

Abstract
BACKGROUND AND PURPOSE
One key mechanism for endothelial dysfunction is endothelial NOS (eNOS) uncoupling, whereby eNOS generates superoxide (O2•−) rather than NO. We explored the effect of pyridoxine on eNOS uncoupling induced by oxidized low-density lipoprotein (ox-LDL) in human umbilical vein endothelial cells (HUVECs) and the potential molecular mechanism.
EXPERIMENTAL APPROACH
HUVECs were incubated with ox-LDL with/without pyridoxine, NG-nitro-L-arginine methylester (L-NAME), chelerythrine chloride (CHCI) or apocynin. Endothelial O2•− was measured using lucigenin chemiluminescence, and O2•−-sensitive fluorescent dye dihydroethidium (DHE). NO levels were measured by chemiluminescence, PepTag Assay for non-radioactive detection of PKC activity, depletion of PKCα and p47phox by siRNA silencing and the states of phospho-eNOS Thr495, total-eNOS, phospho-PKCα/βII, total PKC, phospho-PKCα, total PKCα and p47phox were measured by Western blot.
KEY RESULTS
Ox-LDL significantly increased O2•− production and reduced NO levels released from HUVECs; an effect reversed by eNOS inhibitor, L-NAME. Pyridoxine pretreatment significantly inhibited ox-LDL-induced O2•− generation and preserved NO levels. Pyridoxine also prevented the ox-LDL-induced reduction in phospho-eNOS Thr495 and PKC activity. These protective effects of pyridoxine were abolished by the PKC inhibitor, CHCI, or siRNA silencing of PKCα. However, depletion of p47phox or treatment with the NADPH oxidase inhibitor, apocynin, had no influence on these effects. Also, cytosol p47phox expression was unchanged by the different treatments.
CONCLUSIONS AND IMPLICATIONS
Pyridoxine mitigated eNOS uncoupling induced by ox-LDL. This protectant effect was related to phosphorylation of eNOS Thr495 stimulated by PKCα, not via NADPH oxidase. These results provide support for the use of pyridoxine in ox-LDL-related vascular endothelial dysfunction.
Keywords: pyridoxine, eNOS uncoupling, oxidized low-density lipoprotein (ox-LDL), protein kinase C
Introduction
Endothelial NOS (eNOS) is a critical enzyme that converts L-arginine (L-Arg) to L-citrulline and NO, which is a critical signalling molecule in vascular homeostasis (Sessa, 2004). As NO is an important molecule in the function of endothelial cells, multiple pathways are involved in regulating the enzymatic function of eNOS (Kone et al., 2003). Under several pathophysiological conditions, eNOS becomes dysfunctional and produces superoxide (O2•−) rather than NO (i.e. uncoupled eNOS). Recent experiments and clinical studies have showed that eNOS, as a superoxide-producing enzyme, plays a crucial role in atherosclerosis, hypertension, congestive heart failure, diabetes mellitus and nitrate tolerance (Förstermann and Münzel, 2006). Therefore, tight coupling of eNOS is likely to be important to maintain normal cardiovascular function and prevent the development of cardiovascular disease.
A number of potential mechanisms are responsible for uncoupling eNOS, although the most consistent evidence suggests that is caused by a deficiency in tetrahydrobiopterin (BH4) (Bendall et al., 2005; Bevers et al., 2006). eNOS uncoupling also occurs in the following situations: (i) a shortage of substrate, L-Arg (Vergnani et al., 2000); (ii) an increase in asymmetric dimethyl-L-arginine (ADMA) concentration (Antoniades et al., 2009); (iii) dysregulation of protein–protein interactions (Pritchard et al., 2001); (iv) dephosphorylation of eNOS on threonine residue 495 (Lin et al., 2003); and (v) eNOS redistribution to the cytosolic fraction of the cell. Several studies have postulated that the dephosphorylation of threonine 495 on eNOS may act as a ‘switch’ that uncouples eNOS activity (Lin et al., 2003; Chen et al., 2008). The constitutively active kinase that phosphorylates eNOS Thr495, is most probably PKC, as PKC inhibitors as well as down-regulation of PKC attenuates the phosphorylation of this residue (Fulton et al., 2001; Michell et al., 2001). Oxidized low-density lipoprotein (ox-LDL) causes abnormalities of endothelial function by decreasing the activity of endothelium-derived NO and increasing O2•− production in endothelial cells (Heinloth et al., 2000; Cominacini et al., 2001). Moreover, Fleming et al. have demonstrated that ox-LDL increased O2•− production in endothelial cells in which eNOS is blocked with an eNOS inhibitor and associated this with a decrease in phosphorylation of eNOS at threonine 495; this was attributed to ox-LDL-induced decrease in PKC activity (Fleming et al., 2005).
Pyridoxine, a water-soluble vitamin, has been reported to scavenge reactive oxygen species (Matxain et al., 2006); an effect possibly related to co-enzymatic activities although the antioxidant mechanisms are unclear. We have previously reported that pyridoxine can improve endothelium-dependent relaxation in rabbit aortic rings exposed to ox-LDL (Ji et al., 2003) and prevent ox-LDL-induced dysfunction of NO generation in endothelial cells (Ji et al., 2006). We therefore hypothesized that pyridoxine may prevent ox-LDL-induced eNOS uncoupling through PKC-induced phosphorylation at threonine 495 of eNOS in HUVECs.
Methods
Isolation and culture of human umbilical vein endothelial cells (HUVECs)
HUVECs were isolated from umbilical cords donated by healthy mothers following uncomplicated deliveries, and cultured according to a previously described method (Ferro et al., 1999). Cells were cultured in M199 (Gibco), supplemented with 10% fetal bovine serum (Gibco), 10% newborn calf serum (Gibco), 100 U·mL−1penicillin, 100 µg·mL−1 streptomycin and 5 ng·mL−1 endothelial cell growth supplement under 37°C at 5% CO2 and 95% air. Confluent cells were used for experiments between passages 3 and 6. When HUVECs had grown to 85–90% confluence in 6-well plates, they were incubated with different concentrations (10, 20, 40 mg·L−1) of ox-LDL with or without L-NAME (100 µmol·L−1), pyridoxine (10−7 mol·L−1), CHCI (5 µmol·L−1) or apocynin (10 µmol·L−1) for 24 h. Informed consent was obtained from all subjects donating cords, and the study was approved by the Institutional Review Board for Human Studies of Nanjing Medical University (Nanjing, China).
Transfection with small interference RNAs
HUVECs were cultured in 12-well plates (for DHE assay) or 6-well plates (for Western blot) to 80% confluence, at which point they were transfected with siRNA oligonucleotide against PKCα (5′-GGCUUCCAGUGCCAAGUUUtt-3′, Genepharma, Shanghai, China), p47phox (5′-UGUUCCCUAUUGAGGCAGGtt-3′ Genepharma) or non-specific pooled duplex negative control siRNA (5′-UUCUCCGAACGUGUCACGUtt-3′ Genepharma) using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the protocols recommended by the manufacturer. The cells were used for experiments 24 h after transfection and gene silencing was detected by analysis of PKCα and p47phox protein expression with Western blot.
