

Abstract
Human N-formyl peptide receptor 1 (FPR1) is a G protein-coupled receptor (GPCR) involved in host defense and sensing cellular damage. Since structure-based ligand design for many GPCRs, including FPR1, is restricted by the lack of experimental three dimensional structures, homology modeling has been widely used to study GPCR-ligand binding. Indeed, receptor-ligand binding mode predictions can be derived from homology modeling with supporting ligand information. In the present work, we report comparative docking studies of 2-(benzi-midazol-2-ylthio)-N-phenylacetamide derived FPR1 agonists, identified here and previously, with several known FPR1 peptide agonists in a FPR1 homology model that is based on the crystal structure of bovine rhodopsin. We found that the binding pocket of the most active molecules shares some common features with high affinity FPR1 peptide agonists, suggesting that they may bind to similar binding sites. Classification tree analysis led to the derivation of a good recognition model based on four amino acid descriptors for distinguishing FPR1 ligands from inactive analogs. Hence, the corresponding residues (Thr199, Arg201, Gly202, and Ala261) can be considered as markers of important areas in the ligand binding site. Concurrently, we identified several unique binding features of benzimidazole derivatives and showed that alkoxy-substituents of the benzimidazole ring are located within a FPR1 hole bounded by Thr199, Thr265, Ile268, and Leu271 or in a groove in the vicinity of Leu198, Arg201, Gly202, and Arg205. The understanding of these molecular features will likely prove beneficial in future design of novel FPR1 agonists based on the benzimidazole scaffold.
Keywords: Benzimidazole derivatives, Binding site, FPR1 agonist, G protein-coupled receptor (GPCR), Homology model, Molecular docking, N-formyl peptide receptor 1 (FPR1)
Introduction
N-formyl peptides are potent agonists of innate immune cells, including neutrophils, monocyte/macrophages, and dendritic cells [1]. These peptides are recognized by G protein-coupled receptors known as formyl peptide receptors or FPRs [1]. FPRs contribute to a variety of physiological processes, including host defense against bacterial infection and resolving inflammation [2–4]. Three FPR subtypes are present in humans (designated as FPR1, FPR2, and FPR3), and all three are coupled to the Gαi family of G proteins [1, 5]. Activation of FPRs induces a variety of responses, which are dependent on the agonist, cell type, receptor subtype, and species involved. For example, N-formyl-Met-Leu-Phe (fMLF), an FPR1 agonist, activates human phagocyte inflammatory responses, such as intracellular calcium mobilization, production of cytokines, generation of reactive oxygen species (ROS), and chemotaxis [5]. In contrast, some mediators shown to promote resolution of inflammatory processes have been found to be FPR2 agonists [6], although the exact receptor specificity has recently been questioned [7, 8].
In addition to phagocytes, FPRs are expressed on a variety of non-leukocyte cell types, including epithelial cells, hepatocytes, fibroblasts, and astrocytes (reviewed in [1]). The diverse tissue expression of these receptors suggests the possibility of as-yet unappreciated complexity in the innate response and perhaps other unidentified functions for FPR family members. For example, it was recently found that fMLF promotes osteoblast differentiation from human bone marrow mesenchymal stem cells, suggesting a role for FPR1 in this process [9]. Similarly, the FPR2 peptide agonist Trp-Lys-Tyr-Met-Val-D-Met (WKYMVm) protected against death by enhancing bactericidal activity and inhibiting vital organ inflammation and apoptosis in a sepsis mouse model [10]. Indeed, it has been suggested that FPR agonists have potential for therapeutic development (reviewed in [11, 12]). The list of structurally diverse FPR1/FPR2 agonists has steadily grown in recent years and includes compounds with EC50 values in the low micromolar to nanomolar range [13, 14]. However, attempts to create specific and efficacious therapeutics targeting these receptors have been hindered by the lack of a three dimensional FPR structure.
In the absence of experimental structural data for FPRs, one of the few ways to investigate the binding modes of FPR agonists is through molecular modeling. An understanding of the binding modes of FPR agonists may assist the design of novel non-peptide FPR agonists with high affinity. Indeed, a homology modeling method based on the crystal structure of bovine rhodopsin has been successfully applied to different GPCRs [15] including FPR1 [7, 16, 17] to further understand the ligand–receptor interactions and to identify new ligands. For example, a homology model of angiotensin II type 1 (AT1) receptor was used to explore the binding sites of several nonpeptide AT1 receptor antagonists [18], and a homology model of the M1 muscarinic acetylcholine receptor was applied to understand the mechanism of activation by the agonist–receptor complex [19]. The recent publication of several GPCR structures has increased the information available for homology modeling, including X-ray crystal structures for bovine rhodopsin, turkey β1 and human β2 adrenergic receptors, human histamine H1 receptor, human A2A adenosine receptor, human C-X-C chemokine receptor 4, and human D3 dopaminergic receptor [20, 21].
In recent studies, we demonstrated that 2-(benzimidazol-2-ylthio)-N-phenylacetamides represent a unique chemical scaffold for FPR1 agonists [13]. Since these compounds represent a potentially novel molecular design template for FPR1, we hypothesized that comparison of their binding modality with those of other known FPR1 agonists should be instructive regarding the design of next generation agonists. Hence, we selected 26 additional 2-(benzimida-zol-2-ylthio)-N-phenylacetamide derivatives for testing agonist activity in human neutrophils and HL-60 cells transfected with human FPR1 and FPR2 and identified 11 additional FPR1-specific agonists, and 10 mixed FPR1/FPR2 agonists. To further define the FPR1 ligand–binding site(s) for these and previously described benzimidazole derivatives and several peptide agonists, we performed a precise ligand docking simulation and have successfully identified the putative binding site. Analysis of docking poses based on the homology FPR1 model effectively differentiated agonists from non-active compounds. Overall, unique binding features of benzimidazole derivatives were identified, which will likely prove beneficial in future design of novel FPR1 agonists based on the benzimidazole scaffold.
Materials and methods
Dimethyl sulfoxide (DMSO), fMLF, and Histopaque 1077 were purchased from Sigma Chemical Co. (St. Louis, MO). WKYMVm was from Calbiochem (San Diego, CA). Hanks’ balanced salt solution (HBSS; 0.137 M NaCl, 5.4 mM KCl, 0.25 mM Na2HPO4, 0.44 mM KH2PO4, 4.2 mM NaHCO3, 5.5 6 mM glucose, and 10 mM HEPES, pH 7.4) was from Invitrogen (Carlsbad, CA). HBSS containing 1.3 mM CaCl2 and 1.0 mM MgSO4 is designated HBSS+. Screening compounds were purchased from Princeton BioMolecular Research (Monmouth Junction, NJ) and InterBioScreen (Moscow, Russia). The purity and identity of the compounds were verified using NMR spectroscopy, elemental analysis, and mass spectroscopy, as performed by the suppliers. The compounds were diluted in DMSO at a concentration of 20 mM and stored at −20°C.
