

Abstract
A number of therapeutic strategies targeting high-density lipoprotein (HDL) cholesterol and reverse cholesterol transport are being developed to halt the progression of atherosclerosis or even induce regression. However, circulating HDL cholesterol levels alone represent an inadequate measure of therapeutic efficacy. Evaluation of the potential effects of HDL-targeted interventions on atherosclerosis requires reliable assays of HDL function and surrogate markers of efficacy. Promotion of macrophage cholesterol efflux and reverse cholesterol transport is thought to be one of the most important mechanisms by which HDL protects against atherosclerosis, and methods to assess this pathway in vivo are being developed. Indexes of monocyte chemotaxis, endothelial inflammation, oxidation, nitric oxide production, and thrombosis reveal other dimensions of HDL functionality. Robust, reproducible assays that can be performed widely are needed to move this field forward and permit effective assessment of the therapeutic potential of HDL-targeted therapies.
A number of therapeutic strategies (1,2) are being developed to target high-density lipoprotein cholesterol (HDL-C) in an attempt to halt the progression or induce regression of atherosclerosis and reduce cardiovascular events. The fidelity of the inverse correlation between HDL-C levels and cardiovascular risk noted in observational studies remains uncertain in the setting of pharmacotherapy. Early assessment of changes in high-density lipoprotein (HDL) functionality in response to a new HDL-targeted therapy is critical. This article aims to review the limitations of circulating HDL-C as a therapeutic end point and to discuss emerging metrics of HDL functionality.
Limitations of HDL-C as a Surrogate End Point
Prospective cohort studies, as well as randomized controlled trials of antidyslipidemic therapies, support a powerful inverse correlation between circulating HDL-C levels and coronary risk among patients with elevated, normal, or low low-density lipoprotein cholesterol (LDL-C) (3-6). However, the inverse relationship falls short when applied to particular subgroups with altered lipoprotein structure and metabolism. Naturally occurring genetic conditions in humans indicate that lower HDL-C values need not impart excess cardiovascular risk, as in individuals carrying the apolipoprotein (apo) A-I Milano variant, and that higher HDL-C levels may not always confer a protective benefit, as suggested by the ongoing debate regarding the clinical significance of cholesteryl ester transfer protein deficiency (7-10). The recently published results of imaging studies and an outcome trial with the cholesteryl ester transfer protein (CETP) inhibitor torcetrapib (11-14) showed no impact on atherosclerosis and increased morbidity and mortality despite substantial raising of HDL-C levels, further complicating the picture. Elevation in blood pressure and aldosterone levels, which are not mechanism-based effects, suggest that off-target activities particular to torcetrapib may have increased cardiovascular risk (12). New CETP inhibitors that do not cause elevated blood pressure are in clinical development (15), and definitive answers regarding the potential therapeutic benefit of CETP inhibitors await further study. However, the experience with torcetrapib has fueled the interest in better assessment of HDL functionality to complement measures of HDL mass.
Determined simply as the amount of cholesterol in HDL particles per 100 ml of plasma, HDL-C suffers from limitations intrinsic to its static, mass-based measurement. First, as a snapshot of the steady-state cholesterol pool, HDL-C does not directly assess the rate of centripetal cholesterol flux from peripheral foam cells to the liver, which is influenced by many factors beyond the mass of HDL-C alone. Second, circulating HDL-C values fail to convey information regarding the antiinflammatory, antioxidant, antithrombotic, and endothelial function promoting benefits of HDL, despite mounting evidence supporting the clinical significance of these pleiotropic functions. Importantly, the limitations of cholesterol as a proxy for lipoprotein atherogenicity extend to LDL-C as well (16). By acknowledging particle heterogeneity, measures of apolipoprotein B (17), small dense low-density lipoprotein (LDL) (18), and oxidized LDL (19) provide additional prognostic information above and beyond LDL-C. Newer HDL assays address the limitations of HDL-C in an attempt to provide a more accurate assessment of HDL functionality in the setting of pharmacotherapy.
Cholesterol Efflux and Reverse Cholesterol Transport
Promotion of cholesterol efflux from macrophages and its return to the liver, bile, and feces, completing the pathway of “reverse cholesterol transport” (RCT), is thought to be one of the most important mechanisms by which HDL protects against atherosclerosis (Fig. 1). Evaluating the flux of cholesterol (the rate and magnitude of intercompartmental shifts) provides a dynamic measure of RCT effectiveness and, potentially, a more informative way of assessing the efficacy of a novel HDL-targeted intervention.
Figure 1. Reverse Cholesterol Transport.
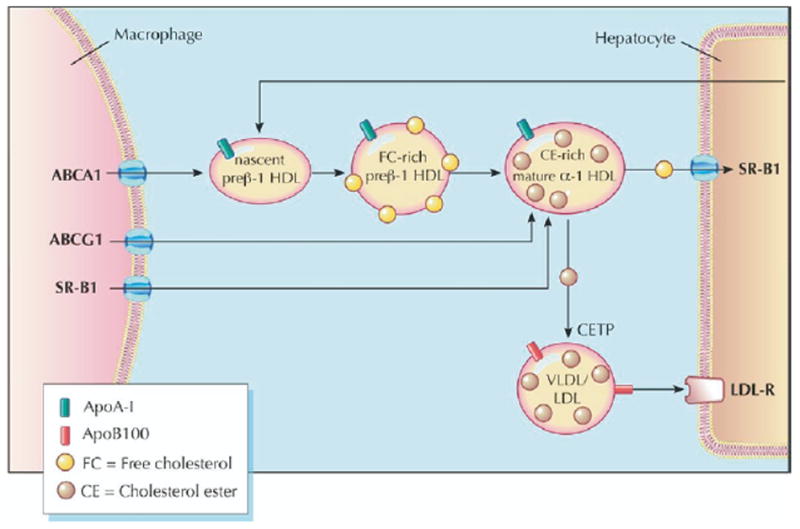
High-density lipoprotein (HDL) cholesterol promotes and facilitates the process of reverse cholesterol transport (RCT), whereby excess macrophage cholesterol is effluxed to HDL and ultimately returned to the liver for excretion. Efflux to nascent and mature HDL occurs via the transporters ABCA1 and ABCG1, respectively. The HDL cholesterol is returned to the liver via the hepatic receptor SR-BI or by transfer to apolipoprotein (apo) B–containing lipoproteins by the action of cholesteryl ester transfer protein (2). Figure illustrations by Rob Flewell. CETP = cholesterol ester transfer protein; LDL = low-density lipoprotein; LDL-R = low-density lipoprotein receptor; SR-BI = scavenger receptor class B-type I; VLDL = very low-density lipoprotein.