Lucigenin-enhanced chemiluminescence assay for production of superoxide (O2•−) in homogenates of HUVECs
Lucigenin is an acridinium compound that emits light upon interaction with O2•−. It was used to measure O2•− production in cell homogenates as described previously (Guzik and Channon, 2005). Briefly, following ox-LDL treatment, 6-well plates of confluent HUVECs were washed in ice-cold sodium PBS (8 g·L−1 NaCl, 2.9 g·L−1 NaH2PO4·12H2O, 0.2 g·L−1 KCl, 0.2 g·L−1 KH2PO4, pH 7.4) and lysed on ice by incubation with 20 mM HEPES buffer containing 1 mM EDTA, protease inhibitors (complete; Roche, Branford, CT, USA) and PMSF (1 mM). The cell debris was removed by centrifugation at 800× g for 10 min and the supernatant retained for further experiments. Thereafter, 50 µL of the cell homogenate was transferred into a 96-well plate and exposed to a Krebs-HEPES buffer (99 mmol·L−1 NaCl, 4.7 mmol·L−1 KCl, 1.2 mmol·L−1 MgSO4, 1 mmol·L−1 KH2PO4, 1.9 mmol·L−1 CaCl2, 25 mmol·L−1 NaHCO3, 11.1 mmol·L−1 glucose, and 20 mmol·L−1 HEPES, pH 7.44) containing dark-adapted lucigenin (5 µmol·L−1). Light emission was detected using a Glomax microplate Luminometer (Glomax, Berlin, Germany). Final values are expressed as relative light units normalized to protein concentration. Results are expressed as percentage inhibition of O2•− production by each dose of drug as compared with the relevant vehicle.
Dihydroethidium (DHE) assay for detection of superoxide
DHE was used to detect intracellular O2•−. DHE enters the cells and is oxidized by O2•− to yield ethidium, which binds to DNA to produce bright red fluorescence. DHE has been widely used for the fluorescence microscopic and flow-cytometric measurement of intracellular O2•− production. Following ox-LDL treatment, HUVECs were washed with PBS, and then incubated for 1 h at 37°C with 5 µmol·L−1 DHE in Krebs-HEPES buffer. After that, cells were washed with PBS again, and the red fluorescence was measured with a Nikon TE2000 Inverted Microscope (Nikon, Tokyo, Japan), using excitation and emission wavelengths of 480 and 610 nm, respectively. Meanwhile, cells were harvested with 0.02% Trypsin/EDTA and adjusted to 2 × 10−5 cells per FACS tube. The fluorescence was measured using the FL2 channel in a FACSCalibur flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA). In each experiment, at least, 10 000 events were analysed.
Chemiluminescence method for NO production measurement
NO, released from HUVECs, was determined by measuring the accumulation of its stable degradation products, nitrite and nitrate, in cell culture medium using a NO quantification kit (Beyotime Institute of Biotechnology, Haimen, China) and following the manufacturer's instructions. After reduction of nitrate with nitrate reductase, total nitrite was determined by chemiluminescence. Absorbance of the samples was determined at 540 nm and compared with the standard curve. The amount of nitrite formed was normalized to the protein content and presented as the % of control.
PepTag assay for non-radioactive detection of PKC activity
The PepTag assay utilizes a brightly coloured, fluorescent peptide substrate (PepTag® C1 Peptide, is P-L-S-R-T-L-S-V-A-A-K) that is highly specific to PKC (Promega, Madison, WI, USA). Phosphorylation by PKC changes the net charge of the substrate from +1 to −1, thereby allowing the phosphorylated and non-phosphorylated versions of the substrate to be separated on an agarose (0.8%) gel. The phosphorylated species migrates towards the positive electrode, whereas the non-phosphorylated substrate migrates towards the negative electrode. The phosphorylated peptide in the band can then be visualized under UV light. HUVECs, plated in 6-mm dishes, were grown to 85–90% confluence followed by incubation with ox-LDL (20 mg·L−1) with or without L-NAME (100 µmol·L−1), pyridoxine (10−7 mol·L−1) for another 24 h. The cells were washed with ice-cold PBS, pelleted by microfuging for 5 min at 1000×g, and then lysed in 200 µL of PKC extraction buffer (50 mM Tris–HCl, pH 7.4, 0.5 mM EDTA, 0.5 mM EGTA, 0.05% Triton® X-100, 10 mM β-mercaptoethanol, 1 mM PMSF, 1 µg·mL–1 aprotinin and 1 µg·mL–1 leupeptin). The lysates were centrifuged at 14 000×g for 10 min. The resulting supernatants were collected, and the protein concentrations were determined in each sample by the Bradford method using Pierce BCA Protein Assay (Thermo Fisher Scientific Inc., Rockford, IL, USA). Equal amounts of protein were incubated with PKC reaction mixture (25 µL) according to the manufacturer's protocol (Promega) at 30°C for 30 min. The reactions were stopped by heating to 95°C for 10 min. After addition of 80% glycerol (1 µL), the samples were loaded onto an agarose gel (0.8% agarose in 50 mM Tris-HCl, pH 8.0). The samples were separated on the agarose gel in the same buffer at 100 V for 20 min, The phosphorylated, negatively charged peptide was separated from the non-phosphorylated, and positively charged peptide were visualized under UV light. Resulting bands were excised, heated and 125 µL of the hot agarose transferred to a tube containing 75 µL of Gel Solubilization Solution (pre-warmed to room temperature and mixed well) and 50 µL of glacial acetic acid. The medium was vortexed and 250 µL transferred to a well in a 96-well plate The absorbance was read at 570 nm using a 96 well plate reader (Biotek Instruments, Winooski, VT, USA) and results normalized to control.
Western blot
Following incubations performed as mentioned earlier, confluent HUVECs were scraped from 6 well plates and lysed on ice with a lysis buffer (20 mmol·L−1 Tris-HCl, 100 mmol·L−1 NaCl, 2.5 mmol·L−1 EDTA, 1% Triton X-100, 1% sodium deoxycholate, 50 mmol·L−1 NaF, 1 mmol·L−1 Na3VO4; pH 7.4) containing a cocktail of protease inhibitors (Roche) plus 1 mM PMSF and thereafter centrifuged at 12 000×g for 15 min 4°C. The BCA Protein Assay (Thermo Fisher Scientific Inc.) was used (following manufacturer's instructions) to quantify the level of protein in each sample to ensure equal protein loading. Proteins in the cell were heated with SDS-PAGE sample buffer and separated by SDS-PAGE according to their molecular weight. Briefly, 40 µg proteins along with a molecular weight protein marker (Thermo Fisher Scientific Inc.) were subjected to SDS-PAGE using 7.5% acrylamide gel, electroblotted onto polyvinylidene fluoride membranes. The membrane was blocked with 5% non-fat milk in TBS containing 0.1% Tween 20 (TBS-T) and then probed overnight at 4°C with anti-phospho-eNOS (Thr495) (rabbit polyclonal, dilution 1:500, Cell Signaling Technology Inc., Danvers, MA, USA), anti-eNOS (rabbit polyclonal, dilution 1:1000, Santa Cruz Biotechnology Inc., Sta. Cruz, CA, USA), anti-phospho-PKCα/βII (rabbit polyclonal, dilution 1:500, Cell Signaling Technology Inc.), anti-PKC (rabbit polyclonal, dilution 1:500, Abcam, Cambridge, MA, USA), anti- PKCα (mouse monoclonal, 1:200 dilution, Santa Cruz Biotechnology), anti-Phospho-PKCα (mouse monoclonal, 1:100 dilution, Santa Cruz Biotechnology), p47phox (rabbit polyclonal, dilution 1:500, Cell Signaling Technology Inc.) or anti-β-actin antibody (mouse monoclonal, 1:5000 dilution) After incubation with peroxidase-conjugated secondary antibody (1:4000 dilution for goat anti-rabbit and 1:4000 dilution for goat anti-mouse) for 2 h at room temperature, membranes were developed using enhanced chemiluminescence substrate (ECL; Thermo Fisher Scientific Inc.). Band intensities were analysed using Image J 1.25 software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are expressed as mean ± SD, and were analysed using Student's paired t-test or one-way anova followed by Newman–Keuls multiple comparison test as appropriate (GraphPad Prism version 4 software). A value of P < 0.05 was considered statistically significant.
