

Abstract
Vinyl ethers selectively undergo the combined C—H functionalization/Cope rearrangement reaction (CHCR) via an s-cis/boat transition. With chiral dirhodium catalysts, products are generated in a highly diastereoselective and enantioselective fashion. This reaction can be considered as a surrogate to the traditional vinylogous Mukaiyama aldol reaction. Effective kinetic resolution has been achieved, leading to the recovery of a cyclic vinyl ether with axial chirality of high enantiomeric purity.
C—H functionalization has the potential to revolutionize the synthetic strategies used for making natural products and pharmaceutical targets.1 Rhodium-catalyzed carbenoid C—H insertion is one of the most effective methods for stereoselective C—H functionalization.2 Of particular interest is the possibility of using the C—H functionalization as a complementary disconnection strategy to the conventional transformations used in organic synthesis.1 Intermolecular C—H functionalization by means of carbenoid-induced C—H insertion has been shown to be complementary to the aldol reaction,3 Mannich reaction,4 Claisen condensation,5 Michael addition6 and the Claisen rearrangement.3a In this paper we describe how the combined C—H functionalization/Cope rearrangement (CHCR) can be applied as a surrogate to the vinylogous Mukaiyama aldol reaction.
The vinylogous Mukaiyama aldol reaction has been widely used in organic synthesis.7 It has the potential of generating highly functionalized products containing two newly-formed stereogenic centers. In recent years, several protocols have been developed to achieve this transformation in an enantioselective manner.8 Even with these advances, controlling both the diastereoselectivity and enantioselectivity of the vinylogous Mukaiyama aldol reaction is still a challenge. Recently, Panek reported a carbenoid approach to generate the typical syn-products of a vinylogous Mukaiyama aldol reaction through a two-steps sequence, a rhodium-catalyzed asymmetric Si—H insertion between vinyldiazoacetates and silanes followed by a Lewis acid-catalyzed crotylation (Scheme 1).9 The C—H functionalization approach described herein, complements the Panek approach, leading to a highly stereoselective method for the formation of the typical anti-products of the vinylogous Mukaiyama aldol.
Scheme 1.

Two carbenoid approaches to vinylogous aldol products
The new methodology arises from the CHCR reaction between allylic C—H bonds and vinyldiazoacetates.10 This reaction typically generates products with two new stereocenters. With suitable substrate design, the reaction can proceed through either s-cis/chair10a–f or s-cis/boat10g transition states to generate independently the two possible diastereomers (eq 1 and 2). Recent computational studies revealed that the reaction proceeds through a concerted, but highly asynchronous hydride transfer/C—C bond forming process and there is a potential energy surface bifurcation between the CHCR reaction and the competing direct C—H insertion reaction.11 As a result, controlling reactions to undergo selectively a CHCR reaction over a direct C—H insertion is challenging. Only a few substrate systems to date favor a clean CHCR reaction, and the majority of these are cyclohexadiene and dihydronaphthalene derivatives.10a–b,10c–f Therefore, a major challenge in this field is to broaden the scope of substrates that will selectively undergo the CHCR reaction.
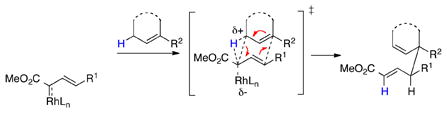 |
(1) |
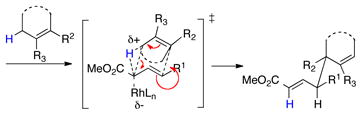 |
(2) |
Recently, we described that disubstituted cyclopentenyl derivatives smoothly undergo the CHCR reaction via a boat transition state.10g Evaluation of the s-cis/boat transition state model11 lead us to the realization that acyclic trisubstituted vinyl ethers might be ideal substrates for CHCR reaction because steric repulsion with the catalyst “wall” would be avoided in a s-cis/boat transition state (Scheme 2). If this is indeed the case the scope of the CHCR reaction will be greatly broadened because vinyl ethers are readily accessible from the corresponding ketones via the Wittig reaction.