Cellular cholesterol efflux ex vivo
To assess cellular efflux, donor cells, such as hepatoma cells, fibroblasts, or macrophages, are first incubated with 3H-cholesterol (20,21). Incubation with a medium containing an “acceptor” (lipid-free apoA-I, isolated HDL, or diluted human serum) is carried out, and after multiple washings, scintigraphy quantifies the radioactivity in the medium and associated with the cells. Cholesterol efflux is then expressed as the amount of label released into the medium divided by the total label present. Acceptors in the medium and donor cells can be manipulated to examine the effects of genetic and pharmacologic manipulation on efflux potential. Manipulation of donor cells may augment cholesterol efflux. Mouse peritoneal macrophages overexpressing ABCA1 efflux cholesterol to apoA-I faster than do wild-type control cells, an effect associated with smaller, less complex aortic valvular atherosclerotic lesions in transgenic ABCA1 mice (22). In humans, investigation of families with ABCA1 mutations demonstrated an inverse correlation between cholesterol efflux from primary fibroblasts and carotid intima-media thickness (23). Administration of a liver X receptor agonist to macrophages results in increased cholesterol efflux, at least in part through the ABCA1 pathway (24,25). However, the most applicable approach of ex vivo efflux assays to drug development would be to use whole serum or isolated HDL from subjects treated with an experimental drug and test its ability to promote cholesterol efflux from a defined cell system ex vivo.
Pre-clinical studies support the concept that augmenting the ability of serum or HDL to promote the efflux of cholesterol from cells may confer an atheroprotective benefit. Administration of recombinant apoA-I Milano or oral D-4F, a small apoA-I mimetic peptide, significantly increased the cholesterol efflux-promoting capacity of plasma taken from treated apoE-deficient mice (26,27). These ex vivo findings were associated with decreased macrophage and lipid content of aortic atheroma, indicating enhanced mobilization of tissue cholesterol (27,28).
Interestingly, treatment with torcetrapib resulted in enhanced efflux capacity of HDL, whether normalized to volume of serum or to HDL mass (29). However, there are few data that link serum efflux capacity to atherosclerosis or cardiovascular events in humans. One relatively small prospective study of men referred for coronary angiography (30) demonstrated that efflux capacity correlated with cardiovascular outcomes and all-cause mortality during a 4.5-year follow-up. For an intervention that is expected to influence circulating HDL or whole serum in such a way as to generate greater “efflux potential,” ex vivo assessment of the cholesterol efflux capacity of isolated HDL or diluted serum may provide important information regarding the efficacy of the intervention. However, a limitation of efflux potential as a surrogate measure is its inability to assess terminal segments of the RCT pathway or effects of interventions that are directed toward the cells themselves. There is also a need to determine the different pathways that contribute to total efflux to serum and how these vary among individuals. Finally, there is a need for additional data linking serum efflux capacity to atherosclerosis progression and clinical outcomes.
Macrophage-specific RCT in vivo
To assay macrophage-specific RCT, the critical atheroprotective pathway, a recently developed technique in mice follows the fate of radiolabeled cholesterol from prepared macrophages after injection (Fig. 2A). Macrophages are cholesterol-loaded with acetylated LDL to become foam cells and labeled with 3H-cholesterol. Loaded and labeled macrophages are then injected intraperitoneally into mice, where they remain in the peritoneal cavity bathed in peritoneal fluid containing HDL acceptor particles. Subsequently, blood is sampled at several time points, feces are collected continuously over 48 h, and plasma and feces are assayed for 3H-steroid. The fecal excretion of 3H-steroid is a measure of macrophage-to-feces RCT. The initial experiments employing this technique used the murine J774 macrophage cell line to examine the effect of apoA-I overexpression achieved with a recombinant adenoviral vector. In contrast to prior studies measuring total centripetal flux (31,32), this macrophage-specific approach indicated increased macrophage RCT with apoA-I overexpression, consistent with the atheroprotective role of apoA-I (33). Compared with control subjects, mice overexpressing apoA-I demonstrated significantly higher levels of 3H-tracer in plasma, liver, and feces after 48 h (Fig. 2B). Importantly, the majority of plasma 3H-cholesterol was detected in the HDL fraction. These results suggest that 3H-cholesterol originating in cholesterol-loaded macrophages was transported through the plasma, via HDL and not migration of intact macrophages, to the liver and then ultimately excreted into the feces, providing direct support for the RCT hypothesis. Subsequent studies demonstrated that apoA-I–deficient mice, on an atherosclerotic background, had markedly reduced macrophage RCT despite similar HDL-C levels, demonstrating the importance of apoA-I in RCT and potentially explaining the increased atherosclerosis in these mice (34).
Figure 2. Macrophage-Specific Reverse Cholesterol Transport In Vivo.
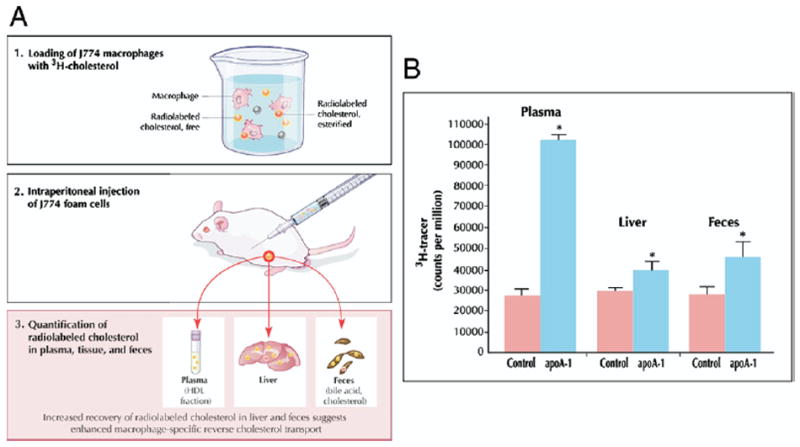
(A) To assay macrophage-specific RCT, the critical atheroprotective pathway, a recently developed technique (33) follows the fate of radiolabeled cholesterol from prepared macrophages after injection. Macrophages are cholesterol-loaded with acetylated low-density lipoprotein (LDL) to become foam cells and labeled with 3H-cholesterol. Loaded and labeled macrophages are then injected intraperitoneally into mice, where they remain in the peritoneal cavity bathed in peritoneal fluid containing HDL acceptor particles. Subsequently, blood is sampled at several time points, and feces and plasma are collected continuously over 48 h and assayed for 3H-steroid. The fecal excretion of 3H-steroid is a measure of macrophage-to-feces RCT. (B) C57BL/6 mice were injected intravenously with apoA-I adenovirus (n = 10) or control (null) adenovirus vector (n = 10). Three days after vector injection, 3H-cholesterol-labeled J774 foam cells were injected. Compared with control subjects, mice overexpressing apoA-I demonstrated significantly higher levels of 3H-tracer in plasma at 48 h and in liver and feces. *p < 0.05 between control and apoA-I adenovirus groups. Reprinted, with permission, from Zhang et al. (33). Figure illustrations by Rob Flewell. Abbreviations as in Figure 1.