Materials
Unless otherwise stated, all reagents used were obtained from Sigma-Aldrich (St. Louis, MO, USA). DHE were dissolved in dimethyl sulphoxide (DMSO) as 5 mM stock solution and stored under nitrogen at −20°C until use. The final concentration of DMSO in the culture medium did not exceed 0.01%, which has no significant pharmacological activity. Lucigenin was dissolved in Krebs-HEPES buffer. Pyridoxine, CHCI, apocynin and L-NAME were dissolved in sterile water.
Results
ox-LDL induced eNOS uncoupling in HUVECs
We first investigated the effect of ox-LDL on O2•− production in HUVECs. HUVECs were treated with ox-LDL (10, 20, 40 mg·L−1) for 24 h. O2•− production was significantly increased by ox-LDL (Figure 1A). Next, we treated HUVECs with L-NAME (100 µmol·L−1), a pharmacological inhibitor of eNOS to investigate whether eNOS affected ox-LDL-induced O2•−production. L-NAME blocked ox-LDL induced O2•− production (Figure 1B). We also measured the nitrite level that indicates the NO concentration. ox-LDL decreased NO levels in HUVECs and the addition of the L-NAME abolished this effect of ox-LDL (Figure 1C). These data suggest that ox-LDL stimulates O2•− production by uncoupling eNOS.
Figure 1.
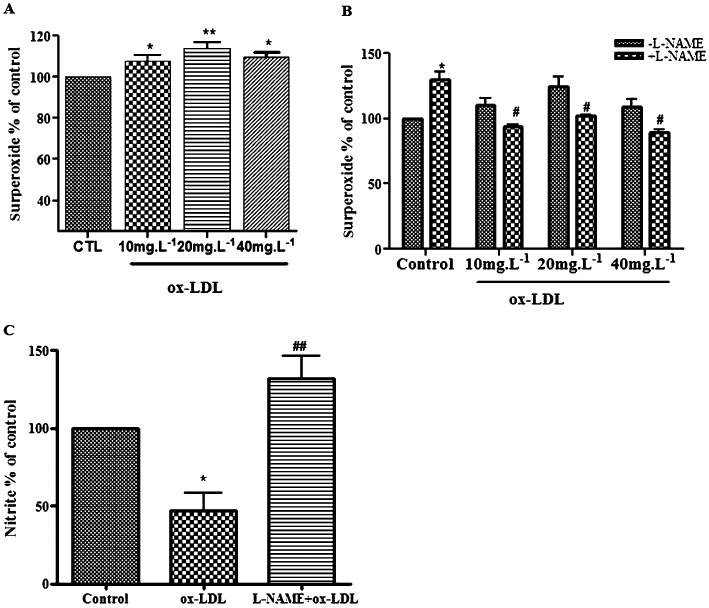
ox-LDL induced eNOS uncoupling in HUVECs. (A) ox-LDL increased the endothelial O2•− production. After incubation of HUVECs with ox-LDL (10, 20, 40 mg·L−1) for 24 h, endothelial O2•− production increased significantly, compared with the control group. Data are expressed relative to control (n = 6 *P < 0.05, ***P < 0.01, vs. control). (B) L-NAME (100 µmol·L−1 for 15 min) significantly decreased ox-LDL-stimulated O2•− production. Data are expressed relative to control (n = 4 *P < 0.05 vs. control, #P < 0.05 vs. ox-LDL alone) (C). L-NAME increased NO levels in ox-LDL-treated HUVECs. ox-LDL (20 mg·L−1 for 24 h) decreased NO release significantly. The decrease in NO was reversed by L-NAME pretreatment. Data are presented as changes relative to control (n = 4 *P < 0.05 vs. control, ##P < 0.01 vs. ox-LDL).
Pyridoxine inhibited O2•− production and preserved NO levels in ox-LDL-treated HUVECs
Next, we tested whether pyridoxine affects eNOS uncoupling. Different concentrations of pyridoxine (10−7, 10−1, 10−5 mol·L−1) decrease ox-LDL-induced O2•− production (data not shown). Therefore, we treated HUVECs with 10−7 mol·L−1 pyridoxine for 15 min before incubation with ox-LDL (20 mg·L−1) for another 24 h. Pyridoxine significantly decreased O2•− production (Figure 2A). Pyridoxine also significantly reduced the ox-LDL-induced decrease in NO levels (Figure 2B). These results demonstrate that pyridoxine effectively inhibits ox-LDL-induced uncoupling of eNOS.
Figure 2.
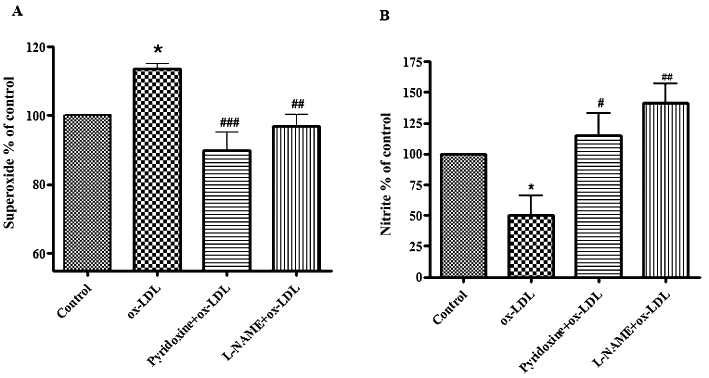
Effect of pyridoxine on ox-LDL-induced O2•− and NO production. (A) Pyridoxine inhibited ox-LDL-induced O2•− generation. Cells were pretreated for 15 min with pyridoxine (10−7 mol·L−1), followed by incubation with 20 mg·L−1 ox-LDL for another 24 h. Data are presented as changes relative to control (n = 6 *P < 0.05 vs. control, ##P < 0.01, ###P < 0.001 vs. ox-LDL). (B) Pyridoxine increased NO level in ox-LDL-treated HUVECs. Pretreatment with pyridoxine significantly prevented the decrease of NO induced by ox-LDL. Data are expressed relative to control (n = 3 *P < 0.05 vs. control, #P < 0.05, ##P < 0.01 vs. ox-LDL).
