 80–90% confluence, adherent cells were removed with 1 ml of trypsin (Clonetics) and subsequently placed in growth media, before seeding in appropriate plates for experiments.
Proliferation assays
Cells (1000 cells/cm2) were seeded on 96-well plates in 100 μl EGM2-MV. Following overnight attachment of the cells, each well was washed with 100 μl PBS, serum starved for 2 h, then incubated with 100 μl of experimental conditions. CellTiter 96 AQ Non-Radioactive Cell Proliferation Assay (Promega Corp., Madison, WI) was performed according to the manufacturer’s instructions. The assay uses a solution of MTS (Owen’s reagent, a tetrazolium compound) and phenazine methosulfate, an electron coupling reagent. Twenty microliters of this solution were added to each well. MTS is bioreduced to soluble formazan by metabolically active cells, which we quantified by ELISA. All experiments were performed in triplicate. Reaction products in each 96-well plate were quantified using the Bio-Rad Laboratories, Inc. Model 3550-UV Microplate Reader. Mean ± se values of the raw absorbance detected at 490 nm were plotted. Many groups have shown that the use of MTS is reliable, and comparable to the gold standard of visualizing and counting cells (28). Furthermore, preliminary studies to provide evidence that the absorbance values generated by the assay are linearly related to growth were performed. Specifically, various numbers of HUVEC (250 cells/well, 500 cells/well, 750 cells/well, 1000 cells/well, 1250 cells/well, 1500 cells/well, 1750 cells/well, 2000 cells/well, 2500 cells/well, and 3000 cells/well) were supplemented with standard growth medium were added to the wells of a 96-well plate, in triplicate. The medium was allowed to equilibrate for 1 h, after which MTS was added. After 1 h, the absorbance at 490 nm was recorded. The correlation coefficient of the line was 0.995, demonstrating that there was a linear response between cell number and absorbance at 490 nm.
Apoptosis assays
Cells (2500 cells/cm2) were seeded on 96-well plates in 100 μl EGM2-MV. Following overnight attachment, cells were washed with PBS and serum starved for 2 h, before incubation for 12 h with 100 μl of experimental conditions. Roche Photometric Cell Death ELISA (Indianapolis, IN) was performed according to the manufacturer’s instructions to quantify histone-associated DNA fragments (mono- and oligo-nucleosomes) generated by apoptotic cells. This immunoassay is based on the sandwich-enzyme principle and used separate mouse monoclonal antibodies directed against DNA fragments and histones. Briefly, cell lysates were placed into a 96-well plate coated with streptavidin-linked, anti-histone antibody. Peroxidase-labeled mouse monoclonal DNA antibodies were used to localize and detect the bound, fragmented DNA by photometry of 2,2′ -azino-bis-[3-ethylbenzathiazoline sulfonate] as the substrate. Calcium ionophore treatment served as the positive control, and serum-free medium (SFM) as the negative control. Each experimental condition was performed in triplicate. Reaction products in each 96-well plate were read using the Bio-Rad Laboratories, Inc. microplate reader. Mean absorbance data at 405 nm (±se) were plotted. Preliminary studies to provide evidence that the absorbance values generated by the assay are linearly related to growth were performed and confirmed in a published paper from our lab (29).
Western immunoblots
Cells (75,000 cells/cm2) were seeded on 6-well plates in EGM2-MV. Following overnight attachment, cells were washed with PBS and serum starved for 2 h, before incubation for 90 min with experimental conditions. All conditions were performed in duplicate. Conditioned media was discarded by gentle aspiration. Cells were lysed in 100 μl RIPA buffer (PBS, Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) on ice for 10 min. Samples were separated by electrophoresis on 10% polyacrylamide-SDS gels overnight at 90 V, then immediately transferred to nitrocellulose membranes for subsequent immunoblots of total and phosphorylated Akt. The blots were subsequently exposed to film for 1 h at −70 C.
Immunofluorescent confocal microscopy
Cells were plated on coverglass slides in EGM2-MV, at a density of 10,000 cells/slide, each slide laying separately at the base of each well in a 12-well plate. After overnight attachment, cells were washed with PBS, then serum starved for 2 h before experimental conditions for 90 min. Conditioned media was removed, and cells were washed once with 1 ml PBS. Cells were then fixed with methanol 0.5 ml/well, at 20 C, for 5 min. Cells were air dried for 5 min, then washed twice with PBS. Cells were incubated for 20 min, on ice, with 0.5 ml of 0.2% TritonX-100 in PBS to permeabilize endothelial cell plasma membranes. After two washes with PBS, cells were incubated with a 1:150 dilution of Akt antibody (5 μg/ml, 83 nm) at 25 C for 60 min, and subsequently washed twice in PBS. Cells were then incubated with the FITC-conjugated secondary antibody at 1:300 (5 μg/ml, 61 nm), for 45 min in darkness at 25 C. After two additional PBS washes, cells were incubated for 2 min at 25 C with DAPI at 1:10,000 (5 μg/ml). Cells were twice washed in ice-cold PBS, and then stored at 4 C under FluoroGuard Antifade Reagent until microscopy.
DAPI binds adenosine-thymidine-rich clusters in the minor groove of DNA and, when excited at 358 nm by the 351–364 nm beam from an argon laser, emits bright blue fluorescence at 461 nm. Fluorescence was detected using the Leica Corp. (Tokyo, Japan) TCS SP Inverted Confocal Microscope equipped with argon, krypton, and helium-neon lasers, courtesy of The Carol Moss Spivak Cell Imaging Facility at UCLA. Images were captured directly by a Himamatsu digital camera and processed with Microscope Control Module Software (QED Imaging, Inc., Pittsburgh, PA).