Additional studies have assessed macrophage-specific RCT in the setting of pharmacologic and genetic manipulation of the host animal. Pharmacologic activation of liver X receptor, which up-regulates the cellular efflux transporters ABCA1 and ABCG1, was shown to promote macrophage RCT (35). Overexpression of the hepatic SR-BI, a receptor mediating selective uptake of cholesterol from HDL enhanced macrophage-specific RCT despite reduced HDL-C levels, whereas hepatic SR-BI deficiency was associated with markedly impaired macrophage RCT in the setting of elevated HDL-C levels (36). ABCA1-knockout mice demonstrated significantly impaired macrophage-specific RCT, manifesting 2- to 3-fold reductions in 3H-tracer in collected feces and liver (37). This approach has also been adapted to use primary mouse peritoneal and bone marrow–derived macrophages, enabling the use of macrophages from knockout mice to test the role of macrophage-specific genes. For example, macrophages deficient in ABCA1 demonstrated impaired RCT in vivo (24,38). Furthermore, increased macrophage expression of ABCG1, a mediator of efflux to mature HDL in vitro, significantly promoted macrophage RCT in vivo, whereas knockdown and knockout of macrophage ABCG1 impaired macrophage RCT in vivo (24). Manipulating macrophage SR-BI expression, on the other hand, did not affect macrophage-specific RCT, highlighting the varied role of lipid transporters across different cell types (24).
Recent experiments examining the effect of CETP in mice, which naturally lack this enzyme, offer new insights into the contrasting roles of CETP in RCT (39). In the apobec-1-null mouse model, which exhibits apoB-lipoprotein metabolism similar to humans, CETP expression achieved via hepatic transduction significantly increased tracer excretion in feces while effecting a substantial reduction in HDL-C. On the other hand, apobec-1/LDL-receptor double-knockout mice failed to exhibit enhanced macrophage-specific RCT in the setting of CETP expression, indicating that the effects of CETP on RCT depend on the efficiency of downstream cholesteryl ester clearance by apoB-lipoprotein pathways. These results suggest that in animals or humans with highly effective LDL clearance, CETP may enhance RCT and serve an atheroprotective role, but when LDL clearance is impaired, CETP may be proatherogenic. Regardless of the ultimate fate of CETP inhibitors, the ability of the macrophage-specific assay to detect changes in RCT arising from perturbations in the metabolic milieu supports the assay’s utility as both a tool for studying RCT pathways and as a viable pre-clinical surrogate for HDL functionality. Macrophage-specific assays have been useful in mice; however, their applicability to humans remains uncertain. Thus, methods for assessing integrated RCT in humans are still needed.
Centripetal cholesterol flux estimated from peripheral synthesis and uptake
A technique to measure net centripetal movement of cholesterol from the periphery in animals was developed on the assumption that peripheral cholesterol egress is equal to peripheral cholesterol acquisition via de novo synthesis and LDL-C uptake (40,41). Dual radioisotope techniques employ labeled cholesterol precursors and LDL-C, administered orally or intravenously, to monitor cholesterol synthesis in and LDL-C transport to extrahepatic organs. Provided no net change occurs within peripheral cholesterol pools, the sum of these 2 input contributions approximates the rate of egress of cholesterol from the periphery. Conclusions drawn from this assay rely on the premise that cholesterol returning to the liver necessarily represents a positive outcome. Increased hepatic delivery may not be atheroprotective, however, if hepatocytes fail to eliminate cholesterol in feces and instead reroute lipid into plasma as LDL and very LDL (42). It is also possible that macrophage cholesterol represents a small compartment sensitive to factors that do not have major effects on the overall transport of cholesterol from the periphery to the liver. Unique attributes of the macrophage cholesterol pool may be the explanation for the failure of this method to detect a difference in centripetal cholesterol flux in mice with markedly different levels of HDL-C and apoA-I (31,32,40,41).
Mass fecal steroid excretion
Measurement of fecal cholesterol excretion, the final common pathway of centripetal flow, theoretically could provide an in vivo proxy of net RCT effectiveness suitable for both pre-clinical and clinical studies. Cholesterol is eliminated via bile into the feces either through direct secretion or following conversion to bile acids in the liver. Quantifying both neutral and acidic steroid fractions in collected fecal samples using gas-liquid chromatography, followed by correction for variability in fecal flow with simultaneous measurement of a nonabsorbable sterol, may yield a measure of total centripetal flux (43). Interventions that augment fecal cholesterol content, by inference, stimulate RCT and may confer an atheroprotective benefit. In support of this surrogate, infusion of full-length human apoA-I not only increased the acceptor particle prebeta HDL (44) but also increased fecal steroid excretion when administered in human studies (45). On the other hand, incongruent results from knockout mice highlight concerns regarding assessment of total centripetal flux via fecal steroid excretion. Although numerous experiments firmly establish the role of ABCA1 as a pivotal mediator of cholesterol efflux, fecal steroid excretion was normal in ABCA1 knockout mice (31,32). Inadequate sensitivity may limit interpretation of fecal steroid excretion as a surrogate for RCT. A critical distinction exists between total centripetal flow and macrophage-specific RCT. Given the minute contribution of atheroma to total exchangeable cholesterol body stores, quantifying fecal cholesterol content may fail to detect even significant changes in cholesterol traffic from macrophage foam cells to hepatocytes (46). Furthermore, multiple factors contribute to fecal steroid excretion, and a drug that promotes RCT may induce counter-regulatory factors that limit fecal steroid excretion in steady state. Thus, although fecal steroid excretion might be useful in an acute intervention such as apoA-I infusion, it is less likely to be useful in a chronic intervention intended to promote RCT.