Cell culture
Human promyelocytic leukemia HL-60 cells stably transfected with human FPR1 or FPR2 were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal calf serum, 10 mM HEPES, 100 μg/ml streptomycin, 100 U/ml penicillin, and G418 (1 mg/ml), as described previously [22]. Wild-type HL-60 cells were cultured under the same conditions but without G418.
Neutrophil isolation
Human neutrophils were isolated from blood collected from healthy donors in accordance with a protocol approved by the Institutional Review Board at Montana State University-Bozeman. Neutrophils were purified from the blood using dextran sedimentation, followed by Histopaque 1077 gradient separation and hypotonic lysis of red blood cells, as described previously [23]. Isolated neutrophils were washed twice and resuspended in HBSS. Neutrophil preparations were routinely >95% pure, as determined by light microscopy, and >98% viable, as determined by trypan blue exclusion.
Ca2+ mobilization assay
Changes in intracellular Ca2+ were measured with a FlexStation II scanning fluorometer using the fluorescent dye Fluo-4AM (Invitrogen). Neutrophils or HL-60 cells, suspended in HBSS, were loaded with Fluo-4AM dye (final concentration, 1.25 μg/ml) and incubated for 30 min in the dark at 37°C. After dye loading, the cells were washed with HBSS, resuspended in HBSS+, separated into aliquots, and deposited into the wells of flat-bottomed, half-area-well black microtiter plates (2×105 cells/well). The compound source plate contained dilutions of test compounds in HBSS+. Changes in fluorescence were monitored (λex= 485 nm, λem=538 nm) every 5 sec for 240 sec at room temperature after automated addition of compounds. Maximum change in fluorescence, expressed in arbitrary units over baseline, was used to determine agonist response. Responses were normalized to the response induced by 5 nM fMLF for HL-60 FPR1 cells and neutrophils or 5 nM WKYMVm for HL-60 FPR2 cells, which were assigned a value of 100%. Curve fitting (at least five to six points) and calculation of median effective concentration values (EC50) were performed by nonlinear regression analysis of the dose-response curves generated using Prism 5 (GraphPad Software, Inc., San Diego, CA). All active compounds were evaluated in wild-type HL-60 cells to verify that the agonists were inactive in non-transfected cells.
Molecular modeling
The FPR1 homology model was created using the crystal structure of bovine rhodopsin, which has a sequence identity of 20% for 348 aligned residues that correspond to the seven transmembrane domains, as reported previously [7]. A PDB file of the homology model for FPR1 was loaded into the Molegro Virtual Docker (MVD) program (MVD 2010.4.2, Molegro ApS), and the MVD “detect cavity” module was applied with probe size 2 Å to identify potential areas of the protein where ligands could be docked. As a result, two cavities were found with volumes of 565 and 33 Å3 (Supplemental Fig. S1). The cavity with lower volume seemed small for incorporating known FPR1 agonists and localization of the hydrophobic pockets that should be present in the binding site [1]. Thus, we focused our attention on the larger cavity, especially because it covered the upper region of the transmembrane (TM) helical bundle comprising TM 2, 5, 6, and 7, determined previously as the region for FPR1 agonist binding by cross-linking and mutagenesis studies [24, 25]. The position of the binding site in this region was recently localized by docking studies of the FPR1 agonist Gln9-Ala10-Trp11-Phe12 (Ac-QAWF) [7]. Because the large cavity did not cover the peptide completely (Supplemental Fig. S1), we utilized another option to define docking search space as a sphere centered at the carbonyl carbon of the Ala residue in Ac-QAWF. The radius of the sphere was adopted to be equal to 11 Å. This search space encompassed the whole Ac-QAWF molecule, most of the larger cavity, and included, at least partially, the following 36 residues of FPR1: Trp91, Trp95, Cys98, Lys99, Leu101, Phe102, Thr103, Val105, Asp106, Phe110, Leu156, Thr157, Leu158, Pro159, Val160, Ile161, Ile162, Asn192, Val193, Ala196, Met197, Leu198, Thr199, Val200, Arg201, Gly202, Arg205, Phe206, Tyr257, Ala261, Ala264, Thr265, Ile268, Arg269, Glu270, and Val283.
Before docking, structures of the compounds were pre-optimized using HyperChem software with MM+ force field and saved in Tripos MOL2 format. The ligand structures were then imported into the MVD with the options “create explicit hydrogens”, “assign charges (calculated by MVD)”, and “detect flexible torsions in ligands” enabled. Selected molecules were docked using the search space indicated above with a rigid receptor structure. Ligand flexibility was accounted for with respect to torsion angles auto-detected in MVD. MolDock score functions were used with 0.3 Å grid resolution. Fifteen docking runs were performed for each molecule, while 30 runs were performed for all peptides. The option “return multiple poses for each run” was enabled. The post-processing option “optimize H-bonds” was applied after docking. Similar poses were clustered at a RMSD threshold of 1 Å.
To evaluate stability of poses obtained in the docking study, molecular dynamics (MD) simulations were performed for the best poses of FPR1 agonists AG-11/03, AG-11/05, and AG-11/10, and the corresponding ligand–receptor complexes were imported into HyperChem 8.0. Ligand and side chains of the residues located within 10 Å of a pose were considered flexible during MD runs, while the remainder of FPR1 was kept rigid. The complexes were heated to 310 K for 20 ps, and the structural data were collected every 25 fs for a run time of 2 ns at 310 K using the OPLS force field [26]. The MD trajectories obtained by HyperChem were analyzed with respect to key interatomic distances to evaluate stability of the docking solutions with time. Average values of these distances over the MD trajectories in production phases were calculated with their standard deviations, and these average distances were compared with corresponding distances in docking poses found by MVD software.
To rationalize docking results, the lowest energy pose of each molecule investigated was described by energies of ligand interaction with different residues of FPR1. Decomposition of total docking score into partial ligand-residue terms was performed using the “energy inspector” tool embedded in MVD. Names of corresponding residues were used in descriptor notation. Descriptors with absolute values >3 kcal mol−1 were considered, all smaller values were zeroed. Another set of variables was produced by H-bonds formed between the receptor and ligands in their lowest-energy poses. Names of these descriptors contained the suffix “H” added to the corresponding FPR1 residues. Energies of each H-bond were assigned to variables of this set.
A 60×46 matrix (46 independent variables obtained as described above for 60 docked compounds) was imported into STATISTICA 8.0 software for further analysis of poses by binary classification tree methodology vs activity class (Active or NA (non-active)) assigned to the compounds according to their biological action and taken as a dependent variable. The classification tree was built with STATISTICA 8.0 using equal prior probabilities and equal misclassification costs for classes [27]. An exhaustive C&RT-style univariate split selection method was used, as described by Breiman et al. [28]. Distribution of molecules among terminal nodes of the classification tree obtained was used for visual inspection of poses for their differences between active and inactive compounds.