Scheme 2.

Rationalization of substrate scope design
In order to test the hypothesis presented above, the Rh2(S-PTAD)4-catalyzed decomposition of siloxyvinyldiazoacetate 1 with vinyl ether 2 was examined. Rh2(S-PTAD)4 is known to be an effective catalyst for the enantioselective reaction of siloxyvinyldiazoacetates such as 1, and when the reaction was conducted at −20 °C with 1 mol % of catalyst, followed by desilylation and diazotization, the diazoacetoacetate 3 was formed as a single diastereomer in 91% overall yield with 94% ee over three steps. The siloxyvinyldiazoacetate 1 is one of the best vinylcarbenoid precursors with regard to favoring the CHCR-reaction over a direct C—H insertion, but the formation of 3 in 91% yield is much higher than the yields obtained in the previous study using cyclopentene substrates.10g
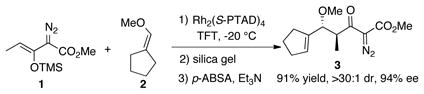 |
(3) |
On the basis of the promising result with 1, we then focused on whether this reaction can be applied to a range of vinyl ethers and vinyldiazoacetates. Previous studies have shown that Rh2(S-DOSP)4 is the optimum chiral catalyst for asymmetric reactions with trans-vinyldiazoacetates (Figure 1).12 The Rh2(S-DOSP)4-catalyzed reaction of trans-phenyl vinyldiazoacetate 8 with vinyl ether 2 generated the CHCR product 13 as a single diastereomer in good yield with extremely high asymmetric induction (entry 1, Table 1). This transformation was then successfully applied to a variety of trans-vinyldiazoacetates and vinyl ethers as summarized in Table 1. In all cases, a single diastereomer of the CHCR product was produced in good yields (67–89%) and with excellent enantioselectivity, in the majority cases greater than 98% ee. Traces of the direct C—H insertion products were observed in the crude reaction mixtures in some cases, but the amounts were never more than 10% of the total C—H functionalization products. The absolute configuration of product 14 was unambiguously assigned by X-ray crystallography.13 The stereo-chemical outcome is consistent with the CHCR reaction proceeding via a boat transition state, The stereochemical configuration of the other CHCR products 13,15–21 were tentatively assigned on the assumption that all the substrates reacted through a similar transition state and trajectory of approach.
Figure 1.
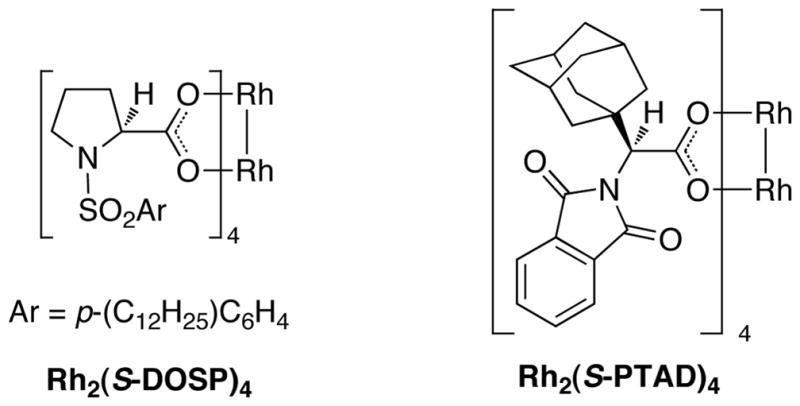
Structures of Rh2(S-DOSP) 4 and Rh2(S-PTAD)4.
Table 1.