Densitometric and statistical analysis
Densitometric measurement of immunoblots was performed using a Bio-Rad Laboratories, Inc. (Melville, NY) GS-670 Imaging Densitometer. Protein levels were estimated by comparing the optical density of each specific protein band from control conditions to that of the treated conditions. All experiments were repeated several times. When applicable, means ± sem are shown. Student’s t tests and ANOVA were used for statistical analysis.
Results
IGFBP-3 inhibits VEGF-mediated HUVEC proliferation
To determine the minimal effective dose of VEGF required for stimulating proliferation, we treated HUVEC for 24 h, in the presence of 0–100 ng/ml (0–3600 μm) VEGF. HUVEC proliferation decreased significantly after 24 h of serum starvation, whereas the number of metabolically active cells remained constant, or increased, in a dose-dependent manner in response to VEGF (Fig. 1A). VEGF significantly induced HUVEC proliferation by 76% above SFM (mean A490nm by MTS ELISA increased from 0.100 ± 0.014 to 0.176 ± 0.017), at the lowest dose used, 10 ng/ml, 360 μm (P < 0.01), and we therefore used VEGF at 10 ng/ml, 360 μm for subsequent experiments. Although the 10 ng/ml, 360 μm, dose was the lowest dose tested, it is supraphysiologic compared with serum levels of healthy controls. However, the treatment dose does correspond to tissue levels found in disease states such as diabetic retinopathy (30), and therefore reflects a phenomenon that occurs within the physiologic range of tissue specific disease states where VEGF concentrations have been shown to be elevated.
Fig. 1.

IGFBP-3 inhibits VEGF-mediated HUVEC proliferation and survival via the induction of apoptosis. Cells were seeded at 1000 cells/cm2 (A–E) or 2500 cells/cm2 (F), in 96-well plates. Cells were grown in 100 μl of EGM2-MV overnight, serum starved for 2 h, and further treated. A, HUVEC were treated in SFM for 24 h with 0–100 ng/ml (0–3600 μm) VEGF. Mean absorbance at 490 nm + sem is shown as percent above growth in SFM. *, P < 0.001 by ANOVA. B, HUVEC were treated for 24 h with in serum free (SF), 5% FBS (serum), VEGF (10 ng/ml, 360 μm), or IGF-I (250 ng/ml, 34.5 nm). Also shown is growth with 250 ng/ml of IGF-I and 1.0 μg/ml of IGFBP-3 (equimolar dose). In addition, HUVEC were pretreated with a dose titration of IGFBP-3 (maximal dose 1 μg/ml, or 34.5 nm) 30 min before exposure VEGF (10 ng/ml, 360 μm). Mean absorbance at 490 nm + sem is shown as percent growth, relative to SFM. *, P < 0.01; #, P < 0.01 relative to IGF-I; ##, P < 0.01 relative to VEGF. C, HUVEC were treated with IGFBP-3 (1 μg/ml, 34.5 nm) or wortmannin (100 nm) alone or 30 min before VEGF stimulation for 24 h. Mean absorbance at 490 nm + sem is shown as percent growth, relative to SFM. *, P < 0.01. BP, IGFBP-3; V, VEGF; W, wortmannin. D, Cell death detection ELISA immuno-assay was performed to quantitate apoptosis. HUVEC were treated with IGFBP-3, at 250-1000 ng/ml (8.6–34.5 nm), for 30 min, before VEGF (10 ng/ml, 360 μm) stimulation for 12 h. Mean absorbance at 405 nm + sem is shown as percent above VEGF alone-treated HUVEC conditions. *, P < 0.05.
To identify the effects of mitogens, HUVEC were treated for 24 h with SFM, 5% bovine serum, and SFM containing IGF-I (250 ng/ml, 34.5 nm), or VEGF (10 ng/ml, 360 μm). IGF-I and VEGF stimulated HUVEC proliferation to 41%, and 47% above serum-free conditions, respectively (Fig. 1B). Mean A490nm by MTS ELISA increased from 0.023 ± 0.001 (SFM) to 0.960 ± 0.008 (IGF-I), and 1.101 ± 0.002 (VEGF) (P < 0.01 vs. SFM for each condition). To test our hypothesis that IGFBP-3 inhibits VEGF-mediated HUVEC proliferation, HUVEC were treated with a dose titration of IGFBP-3 (maximal dose 1 μg/ml, 34.5 nm) 30 min before exposure VEGF (10 ng/ml, 360 μm) for 24 h. Only one dose was used before exposure to IGF-I, 1 μg/ml, 34.5 nm. IGFBP-3 significantly inhibited VEGF-mediated and IGF-mediated HUVEC proliferation at a dose of 1 μg/ml, 34.5 nm (Fig. 1B); A490nm decreased from 1.101 ± 0.002 to 0.281 ± 0.004 and 0.024 ± 0.002, respectively (P < 0.01).
The PI3-kinase/Akt signal transduction pathway is activated by a number of mitogens, including VEGF, insulin, and IGF-I, and is thought to be responsible for enhancing cell survival through the inhibition of apoptosis. We first compared the inhibitory action of IGFBP-3 on VEGF-induced growth, to a known inhibitor of VEGF-induced Akt phosphorylation, wortmannin. HUVEC were preincubated for 1 h with wortmannin (100 nm) or IGFBP-3 (1 μg/ml, 34.5 nm)) before VEGF (10 ng/ml, 360 μm) stimulation for 24 h. VEGF increased growth of HUVEC by 60% from baseline (P < 0.01). The addition of wortmannin, or IGFBP-3, inhibited VEGF-mediated growth, allowing only 4% and 7% stimulation, respectively (not significantly different from SFM, P < 0.01 relative to VEGF alone) (Fig. 1C); A490nm decreased from 1.110 ± 0.115 with VEGF alone to 0.519 ± 0.007 in the presence of IGFBP-3 (P < 0.01), and to 0.484 ± 0.012 in the presence of wortmannin (P < 0.01).