Results and discussion
Identification of novel FPR1/FPR2 agonists with a 2-(benzimidazol-2-ylthio)-N-phenylacetamide scaffold
Previously, we identified six FPR1-specific agonists and six mixed FPR1/FPR2 agonists among 27 2-(benzimidazol-2-ylthio)-N-phenylacetamide derivatives analyzed and noticed that FPR1 agonists with the benzimidazole scaffold contained either a para methoxy or ethoxy group in the benzene moiety of the benzimidazole cycle [13]. To increase the set of active compounds for molecular docking studies we selected 26 additional derivatives, which contained either methoxy or ethoxy groups at the para position of the benzimidazole cycle. These compounds are designated as AG-11/1 through AG-11/26 (Tables 1 and 2).
Table 1.
Structure and activity of 2-(benzimidazol-2-ylthio)-N-phenylacetamide derivatives in human neutrophils and FPR1/FPR2-transfected HL-60 cells
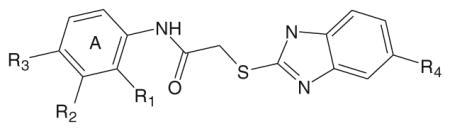
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Ca2+ mobilization EC50 (μM) and efficacy (%)a | ||
| FPR1 | FPR2 | Neutrophils | |||||
| AG-11/01 | Br | H | H | OCH3 | 8.3 (80) | 4.0 (30) | 7.1 (115) |
| AG-11/02 | H | H | OCF3 | OCH3 | 11.6 (60) | N.A. | 3.0 (55) |
| AG-11/03 | H | H | SCH3 | OCH3 | 2.0 (105) | N.A. | 0.5 (120) |
| AG-11/04 | OCH2CH3 | H | H | OCH3 | 4.1 (80) | N.A. | 3.7 (90) |
| AG-11/05 | H | H | COOCH3 | OCH3 | 1.5 (85) | N.A. | 0.9 (100) |
| AG-11/06 | H | COCH3 | H | OCH3 | 2.2 (135) | 6.0 (100) | 0.8 (105) |
| AG-11/07 | H | H | SO2NH2 | OCH3 | 14.9 (60) | N.A. | 17.2 (60) |
| AG-11/08 | H | H | CH2CH3 | OCH3 | 5.5 (90) | 3.2 (45) | 0.8 (105) |
| AG-11/09 | NO2 | H | H | OCH3 | 14.5 (70) | 24.5 (70) | 9.0 (100) |
| AG-11/10 | H | NHCOCH3 | H | OCH3 | 13.2 (90) | 7.3 (70) | 8.8 (105) |
| AG-11/11 | H | H | Cl | OCH2CH3 | 8.1 (35) | N.A. | 1.3 (70) |
| AG-11/12 | H | H | Br | OCH2CH3 | 13.2 (90) | N.A. | 11.2 (120) |
| AG-11/13 | H | H | OCH2CH3 | OCH2CH3 | N.A. | N.A. | N.A. |
| AG-11/14 | H | H | SO2NH2 | OCH2CH3 | N.A. | N.A. | N.A. |
| AG-11/15 | H | H | CH2CH3 | OCH2CH3 | 6.8 (55) | N.A. | 1.5 (70) |
| AG-11/16 | H | H | COCH3 | OCH2CH3 | 6.3 (90) | N.A. | 2.4 (80) |
| AG-11/17 | H | H | NO2 | OCH2CH3 | N.A. | N.A. | 3.6 (90) |
| AG-11/18 | H | NO2 | H | OCH2CH3 | N.A. | N.A. | 6.4 (100) |
| AG-11/19 | H | H | OCF3 | OCH2CH3 | N.A. | N.A. | N.A. |
| AG-11/20 | SCH3 | H | H | OCH2CH3 | 10.4 (90) | 8.0 (35) | 12.2 (80) |
| AG-11/21 | OCH3 | H | H | OCH2CH3 | 9.9 (70) | N.A. | 10.7 (60) |
Median effective concentration values (EC50) were determined by nonlinear regression analysis of the dose-response curves 5–6 points) generated using GraphPad Prism 5 with 95% confidential interval (p<0.05). EC50 values are presented as the mean of three independent experiments. Efficacy (in parentheses) is expressed as % of the response induced by 5 nM fMLF (FPR1) or 5 nM WKYMVm (FPR2). N.A., very low response (efficacy <20% of positive control) or no activity (no Ca2+ flux response was observed during the 3 min after addition of compounds under investigation)
Table 2.
Structure and activity of 2-(5-alkoxybenzimidazol-2-ylthio)-N-phenylacetamide derivatives in human neutrophils and FPR1/FPR2-transfected HL-60 cells
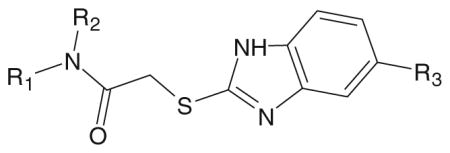
| ||||||
|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | Ca2+ mobilization EC50 (μM) and efficacy (%)a | ||
| FPR1 | FPR2 | Neutrophils | ||||
| AG-11/22 |
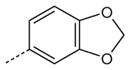
|
H | OCH2CH3 | 6.9 (50) | 14.3 (35) | 3.9 (60) |
| AG-11/23 |
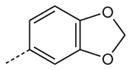
|
H | OCH3 | 2.2 (70) | N.A. | 2.6 (80) |
| AG-11/24 |
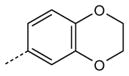
|
H | OCH2CH3 | 14.2 (65) | 3.7 (40) | 5.6 (75) |
| AG-11/25 |
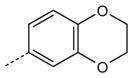
|
H | OCH3 | 2.1 (110) | 10.4 (70) | 1.5 (105) |
| AG-11/26 |
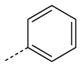
|
CH3 | OCH3 | 7.5 (90) | 11.0 (45) | 17.8 (75) |
See legend for Table 1
Screening of these compounds for their ability to induce Ca2+ mobilization in human neutrophils and HL-60 cells transfected with FPR1 and FPR2 demonstrated that 21 such derivatives (80.8%) were agonists, supporting the significance of these groups for the agonist activity. Eleven compounds were FPR1-specific agonists, and ten were mixed FPR1/FPR2 agonists. Two of the compounds identified, AG-11/03 and AG-11/05, represent the most potent and specific FPR1 agonists among all benzimidazole derivatives evaluated to date. It should be noted, that no response was observed in control, untransfected HL-60 cells treated with each of the active compounds. All compounds that activated Ca2+ mobilization in HL-60 transfected cells also activated Ca2+ flux in human neutrophils. Note, however, that two compounds, AG-11/17 and AG-11/18, activated Ca2+ flux in neutrophils but did not activate FPR1- or FPR2-transfected cells. The reason for this anomalous response pattern is not clear, so these compounds were not included in docking analyses. Further studies will be necessary in the future to evaluate the specificity of these two compounds.