Substrate Scope between vinyl ethers and vinyldiazoacetates
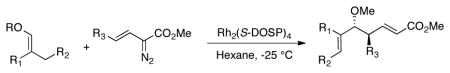 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | ethers | R3 | producta | yield (%) | ee (%) | |||
| 1 |
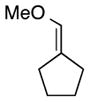
|
2 | Ph | 8 |
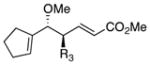
|
13 | 78 | 99 |
| 2 | 2-Naph | 9 | 14 | 88 | 99 | |||
| 3 | p-OMePh | 10 | 15 | 77 | 98 | |||
| 4 | Et | 11 | 16 | 87 | 98 | |||
| 5 | Me | 12 | 17 | 67 | 93 | |||
| 6 |
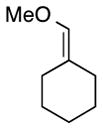
|
4 | Ph | 8 |
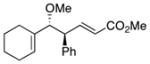
|
18 | 88 | 99 |
| 7 |
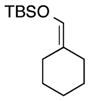
|
5 | Ph | 8 |
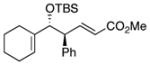
|
19 | 75 | 98 |
| 8 |
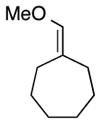
|
6 | Ph | 8 |
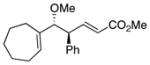
|
20 | 89 | 99 |
| 9 |
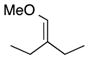
|
7 | Ph | 8 |
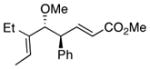
|
21 | 74 | 99 |
The diastereoselective ratio is greater than 30:1 in all reactions
The CHCR reaction with acyclic ether 7 deserves further comment. In previous studies, effective CHCR reactions were limited to cyclic substrates as otherwise a significant amount of competing direct C-H insertion products occurred.14 The ether 7 represents nice examples of effective CHCR reactions occurring in acyclic systems. The geometry of the newly formed trisubstituted double bonds in 21 is consistent with the hypothesis that the reaction is proceeding through an s-cis/boat transition state (Scheme 3).11 The resulting geometry of the enoate is characteristic of whether the reaction is occurring on the s-cis vinylcarbenoid (to form the E-enoate) or on the s-trans vinylcarbenoid (to form the Z-enoate).14 The anti diastereoselectivity is indicative of a boat transition state11 and the geometry of the trisubstituted double is set through positioning of the methyl group away from the rhodium “wall” in the transition state.
Scheme 3.

Transition state analysis with acyclic substrate
The reactions of donor/acceptor carbenoids are very sensitive to steric and electronic effects2b and this is also the case with the CHCR reaction. These controlling influences were readily seen in the reaction of 8 with the isomeric mixture of 22a and 22b (eq 4). Each isomer has two possible allylic sites for C—H functionalization, yet product 23 was formed as the major product in 60% yield. Less than 10% of other CHCR products were observed. The methoxy group blocks attacks at the allylic site cis to it,2a which means the methyl site (H2) in 22a and the methylene site (H4) in 22b are not prone to C—H functionalization. The CHCR reaction is initiated by a hydride transfer event.11 Thus, the methylene site (H1) in 22a is more reactive than the primary methyl group (H3) in 22b because the methylene site would be better at stabilizing positive charge build-up in the transition state. Once again the C—H functionalization is highly stereoselective as 23 was formed with >30: 1 dr and 94% ee.
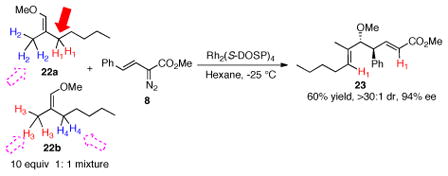 |
(4) |
A few examples have been reported of substrates capable of undergoing two carbenoid reactions, resulting in the rapid generation of synthetic complexity.15 We recognized that if the CHCR reaction was initiated at a primary methyl site of a vinyl ether such as 24, then the resulting 1,2-disubstituted alkene 25 would be sterically accessible for a subsequent cyclopropanation (eq 5). In order to test this possibility, the Rh2(S-DOSP)4-catalyzed reaction of ether 24 with excess of trans-styryldiazoacetate 8 was examined. This resulted in the formation of 26, containing four newly formed contiguous stereo-centers, two of which are quaternary. 26 was formed in 53% yield as a single diastereomer in 99% ee. The relative configuration of the cyclopropane in 26 was determined by nOe, while the absolute configuration of 26 was tentatively assigned on the basis of the expected s-cis/boat transition state model and the predictive transition state model for the cyclopropanation.16 This reaction indicated that both the CHCR and the cyclopropanation reactions occurred in a highly stereoselective fashion as only one diastereomer of the product was observed. The reaction represents the first example of the CHCR reaction being initiated by attack at a methyl C—H bond.