VEGF is known to activate the PI3-kinase/Akt signal transduction pathway, thereby inhibiting cell apoptotic signaling and enhancing HUVEC survival. We therefore hypothesized that IGFBP-3 inhibits VEGF-mediated mitogenesis through the induction of apoptosis. The addition of IGFBP-3 to HUVEC, treated with VEGF, increased apoptosis in a dose-dependent trend, with a significant effect at 1 μg/ml. The addition of IGFBP-3 induced apoptosis to 50% above baseline (Fig. 1D), and therefore reversed VEGF-induced survival; A405nm increased from 0.239 ± 0.003 (VEGF alone) to 0.577 ± 0.002 in the presence of IGFBP-3 (P < 0.05).
IGFBP-3 antagonizes VEGF actions via an IGF-independent mechanism
To determine whether IGFBP-3 inhibition of VEGF-induced survival required the IGF1R, we pretreated cells with the αIR-3 antibody, which blocks IGF binding to the IGF1R. This antibody fully blocked IGF-induced proliferation, decreasing proliferation from 160% to 16% above SFM (P < 0.01), but had no effect on VEGF-induced proliferation (150% vs. 100% above SFM, P > 0.05.), shown in Fig. 2A. IGFBP-3 inhibited both IGF-I- (160% above SFM vs. 16% below SFM) and VEGF-mediated (150% above SFM vs. 16% below SFM) HUVEC proliferation, (Fig. 2A); A490nm decreased from 0.428 ± 0.038 (with IGF-I alone) to 0.138 ± 0.055 in the presence of IGFBP-3 (P < 0.01); A490nm decreased from 0.412 ± 0.038 (with VEGF alone) to 0.138 ± 0.033 in the presence of IGFBP-3 (P < 0.01). αIR-3 did not abolish VEGF-induced proliferation (A490nm = 0.412 ± 0.0375 vs. 0.329 ± 0.1304; P > 0.05) but did abolish IGF-I-induced proliferation (A490nm = 0.428 ± 0.0375 vs. 0.145 ± 0.0217; P < 0.01). These results demonstrate that blocking the type 1 IGF receptor has no effect on IGFBP-3 inhibition of VEGF mitogenesis, suggesting that IGFBP-3 does not require the type 1 IGF receptor system to inhibit VEGF action.
Fig. 2.
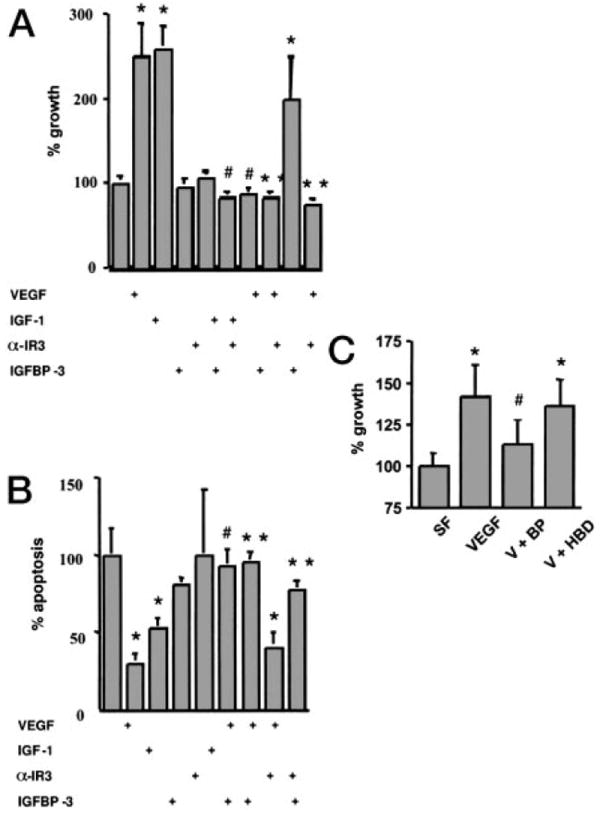
IGFBP-3 abolishes survival induction by VEGF in a type 1 receptor-independent manner. A, Cells were seeded at 1000 cells/cm2 in 96-well plates and were grown in 100 μl of EGM2-MV overnight, serum starved for 2 h, and incubated with 100 μl of treated SFM. Growth was determined by MTS assay. HUVEC were treated with VEGF (10 ng/ml, 360 μm), IGF-I (250 ng/ml, 34.5 nm), IGFBP-3 (1 μg/ml, 34.5 nm), or α-IR3 (1 μg/ml), or combinations thereof. Mean absorbance at 490 nm + sem is shown as percent of growth in SFM. *, P < 0.01 in comparison with SFM. **, P < 0.1, in comparison with VEGF. #, P < 0.01, in comparison with IGF-I. B, Cells were seeded at 2500 cells/cm2 in 96-well plates for apoptosis assays and were grown in 100 μl of EGM2-MV overnight, serum starved for 2 h, and incubated with 100 μl of treated SFM. HUVEC were treated with VEGF (10 ng/ml, 360 μm), IGF-I (250 ng/ml, 34.5 nm), IGFBP-3 (1 μg/ml, 34.5 nm), α-IR3 (1 μg/ml), or combinations thereof for 12 h. Cell death detection ELISA immunoassay was performed to quantitate apoptosis. Mean absorbance at 405 nm + sem is shown as percent relative to SFM. *, P < 0.01 in comparison with SFM. **, P < 0.01, in comparison with VEGF. #, P < 0.01, in comparison with IGF-I. C, HUVEC were treated in SFM with VEGF (10 ng/ml, 360 μm) alone, VEGF (10 ng/ml, 360 μm) + IGFBP-3 (1 μg/ml, 34.5 nm). Mean absorbance at 490 nm + sem is shown as percent of growth in SFM. W, Wortmannin; BP, IGFBP-3; HBD, IGFBP-3 mutant for the HBD. *, P < 0.01 in comparison with SFM. #, P < 0.01 in comparison with VEGF.