Structure–activity relationship (SAR) analysis of benzimidazole derivatives
SAR analysis of the new benzimidazole derivatives, together with previously published compounds [13], provides additional information regarding the role of different substituents for FPR1 agonist activity. As demonstrated previously [13], active benzimidazole derivatives required either a para methoxy or para ethoxy group in the benzene moiety of the benzimidazole group (see R4 substituents in Table 1). Thus, we varied substituents in benzene ring A on the other end of the molecules. Substitution of a methoxy with a methylthio group at either the para or meta positions of benzene ring A did not significantly alter agonist activity (compare AG-11/02 and AG-11/03 or AG-11/20 and AG-11/21, respectively) (Table 1). However, substitution of hydrogen atoms in the para methoxy group with fluorine atoms led to total or partial loss of FPR1 agonist activity (compare AG-09/13 and AG-11/19, and AG-09/16 and AG-11/02, respectively). Analogs with a para ethoxy group in the benzimidazole cycle were less tolerant to modification of benzene ring A by different substituents (Table 1). For example, compound AG-11/13 with a para ethoxy group in benzene ring A was inactive (compare with active compound AG-09/2) in this series. However, all tested only AG-11/23 was specific for FPR1, and the other derivatives in which benzene ring A contained fused 1,3- compounds tested with this feature were mixed FPR1/dioxolane or 1,4-dioxane rings were FPR agonists, although FPR2 agonists (Table 2).
Binding site in the homology model of FPR1
Molecular docking studies have demonstrated that ~20% sequence identity between a template and target GPCR is necessary for a robust homology model [21]. On the other hand, slightly higher sequence identity between a given target and template alone does not always justify the choice of GPCR structure for the optimal homology modeling template, and additional considerations are important [21]. In previous studies, the crystal structure of bovine rhodopsin was successfully applied to homology modeling of FPR1 [7, 16, 17]. The position of the ligand binding site in these models, based on cross-linking and mutagenesis studies, was found to be located in the upper region of a helical bundle comprising TM 2, 5, 6, and 7 [24, 25]. Indeed, the FPR1 ligand binding site location was recently confirmed by docking studies with Ac-QAWF, which is the shortest core structure of the annexin AI-derived peptide Ac9–25 [29]. Since our pre-docking studies indicated that this region is coincident with the ligand binding site, we propose this feature is relevant to the modeling process [20] and justifies use of the rhodopsin-based model as a template for the docking of our novel small-molecule FPR1 agonists vs. the other known GPCR crystal structures currently available.
Recently, Movitz et al. [7] found that the Ala carbonyl carbon of Ac-QAWF is located approximately in the geometric center of the docked peptide. In the present work, this carbon atom was chosen as the center of spherical search space for docking of an 11 Å radius sphere. Such an area (indicated in green on Fig. 1a) completely encompassed the peptide molecule, as well as 36 neighboring residues of FPR1 (see Materials and methods). Among these residues, six residues (Asp106, Arg201, Arg205, Trp254, Tyr257, and Phe291) were identified previously by mutagenesis experiments as important for binding of FPR1 agonists [25].
Fig. 1.

FPR1 binding site with docked peptides Ac-QAWF and fMLF. Panel a: Position of the spherical search space in the FPR1 selected as a sphere with 11 Å radius around the Ac-QAWF Ala carbonyl carbon. Panel b: Key features of the FPR1 binding site with docking poses of Ac-QAWF reported by Movitz et al. [7] (thin sticks) and obtained by us (purple) compared with the pose of fMLF peptide (white). Arrows indicate: channel A; curved cavity B located behind the blue-colored ledge; channel C; “bottom” D of the binding site between channels A and C; and large cavity E, located between channel C and larger blue-colored ledge. See text for further details or regions A–F. Surface coloring was made according to electrostatic properties – negatively and positively charged areas are shown in red and blue, respectively
A visual inspection allows partitioning the binding site within the spherical search space into several key sub-areas important for subsequent analysis of the docking results. These key features include two channels (A and C), two cavities (B and E), and the bottom (D) of the binding site (Fig. 1b). Channel A is located in the vicinity of Asn192, Thr199, Thr265, Ile268, and Leu271. Channel C is bounded by Leu101, Val105, Tyr257, Ser287, and Phe291. Cavity B is a curved groove, located behind the hydrophobic ledge formed by the isobutyl group of Leu198. This cavity is restricted by Val160, Leu198, Arg201, Gly202, and Arg205. Large cavity E is located near Trp91, Trp95, Cys98, and Lys99, between channel C and a larger ledge. The bottom D of the binding site is bounded by channels A and C and is associated with Ala261, Ala264, and Val283.
It should be noted that the docking methodology utilized by Movitz et al. [7] was different from the algorithm implemented in MVD software. Thus, we performed an additional run of MVD and found a pose of Ac-QAWF that is quite similar to that reported previously [7] (Fig. 1b). However, this pose had a docking score of −130.0 kcal mol−1, while for the reported literature pose [7] we obtained a higher-energy score of −100.2 kcal mol−1, as evaluated by MVD MolDock. Comparison of docked poses of fMLF and Ac-QAWF shows that the skeletons of both peptides overlap significantly; however, Ac-QAWF does not have a protrusion reaching the inlet of channel A. On the other hand, the phenyl ring of Ac-QAWF is located near cavity E. Perhaps, such peculiarities of the binding mode are the reason for low FPR1 agonist activity of Ac-QAWF versus fMLF, which is a more potent FPR1 agonist [7, 29].
Figure 2a shows that the H-bond between fMLF and Arg205 is formed with participation of the peptide terminal carboxyl group and the guanidine group of Arg205. Moreover, there is a significant ion pair interaction between these moieties, with energy of 3.0 kcal mol−1, as determined by the MVD “energy inspector” tool. This observation is supported by previous studies of Mills and co-authors [25] who reported a strong interaction of the fMLF peptide carboxyl terminus with the Arg205 guanidine. They also mentioned that the fMLF formyl group forms a H-bond with Arg201 of FPR1. In contrast, we did not observe a H-bond between the formyl group and Arg201, although these moieties are located close to each other (the distance between a guanidine nitrogen atom in Arg201 and the fMLF formyl oxygen is 4.05 Å).
Fig. 2.

Docking poses of fMLF and the two most potent benzimid-azole FPR1 agonists. Panel a. Specific interaction between Arg205 and fMLF (thick sticks) docked into the FPR1 binding site. H-bond is shown by dashed blue lines. Electrostatic ion–pair interaction is indicated by the green marker. Panel b. Docking poses of benzimidazole-derived FPR1 agonists AG-11/03 (purple) and AG-11/05 (green). H-bonds are indicated with dash lines
Docking of multiple FPR1 agonists
Docking of FPR1 agonists and their inactive analogues is helpful in elucidating key features of ligand–receptor interactions between small molecules and the FPR1 binding site. Such a systematic docking study of multiple FPR1 agonists has not been previously performed. The set of FPR1-specific and mixed FPR1/FPR2 agonists included 33 benzimidazole derivatives reported here (Tables 1 and 2) and previously [13] with EC50<22 μM. These compounds are designated as “active” in our post-docking classification analysis. Inactive members of the data set (designated as “NA”) included 17 benzimidazole derivatives (Tables 1 and 2 and [13]).