Kinetic modeling of cholesterol efflux from tissues in vivo by isotope dilution
A recently developed approach (47,48) may permit in vivo assessment of individual components of intact RCT in humans. The steady-state isotope dilution principle, a method initially applied to the study of amino acid metabolism, provides the conceptual foundation. In brief, the kinetic model considers 2 distinct cholesterol compartments: a slowly exchanging extrahepatic tissue cholesterol pool and a rapidly exchanging plasma-hepatic cholesterol pool (Fig. 3). The flow of cholesterol from tissue to plasma, which encompasses the first step in RCT, can be assessed in vivo by noting the dilution of exogenous labeled cholesterol infused directly into the plasma compartment until steady-state levels are attained. The tracer abundance in plasma is diluted by the efflux of endogenous (unlabeled) cholesterol from tissues into plasma. At the isotopic plateau, the rate of appearance of endogenous cholesterol, representing tissue cholesterol efflux, equals the rate of tracer infusion divided by the plasma molar percent enrichment with labeled cholesterol.
Figure 3. Kinetic Modeling of Cholesterol Efflux From Tissues In Vivo by Isotope Dilution.
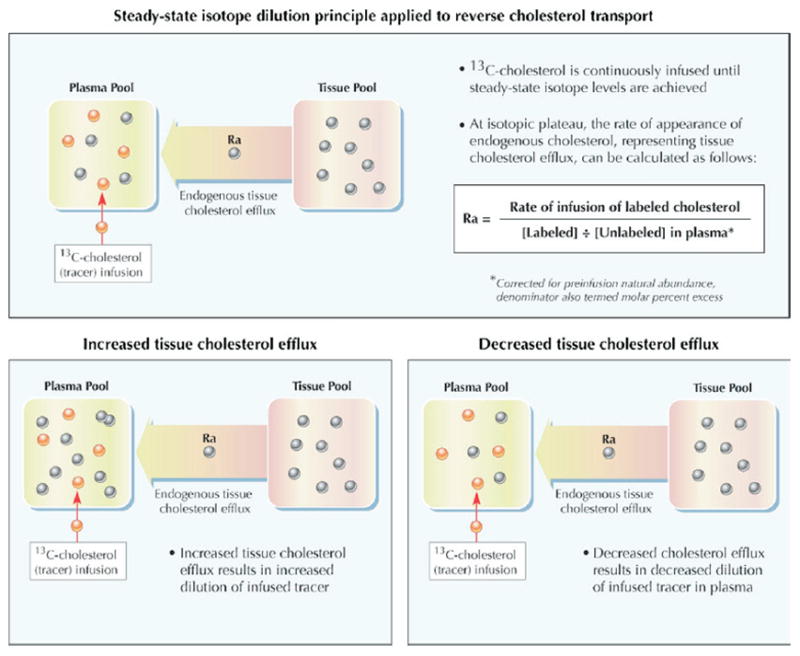
The flow of cholesterol from tissue to plasma, which encompasses the first step in RCT, can be assessed in vivo by noting the dilution of exogenous labeled cholesterol infused directly into the plasma compartment until steady-state levels are attained (47,48). The tracer abundance in plasma is diluted by the efflux of endogenous (unlabeled) cholesterol from tissues into plasma. At the isotopic plateau, the rate of appearance of endogenous cholesterol, representing tissue cholesterol efflux, equals the rate of tracer infusion divided by the plasma molar percent enrichment with labeled cholesterol. Figure illustrations by Rob Flewell. Abbreviations as in Figure 1.
Presentation of preliminary human studies using 13C-cholesterol demonstrated feasibility, revealing that an 18-h infusion period was required to achieve plateau tracer levels and reproducible measurements of tissue cholesterol efflux (48). Although this method requires validation and has not yet been published in a peer-reviewed journal, it may have utility in assessing RCT following pharmacologic intervention. However, its ability to specifically assess peripheral tissue cholesterol efflux depends on successful equilibration of the infused tracer with hepatic cholesterol, an issue difficult to prove in humans. Otherwise, efflux of unlabeled cholesterol from the liver could contribute to the dilution of the tracer in plasma. Furthermore, this method measures total peripheral efflux rather than the small pool from macrophages. Whether a consistent relationship exists between total tissue cholesterol flux and macrophage-specific RCT remains to be determined.
Indexes of Anti-Inflammatory and Antioxidant Activity
High-density lipoprotein cholesterol exerts a number of potentially antiatherogenic effects independent of cholesterol efflux and centripetal transport, including inhibiting lipid oxidation, impairing leukocyte adhesion and monocyte activation, promoting nitric oxide (NO) production and flow-induced vasodilation, preventing endothelial cell damage and death, and inhibiting activation of platelets and the coagulation cascade (Fig. 4) (49,50). However, the clinical significance of these varied functions remains unclear. The anti-inflammatory activities of HDL have generated the most excitement, prompted by conclusive evidence implicating LDL modification and monocyte migration, 2 interrelated processes, in the critical first steps of atherogenesis.
Figure 4. HDL Functions Other Than Reverse Cholesterol Transport.
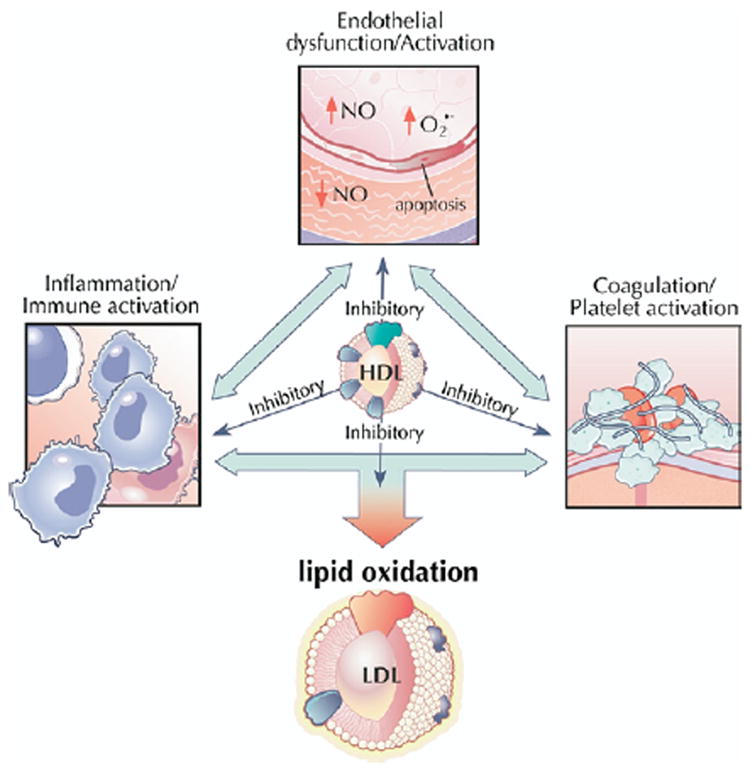
High-density lipoprotein exerts a number of potentially antiatherogenic effects independent of cholesterol efflux and centripetal transport, including inhibiting lipid oxidation, impairing leukocyte adhesion and monocyte activation, promoting nitric oxide (NO) production and flow-induced vasodilation, preventing endothelial cell damage and death, and inhibiting activation of platelets and the coagulation cascade. The clinical significance of these varied functions remains unclear. Figure illustrations by Rob Flewell. Abbreviations as in Figures 1 and 2.