Complementary apoptosis assays are depicted in Fig. 2B. αIR-3 fully blocked IGF inhibition of apoptosis, increasing apoptosis in the presence of IGF-I from 53–93% of SFM (A405nm = 0.880 ± 0.008 vs. 1.550 ± 0.049; P < 0.01) but did not prevent VEGF inhibition of apoptosis (30% vs. 40% of SFM; A405nm = 0.504 ± 0.056 vs. 0.672 ± 0.126; P > 0.05). In comparison, IGFBP-3 was able to inhibit the antiapoptotic effects of both IGF-I and VEGF; A405 nm increased from 0.880 ± 0.008 (with IGF-I alone) to 1.520 ± 0.010 in the presence of IGFBP-3, and from 0.504 ± 0.056 (with VEGF alone) to 1.590 ± 0.118 in the presence of IGFBP-3 (P < 0.01).
IGFBP-3 is noted to have a mid-region domain, which allows it to interact with several molecules including heparin and is known as the HBD (5). IGFBP-3, in which the HBD sequence was substituted with the corresponding region from IGFBP-1, was used to further demonstrate the IGF independent nature of IGFBP-3 on VEGF-induced growth. This substitution does not change the molecule’s ability to bind IGFs, but interferes with interactions with other molecules, such as retinoic X receptor. HUVEC were treated with IGFBP-3 (1 μg/ml, 34.5 nm) or the IGFBP-3 HBD mutant (1 μg/ml 34.5 nm) 30 min before stimulation with VEGF (10 ng/ml, 360 μm) for 24 h. Recombinant IGFBP-3, but not the HBD mutant, decreased VEGF-induced HUVEC proliferation (Fig. 2C). VEGF induced proliferation to 40% above SFM; A490 nm increased from 0.397 ± 0.023 to 0.556 ± 0.018 (P < 0.01). The addition of IGFBP-3 to VEGF treatment inhibited VEGF-induced growth, allowing an increase to 11% above SFM (mean A490 nm 0.443 ± 0.020; P < 0.01 relative to VEGF alone, but not significantly different from SFM), whereas the HBD mutant was unable to inhibit VEGF induction of proliferation, allowing VEGF-induced growth to increase 36% above SFM (mean A490 nm 0.539 ± 0.023; not significantly different from VEGF alone). The HBD mutant is fully capable of binding IGF, providing further evidence that the effect seen in our experiments is truly IGF system independent.
IGFBP-3 inhibits VEGF-induced phosphorylation of Akt by PI3-kinase
VEGF induces the phosphorylation of Akt, therefore inhibiting apoptosis. Our group has shown that IGFBP-3 induces apoptosis of cancer cells, independently of the IGF/IGF1R (24). The effect of IGFBP-3 on VEGF in our system appears to also be an IGF/IGF1R independent induction of apoptosis. Therefore, we hypothesized that IGFBP-3 inhibits VEGF-induced phosphorylation of Akt. HUVEC were treated in SFM with wortmannin (100 nm), IGFBP-3 (1 μg/ml, 34.5 nm), VEGF (10 ng/ml, 360 μm), insulin (100 nm), and IGF-I (250 ng/ml, 34.5 nm) for 90 min. HUVEC were also preincubated with wortmannin (100 nm) or IGFBP-3 (1 μg/ml, 34.5 nm) for 30 min before stimulation with VEGF (10 ng/ml, 360 μm) for 90 min. Western immunoblots for total and phosphorylated Akt demonstrated that insulin, IGF-I and VEGF all rapidly stimulated phosphorylation of Akt. Wortmannin and IGFBP-3 fully inhibited VEGF-induced phosphorylation of Akt (P < 0.01, Fig. 3A). Of note is that IGFBP-3 caused a small nonsignificant increase in phospho-Akt levels. To further assess Akt in living cells, we used confocal microscopy (Fig. 3BI-IV). Figure 3BI demonstrates baseline staining for total Akt in serum-free conditions. Very little staining for phospho-Akt in serum free conditions was visualized (data not shown). As can be seen in Fig. 3BII, VEGF (10 ng/ml, 360 μm) induced the phosphorylation of Akt. In Fig. 3BIII, we demonstrate that when IGFBP-3 (1 μg/ml, 34.5 nm) is added 30 min before stimulation with VEGF (10 ng/ml, 360 μm), there is a dramatic reduction in staining for phosphorylated Akt, thus corroborating the Western blot results shown in Fig. 3A for the direct and rapid effects of IGFBP-3 on the Akt signal transduction pathway. Lastly, in Fig. 3BIV, we show that the HBD mutant, capable of binding IGF, does not demonstrate the same effect as recombinant whole IGFBP-3 in the prevention of VEGF-induced phosphorylation of Akt. These results were observed in all fields visualized in multiple slides and similar results were observed using immunoblot data (data not shown). These results illustrate the importance of the HBD sequence in the interaction of IGFBP-3 and Akt, suggesting that there is a mechanism independent of the IGF pathway.
Fig. 3.