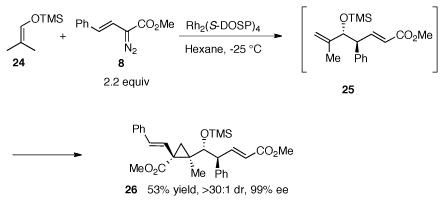 |
(5) |
Donor/acceptor carbenoids have been shown to be capable of kinetic resolution, desymmetrization and enantiomer differ-differentiation.10a–b,10g,17 In order to explore the possibility of kinetic resolution in the CHCR reaction, the reaction between phenyl-substituted substrate 27a and 0.6 equiv of diazo compound 8 in the presence of Rh2(S-DOSP)4 was examined (eq 6). This generated the CHCR product 28a as a single diastereomer in 99% ee in good isolated yield (94% yield based on one enantiomer of ether 27a) (eq 6). The unreactive vinyl ether 27a was recovered in 40% isolated yield with 98% ee. Thus, the Rh2(S-DOSP)4-catalyzed reaction between 27a and 8 displays a very high level of kinetic resolution. A similar kinetic resolution was achieved with the methyl-substituted substrate 27b, but the stereoselectivity was not as pronounced. The CHCR product 28b was produced in 92% ee, and the recovered vinyl ether 27b was obtained in 60% ee. This kinetic resolution approach provides a convenient way of making enantio-pure cyclic vinyl ethers with axial chirality. The asymmetric synthesis of this type of compounds is typically challenging and the only successful previous approach required the use of chiral Wittig reagents as stoichiometric reagents.18 The absolute and relative configuration of products 28a and 28b were unambiguously assigned by X-ray crystallography13 and are consistent with the CHCR reaction proceeding though a s-cis/boat transition state.11
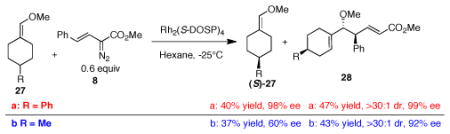 |
(6) |
A reasonable mechanism that would be consistent with the kinetic resolution is shown in Scheme 4. The Rh2(S-DOSP)4 is proposed to adopt a D2-symmetry and the substrate would only approach from the front face.12 The CHCR reaction proceeds through a boat transition state as shown.11 The R group in the substrate will need to point away from the carbenoid to avoid steric interference. This controlling element must be severe for 27a when R = phenyl, as only one enantiomer of the substrate reacts, leading to products with high stereochemical control. When the R group is methyl, the steric interference is not as dramatic and this would account for the moderate kinetic resolution with 27b.
Scheme 4.

Proposed transition state for the kinetic resolution
In conclusion, vinyl ethers have been demonstrated to be excellent substrates for the CHCR reaction. The reaction proceeds through an s-cis boat transition state, leading to the formation of products of defined stereochemistry that might typically be approached by the vinylogous Mukaiyama aldol reaction. These studies demonstrate that asymmetric C—H functionalization by the CHCR reaction can compete with a classic strategic reaction for organic synthesis. Furthermore, the studies underscore the subtle steric and electronic influences that allow the CHCR reaction to be highly regio, di-astereo- and enantioselective.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation under the Center for Chemical Innovation in Stereoselective C–H Functionalization (CHE-0943980) and by the National Institutes of Health (GM080337). We thank Dr. Ken Hardcastle for the X-ray crystallographic structure determination.