Endothelial adhesion molecule expression
To gain entry into the subendothelial space, monocytes must first arrest their passage through the circulation. Adhesion molecules presented on the luminal surface of endothelial cells slow neighboring monocytes, first via loose tethering mediated by E-selectin and then by tight association of endothelial intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 with leukocyte integrins (51,52). Expression of adhesion molecules represents a dynamic process, activated by inflammatory cytokines (53) and vascular injury (54) and up-regulated in atherosclerosis in both animals (55) and humans (56,57). As data implicate increased endothelial expression of adhesion molecules in atherosclerotic disease (58), in vitro assays have been developed to evaluate the effect of HDL on this particular facet of endothelial activation (59). In one model, endothelial cells are incubated with test HDL, followed by stimulation with a proinflammatory agent such as tumor necrosis factor (TNF)-alpha. Labeled antibodies specific for each adhesion molecule of interest are then added and flow cytometry analysis performed to determine levels of expression. The original experiment using human umbilical endothelial cells (HUVECs) demonstrated the ability of HDL obtained from healthy donors to inhibit cytokine-mediated protein expression of VCAM-1, ICAM-1, and E-selectin (59). Subsequent in vitro studies have established the ability of native and reconstituted HDL to down-regulate inducible expression of adhesion molecules (60-62), with a few exceptions (63). Reasons for these conflicting results remain unclear; however, the majority of in vitro data support a role of HDL in the inhibition of endothelial activation.
In vivo pre-clinical studies of endothelial expression of adhesion molecules may provide additional methods for evaluating HDL-directed therapeutics. Following carotid injury by surgical placement of a periadvential cuff, HDL infusion decreased local ICAM- and VCAM-1 expression in both rabbits (64) and mice (65). Suppression of adhesion molecules was associated with decreased leukocyte infiltration (64) and neointimal hyperplasia (65), recognized steps in atherogenesis. Importantly, suppression of endothelial expression of adhesion molecules in vivo, which has been associated with decreased atherosclerosis (66,67), appears to correlate with in vitro findings. Reconstituted HDL inhibited expression of interleukin-1–induced E-selectin both in vitro by porcine aortic endothelial cells and in vivo by subcutaneous microvascular endothelial cells, as measured by radiolabeled antibodies specific for E-selectin (68). Administration of the apoA-I mimetic peptide L-4F, associated with reduced atheroma formation in dyslipidemic mouse models prone to atherosclerosis (69), reduced HUVEC expression of VCAM-1 in a dose-dependent fashion (70). In a clinical study, consumption of saturated fat, a known atherogenic diet (71), significantly impaired the ability of HDL to suppress TNF-alpha–induced expression of ICAM- and VCAM-1 on HUVECs (72). Conversely, polyunsaturated fat, which population studies suggest may offer protection against atherosclerosis (71,73), enhanced HDL-mediated endothelial inhibition. However, the reliability and reproducibility of the in vitro assay of inhibition of endothelial adhesion molecule expression as a surrogate measure of the antiatherogenic functionality of HDL remains to be established, and standardized and validated assays are needed.
Monocyte chemotaxis
To characterize the anti-inflammatory or proinflammatory potential of HDL, a cell-based assay measures the effect of HDL on monocyte chemotactic activity, the propensity of monocytes to migrate into the subendothelial space (74). This coculture model provides a simulated arterial wall consisting of a confluent human aortic endothelial cell monolayer and smooth muscle cell multilayer separated by a layer of collagen. The endothelial cell monolayer is first incubated with standard control LDL or oxidized phospholipids, stimulants of monocyte chemotaxis, in the presence or absence of test HDL. Human peripheral blood monocytes are subsequently added to the endothelial side of the coculture and visualized by the addition of antimonocyte antibodies. Finally, monocytes that migrated into the subendothelial space are counted. Results obtained from coculture incubation with test HDL can be divided by those following addition of control LDL alone to yield an “HDL inflammatory index,” with values above 1 indicating proinflammatory HDL and values below 1 denoting anti-inflammatory HDL (75).
Studies of pharmacologic intervention and genetic manipulation in mouse models support the utility of indexes of monocyte migration. In LDL receptor and apoE knockout mice, orally administered D-4F, an apoA-I mimetic peptide, significantly decreased both monocyte migration and atherosclerotic burden (76,77). A peptide derived from apoJ, an inhibitor of LDL oxidation and the terminal complement cascade, yielded similar outcomes in mouse models and provided additional support to an association between impaired monocyte chemotactic activity and reduced atherosclerosis (78). Absence of phospholipid transfer protein attenuates the proatherogenic effect of LDL receptor deficiency in mice in part through enhancing the ability of the HDL to reduce monocyte chemotaxis (79).
An interesting human study (75) compared HDL inflammatory indexes in healthy control subjects and patients with coronary artery disease (CAD) before and after statin therapy. High-density lipoprotein cholesterol taken from age- and gender-matched healthy control patients decreased monocyte chemotaxis, yielding a mean inflammatory index of 0.38. Patients with CAD, however, exhibited proinflammatory HDL augmenting monocyte transmigration more than control LDL alone, with a mean index of 1.38. Administration of simvastatin significantly reduced the inflammatory index to 1.08, suggesting improvement of HDL anti-inflammatory function with statin therapy. In addition, the study evaluated monocyte chemotaxis in CAD patients with elevated HDL-C levels (mean 95 mg/dl) in an attempt to explain the apparent discrepancy. The HDL obtained from this group was pro-inflammatory with an index of 1.28, highlighting the limitations of HDL-C alone and the importance of functional metrics. Although this assay is conceptually attractive and has been validated in several settings, it may be cumbersome to use as a routine assay for assessing HDL anti-inflammatory function.