IGFBP-3 inhibits VEGF-induced phosphorylation of Akt. A, 75,000 cells/cm2 were seeded on 6-well plates in EGF2-MV overnight and serum starved for 2 h before incubation for 90 min with experimental conditions. Cells were lysed in 100 μl iced RIPA buffer and samples were separated by electrophoresis on 10% polyacrylamide-SDS gels overnight and transferred to nitrocellulose membranes for immunoblots of total and phosphorylated Akt. HUVEC were treated in serum-free (SF) conditions or with 100 nm wortmannin (W), 1 μg/ml, or 34.5 nm IGFBP-3 (B), 10 ng/ml or 360 μm VEGF (V), 100 nm insulin (In), and 250 ng/ml or 34.5 nm IGF-I (I) or stimulated with VEGF for 90 min after preincubation with wortmannin (V + W) or IGFBP-3 (V + B) for 30 min. Mean densitometric values are shown. *, P < 0.01. B, HUVEC were plated on coverglass slides in EGF2-MV, at a density of 10,000 cells/slide. After overnight attachment, cells were serum starved for 2 h before experimental conditions for 90 min. Cells were then fixed, air dried, and stained with 1:150 dilution of Akt antibody (5 μg/ml, 83 nm) and then incubated with the FITC-conjugated secondary antibody (5 μg/ml, 61 nm) at 1:300 as well as DAPI (5 μg/ml, 10.9 μm at 1:10,000. Fluorescence was detected using the Leica TCS SP Inverted Confocal Microscope. I, HUVEC were incubated in SMF for 90 min before staining for total Akt. II, HUVEC were treated with VEGF (10 ng/ml, 360 μm) for 90 min before staining for phosphorylated Akt, and subsequent visualization by confocal microscopy. III, HUVEC were serum starved for 30 min and pretreated IGFBP-3 (1 μg/ml, 34.5 nm) for 30 min before stimulation with VEGF (10 ng/ml), 360 μm) for 90 min. IV, HUVEC were serum starved for 30 min, pretreated with the HBD mutant for IGFBP-3 (1 μg/ml, 34.5 nm) for 30 min before stimulation with VEGF (10 ng/ml, 360 μm) for 90 min. Staining for phosphorylated Akt was subsequently visualized with confocal microscopy.
Discussion
We have demonstrated for the first time that IGFBP-3 is a potent inhibitor of VEGF-stimulated proliferation via a mechanism independent of the IGF-IGF1R. Our findings add support to the rapidly evolving role of IGFBP-3 as an independent regulator of cell survival. First, we have demonstrated that VEGF induces HUVEC proliferation. Second, we have shown that VEGF-induced survival of HUVEC is inhibited by IGFBP-3, via the induction of apoptosis. Third, we show that the inhibitory activity of IGFBP-3, in this system, is independent of IGF1R. Fourth, we show that IGFBP-3 inhibits VEGF-mediated activation of the PI3-kinase/Akt signal transduction pathway. Furthermore, we have demonstrated a rapid effect of IGFBP-3 in the regulation of cell survival.
The detailed mechanism by which IGFBP-3 inhibits VEGF-induced Akt phosphorylation is not known and is currently under active investigation in our laboratory. The involvement of the PI-3-kinase pathway in VEGF signaling in endothelial cells has been previously demonstrated. IGF-I has also been shown to stimulate Akt phosphorylation in cultured bovine fibroblasts; in addition, it does so in a manner that is potentiated by IGFBP-3. It is clear, however, from the results from this current work and from a growing number of observations from other research groups, that the effects of IGFBP-3 are mediated in an IGF-I-independent manner. The fact that the HBD mutant, which retains the ability to bind to IGF-I, does not inhibit the actions of VEGF indicates that IGF-I binding is not critical for the observed inhibition of Akt phosphorylation. The HBD domain is either directly involved in mediating the observed effects, or that its substitution induces a change in tertiary structure that is critical for mediating these effects.
Although there is evidence that IGFBP-3 can enter the nucleus in certain cell lines, it is not clear whether the observed effects on Akt phosphorylation in endothelial cells is also mediated by such nuclear interactions. Thus, the nature of the interactions between Akt and IGFBP-3 at the molecular level remains to be elucidated. Further investigation will be necessary to ascertain whether the inhibition of Akt phosphorylation reflects a decreased activation of upstream signaling intermediates, such as PI-3-kinase or insulin receptor substrates. Conversely, it is also possible that IGFBP-3 is inducing the activity of cellular phosphatases, a mechanism that has been recently supported.
Cell culture systems have commonly been used to study mechanisms implicated in the pathogenesis of diabetic complications, but the great majority of cell preparations used have been either of nonhuman origin or nonorgan-specific human origin. Because of questions of species and organ specificity in the function of cells of vascular origin, many groups have looked at the differences in cells in different vascular beds, confirming the suspicion that there are differences between endothelial cells with respect to secretory functions and their modulation by glucose, indicating regional specificity of these functions. Extrapolation to all vascular cells from experiments using cells from heterologous vascular beds to draw inferences about the pathophysiology of diabetic complications should be performed with caution. The number of adequate tissue specific available models is limited, and therefore makes the applicability of the findings difficult. The results must therefore be confirmed in tissue specific models in vitro, or animal models in vivo, before specific conclusions can be drawn.
Angiogenesis, the development of new blood vessels from preexisting endothelium, is a major component of diverse biological processes including embryogenesis, wound healing, and organ regeneration (31). Mounting evidence in vitro and in vivo suggests that IGFs and their binding proteins are important participants in angiogenesis. In vitro, IGF-I has repeatedly been shown to enhance endothelial cell survival (32). Furthermore, IGF-I has been shown to be synergistic with VEGF in the activation of Akt (33). In vivo, during the menstrual cycle, extensive angiogenesis accompanies luteinization. During luteolysis, endothelial cells die resulting in vascular collapse. In the rodent and primate corpus luteum, endothelial cells express high concentrations of IGFBP-3 during luteolysis, suggesting a proapoptotic role for IGFBP-3 during rat and primate endothelial cell apoptosis (34). Expanding this discovery to humans, Fraser et al. and others (35, 36) have shown that IGFBP-3 messenger RNA is expressed in the endothelium of the human corpus luteum, raising the possibility that regulation of IGFBP-3 expression in endothelia can control angiogenesis and cellular responses in human corpus luteum via autocrine/paracrine mechanisms.