The most active benzimidazole derivatives AG-11/03 and AG-11/05 did not interact strongly with Arg205. While AG-11/05 formed a H-bond of 1.5 kcal mol−1 with Arg205 (Fig. 2b), this interaction was weaker than the H-bond formed between Arg205 and fMLF, which has been suggested to contribute to the very high agonist activity of fMLF (see above). On the other hand, AG-11/03 and AG-11/05 both had very similar benzimidazole orientations with their methoxy substituents H-bonded to Thr199 (Fig. 2b).
We evaluated stability of the docking poses over time by performing MD simulations at 310 K for selected FPR1 agonists starting from their best docking poses within the FPR1 binding site. Three agonists (AG-11/03, AG-11/05, and AG-11/10), which belong to the most populated “active” nodes of the classification tree (see below) were chosen for the MD study. Distances from the carbonyl carbons in FPR1 residues to heteroatoms in different regions of the agonist molecules (e.g., sulfur, alkoxy oxygen, or acetylamino nitrogen) are indicated in Table 3. As is evident, distance changes over time were not significant (i.e., molecules oscillated near their positions obtained with the docking calculations).
Table 3.
Distances between heteroatoms in FPR1 agonists and carbonyl carbon atoms in nearest residues of the FPR1 binding site for docking poses and molecular dynamics (MD) trajectories for ligand–receptor complexes
| Compound | Position of the atoms in the ligand–receptor complex | Distance (Å)
|
|
|---|---|---|---|
| Docking pose | MD trajectorya | ||
| AG-11/03 | SCH3 sulfur – Ala264 | 4.80 | 5.0±0.9 |
| OCH3 oxygen – Leu198 | 4.66 | 4.9±1.0 | |
| Sulfur – Gly202 | 5.86 | 5.6±1.1 | |
| AG-11/05 | OCH3 oxygen (ester) – Val160 | 5.90 | 6.1±1.0 |
| OCH3 oxygen (attached to benzimidazole) – Leu198 | 3.78 | 4.1±0.8 | |
| Sulfur – Ala261 | 4.42 | 4.6±0.9 | |
| AG-11/10 | OCH3 oxygen – Cys98 | 3.85 | 4.0±0.7 |
| CH3CONH nitrogen – Leu198 | 4.28 | 4.1±0.8 | |
| Sulfur – Val160 | 6.70 | 6.3±1.0 | |
The ligand–receptor complexes were heated to 310 K for 20 ps, structural data were collected for the next 2 ns at 310 K, and the MD trajectories obtained by HyperChem were analyzed, as described under Materials and methods. Average distances are indicated with their standard deviations over the MD trajectory
As mentioned above, one of the most important molecular features necessary for the FPR1 agonist activity of 2-(benzimidazol-2-ylthio)-N-phenylacetamide derivatives is the presence of an alkoxy group in the benzimid-azole moiety. Hence, we analyzed the positions of alkoxy substituents in the docking poses with respect to the FPR1 binding site. We determined all FPR1 residues located within 3.5 Å of the oxygen atom in alkoxy groups of all benzimidazole derivatives containing such groups. Using the assignments of the residues to certain areas of the binding site (Fig. 1b), it was possible to describe the positions of alkoxy substituents in terms of these key regions. We found that 37 of 43 alkoxy-substituted benzimidazoles had methoxy or ethoxy oxygen atoms embedded in channel A or curved groove B. Thirty-four of these benzimidazole compounds were active FPR1 agonists, demonstrating the importance of the ligand–receptor interactions in regions A and B for FPR1 agonist activity (see examples in Fig. 3). For the remaining active compounds (AG-09/17, AG-09/20, and AG-11/10), the alkoxy groups occupied large cavity E.
Fig. 3.

Docking poses of alkoxy-substituted benzimidazole derivatives AG-11/03, AG-11/05, and AG-09/01 relative to the surface of the FPR1 binding site (Panel a) and amino acids Thr199, Arg201, Gly202, and Ala261 with the protein surface removed (Panel b). In Panel b, AG-11/03, AG-11/05, and AG-09/01 are shown in pink, green, and blue, respectively
Recognition model for FPR1 agonists
The analysis of docking results given above was based on spatial arrangement of specific molecular moieties in chemically related compounds. Below we perform analysis of docking poses in a more general way with additional molecules included. Nine peptide derivatives were added to obtain a diversity set of molecules for docking analysis. Among these, fMLF and three peptide derivatives (N-formyl-Met-Leu-Phe-OMe [fMLF-OMe], boc-cis-4-mer-captomethyl-Pro-Leu-Phe-OMe [boc-cis-4-mercapto-methyl-PLF-OMe] [30], and N-formyl-Met-Leu-Ψ(COO) Phe-NHBzl [fMLΨ(COO)F-NHBzl] [31]) were selected as known FPR1 agonists. Inactive fMLF-OMe analogs included N-formyl-Met-Ψ(COO)Aib-Phe-OMe [fMΨ(COO) Aib-F-OMe] [31], N-formyl-Met-(NMe)Leu-Phe-OMe [fM-(NMe)LF-OMe] [32], N-formyl-Met-azaPro-Phe-OMe [fM-azaPF-OMe] [33], [Pheol]3 [33], and N-formyl-Cys-Leu-Phe-Cys-OMe [fCLFC-OMe] [34].
Analysis of the entire pool of molecules, including peptides, required a more systematic approach to compare their docking poses. For this analysis, the lowest-energy poses of docked molecules were analyzed computationally in terms of partial ligand–residue interactions because direct visual comparison of docking results for active and inactive compounds is very difficult for such a large data set. The total docking score for a pose of each molecule was decomposed into ligand–residue components using the MVD “energy inspector” feature. The corresponding partial terms >3 kcal mol−1 in absolute values served as molecular descriptors for each residue. Along with these variables, H-bond energy terms were also taken as independent descriptors. Such descriptors are designated with additional letter “H” (e.g., Thr199H).
To find a subset of descriptors giving a good recognition model for distinguishing biologically-active from inactive compounds, various data mining techniques can be used, including, classification tree analysis [28], artificial neural networks [35], and linear discriminant analysis [36]. We found that binary classification tree methodology leads to satisfactory results based on four descriptors in a recognition model when all 46 variables were considered initially. The optimal classification tree (Fig. 4) contained two descriptors, Thr199H and Ala261H, corresponding to H-bonding interactions with Thr199 in channel A and with Ala261 in the “bottom” area D.
Fig. 4.

Classification tree obtained based on 46 initial descriptors taken from docking results
Additionally, two other descriptors were involved in the model (Arg201 and Gly202). This pair of residues lies in curved groove B (Fig. 1b). The binary tree has five terminal nodes, with one node corresponding to the classification of compounds as inactive (class “NA”, node 9). The other terminal nodes (2, 4, and 6) classify compounds as active FPR1 agonists. Detailed results of individual compound recognition, along with terminal nodes, descriptor values, and experimental activity classes are reported in Table 4.
Table 4.