Antioxidant activity
The interplay between LDL oxidation and atherogenesis may provide additional opportunities for functional metrics of HDL. Oxidized LDL has a variety of proatherogenic effects (80). High-density lipoprotein cholesterol has been shown to inhibit LDL oxidation in vitro (81,82). The HDL-associated enzymes paraoxonase and glutathione selenoperoxidase have been suggested to play a role in this activity of HDL (83). As with its ability to impede monocyte chemotaxis, the antioxidant functionality of HDL is dynamic and can be enhanced or diminished by altering the metabolic milieu (84).
One recently developed assay (85) measures integrated HDL antioxidant potential, using physiologic phospholipids known to participate in LDL modification (Fig. 5A). In the presence of dichlorofluorescein, a fluorescent marker of lipid oxidation products, HDL is added to various phospholipids found in oxidized LDL to determine its ability to inactivate or prevent the formation of biologically active oxidized phospholipids. After the reagents are combined, spectroscopy permits quantification of net oxidation, with diminished fluorescence intensity signaling fewer oxidized phospholipids and suggesting a more antiatherogenic HDL. Evidence for an inverse association between atherosclerosis and net antioxidant activity determined by this assay comes from several studies. High-density lipoprotein from CAD patients yields significantly higher levels of oxidation products compared with that of healthy control patients (85). As with indexes of monocyte chemotaxis, treatment with simvastatin attenuates the oxidation activity of HDL in CAD patients but fails to restore its antioxidant ability (Fig. 5B) (75). Similarly, studies of oral D-4F as well as of an apoJ peptide in mice (76-78) reveal concordance between oxidation and monocyte migration and a negative association between these surrogates and atherosclerosis. This relatively simple cell-free assay is an interesting candidate for validation and application to high-throughput assessment of HDL antioxidant function in response to therapeutic intervention.
Figure 5. Lipid Oxidation.
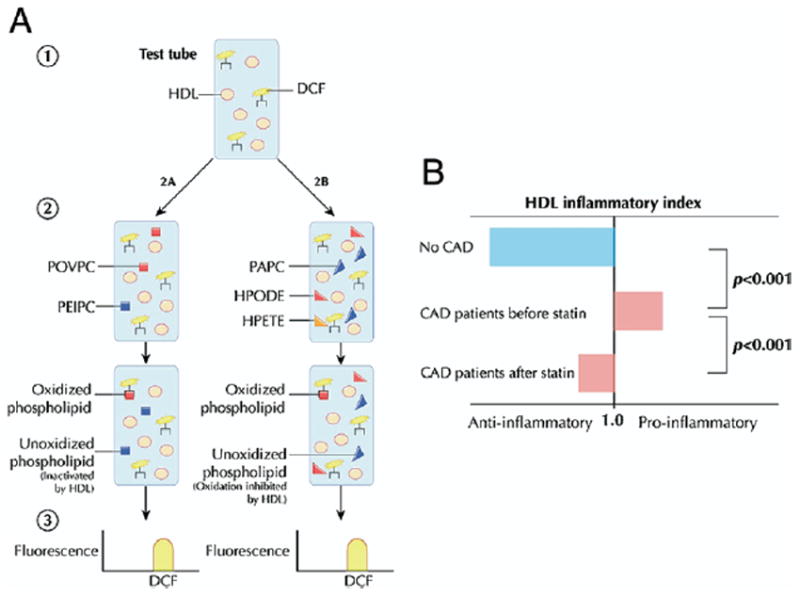
(A) Dichlorofluorescein (DCF), a fluorescent marker of lipid oxidation products, is added to test HDL isolated from patient serum (85). Oxidized phospholipids found in mildly oxidized HDL, such as 1-palmitoyl-2(5-oxovaleroyl)-sn-glycero-3-phosphorylcholine (POVPC) and 1-palmitoyl-2(5,6-epoxyisoprostane E2)-sn-glycero-3-phosphorylcholine (PEIPC), are added to assess the ability of HDL to inactivate biologically active phospholipids. To assess the ability of HDL to prevent phospholipid oxidation, HDL is added to a mixture of 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine (PAPC) from unoxidized LDL and the endothelium-derived oxidants hydroperoxyoctadecadienoic acid (HPODE) or hydroperoxyeicosatetraenoic acid (HPETE). After the reagents are combined, spectroscopy permits quantification of net oxidation, with diminished fluorescence intensity signaling fewer oxidized phospholipids and suggesting a more antiatherogenic HDL. (B) The “inflammatory index” is derived by dividing net antioxidant activity in the presence of HDL by that observed in the absence of HDL. The HDL obtained from coronary artery disease (CAD) patients exhibits an impaired ability to antagonize monocyte chemotaxis and lipid oxidation compared with control subjects (75). The atheroprotective effect of HDL is partially restored following statin therapy. Figure illustrations by Rob Flewell. Abbreviations as in Figures 1 and 2.
NO Production and Endothelial Function
Quiescent endothelium produces NO, which acts to silence cellular pathways of inflammation, proliferation, and thrombosis. Activated endothelial cells, on the other hand, produce reactive oxygen species, such as superoxide ( ), which promote atheroma formation via leukocyte migration and infiltration, neointimal hyperplasia, and platelet aggregation (86). High-density lipoprotein cholesterol has been shown to promote endothelial generation of NO in vitro and improve endothelial function and arterial vasoreactivity in vivo (50), providing another potential antiatherogenic mechanism and a basis for assessing the effects of HDL-targeted therapies.
Studies (50,87) have clearly demonstrated the ability of HDL added to endothelial cells in culture to significantly enhance NOS activity in a manner that is dependent on SR-BI. Assaying endothelial NO production in response to HDL could provide the basis of an in vitro proxy of endothelial function and permit assessment of this potentially important function of HDL. In principle, basal endothelial production of nitrite and nitrate is generated by incubating endothelial cells with L-arginine, the substrate of nitric oxide synthase (NOS), and stimulated production is achieved through simultaneous addition of an NOS agonist (Fig. 6A). The NO synthase activity may be determined by measuring NOS conversion of [3H]L-arginine to [3H]L-citrulline via scintillation counting (88) or by quantifying production of nitrite and nitrate via ozone chemiluminescence (89). To stimulate endothelial production of the oxygen free radical , endothelial cells are incubated with a NOS inhibitor, thereby inhibiting free-radical scavenging capacity, and the concentration of is measured (90). Using this cell-based assay, enhanced NOS activity, NO production, and diminished generation achieved by the test agent suggest restorative effects on endothelium and thus an antiatherogenic benefit.