Major diseases including rheumatoid arthritis (37), tumor development (38), and proliferative diabetic retinopathy (39) are driven by persistent, unregulated angiogenesis (40). In particular, diabetic retinopathy is characterized by progressive alterations in the retinal microvasculature, starting with regional nonperfusion, increased vasopermeability, and pathologic, intraocular proliferation of retinal vessels. Uncontrolled neovascularization can result in severe or permanent visual loss in diabetic patients (41). Although the mechanisms of ocular neovascularization are not fully understood, several growth factors, particularly VEGF, IGF-I, and GH are considered major contributors (42). Increased intraocular VEGF levels in diabetic patients correlate with the development of neovascularization (43, 44). Intravitreal injection of VEGF into the eyes of normal, nonhuman primates induces retinal edema, hemorrhage, intraretinal microvascular abnormalities, ischemia, microaneurysms, and intraretinal neovascularization (45). Specific inhibition of VEGF bioactivity prevents neovascularization in animal models (46). In addition, insulin, IGF-I, and GH, may participate in the pathogenesis of diabetic retinopathy through their ability to increase VEGF production (47). Thus, the discovery of endothelial growth inhibitors, such as IGFBP-3, not only increases our understanding of the interplay of these molecules in physiological and pathological angiogenesis but may also provide a novel therapeutic strategy for treatment of angiogenesis-dependent diseases, including cancer and chronic inflammation.
The field of angiogenesis inhibitors continues to evolve rapidly from benchside discovery through clinical application. Currently, diverse compounds are in clinical trials, and new compounds are continually joining the list (48, 49). Despite the promising outlook for antiangiogenic proteins, their production as functional recombinant proteins has proven difficult. Disadvantages of antiangiogenic protein therapy include repeated injections, prolonged treatment, transmission of toxins and infectious particles, and high cost for manufacturing large amounts of protein molecules. Thus, alternative strategies need to be developed to improve the clinical utility of antiangiogenic therapy. Developments of these strategies are ongoing and they include identification of more potent inhibitors, antiangiogenic gene therapy (50), improvement of protein/compound half-lives in the circulation, increase of their concentrations at the disease location, and combinatorial therapies with approaches including chemotherapy, radiotherapy, and immunotherapy. With the identification of IGFBP-3 as a potent inhibitor of VEGF-induced endothelial proliferation, we have come one step closer to improving the understanding of the underlying cellular mechanisms to further the possibility of different therapeutic targets for disease states involving unregulated angiogenesis, such as diabetes.
Acknowledgments
This work was supported in part by NIH Grants 2R01-DK-47591, 1RO1-AI-40203, 1R01-AG-20954, and 1UO1-CA-84128 (to P.C.); 5T32-DK-007688 (to S.L.F.); K08-DK-02876 and 5P30-HD-34610 (to R.F.); grants from the Juvenile Diabetes Foundation; and a Pharmacia GEM grant (to P.C.) as well as a fellowship award from Eli Lilly (to S.L.F.).
Abbreviations
- DAPI
4′,6-Diamidino-2-phenylindole
- EGM-2-MV Bullet Kit
Microvascular Endothelial Cell Growth Medium Bullet Kit-2
- FITC
fluorescein isothiocyanate
- HBD
heparin-binding domain
- HUVEC
human macrovascular umbilical vein endothelial cells
- IGF1R
type 1 IGF receptor
- IGFBP-3
IGF binding protein-3
- PI3-kinase
phosphatidylinositol 3′-kinase
- SDS
sodium dodecyl sulfate
- SFM
serum-free medium
- VEGF
vascular endothelial-derived growth factor
References
- 1.Gibbons GH. Endothelial functions as a determinant of vascular function and structure: a new therapeutic target. Am J Cardiol. 1997;79:3–8. doi: 10.1016/s0002-9149(97)00122-7. [DOI] [PubMed] [Google Scholar]
- 2.Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Prober JS, Wick TM, Konkle BA, Schwartz BS, Barnathan ES, McCrae KR, Hug BA, Schmidt AM, Stern DM. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–3561. [PubMed] [Google Scholar]
- 3.Stehouwer CD, Lambert J, Donker AJ, van Hinsbergh VW. Endothelial dysfunction and pathogenesis of diabetic angiopathy. Cardiovasc Res. 1997;34:55–68. doi: 10.1016/s0008-6363(96)00272-6. [DOI] [PubMed] [Google Scholar]
- 4.Miller JW, Adamis AP, Aiello LP. Vascular endothelial growth factor in ocular neovascularization and proliferative diabetic retinopathy. Diabetes Metab Rev. 1997;13:37–50. doi: 10.1002/(sici)1099-0895(199703)13:1<37::aid-dmr174>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 5.Yokayama H, Deckert T. Central role of TGF-β in the pathogenesis of diabetic nephropathy and macrovascular complications: a hypothesis. Diabetes Med. 1996;13:313–320. doi: 10.1002/(SICI)1096-9136(199604)13:4<313::AID-DIA56>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 6.Minshall C, Arkins S, Freund GG, Kelley KW. Requirement for phosphatidylinositol 3[prime]-kinase to protect hemopoietic progenitors against apoptosis depends upon the extracellular survival factor. J Immunol. 1996;156:939–947. [PubMed] [Google Scholar]