Footnotes
Supporting Information Available: Full experimental data for the compounds described in the paper and X-ray crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gutekunst WR, Baran PS. Chem Soc Rev. 2011;40:1976. doi: 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]; (c) Colby DA, Bergman RG, Ellman JA. Chem Rev. 2010;110:624. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a. [DOI] [PubMed] [Google Scholar]; (e) Zalatan DN, Du Bois J. In: C-H Activation. Yu J-Q, Shi Z, editors. Vol. 292. WILEY-VCH Verlag; Heidelberg: 2010. pp. 347–379. [Google Scholar]; (f) Che CM, Lo VK, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]; (g) Lyons TW, Sanford MS. Chem Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Collet F, Lescot C, Dauban P. Chem Soc Rev. 2011;40:1926. doi: 10.1039/c0cs00095g. [DOI] [PubMed] [Google Scholar]; (i) McMurray L, O’Hara F, Gaunt MJ. Chem Soc Rev. 2011;40:1885. doi: 10.1039/c1cs15013h. [DOI] [PubMed] [Google Scholar]; (j) Zhou M, Crabtree RH. Chem Soc Rev. 2011;40:1875. doi: 10.1039/c0cs00099j. [DOI] [PubMed] [Google Scholar]; (k) Newhouse T, Baran PS. Angew Chem, Int Ed. 2011;50:3362. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Davies HML, Denton JR. Chem Soc Rev. 2009;38:3061. doi: 10.1039/b901170f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (c) Davies HML, Morton D. Chem Soc Rev. 2011;40:1857. doi: 10.1039/c0cs00217h. [DOI] [PubMed] [Google Scholar]; (d) Slattery CN, Ford A, Maguire AR. Tetrahedon. 2010;66:6681. [Google Scholar]; (e) Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]; (f) Lu H, Zhang XP. Chem Soc Rev. 2011;40:1899. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]; (g) Díaz-Requejo MM, Pérez PJ. Chem Rev. 2008;108:3379. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]; (h) Doyle MP, Forbes CS. Chem Rev. 1998;98:911. doi: 10.1021/cr940066a. [DOI] [PubMed] [Google Scholar]
- 3.(a) Davies HML, Beckwith REJ, Antoulinakis EG, Jin Q. J Org Chem. 2003;68:6126. doi: 10.1021/jo034533c. [DOI] [PubMed] [Google Scholar]; (b) Davies HML, Antoulinakis EG, Hansen T. Org Lett. 1999;1:383. [Google Scholar]; (c) Davies HML, Antoulinakis EG. Org Lett. 2000;2:4153. doi: 10.1021/ol006671j. [DOI] [PubMed] [Google Scholar]
- 4.Davies HML, Venkataramani C, Hansen T, Hopper DW. J Am Chem Soc. 2003;125:6462. doi: 10.1021/ja0290072. [DOI] [PubMed] [Google Scholar]
- 5.(a) Davies HML, Ren P, Jin Q. Org Lett. 2001;3:3587. doi: 10.1021/ol0167255. [DOI] [PubMed] [Google Scholar]; (b) Davies HML, Yang J, Nikolai J. J Organomet Chem. 2005;690:6111. [Google Scholar]
- 6.Davies HML, Ren P. J Am Chem Soc. 2001;123:2070. doi: 10.1021/ja0035607. [DOI] [PubMed] [Google Scholar]
- 7.For some reviews, see: Casiraghi G, Battistini L, Curti C, Rassu G, Zanardi F. Chem Rev. 2011;111:3076. doi: 10.1021/cr100304n.Denmark SE, Heemstra JR, Jr, Beutner GL. Angew Chem, Int Ed. 2005;44:4682. doi: 10.1002/anie.200462338.Casiraghi G, Zanardi F, Appendino G, Rassu G. Chem Rev. 2000;100:1929. doi: 10.1021/cr990247i.Kalesse M. Top Curr Chem. 2005;244:43.