Figure 6. Endothelial Cell NO Generation and Vasodilation.
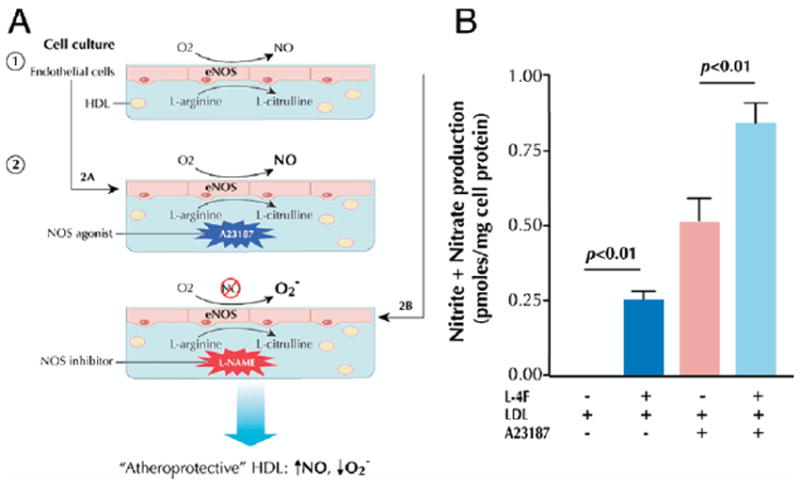
(A) Basal endothelial production of nitrite and nitrate is generated by incubating endothelial cells with L-arginine, the substrate of nitric oxide synthase (NOS), in the presence of test serum/HDL (50,88-90). Stimulated production is achieved through simultaneous addition of an NOS agonist, such as A23187. To stimulate endothelial production of free radicals, endothelial cells are incubated with an NOS inhibitor, such as L-nitroarginine-methylester (L-NAME), thereby inhibiting free-radical scavenging capacity, and the concentration of is measured. Using this cell-based assay, enhanced NOS activity, nitric oxide (NO) production, and diminished generation achieved by the test serum/HDL suggest restorative effects on endothelium and thus an antiatherogenic benefit. (B) The apoA-I mimetic L-4F restores NO production in cultured endothelial cells coincubated with LDL in the presence or absence of the NOS agonist A23187 (90). Reprinted, with permission, from Ou et al. (90). Figure illustrations by Rob Flewell. eNOS = endothelial nitric oxide synthase; other abbreviations as in Figures 1 and 2.
This approach has been used to assess the efficacy of HDL-based interventions. The apoA-I mimetic L-4F increased endothelial cell NO generation under basal and stimulated conditions and attenuated LDL-induced increases in endothelial cell generation (Fig. 6B) (90). A subsequent study (91) correlated these in vitro findings with an improved vasodilatory response to acetylcholine in hypercholesterolemic, LDL receptor-null mice. Administration of the apoA-I mimetic oral D-4F in the same mouse model restored NO and balance in the presence of the proatherogenic oxysterol 22(R)-hydroxycholesterol, which was again shown to reflect enhanced vasodilatory activity (91).
Vasoreactivity is the most common method of assessing endothelial function in vivo and is usually studied in humans using ultrasound imaging of the brachial artery at baseline and during reactive hyperemia (92). Longitudinal images are initially obtained after a 10-min equilibration period with patients in a fully recumbent position. A blood-pressure cuff is then placed proximal to the transducer on the upper arm and inflated to suprasystolic pressure. After 5 min of occlusion, the blood-pressure cuff is deflated and the brachial artery scanned continuously for 60 s. Finally, flow-mediated dilation (FMD) is calculated as the maximum percent increase in brachial artery diameter after 1 min of reactive hyperemia. In observational studies of patients with established CAD (93,94), patients with atherosclerotic risk factors (95,96), and healthy subjects (97), the extent of FMD has consistently correlated with HDL-C levels. Measurement of brachial artery vasodilatation has been used to evaluate various antidyslipidemic therapies (98). Studies of HDL-directed therapies have utilized FMD as a proxy of endothelial protection. In hypercholesterolemic patients, reconstituted HDL augmented both FMD assessed by ultrasound and acetylcholine-induced increases in forearm blood flow, another measure of vasoreactivity (99). Treatment with niacin augmented FMD among dyslipidemic patients with CAD compared with control subjects (92,100).
Although FMD has been suggested to have prognostic value (101), change in FMD following pharmacotherapy has never been correlated with cardiovascular outcomes. Widespread application of FMD as a surrogate measure in drug development, including HDL-targeted therapies, will require further validation in intervention studies to ensure its clinical relevance following pharmacotherapy.
The complexity of endothelial pathophysiology in atherosclerosis necessitates additional measures of endothelial activities and function. Developments in the vascular laboratory promise a rich array of techniques for the assessment of vasoreactivity in large conduit vessels and the microvasculature, including pulse-wave analysis, contour analysis, and amplitude tonometry. Regarding in vitro assays, evaluating upstream effects of isolated HDL after an intervention on NOS activation and localization in caveolae, specialized plasma membrane microdomains containing a variety of signal transduction molecules, may also prove informative (102). Finally, apart from preserving NO and balance, HDL exerts other putatively atheroprotective effects on endothelial cells, promoting their proliferation and migration (103) and inhibiting their apoptosis (104). Ongoing studies will further elucidate techniques to assess endothelial effects mediated by HDL and clarify their utility as reliable biomarkers in the setting of pharmacotherapy.
Metrics of Antiplatelet and Antithrombotic Activity
High-density lipoprotein cholesterol may provide additional cardiovascular benefits by antagonizing platelet activity and inactivating the coagulation cascade, both through direct inhibition and via restoration of endothelial function (50). Early in vitro studies (105-108) demonstrated the ability of purified or reconstituted HDL to inhibit serotonin release and aggregation of platelet preparations, contrary to the prothrombotic effect of LDL. A recent experiment (109) recapitulated these findings in a novel setting, showing that HDL adsorbed onto synthetic polymeric surfaces inhibited thrombin generation and formation at the blood-biomaterial interface. More limited animal and human data exist regarding the role of HDL in thrombosis. Employing a rat model of aortic thrombosis, one pre-clinical trial (110) demonstrated that administration of apoA-I Milano significantly delayed time to thrombus formation, inhibited platelet aggregation, and reduced the weight of thrombus produced. In a human study of hyperlipidemic and normolipidemic patients without significant coronary risk factors (111), high HDL-C levels were associated with reduced thrombus in an ex vivo model. Clot formation, evaluated by exposing porcine aortic intima to flowing, nonanticoagulated venous blood from test subjects, varied inversely to HDL-C in univariate and multivariable analyses. Human studies (112) have demonstrated a significant association between HDL deficiency and venous thromboembolism.