- 7.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–2005. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 8.Songyang Z, Baltimore D, Cantley LC, Kaplan DR, Franke TF. Interleukin 3-dependent survival by the Akt protein kinase. Proc Natl Acad Sci USA. 1997;94:11345–11350. doi: 10.1073/pnas.94.21.11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerber HP, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3[prime]-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 10.Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J, Evan G. Suppression of c-Myc-induced apoptosis by Ras signaling through PI(3)K and PKB. Nature. 1997;385:544–548. doi: 10.1038/385544a0. [DOI] [PubMed] [Google Scholar]
- 11.Nicolaou KC, Trujillo JI, Jandeleit B, Chibale K, Rosenfeld M, Diefenback B, Cheresh DA, Goodman SL. Design, synthesis and biological evaluation of nonpeptide integrin antagonists. Bioorg Med Chem. 1998;6:1185–1208. doi: 10.1016/s0968-0896(98)00090-x. [DOI] [PubMed] [Google Scholar]
- 12.Carpenter CL, Cantley LC. Phosphoinositide kinases. Curr Opin Cell Biol. 1996;8:153–158. doi: 10.1016/s0955-0674(96)80060-3. [DOI] [PubMed] [Google Scholar]
- 13.Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3, 4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- 14.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phophoinositide-dependent protein kinase which phosphorylates and activates protein kinase alpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 15.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 16.Punglia RS, Lu M, Hsu J, Kuroki M, Tolentino MJ, Keough K, Levy AP, Levy Goldberg MA, D’Amato RJ, Adamis AP. Regulation of vascular endothelial growth factor expression by insulin-like growth factor 1. Diabetes. 1997;46:1619–1626. doi: 10.2337/diacare.46.10.1619. [DOI] [PubMed] [Google Scholar]
- 17.Dahlfors G, Arnqvist HJ. Vascular endothelial growth factor and transforming growth factor-β1 regulate the expression of insulin-like growth factorbinding protein-3, -4, and -5 in large vessel endothelial cells. Endocrinology. 2000;141:2062–2067. doi: 10.1210/endo.141.6.7481. [DOI] [PubMed] [Google Scholar]
- 18.Erondu NE, Dake BL, Moser DR, Lin M, Boes M, Bar RS. Regulation of endothelial IGFBP-3 synthesis and secretion by IGF-1 and TGF-beta. Growth Regul. 1996;6:1–9. [PubMed] [Google Scholar]
- 19.Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr Rev. 1999;20:761–787. doi: 10.1210/edrv.20.6.0382. [DOI] [PubMed] [Google Scholar]
- 20.Oh Y, Muller HL, Pham H, Lamson G, Rosenfeld RG. Non-receptor mediated, post-transcriptional regulation of insulin-like growth factor binding protein (IGFBP)-3 inHs578T human breast cancer cells. Endocrinology. 1992;131:3123–3125. doi: 10.1210/endo.131.6.1280212. [DOI] [PubMed] [Google Scholar]
- 21.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 22.Valentinis B, Bhala A, DeAngelis T, Baserga R, Cohen P. The human insulin-like growth factor (IGF) binding protein-3 inhibits the growth of fibroblasts with a targeted disruption of the IGF-1 receptor gene. Mol Endocrinol. 1995;9:361–367. doi: 10.1210/mend.9.3.7539889. [DOI] [PubMed] [Google Scholar]
- 23.Schedlich LJ, Le Page SL, Firth SM, Briggs LJ, Jans DA, Baxter RC. Nuclear import of insulin-like growth factor-binding protein-3 and -5 is mediated by the importinβ subunit. J Biol Chem. 2000;275:23462–23470. doi: 10.1074/jbc.M002208200. [DOI] [PubMed] [Google Scholar]
- 24.Liu B, Lee HY, Weinzimer SA, Powell DR, Clifford JL, Kurie JM, Cohen P. Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-α regulate transcriptional signaling and apoptosis. J Biol Chem. 2000;275:33607–33613. doi: 10.1074/jbc.M002547200. [DOI] [PubMed] [Google Scholar]
- 25.Weinzimer SA, Gibson TB, Collett-Solberg PF, Khare A, Liu B, Cohen P. Transferrin is an insulin-like growth factor-binding protein-3 binding protein. J Clin Endocrinol Metab. 2001;86:1806–1813. doi: 10.1210/jcem.86.4.7380. [DOI] [PubMed] [Google Scholar]
- 26.Yamanaka Y, Fowlkes JL, Wilson EM, Rosenfeld RG, Oh Y. Characterization of insulin-like growth factor binding protein-3 (IGFBP-3) binding to human breast cancer cells: kinetics of IGFBP-3 binding and identification of receptor binding domain on the IGFBP-3 molecule. Endocrinology. 1999;140:1319–1328. doi: 10.1210/endo.140.3.6566. [DOI] [PubMed] [Google Scholar]
- 27.Knedler A, Ham RG. Optimized medium for clonal growth of human microvascular endothelial cells with minimal serum. In Vitro Cell Dev Biol. 1987;23:481–491. doi: 10.1007/BF02628418. [DOI] [PubMed] [Google Scholar]
- 28.Cory AH, Owen TC, Barltrop JA, Cory JG. Use of an aqueous soluble tetrazolium/formazan assay for cell growth assays in culture. Cancer Commun. 1991;3:207–212. doi: 10.3727/095535491820873191. [DOI] [PubMed] [Google Scholar]