Pyridoxine increased phosphorylation of eNOS at Thr495 in ox-LDL-treated HUVECs
eNOS uncoupling is tightly regulated by the phosphorylation status of eNOS Thr495. To determine whether pyridoxine also affects phosphorylation of eNOS at threonine 495, HUVECs were treated with pyridoxine (10−7 mol·L−1) for 15 min and then exposed to 20 mg·L−1 ox-LDL for another 24 h. eNOS phosphorylation was examined. ox-LDL significantly reduced the phosphorylation of eNOS Thr495 and pyridoxine treatment restored the eNOS phosphorylation (Figure 3). These data strongly suggest that pyridoxine prevents the ox-LDL-evoked decrease in phosphorylation of eNOS at Thr495 in HUVECs.
Figure 3.
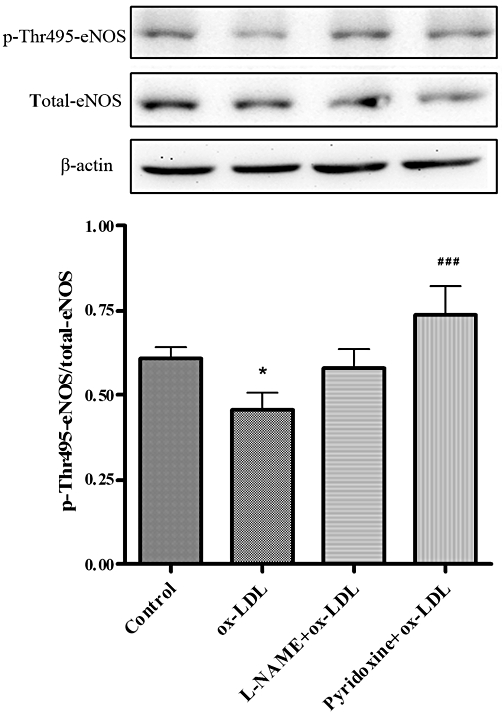
Pyridoxine increased the ox-LDL-reduced phosphorylation of eNOS Thr495. Cells were pretreated for 15 min with pyridoxine (10−7 mol·L−1), followed by incubation with 20 mg·L−1 ox-LDL for another 24 h. The expression of phosphorylated eNOS Thr495, total-eNOS and β-actin was measured. Pyridoxine increased ox-LDL-reduced phosphorylation of eNOS Thr495. Phosphorylation was quantified densitometrically as the ratio of phospho-Thr495-eNOS/total-eNOS (n = 5 *P < 0.05 vs. control, ###P < 0.001 vs. ox-LDL).
Role of PKC in the effect of pyridoxine on ox-LDL-treated HUVECs
PKC is known to be a major kinase phosphorylating eNOS Thr495 (Fulton et al., 2001; Michell et al., 2001). Thus, we examined the possibility that phosphorylation of eNOS by PKC contributes to the inhibitory effect of pyridoxine on eNOS uncoupling. We firstly measured the effect of ox-LDL and pyridoxine on PKC activity using the PepTag Non-Radioactive Detection Assay. Stimulation of HUVECs with ox-LDL (20 mg·L−1) for 24 h caused a decrease in PKC activity, and this decrease was prevented by pretreatment with pyridoxine (Figure 4A). Western blot results also confirmed that pretreatment of pyridoxine prevented the decrease in PKCα/βII phosphorylation induced by ox-LDL, no difference in the expression of total PKC or β-actin was noted between all the groups (Figure 4B).
Figure 4.
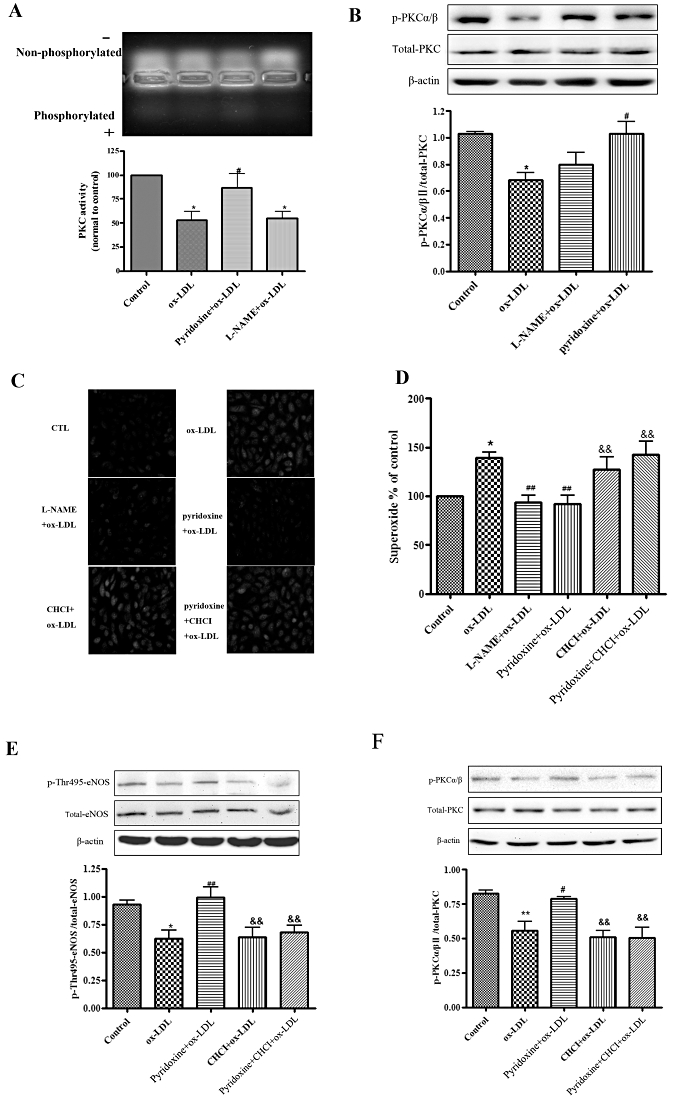
PKC regulated the effect of pyridoxine in ox-LDL-treated HUVECs. (A) Representative agarose gel separation of phosphorylated PepTag showed fluorescent phosphorylated Peptag (upper panel). The bands corresponding to the phosphorylated peptide were quantified and normalized to controls (lower panel). (B) Western blot analysis showed the expression of phosphorylated PKCα/β, total-PKC and β-actin. Phosphorylation was quantified densitometrically as the ratio of p-PKCα/β/total-PKC. (n = 6 *P < 0.05 vs. control, #P < 0.05 vs. ox-LDL). (C, D) Cells were treated or untreated with pyridoxine(10−7 mol·L−1), CHCI (5 µmol·L−1), L-NAME (100 µmol·L−1) for 15 min followed by incubation with 20 mg·L−1 ox-LDL for another 24 h. Endothelial O2•− production was measured by DHE using fluorescence microscope and flow cytometric. (n = 7 *P < 0.05 vs. control, ##P < 0.01 vs. ox-LDL, &&P < 0.01 vs. pyridoxine + ox-LDL). (E) Western blot analysis showed the expression of phosphorylated eNOS Thr495, total-eNOS and β-actin. Phosphorylation was quantified densitometrically as the ratio of p-Thr495-eNOS/total-eNOS (n = 5 *P < 0.05 vs. control, ##P < 0.01 vs. ox-LDL, &&P < 0.01 vs. pyridoxine + ox-LDL). (F) Western blot analysis showed that the expression of phosphorylated PKCα/βII, total-PKC and β-actin. Phosphorylation was quantified densitometrically as the ratio of p-PKCα/βII/total-PKC (n = 5 **P < 0.01 vs. control, #P < 0.05 vs. ox-LDL, &&P < 0.01 vs. pyridoxine + ox-LDL).