Results of binary classification tree analysis based on partial energetic characteristics of docking poses of compounds under investigation
| Compound | Absolute values of descriptors (kcal/mol)a
|
Observed value | Predicted value | Terminal node | |||
|---|---|---|---|---|---|---|---|
| Arg201 | Gly202 | Ala261 | Thr199H | ||||
| AG-11/01 | 10.63 | 9.85 | 4.10 | 2.5 | Active | Active | 2 |
| AG-11/02 | 13.70 | 8.52 | 3.86 | 2.16 | Active | Active | 2 |
| AG-11/03 | 0 | 5.03 | 9.13 | 2.5 | Active | Active | 2 |
| AG-11/04 | 0 | 7.44 | 20.04 | 0 | Active | Active | 6 |
| AG-11/05 | 0 | 7.57 | 3.38 | 2.5 | Active | Active | 2 |
| AG-11/06 | 7.01 | 0 | 8.66 | 1.97 | Active | Active | 2 |
| AG-11/07 | 0 | 4.02 | 3.65 | 1.92 | Active | Active | 2 |
| AG-11/08 | 11.11 | 9.97 | 3.22 | 0 | Active | Active | 8 |
| AG-11/09 | 10.62 | 4.01 | 9.30 | 0 | Active | Active | 6 |
| AG-11/10 | 0 | 9.10 | 0 | 0 | Active | Active | 8 |
| AG-11/11 | 8.45 | 7.65 | 5.09 | 2.5 | Active | Active | 2 |
| AG-11/12 | 5.90 | 9.07 | 3.28 | 0 | Active | Active | 8 |
| AG-11/13 | 0 | 8.07 | 10.39 | 0 | NA | NA | 9 |
| AG-11/14 | 0 | 0 | 0 | 0 | NA | NA | 9 |
| AG-11/15 | 5.85 | 5.02 | 7.76 | 0 | Active | NA | 9 |
| AG-11/16 | 3.89 | 5.09 | 4.54 | 2.5 | Active | Active | 2 |
| AG-11/19 | 6.27 | 7.67 | 4.24 | 0 | NA | NA | 9 |
| AG-11/20 | 0 | 9.22 | 0 | 0 | Active | Active | 8 |
| AG-11/21 | 0 | 6.43 | 0 | 0 | Active | NA | 9 |
| AG-11/22 | 0 | 9.78 | 5.29 | 0 | Active | Active | 8 |
| AG-11/23 | 0 | 4.09 | 12.82 | 0.79 | Active | Active | 2 |
| AG-11/24 | 0 | 7.81 | 9.89 | 2.5 | Active | Active | 2 |
| AG-11/25 | 10.28 | 7.10 | 9.42 | 0 | Active | NA | 9 |
| AG-11/26 | 0 | 9.23 | 4.58 | 0 | Active | Active | 8 |
| AG-09/1 | 15.63 | 8.17 | 3.54 | 2.5 | Active | Active | 2 |
| AG-09/2 | 4.73 | 9.99 | 3.05 | 1.29 | Active | Active | 2 |
| AG-09/11 | 11.98 | 10.53 | 4.22 | 0 | NA | Active | 8 |
| AG-09/12 | 0 | 0 | 5.54 | 0 | NA | NA | 9 |
| AG-09/13 | 14.09 | 11.35 | 0 | 2.5 | Active | Active | 2 |
| AG-09/14 | 8.00 | 5.59 | 3.34 | 2.5 | Active | Active | 2 |
| AG-09/15 | 0 | 3.35 | 11.85 | 0 | NA | NA | 9 |
| AG-09/16 | 0 | 7.18 | 3.20 | 2.5 | Active | Active | 2 |
| AG-09/17 | 17.54 | 0 | 0 | 0 | Active | Active | 4 |
| AG-09/18 | 11.20 | 10.08 | 4.14 | 2.5 | Active | Active | 2 |
| AG-09/19 | 9.41 | 7.88 | 4.12 | 2.5 | Active | Active | 2 |
| AG-09/20 | 0 | 0 | 0 | 0 | Active | NA | 9 |
| AG-09/21 | 0 | 5.49 | 4.08 | 2.0 | Active | Active | 2 |
| AG-09/22 | 0 | 6.46 | 4.64 | 2.4 | Active | Active | 2 |
| AG-09/23 | 5.16 | 4.46 | 10.69 | 0 | Active | Active | 6 |
| AG-09/24 | 7.02 | 0 | 4.48 | 0 | Active | NA | 9 |
| AG-09/25 | 11.89 | 8.01 | 3.39 | 0 | NA | NA | 9 |
| AG-09/26 | 0 | 5.24 | 13.56 | 0 | NA | NA | 9 |
| AG-09/27 | 11.92 | 9.12 | 0 | 0 | NA | Active | 8 |
| AG-09/28 | 0 | 5.82 | 10.21 | 2.72 | NA | Active | 2 |
| AG-09/29 | 12.46 | 9.04 | 4.86 | 2.25 | NA | Active | 2 |
| AG-09/30 | 13.65 | 7.91 | 0 | 0 | NA | NA | 9 |
| AG-09/31 | 0 | 4.73 | 8.30 | 0 | NA | NA | 9 |
| AG-09/32 | 12.86 | 3.18 | 0 | 2.05 | NA | Active | 2 |
| AG-09/33 | 9.03 | 3.93 | 0 | 0 | NA | NA | 9 |
| AG-09/34 | 4.96 | 3.34 | 6.23 | 0 | NA | NA | 9 |
| AG-09/35 | 12.01 | 8.56 | 3.58 | 0 | NA | NA | 9 |
| fMLF-OMe | 14.18 | 7.4 | 4.67 | 0 | Active | Active | 4 |
| fMLΨ(COO)F-NHBzl | 10.04 | 3.18 | 5.17 | 0 | Active | NA | 9 |
| [Pheol]3 | 3.95 | 3.36 | 13.82 | 0 | NA | NA | 9 |
| fCLFC-OMe | 0 | 0 | 0 | 0 | NA | NA | 9 |
| fM-azaPF-OMe | 11.5 | 7.05 | 0 | 0 | NA | NA | 9 |
| boc-cis-4-mercaptomethyl-PLF-OMe | 17.89 | 8.75 | 0 | 0 | Active | Active | 4 |
| fMLF | 15.72 | 5.54 | 6.91 | 0 | Active | Active | 4 |
| fMΨ(COO)Aib-F-OMe | 0 | 0 | 0 | 0 | NA | NA | 9 |
| fM-(NMe)LF-OMe | 0 | 6.24 | 3.66 | 0 | NA | NA | 9 |
Partial terms of the docking score reflecting interactions with the corresponding three residues or H-bond energy with Thr199 residue. All terms were negative, and their absolute values are presented. A H-bond in MVD MolDock has an upper energy limit of 2.5 kcal mol-1. For molecule AG-09/28, a sum of energies for two H-bonds with Thr199 is presented. Such a limit did not influence accuracy of the binary classification tree model, which is based on simple splitting rules applied to the descriptors
According to the simple classification rules encoded in the tree, correct recognition was obtained for 85.3% (29 of 34) active and 70.6% (12 of 17) inactive benzimidazole derivatives. Only one peptide agonist of the nine docked peptide derivatives, fMLΨ(COO)F-NHBzl, was classified incorrectly (Table 4). Thus, the total fraction of correct classifications was 81.7% (49 of 60 compounds investigated). Four descriptors (Thr199H, Arg201, Gly202, and Ala261H) constitute a basis for this satisfactory model, and the corresponding residues can be considered as markers of some important regions of the FPR1 ligand-binding site. Although there exist other descriptors with high average values (e.g., Phe102, Leu198, Arg205, Lys99, Val160, Tyr 257, and Thr265) most of these variables reflect strong interactions between ligands and corresponding residues, but their values do not show statistically significant differences between FPR1 agonists and non-agonists. Interactions with these residues can serve for anchoring both active and inactive compounds within the binding site, whereas only the four selected descriptors correspond to regions of FPR1 statistically important for molecular recognition of agonists.