- 8.For some recent examples, see: Hassan A, Zbieg JR, Krische MJ. Angew Chem, Int Ed. 2011;50:3493. doi: 10.1002/anie.201100646.Gieseler MT, Kalesse M. Org Lett. 2011;13:2430. doi: 10.1021/ol2006727.Luo J, Wang H, Han X, Xu LW, Kwiatkowski J, Huang KW, Lu Y. Angew Chem, Int Ed. 2011;50:1861. doi: 10.1002/anie.201006316.Ratjen L, Garcia-Garcia P, Lay F, Beck ME, List B. Angew Chem, Int Ed. 2011;50:754. doi: 10.1002/anie.201005954.Matsui R, Seto K, Sato Y, Suzuki T, Nakazaki A, Kobayashi S. Angew Chem, Int Ed. 2011;50:680. doi: 10.1002/anie.201006230.Ube H, Shimada N, Terada M. Angew Chem, Int Ed. 2010;49:1858. doi: 10.1002/anie.200906647.Singh RP, Foxman BM, Deng L. J Am Chem Soc. 2010;132:9558. doi: 10.1021/ja103331t.
- 9.Wu J, Chen Y, Panek JS. Org Lett. 2010;12:2112. doi: 10.1021/ol100604m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485. doi: 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]; (b) Davies HML, Walji AM. Angew Chem Int Ed. 2005;44:1733. doi: 10.1002/anie.200462227. [DOI] [PubMed] [Google Scholar]; (c) Davies HML, Jin Q. Proc Natl Acad Sci U S A. 2004;101:5472. doi: 10.1073/pnas.0307556101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Davies HML, Jin Q. J Am Chem Soc. 2004;126:10862. doi: 10.1021/ja047185k. [DOI] [PubMed] [Google Scholar]; (e) Davies HML, Manning JR. J Am Chem Soc. 2006;128:1060. doi: 10.1021/ja057768+. [DOI] [PubMed] [Google Scholar]; (f) Davies HML, Stafford DG, Hansen T. Org Lett. 1999;1:233. doi: 10.1021/ol9905699. [DOI] [PubMed] [Google Scholar]; (g) Lian Y, Hardcastle KI, Davies HML. Angew Chem Int Ed. submitted. [Google Scholar]
- 11.Hansen JH, Gregg TM, Ovalles SR, Lian Y, Autschnach J, Davies HML. J Am Chem Soc. 2011;133:5076. doi: 10.1021/ja111408v. [DOI] [PubMed] [Google Scholar]
- 12.Hansen JH, Davies HML. Coord Chem Rev. 2008;252:545. doi: 10.1016/j.ccr.2007.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The crystal structures of 14, 28a, 28b have been deposited at the Cambridge Crystallographic Data Centre, and the deposition number CCDC 827302, 827300, 827301 have been allocated correspondingly.
- 14.Davies HML, Beckwith REJ. J Org Chem. 2004;69:9241. doi: 10.1021/jo048429m. [DOI] [PubMed] [Google Scholar]
- 15.(a) Davies HML, Jin Q. Org Lett. 2004;6:1769. doi: 10.1021/ol0495467. [DOI] [PubMed] [Google Scholar]; (b) Yan M, Jacobsen N, Hu W, Gronenberg LS, Doyle MP, Colyer JL, Bykowski D. Angew Chem, Int Ed. 2004;43:6713. doi: 10.1002/anie.200461722. [DOI] [PubMed] [Google Scholar]
- 16.Davies HML, Bruzinski P, Hutcheson DK, Kong N, Fall MJ. J Am Chem Soc. 1996;118:6897. [Google Scholar]
- 17.(a) Nadeau E, Ventura DL, Brekan JA, Davies HML. J Org Chem. 2010;75:1927. doi: 10.1021/jo902644f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lian Y, Miller LC, Born S, Sarpong R, Davies HML. J Am Chem Soc. 2010;132:12422. doi: 10.1021/ja103916t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deng L, Giessert AJ, Gerlitz OO, Dai X, Diver ST, Davies HML. J Am Chem Soc. 2005;127:1342. doi: 10.1021/ja045173t. [DOI] [PubMed] [Google Scholar]
- 18.(a) Hanessian S, Delorme D, Beaudoin S, Leblanc Y. J Am Chem Soc. 1984;106:5754. [Google Scholar]; (b) Denmark SE, Rivera I. J Org Chem. 1994;59:6887. [Google Scholar]; (c) Dai W, Wu A, Wu H. Tetrahedron: Asymmetry. 2002;13:2187. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.