To determine whether activation of PKC is required for the effect of pyridoxine on O2•− production, we used a general PKC inhibitor CHCI, which affects the translocation of PKC from cytosol to the plasma membrane, and monitored the generation of O2•−. As shown in Figure 4C and D, when cells were pre-incubated with 5 µmol·L−1 CHCI for 15 min, pyridoxine failed to reduce O2•− production in response to ox-LDL. In addition, pyridoxine failed to prevent the dephosphorylation of eNOS Thr495 and decrease in PKC phosphorylation induced by ox-LDL after CHCI pretreatment (Figure 4E and F). These results suggest that PKC activity is responsible for the inhibitory effects of pyridoxine on ox-LDL-induced eNOS uncoupling in endothelial cells.
As chelerythrine has broad specificity against PKC isoforms, additional experiments were performed to confirm the involvement of PKCα in the protective effect of pyridoxine, using siRNAs specific for PKCα. An unrelated siRNA was used as a negative control. Western blot analysis of PKCα siRNA transfection showed a significant decrease in PKCα protein expression in HUVECs as compared with control siRNA, whereas β-actin expression was unchanged (Figure 5A). As shown in Figure 5B and C, transfection of PKCα siRNA, but not control siRNA, prevented the pyridoxine-induced decrease in O2•− production stimulated by ox-LDL. Furthermore, similar to pharmacological inhibition of PKC by CHCI, gene knockdown of PKCα markedly reduced the pyridoxine-induced increase in phosphorylation of eNOS Thr495 (Figure 5D). These data indicated that PKCα appears to be the isoform that phosphorylates eNOS Thr495 and is targeted by pyridoxine and ox-LDL.
Figure 5.
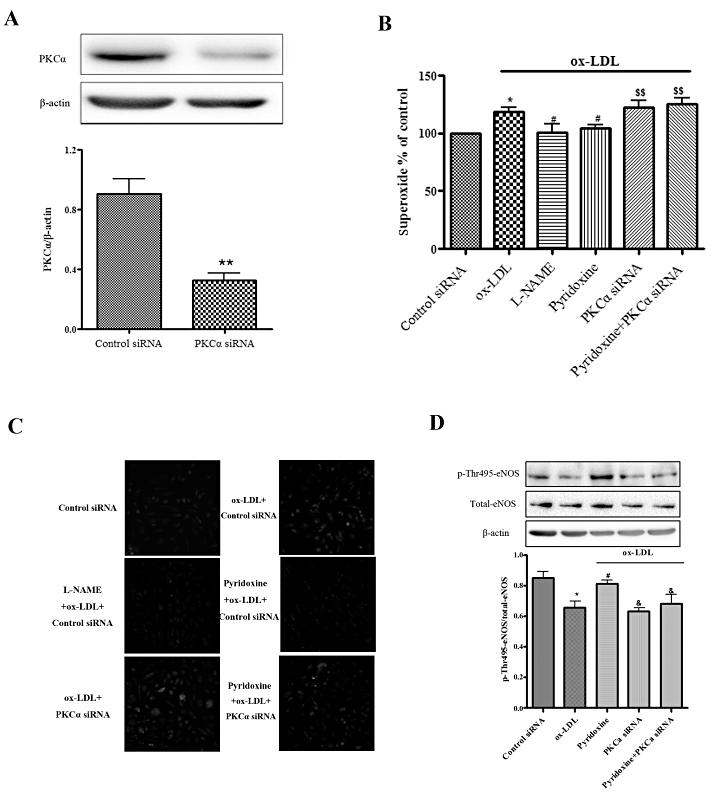
PKCα isoform regulated the effect of pyridoxine in ox-LDL-treated HUVECs. (A) Western blot analysis showed the protein expression levels of PKCα in control siRNA or PKCα siRNA. (B, C) The effect of combination treatment of knockdown PKCα, pyridoxine(10−7 mol·L−1) and L-NAME (100 µmol·L−1) on ox-LDL-treated HUVECs and endothelial O2•− production was measured by DHE using flow cytometric (n = 7–10 *P < 0.05 vs. control siRNA, #P < 0.05 vs. ox-LDL, &&P < 0.01 vs. pyridoxine + ox-LDL) and fluorescence microscope. (D) Western blot analysis showed the expression of phosphorylated eNOS Thr495, total-eNOS and β-actin. Phosphorylation was quantified densitometrically as the ratio of p-Thr495-eNOS/total-eNOS (n = 4 *P < 0.05 vs. control siRNA, #P < 0.05 vs. ox-LDL, &P < 0.05 vs. pyridoxine + ox-LDL).
NADPH oxidase is not involved in the effect of pyridoxine in ox-LDL-treated HUVECs
To test whether NADPH oxidase is the primary source of ox-LDL-stimulated O2•− involved in the effect of pyridoxine on eNOS uncoupling, we treated HUVECs with the NADPH oxidase inhibitor apocynin. Apocynin did not affect ox-LDL-induced O2•− production in HUVECs treated with pyridoxine (Figure 6A and B). Accordingly, we conducted an experiment to measure expression of the cytosolic p47phox subunits of the NADPH oxidase. The data show that protein expression of p47phox in HUVECs was not different between treatments (Figure 6C). We also knocked down p47phox using siRNA to further investigate a possible role of NADPH oxidase in the effect of pyridoxine in ox-LDL-treated HUVECs (Figure 6D). Inhibition of p47phox did not change the effect of pyridoxine or ox-LDL on O2•− production (Figure 6E and F). Together, these data suggested that NADPH oxidase is not involved in the effect of pyridoxine on ox-LDL-induced O2•−.
Figure 6.