Results of the binary tree modeling allowed us to perform a more rational visual analysis of the docking poses obtained. The most populated terminal nodes 2 and 9, each with 19 compounds correspond to classes “active” and “NA”, respectively. Node 2 contains molecules with H-bonding to Thr199, i.e., the Thr199H descriptor value is more negative than 0.395 kcal mol-1. Superimposition of poses for correctly classified active FPR1 agonists associated with node 2 is presented in Fig. 5a. Molecules from this node occupy channel A with their tails, as they form H-bonds with Thr199 located deeply in the channel. Other parts of the molecules lay compactly along curved groove B (Fig. 5a). Poses of active molecules from terminal nodes 4, 6, and 8 are given in Fig. 5b, c, and d, respectively. As is evident, these docked FPR1 agonists are also oriented near channel A, curved groove B, and (in node 6) close to “bottom” D. Several correctly classified inactive molecules from terminal node 9 (Fig. 5e) noticeably shield channel C of the binding site and enter large cavity E in the vicinity of Trp91, Trp95, Cys98, and Lys99. The differences between docking poses associated with the most populated nodes of types “active” and “NA” are also demonstrated in the right panels of Fig. 5a and e, where residues Thr199, Arg201, Gly202, and Ala261 are shown, while the protein surface is removed. However, we have found that shielding of channel C is produced mainly by peptide fMΨ(COO)Aib-F-OMe and benzimidazole derivative AG-11/14. Additionally, cavity E is noticeably occupied by fMΨ(COO)Aib-F-OMe and AG-09/15. Docking poses for all of the other inactive compounds from node 9 look similar to poses found for active agonists from other nodes. Nevertheless, their main underlying characteristic consists of weak interactions with Thr199, Arg201, Gly202, and Ala261 located in channel A, groove B, and “bottom” D.
Fig. 5.

Docking poses for FPR1 agonists (panels a–d) and non-active analogs (panel e) relative to the surface of the FPR1 binding site (left part) and amino acids Thr199, Arg201, Gly202, and Ala261 with the protein surface removed (right part)
Three peptides correctly classified as active FPR1 agonists (fMLF, fMLF-OMe, and Boc-cis-4-mercaptomethyl-PLF-OMe) passed to terminal node 4 of the classification tree together with one non-peptide molecule AG-09/17 (Fig. 5b). It can be seen that poses of peptides within the binding site look very similar to those of benzimidazole-derived FPR1 agonists (Fig. 5a, c, and d), i.e., they are located mainly in groove B and near the inlet of channel A. In spite of visual similarity between the docking poses of fMLF and other agonists, the activity of fMLF is very high (EC50~0.5 nM) in human neutrophils [14], while the docking score for this peptide is on a similar scale as the other compounds investigated. To evaluate this issue, we examined energies of ligand-residue interactions obtained with the MVD “energy inspector” tool and found that fMLF had much higher absolute values of Arg205 and Arg205H descriptors (32.36 and 4.07 kcal mol−1, respectively) compared to all of the compounds. Again, these results support the role of Arg205 interactions in the very high agonist activity of this peptide. Indeed, site-directed mutagenesis demonstrated that residue Arg205 plays an important role in positioning of fMLF in the FPR1 binding pocket [25]. Furthermore, it has been shown that the region corresponding residues to FPR1 201RGIIR205 in TM 5 is involved in binding sites of several other GPCR, such as the β2 adrenergic receptor [37].
Conclusions
The identification of additional benzimidazole derivatives with FPR1 agonist activity further emphasized the importance of alkoxy substituents in the benzimidazole moiety and allowed us to perform molecular docking studies to expand our understanding to the FPR1 ligand-binding site. These docking studies suggested that the most important areas of the FPR1 binding site for receptor activation were channel A and cavity B. On the other hand, positioning of a ligand in cavity E or in the vicinity of channel C was in most cases unfavorable for inducing the biological activity under investigation. Analysis of docking poses based on the FPR1 homology model effectively differentiated agonists from non-active compounds, and activity classification involved four descriptors (Thr199H, Arg201, Gly202, and Ala261H) that had high statistical impact for distinguishing FPR1 agonists from non-agonists. Importantly, these descriptors are directly associated with the above-mentioned key areas of the ligand binding site. Overall, the model described here will be useful in assessment of the interaction of putative FPR1 agonists with the receptor and in virtual search for novel and effective FPR1 agonists.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health grant P20 RR-020185, National Institutes of Health contract HHSN266200400009C, the Swedish Medical Research Council, the Inga Lisa and Arne Lundberg foundation, the King Gustaf V 80-Year Foundation, an equipment grant from the M.J. Murdock Charitable Trust, and the Montana State University Agricultural Experimental Station. We thank Dr. Marie-Josèphe Rabiet and Francois Boulet (CEA, DSV, iRTSV, Laboratoire Biochimie et Biophysique des Systèmes Intégrés, Grenoble, France) for kindly providing FPR-transfected HL-60 cells.
Abbreviations
- FPR
N-formyl peptide receptor
- FPR1
N-formyl peptide receptor 1
- FPR2
N-formyl peptide receptor 2
- GPCR
G protein-coupled receptor
- HBSS
Hank’s balanced-salt solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- SAR
Structure-activity relationship
- TM
Transmembrane
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00894-011-1307-x) contains supplementary material, which is available to authorized users.
Contributor Information
Andrei I. Khlebnikov, Department of Chemistry, Altai State Technical University, Barnaul, Russia
Igor A. Schepetkin, Department of Immunology and Infectious Diseases, Montana State University, Bozeman, MT, USA
Liliya N. Kirpotina, Department of Immunology and Infectious Diseases, Montana State University, Bozeman, MT, USA
Lars Brive, Department of Cell and Molecular Biology, University of Gothenburg, Gothenburg, Sweden.
Claes Dahlgren, Department of Rheumatology and Inflammation Research, University of Gothenburg, Gothenburg, Sweden.