- 29.Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induced apoptosis and mediates effects of transforming growth factor-beta1 on programmed cell death through p53- and IGF-independent mechanisms. J Biol Chem. 1997;272:12181–12188. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- 30.Chiarelli F, Spagnoli A, Basciani F, Tumini S, Mezzetti A, Cipollone F, Cuccurullo F, Morgese G, Verrotti A. Vascular endothelial growth factor (VEGF) in children, adolescents and young adults with Type1 diabetes mellitus: relation to glycaemic control and microvascular complications. Diabet Med. 2000;17:650–656. doi: 10.1046/j.1464-5491.2000.00350.x. [DOI] [PubMed] [Google Scholar]
- 31.Folkman J. Angiogenesis and angiogenesis inhibition: an overview. EXS. 1997;79:1–8. doi: 10.1007/978-3-0348-9006-9_1. [DOI] [PubMed] [Google Scholar]
- 32.Wilson SH, Davis MI, Caballero S, Grant MB. Modulation of retinal endothelial cell behavior by insulin-like growth factor I and somatostatin analogues: implications for diabetic retinopathy. Growth Horm IGF Res. 2001;11:S53–S59. doi: 10.1016/s1096-6374(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 33.Hellstrom A, Perruzzi C, Ju M, Engstrom E, Hard AL, Liu JL, Albersson-Wikland K, Carlsson B, Niklasson A, Sjodell L, LeRoith D, Senger DR, Smith LE. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: direct Correlation with clinical retinopathy of prematurity. Proc Natl Acad Sci USA. 2001;98:5804–5808. doi: 10.1073/pnas.101113998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fraser HM, Lunn SF, Kim H, Erickson GF. Insulin-like growth factor binding protein-3 mRNA expression in endothelial cells of the primate corpus luteum. Hum Reprod. 1998;13:2180–2185. doi: 10.1093/humrep/13.8.2180. [DOI] [PubMed] [Google Scholar]
- 35.Fraser HM, Lunn SF, Kim H, Duncan WC, Rodger FE, Illingworth PJ, Erickson GF. Changes in insulin-like growth factor-binding protein-3 messenger ribonucleic acid In endothelial cells of the human corpus luteum: a possible role in luteal development and rescue. J Clin Endocrinol Metab. 2000;85:1672–1677. doi: 10.1210/jcem.85.4.6497. [DOI] [PubMed] [Google Scholar]
- 36.Klein NA, Battaglia DE, Woodruff TK, Padmanabhan V, Giudice LC, Bremner WJ, Soules MR. Ovarian follicular concentrations of activin, follistatin, inhibin, insulin-like growth factor I (IGF-I), IGF-II, IGF-binding protein-2 (IGFBP-2), IGFBP-3, and vascular endothelial growth factor in spontaneous Menstrual cycles of normal women of advanced reproductive age. J Clin Endocrinol Metab. 2000;85:4520–4525. doi: 10.1210/jcem.85.12.7056. [DOI] [PubMed] [Google Scholar]
- 37.Firestein GS. Starving the synovium: angiogenesis and inflammation in rheumatoid arthritis. J Clin Invest. 1999;103:3–4. doi: 10.1172/JCI5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folkman J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65–71. [PubMed] [Google Scholar]
- 39.Aiello LP, Gardner TW, King GL, Blankenship G, Cavallerno JD, Ferris LP, Klein R. Diabetic retinopathy. Diabetes Care. 1998;21:143–156. doi: 10.2337/diacare.21.1.143. [DOI] [PubMed] [Google Scholar]
- 40.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 41.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–10934. [PubMed] [Google Scholar]
- 42.Lu M, Amano S, Miyamoto K, Garland R, Keough K, Pin W, Adamis AP. Insulin-induced vascular endothelial growth factor expression in the retina. Invest Ophthalmol Vis Sci. 1999;40:3281–3286. [PubMed] [Google Scholar]
- 43.Adamis AP, Miller JW, Bernal M, D’Amico DJ, Folkman J, Yeo TK, Yeo KT. Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthal. 1994;118:445–450. doi: 10.1016/s0002-9394(14)75794-0. [DOI] [PubMed] [Google Scholar]
- 44.Aiello LP, Northrup JM, Keyt BA, Takagi H, Iwamoto MA. Hypoxic regulation of vascularendothelial growth factor in retinal cells. Arch Ophthalmol. 1995;113:1538–1544. doi: 10.1001/archopht.1995.01100120068012. [DOI] [PubMed] [Google Scholar]
- 45.Tolentino MT, Miller JW, Gragoidas ES, Jakobiec FA, Flynn E, Chatzistefanou K, Ferrara N, Adamis AP. Intravitreal injections of vascular endothelial growth factor (VEGF) produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology. 1996;103:1820–1828. doi: 10.1016/s0161-6420(96)30420-x. [DOI] [PubMed] [Google Scholar]
- 46.Adamis AP, Shima DT, Tolentino MJ, Gragoudas ES, Ferrera N, Folkman J, D’Amore PA, Miller JW. Inhibition of VEGF prevents retinal ischemiaassociated iris neovascularization in a primate. Arch Ophthalmol. 1996;114:66–71. doi: 10.1001/archopht.1996.01100130062010. [DOI] [PubMed] [Google Scholar]
- 47.Lu M, Kuroki M, Amano S, Amano S, Tolentino M, Keough K, Kim I, Bucala R, Adamis AP. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–1224. doi: 10.1172/JCI1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol. 2001;33:357–369. doi: 10.1016/s1357-2725(01)00023-1. [DOI] [PubMed] [Google Scholar]
- 49.Rosen L. Antiangiogenic strategies and agents in clinical trials. Oncologist. 2000;5:20–27. doi: 10.1634/theoncologist.5-suppl_1-20. [DOI] [PubMed] [Google Scholar]
- 50.Nesbit M. Abrogation of tumor vasculature using gene therapy. Cancer Metastasis Rev. 2000;19:45–49. doi: 10.1023/a:1026588028780. [DOI] [PubMed] [Google Scholar]