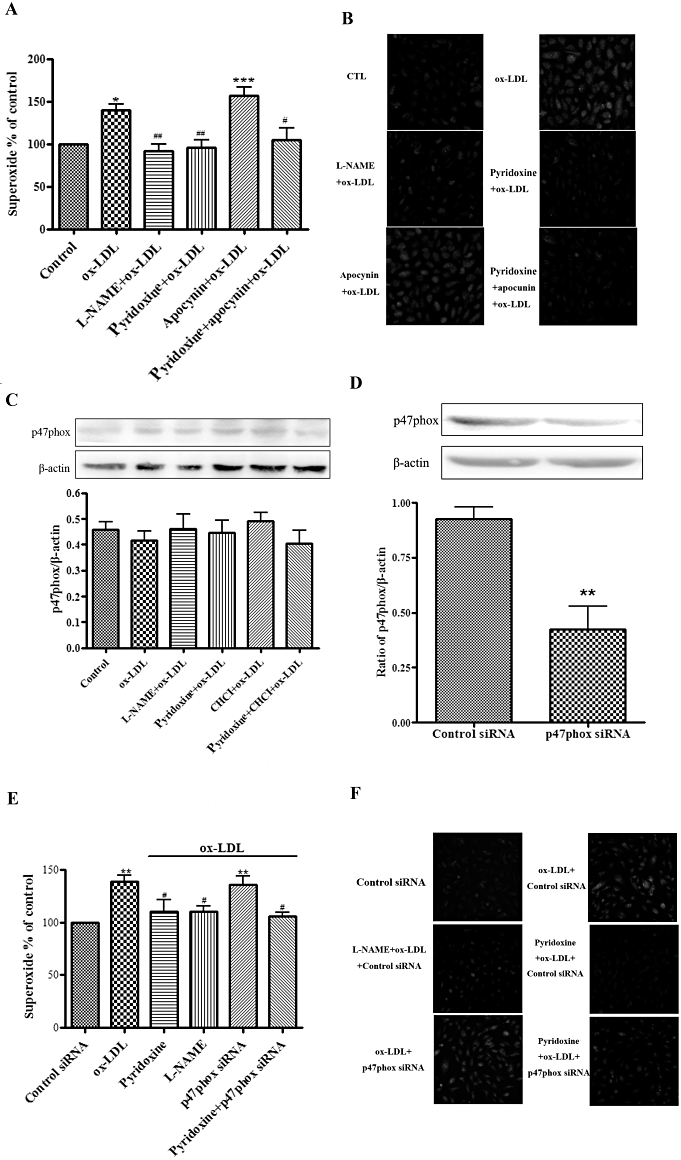
NADPH oxidase is not involved in the effect of pyridoxine in ox-LDL-treated HUVECs. (A) and (B) Cells were pretreated for 15 min with or without pyridoxine (10−7 mol·L−1), apocynin (10 µmol·L−1) or L-NAME (100 µmol·L−1) followed by incubation with 20 mg·L−1 ox-LDL for another 24 h. Endothelial O2•− production was measured by DHE using flow-cytometry (A) and fluorescence microscope (n = 7, *P < 0.05 vs. control, ##P < 0.01 vs. ox-LDL). (C) Western blot analysis showed the expression of p47phox and β-actin. The expression of p47phox was quantified densitometrically as the ratio of p47phox/β-actin (n = 6). (D) Western blot analysis showed the protein expression levels of p47phox in the control siRNA or p47phox siRNA. (E, F) The combination effect of knockdown of p47phox and pyridoxine (10−7 mol·L−1) or L-NAME (100 µmol·L−1) in ox-LDL-treated HUVECs. Endothelial O2•− production was measured by DHE using flow cytometric (n = 4 **P < 0.01 vs. control siRNA, #P < 0.05 vs. ox-LDL, &&P < 0.01 vs. pyridoxine + ox-LDL) and fluorescence microscope.
Discussion
Endothelial dysfunction is seen in the early development of atherosclerosis before overt vascular and structural changes (Rabelink, 2007). eNOS-derived NO is recognized as one of the major mediators maintaining vascular homeostasis (Anderson, 2003). In the presence of a number of pathological conditions, eNOS becomes uncoupled and produces O2•−, which contributes to the development of cardiovascular disease (Förstermann and Münzel, 2006). Considerable evidence indicates that ox-LDL contributes to many atherogenic steps in the vessel wall including inducing modification of cell protein structure, eliciting ROS generation and peroxidation of cellular lipids, and altering the regulation of various signalling pathways and gene expression (Matsuura et al., 2006; Matsuura et al., 2008). It has been shown that endothelial dysfunction caused by ox-LDL is mainly due to an increase in eNOS-derived O2•− (Heeba et al., 2007). Here, we investigated the effect of ox-LDL on endothelial cells. Consistent with previous studies (Vergnani et al., 2000), ox-LDL significantly reduced the concentration of NO and increased O2•− production. This process was reversed by the eNOS inhibitor, L-NAME. The results presented here strongly demonstrated that ox-LDL induces endothelial dysfuction through eNOS uncoupling. Therefore, it is of critical importance to prevent eNOS uncoupling induced by ox-LDL.
Pyridoxine plays an important role in numerous metabolic processes. Pyridoxine and other B vitamins have been reported to decrease plasma homocysteine and inhibit atherosclerosis (Zhou et al., 2003). In addition, pyridoxine has been shown to ameliorate endothelial dysfunction in vivo in cardiac transplant recipients (Miner et al., 2001). We have recently reported that pyridoxine protects against endothelial dysfunction induced by LDL-stimulated NO production (Ji et al., 2003; 2006). Here, we evaluated the effect of pyridoxine on ox-LDL-induced eNOS uncoupling in HUVECs. We showed that pyridoxine significantly decreased the O2•− generation and subsequently inhibited ox-LDL-diminished NO level. These data suggest that pyridoxine inhibits ox-LDL-induced eNOS uncoupling and protects the function of the endothelium.
However, the mechanism of the effect of pyridoxine on ox-LDL-induced eNOS uncoupling remains unclear. Several pathological effects mediate eNOS uncoupling. The phosphorylation of eNOS Thr495 is a pivotal factor in eNOS uncoupling. Cells expressing T497A eNOS mutant are reported to produce higher superoxide than wild-type eNOS and this effect is reduced by L-NAME treatment (Lin et al., 2003). The non-phosphorylatable T495A mutant produces more O2•− than the phospho-mimetic T495D mutant in COS-7 cells (Fleming et al., 2005). We also showed that ox-LDL significantly reduced the phosphorylation of eNOS Thr495 whereas it did not affect eNOS protein expression. It is still unclear how ox-LDL affects the expression of eNOS. On the one hand, low concentrations of ox-LDL do not alter the total protein expression of eNOS (Fleming et al., 2005). On the other hand, a higher concentration of ox-LDL significantly reduces the protein expression of eNOS (Liao et al., 1995; Lee et al., 2009). The reason for this apparent disparity may be due to the different concentrations of ox-LDL used in different studies. As we expected, pyridoxine prevented the ox-LDL-diminished phosphorylation of eNOS Thr495. These data suggest that the inhibitory effect of pyridoxine on ox-LDL-induced eNOS uncoupling is produced by phosphorylation of eNOS at Thr495.
PKC is a major kinase that phosphorylates eNOS at Thr495 (Fulton et al., 2001; Michell et al., 2001). We found that PMA, an activator of PKC, increased the phosphorylation of eNOS Thr495 (Figure S1). PKC signalling inhibits eNOS activity in endothelial cells (Chen et al., 1999; Michell et al., 2001), but the inhibition PKC was also associated with an increase in NO production. Thr495 is thought to have a negative regulatory role on CaM binding and eNOS activity. However, it has been found that blockade of PKC activity does not prevent the NO-mediated inhibition of eNOS activity (Sheehy et al., 1998). In addition, even though activation of PKC is involved in producing O2•− generation stimulated by angiotensin II (Silva and Garvin, 2008; Herrera et al., 2010) and ox-LDL was found to acutely stimulate PKC activity (Mukherjee et al., 2001), the results of another study clearly demonstrated that ox-LDL decrease the phosphorylation and activity of PKC (Fleming et al., 2005). In the present study we demonstrated that PKC phosphorylates eNOS Thr495, but the elevated phosphorylation of eNOS Thr495, in response to the PKC agonist PMA, was attenuated in ox-LDL-treated cells (Figure S1). Pyridoxine significantly restored the activity and phosphorylation of PKC, which was reduced by ox-LDL, and this protective effect of pyridoxine was abolished by the PKC inhibitor, CHCI. These data suggest that activation of PKC may be involved in preventing eNOS uncoupling.