Mark A. Jutila, Department of Immunology and Infectious Diseases, Montana State University, Bozeman, MT, USA
Mark T. Quinn, Email: mquinn@montana.edu, Department of Immunology and Infectious Diseases, Montana State University, Bozeman, MT, USA
References
- 1.Ye RD, Boulay F, Wang JM, Dahlgren C, Gerard C, Parmentier M, Serhan CN, Murphy PM. Pharmacol Rev. 2009;61:119–161. doi: 10.1124/pr.109.001578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filep JG, El KD. J Cell Biochem. 2009;108:1039–1046. doi: 10.1002/jcb.22351. [DOI] [PubMed] [Google Scholar]
- 4.Kretschmer D, Gleske AK, Rautenberg M, Wang R, Koberle M, Bohn E, Schoneberg T, Rabiet ML, Boulay F, Klebanoff SJ, van Kessel KA, van Strijp JA, Otto M, Peschel A. Cell Host Microbe. 2010;7:463–473. doi: 10.1016/j.chom.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Migeotte I, Communi D, Parmentier M. Cytokine Growth Factor Rev. 2006;17:501–519. doi: 10.1016/j.cytogfr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 6.Serhan CN. Annu Rev Immunol. 2007;25:101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 7.Movitz C, Brive L, Hellstrand K, Rabiet ML, Dahlgren C. J Biol Chem. 2010;285:14338–14345. doi: 10.1074/jbc.M109.080465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forsman H, Onnheim K, Andreasson E, Dahlgren C. Scand J Immunol. 2011;74:227–234. doi: 10.1111/j.1365-3083.2011.02570.x. [DOI] [PubMed] [Google Scholar]
- 9.Shin MK, Jang YH, Yoo HJ, Kang DW, Park MH, Kim MK, Song JH, Kim SD, Min G, You HK, Choi KY, Bae YS. Min dS J Biol Chem. 2011;286:17133–17143. doi: 10.1074/jbc.M110.197772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim SD, Kim YK, Lee HY, Kim YS, Jeon SG, Baek SH, Song DK, Ryu SH, Bae YS. J Immunol. 2010;185:4302–4310. doi: 10.4049/jimmunol.1001310. [DOI] [PubMed] [Google Scholar]
- 11.Dufton N, Perretti M. Pharmacol Ther. 2010;127:175–188. doi: 10.1016/j.pharmthera.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 12.Gavins FN. Trends Pharmacol Sci. 2010;31:266–276. doi: 10.1016/j.tips.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirpotina LN, Khlebnikov AI, Schepetkin IA, Ye RD, Rabiet MJ, Jutila MA, Quinn MT. Mol Pharmacol. 2010;77:159–170. doi: 10.1124/mol.109.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schepetkin IA, Kirpotina LN, Khlebnikov AI, Jutila MA, Quinn MT. Mol Pharmacol. 2011;79:77–90. doi: 10.1124/mol.110.068288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballesteros J, Palczewski K. Curr Opin Drug Discov Devel. 2001;4:561–574. [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards BS, Bologa C, Young SM, Balakin KV, Prossnitz ER, Savchuck NP, Sklar LA, Oprea TI. Mol Pharmacol. 2005;68:1301–1310. doi: 10.1124/mol.105.014068. [DOI] [PubMed] [Google Scholar]
- 17.Ferrari C, Macchiarulo A, Costantino G, Pellicciari R. J Comput Aided Mol Des. 2006;20:295–303. doi: 10.1007/s10822-006-9055-1. [DOI] [PubMed] [Google Scholar]
- 18.Patny A, Desai PV, Avery MA. Proteins. 2006;65:824–842. doi: 10.1002/prot.21196. [DOI] [PubMed] [Google Scholar]
- 19.Hulme EC, Lu ZL, Bee M, Curtis CA, Saldanha J. Life Sci. 2001;68:2495–2500. doi: 10.1016/s0024-3205(01)01044-x. [DOI] [PubMed] [Google Scholar]
- 20.Kufareva I, Rueda M, Katritch V, Stevens RC, Abagyan R. Structure. 2011;19:1108–1126. doi: 10.1016/j.str.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deflorian F, Jacobson KA. J Comput Aided Mol Des. 2011;25:329–338. doi: 10.1007/s10822-011-9423-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christophe T, Karlsson A, Rabiet MJ, Boulay F, Dahlgren C. Scand J Immunol. 2002;56:470–476. doi: 10.1046/j.1365-3083.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 23.Siemsen DW, Schepetkin IA, Kirpotina LN, Lei B, Quinn MT. Methods Mol Biol. 2007;412:21–34. doi: 10.1007/978-1-59745-467-4_3. [DOI] [PubMed] [Google Scholar]
- 24.Miettinen HM, Mills JS, Gripentrog JM, Dratz EA, Granger BL, Jesaitis AJ. J Immunol. 1997;159:4045–4054. [PubMed] [Google Scholar]
- 25.Mills JS, Miettinen HM, Cummings D, Jesaitis AJ. J Biol Chem. 2000;275:39012–39017. doi: 10.1074/jbc.M003081200. [DOI] [PubMed] [Google Scholar]
- 26.Carlsson J, Boukharta L, Aqvist J. J Med Chem. 2008;51:2648–2656. doi: 10.1021/jm7012198. [DOI] [PubMed] [Google Scholar]
- 27.Loh WY, Shih YS. Stat Sin. 1997;7:815–840. [Google Scholar]
- 28.Breiman L, Friedman JH, Olshen RA, Stone CJ. Classification and regression trees. Wadsworth & Brooks; Monterey: 1984. [Google Scholar]
- 29.Karlsson J, Fu HM, Boulay F, Dahlgren C, Hellstrand K, Movitz C. J Leukoc Biol. 2005;78:762–771. doi: 10.1189/jlb.0305153. [DOI] [PubMed] [Google Scholar]
- 30.Mollica A, Paradisi MP, Varani K, Spisani S, Lucente G. Bioorg Med Chem. 2006;14:2253–2265. doi: 10.1016/j.bmc.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Cavicchioni G, Vertuani G, Scatturin A, Spisani S. Bioorg Med Chem Lett. 1994;4:1551–1554. [Google Scholar]
- 32.Cavicchioni G, Breveglieri A, Boggian M, Vertuani G, Reali E, Spisani S. J Pept Sci. 1996;2:135–140. doi: 10.1002/psc.55. [DOI] [PubMed] [Google Scholar]
- 33.Pagani Zecchini G, Paglialunga Paradisi M, Torrini I, Lucente G, Mastropietro G, Paci M, Spisani S. Arch Pharm. 1996;329:517–523. doi: 10.1002/ardp.19963291202. [DOI] [PubMed] [Google Scholar]
- 34.Pagani Zecchini G, Paglialunga Paradisi M, Torrini I, Lucente G, Gavuzzo E, Mazza F, Pochetti G, Paci M, Sette M, Di Nola A. Biopolymers. 1993;33:437–451. doi: 10.1002/bip.360330310. [DOI] [PubMed] [Google Scholar]
- 35.Mehrotra K, Mohan CK, Ranka S. Elements of Artificial Neural Networks. MIT Press; Cambridge: 2000. [Google Scholar]
- 36.McLachlan GJ. Discriminant Analysis and Statistical Pattern Recognition. Wiley; Hoboken: 2004. [Google Scholar]
- 37.Goddard WA, III, Abrol R. J Nutr. 2007;137:1528S–1538S. doi: 10.1093/jn/137.6.1528S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.