PKC is a multi-gene family with at least 12 members. There are three families of PKC: conventional (α, βI, βII, γ), which are Ca2+ and lipid activated, the novel (δ, ε, η, θ) and atypical (ζ, ν, µ, ι), which are Ca2+ independent and activated by distinct lipid moieties (Bynagari-Settipalli et al., 2010). The role of each PKC isoform in cell signalling has not been elucidated. The high homology of the domains targeted by the pharmacological modulators of PKC always leads to non-specific isozyme selectivity. PKCα isoform plays an important role in ox-LDL-induced O2•− (Fleming et al., 2005). Here, we found that the knocked down PKCα isoform markedly inhibited the protective effect of pyridoxine on the increased O2•− production and decreased phosphorylation of eNOS Thr495 in ox-LDL-treated HUVECs. Thus, activation of PKCα plays an important role in producing the effect of pyridoxine on ox-LDL-induced eNOS uncoupling.
NADPH oxidase is a source of O2•−. The increased NADPH oxidase-mediated O2•− production may act as a kindling radical favouring eNOS uncoupling by stimulating increased formation of vascular peroxynitrites (Xu et al., 2006). Statins are reported to prevent eNOS uncoupling by inhibiting NADPH oxidase expression and activity (Vergnani et al., 2000). NADPH oxidase is an enzymatic complex that comprises five components: p40phox; p47phox; p67phox; p22phox; and NOX (Babior, 1999). Under basal conditions, p40phox, p47phox and p67phox are located in the cytosol as a complex. Upon stimulation, p47phox is phosphorylated. The cytosolic complex thereafter translocates to the cell membrane, where it assembles with p22phox and NOX, and generates O2•− (Shiose et al., 2001). p47phox is thus essential for activation of NADPH oxidase. The expression of cytosol p47phox represents the function of NADPH oxidase (Rupin et al., 2004). We showed that the expression of cytosol p47phox was not changed by our different treatments. Apocynin is the most widely studied NADPH oxidase inhibitor (Drummond et al., 2011). We found that apocynin did not affect ox-LDL-stimulated O2•− production or the effect of pyridoxine. As apocynin is not a selective inhibitor for NADPH oxidases and also acts as a pro-oxidant under certain conditions (Heumüller et al., 2008), we knocked down the expression of p47phox by siRNA. The knockdown of p47phox did not change the effect of pyridoxine or of ox-LDL on the O2•−. Together the results indicate that the NADPH oxidase is not involved in ox-LDL-induced O2•− and the effect of pyridoxine.
In conclusion, our study demonstrates that pyridoxine induces phosphorylation of eNOS Thr495, improves NO production and reduces O2•− production by modulating PKCα activity in ox-LDL-treated cells. The effect of pyridoxine on the uncoupling of eNOS may provide a novel strategy to prevent ox-LDL-mediated dysfunction of the vascular endothelium.
Acknowledgments
This work was generously supported by grants from the National Basic Research Program of China (973) (Nos. 2010CB535010, No.2011CB503903) and by the National Natural Science Foundation of China (Nos. 30770891, 30971256, 30730044 and 81170083).
Glossary
- CHCI
chelerythrine chloride
- DHE
dihydroethidium
- eNOS
endothelial NOS
- HUVEC
human umbilical vein endothelial cell
- L-NAME
NG-nitro-L-arginine methylester
- O2•−
superoxide
- ox-LDL
oxidized low-density lipoprotein
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 HUVECs were treated or untreated withpyridoxine (10−7 mol·L−1),PMA (10 nmol·L−1), L-NAME (100μmol·L−1) for 15 min followed byincubation with 20 mg·L−1 ox-LDL for another 24 h. Western blot analysis showed the expression of phosphorylated eNOS Thr495, total-eNOS and β-actin. Phosphorylation was quantified densitometrically as the ratio of p-Thr495-eNOS/total-eNOS (n = 4 *P < 0.05 vs. control,#P < 0.05, ##P < 0.01 vs. ox-LDL).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Anderson TJ. Nitric oxide, atherosclerosis and the clinical relevance of endothelial dysfunction. Heart Fail Rev. 2003;8:71–86. doi: 10.1023/a:1022199021949. [DOI] [PubMed] [Google Scholar]
- Antoniades C, Shirodaria C, Leeson P, Antonopoulos A, Warrick N, Van-Assche T, et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. Eur Heart J. 2009;30:1142–1150. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, et al. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- Bevers LM, Braam B, Post JA, van Zonneveld AJ, Rabelink TJ, Koomans HA, et al. Tetrahydrobiopterin, but not L-arginine, decreases NO synthase uncoupling in cells expressing high levels of endothelial NO synthase. Hypertension. 2006;47:87–94. doi: 10.1161/01.HYP.0000196735.85398.0e. [DOI] [PubMed] [Google Scholar]
- Bynagari-Settipalli YS, Chari R, Kilpatrick L, Kunapuli SP. Protein kinase C-possible therapeutic target to treat cardiovascular diseases. Cardiovasc Hematol Disord Drug Targets. 2010;10:292–308. doi: 10.2174/187152910793743869. [DOI] [PubMed] [Google Scholar]
- Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, et al. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 1999;443:285–289. doi: 10.1016/s0014-5793(98)01705-0. [DOI] [PubMed] [Google Scholar]
- Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. Phosphorylation of endothelial nitric-oxide synthase regulates superoxide generation from the enzyme. J Biol Chem. 2008;283:27038–27047. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cominacini L, Rigoni A, Pasini AF, Garbin U, Davoli A, Campagnola M, et al. The binding of oxidized low density lipoprotein (ox-LDL) to ox-LDL receptor-1 reduces the intracellular concentration of nitric oxide in endothelial cells through an increased production of superoxide. J Biol Chem. 2001;276:13750–13755. doi: 10.1074/jbc.M010612200. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro A, Queen LR, Priest RM, Xu B, Ritter JM, Poston L, et al. Activation of nitric oxide synthase by β2-adrenoceptors in human umbilical vein endothelium in vitro. Br J Pharmacol. 1999;126:1872–1880. doi: 10.1038/sj.bjp.0702512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming I, Mohamed A, Galle J, Turchanowa L, Brandes RP, Fisslthaler B, et al. Oxidized low-density lipoprotein increases superoxide production by endothelial nitric oxide synthase by inhibiting PKCalpha. Cardiovasc Res. 2005;65:897–906. doi: 10.1016/j.cardiores.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818–824. [PubMed] [Google Scholar]
- Guzik TJ, Channon KM. Measurement of vascular reactive oxygen species production by chemiluminescence. Methods Mol Med. 2005;108:73–89. doi: 10.1385/1-59259-850-1:073. [DOI] [PubMed] [Google Scholar]
- Heeba G, Hassan MK, Khalifa M, Malinski T. Adverse balance of nitric oxide/peroxynitrite in the dysfunctional endothelium can be reversed by statins. J Cardiovasc Pharmacol. 2007;50:391–398. doi: 10.1097/FJC.0b013e31811f3fd0. [DOI] [PubMed] [Google Scholar]
- Heinloth A, Heermeier K, Raff U, Wanner C, Galle J. Stimulation of NADPH oxidase by oxidized low-density lipoprotein induces proliferation of human vascular endothelial cells. J Am Soc Nephrol. 2000;11:1819–1825. doi: 10.1681/ASN.V11101819. [DOI] [PubMed] [Google Scholar]
- Herrera M, Silva GB, Garvin JL. Angiotensin II stimulates thick ascending limb superoxide production via protein kinase C(alpha)-dependent NADPH oxidase activation. J Biol Chem. 2010;285:21323–21328. doi: 10.1074/jbc.M110.109157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumüller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schröder K, et al. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- Ji Y, Han Y, Diao J, Huang Y, Chen Q, Ferro A. Rabbit aortic endothelial dysfunction by low-density lipoprotein is attenuated by L-arginine, L-ascorbate and pyridoxine. Br J Pharmacol. 2003;140:1272–1282. doi: 10.1038/sj.bjp.0705545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Diao J, Han Y, Huang Y, Bai H, Chen Q, et al. Pyridoxine prevents dysfunction of endothelial cell nitric oxide production in response to low-density lipoprotein. Atherosclerosis. 2006;188:84–94. doi: 10.1016/j.atherosclerosis.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Kone BC, Kuncewicz T, Zhang W, Yu ZY. Protein interactions with nitric oxide synthases: controlling the right time, the right place, and the right amount of nitric oxide. Am J Physiol Renal Physiol. 2003;285:F178–F190. doi: 10.1152/ajprenal.00048.2003. [DOI] [PubMed] [Google Scholar]
- Lee WJ, Ou HC, Wu CM, Lee IT, Lin SY, Ly L, et al. Sesamin mitigates inflammation and oxidative stress in endothelial cells exposed to oxidized low-density lipoprotein. J Agric Food Chem. 2009;57:11406–11417. doi: 10.1021/jf902876p. [DOI] [PubMed] [Google Scholar]
- Liao JK, Shin WS, Lee WY, Clark SL. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem. 1995;270:319–324. doi: 10.1074/jbc.270.1.319. [DOI] [PubMed] [Google Scholar]
- Lin MI, Fulton D, Babbitt R, Fleming I, Busse R, Pritchard KA, et al. Phosphorylation of threonine 497 in endothelial nitric-oxide synthase coordinates the coupling of L-arginine metabolism to efficient nitric oxide production. J Biol Chem. 2003;278:44719–44726. doi: 10.1074/jbc.M302836200. [DOI] [PubMed] [Google Scholar]
- Matsuura E, Kobayashi K, Tabuchi M, Lopez LR. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog Lipid Res. 2006;45:466–486. doi: 10.1016/j.plipres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Matsuura E, Hughes GR, Khamashta MA. Oxidation of LDL and its clinical implication. Autoimmun Rev. 2008;7:558–566. doi: 10.1016/j.autrev.2008.04.018. [DOI] [PubMed] [Google Scholar]
- Matxain JM, Ristilä M, Strid A, Eriksson LA. Theoretical study of the antioxidant properties of pyridoxine. J Phys Chem A. 2006;110:13068–13072. doi: 10.1021/jp065115p. [DOI] [PubMed] [Google Scholar]
- Michell BJ, Chen Z, Tiganis T, Stapleton D, Katsis F, Power DA, et al. Coordinated control of endothelial nitric-oxide synthase phosphorylation by protein kinase C and the cAMP-dependent protein kinase. J Biol Chem. 2001;276:17625–17628. doi: 10.1074/jbc.C100122200. [DOI] [PubMed] [Google Scholar]
- Miner SE, Cole DE, Evrovski J, Forrest Q, Hutchison S, Holmes K, et al. Pyridoxine improves endothelial function in cardiac transplant recipients. J Heart Lung Transplant. 2001;20:964–969. doi: 10.1016/s1053-2498(01)00293-5. [DOI] [PubMed] [Google Scholar]
- Mukherjee S, Coaxum SD, Maleque M, Das SK. Effects of oxidized low density lipoprotein on nitric oxide synthetase and protein kinase C activities in bovine endothelial cells. Cell Mol Biol (Noisy-le-grand) 2001;47:1051–1058. [PubMed] [Google Scholar]
- Pritchard KA, Jr, Ackerman AW, Gross ER, Stepp DW, Shi Y, Fontana JT, et al. Heat shock protein 90 mediates the balance of nitric oxide and superoxide anion from endothelial nitric-oxide synthase. J Biol Chem. 2001;276:17621–17624. doi: 10.1074/jbc.C100084200. [DOI] [PubMed] [Google Scholar]
- Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. doi: 10.1161/CIRCULATIONAHA.106.652859. Review. [DOI] [PubMed] [Google Scholar]
- Rupin A, Paysant J, Sansilvestri-Morel P, Lembrez N, Lacoste JM, Cordi A, et al. Role of NADPH oxidase-mediated superoxide production in the regulation of E-selectin expression by endothelial cells subjected to anoxia/reoxygenation. Cardiovasc Res. 2004;63:323–330. doi: 10.1016/j.cardiores.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Sessa WC. eNOS at a glance. J Cell Sci. 2004;117((Pt 12)):2427–2429. doi: 10.1242/jcs.01165. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Burson MA, Black SM. Nitric oxide exposure inhibits endothelial NOS activity but not gene expression: a role for superoxide. Am J Physiol Lung Cell Mol Physiol. 1998;274((5 Pt 1)):L833–L841. doi: 10.1152/ajplung.1998.274.5.L833. [DOI] [PubMed] [Google Scholar]
- Shiose A, Kuroda J, Tsuruya K, Hirai M, Hirakata H, Naito S, et al. A novel superoxide-producing NAD(P)H oxidase in kidney. J Biol Chem. 2001;276:1417–1423. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- Silva GB, Garvin JL. Angiotensin II-dependent hypertension increases Na transport-related oxygen consumption by the thick ascending limb. Hypertension. 2008;52:1091–1098. doi: 10.1161/HYPERTENSIONAHA.108.120212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnani L, Hatrik S, Ricci F, Passaro A, Manzoli N, Zuliani G, et al. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production: key role of L -arginine availability. Circulation. 2000;101:1261–1266. doi: 10.1161/01.cir.101.11.1261. [DOI] [PubMed] [Google Scholar]
- Xu J, Xie Z, Reece R, Pimental D, Zou MH. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol. 2006;26:2688–2695. doi: 10.1161/01.ATV.0000249394.94588.82. [DOI] [PubMed] [Google Scholar]
- Zhou J, Møller J, Ritskes-Hoitinga M, Larsen ML, Austin RC, Falk E. Effects of vitamin supplement and hyperhomocysteinemia on atherosclerosis in apoE-deficient mice. Atherosclerosis. 2003;168:255–262. doi: 10.1016/s0021-9150(03)00138-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.