Within the intricate cascade of hemostasis and thrombosis, a multiplicity of interacting factors complicates determination and prioritization of clinically relevant, HDL-specific pathways. Studies thus far (113,114) implicate the serine protease protein C, a major physiologic anticoagulant that inactivates factors Va and VIIIa in plasma. Using a modified prothrombin time clotting assay, one experiment (115) demonstrated that HDL significantly enhanced inactivation of coagulation factor Va by activated protein C and protein S, and levels of apoA-I correlated with the anticoagulant response. Further support of the importance of protein C arose from a rabbit model of atherosclerosis, in which short-term infusion of reconstituted HDL reduced aortic atheroma burden and doubled expression of thrombomodulin, a cofactor for thrombin-induced activation of protein C (116). Evidence suggests that HDL may exert antithrombotic activity via inhibition of tissue factor expression, factor X activation, and plasminogen activator inhibitor secretion as well (117-119).
Surrogate measures of HDL antiplatelet and antithrombotic functionality remain underdeveloped, in large part because of the absence of consensus regarding the clinically relevant atheroprotective pathways, and none have been consistently employed or correlated with plaque burden or morphology. It is important to note the accumulating evidence implicating platelet activation and thrombin generation in the chronic progression of atheroma (120-123), as well as the final, catastrophic event of acute coronary syndrome and other atherothrombotic diseases. These data support a critical role of platelets and the coagulation cascade in the ongoing progression of atherosclerosis and highlight the importance of elucidating HDL-mediated pathways and developing reliable metrics of antiplatelet and antithrombotic activity.
Conclusions
Changes in circulating HDL-C levels in response to HDL-targeted therapeutic interventions represent an inadequate measure of impact on atherosclerotic risk, and better measures of HDL function are needed. Change in the cholesterol efflux capacity of serum or HDL ex vivo or cholesterol flux from tissues in vivo following pharmacotherapeutic interventions may provide one approach to assessing the RCT-promoting effects of a new therapy. Indexes of inflammation, oxidation, monocyte chemotaxis, NO production, endothelial function, and thrombosis reveal other dimensions of HDL functionality. Robust reproducible assays that can be performed easily are needed to move this field forward. Several clinical and animal studies provide hope for a new generation of metrics of HDL functionality. The hope is that detailed compositional assessment of HDL, including both lipid and protein components, coupled with sophisticated measures of HDL function, may yield mass-based biomarkers that can serve as proxies for function and that can then be applied with much greater ease and precision to large populations and clinical trials. Additional data are required to corroborate preliminary associations among novel HDL assays, mass-based proxies of function, and atherosclerotic risk to firmly establish reliable surrogate measures and ultimately facilitate comprehensive assessment of HDL-directed drugs in development.
Acknowledgments
Dr. Rader is supported by grants from the National Heart, Lung, and Blood Institute (HL22633, HL70128) and is a recipient of the Doris Duke Charitable Foundation Distinguished Clinical Scientist Award. Dr. Rader has had research support from and consulted for Abbott Laboratories, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Co., Merck, Merck/Schering-Plough, Novartis, Pfizer, and Wyeth on topics related to highdensity lipoprotein cholesterol. Dr. deGoma has received honoraria from Abbott Laboratories, AstraZeneca, and Merck/Schering-Plough for speaking on the management of dyslipidemia in clinical practice.
Abbreviations and Acronyms
- CAD
coronary artery disease
- CETP
cholesteryl ester transfer protein
- FMD
flow-mediated dilation
- HDL
high-density lipoprotein
- HUVEC
human umbilical endothelial cell
- ICAM
intercellular adhesion molecule
- LDL
low-density lipoprotein
- NO
nitric oxide
- NOS
nitric oxide synthase
- RCT
reverse cholesterol transport
- TNF
tumor necrosis factor
- VCAM
vascular cell adhesion molecule
References
- 1.Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090–100. doi: 10.1172/JCI30163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duffy D, Rader DJ. Emerging therapies targeting high-density lipoprotein metabolism and reverse cholesterol transport. Circulation. 2006;113:1140–50. doi: 10.1161/CIRCULATIONAHA.105.593855. [DOI] [PubMed] [Google Scholar]
- 3.Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, antioxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–92. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- 4.Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB. Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986;256:2835–8. [PubMed] [Google Scholar]
- 5.Robins SJ, Collins D, Wittes JT, et al. Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA. 2001;285:1585–91. doi: 10.1001/jama.285.12.1585. [DOI] [PubMed] [Google Scholar]
- 6.deGoma EM, Leeper NJ, Heidenreich PA. Clinical significance of high-density lipoprotein cholesterol in patients with low low-density lipoprotein cholesterol. J Am Coll Cardiol. 2008;51:49–55. doi: 10.1016/j.jacc.2007.07.086. [DOI] [PubMed] [Google Scholar]
- 7.Cuchel M, Rader DJ. Genetics of increased HDL cholesterol levels: insights into the relationship between HDL metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1710–2. doi: 10.1161/01.ATV.0000092947.15939.93. [DOI] [PubMed] [Google Scholar]
- 8.Hirano K, Yamashita S, Nakajima N, et al. Genetic cholesteryl ester transfer protein deficiency is extremely frequent in the Omagari area of Japan. Marked hyperalphalipoproteinemia caused by CETP gene mutation is not associated with longevity. Arterioscler Thromb Vasc Biol. 1997;17:1053–9. doi: 10.1161/01.atv.17.6.1053. [DOI] [PubMed] [Google Scholar]
- 9.Zhong S, Sharp DS, Grove JS, et al. Increased coronary heart disease in Japanese-American men with mutation in the cholesteryl ester transfer protein gene despite increased HDL levels. J Clin Invest. 1996;97:2917–23. doi: 10.1172/JCI118751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borggreve SE, Hillege HL, Wolffenbuttel BH, et al. An increased coronary risk is paradoxically associated with common cholesteryl ester transfer protein gene variations that relate to higher high-density lipoprotein cholesterol: a population-based study. J Clin Endocrinol Metab. 2006;91:3382–8. doi: 10.1210/jc.2005-2322. [DOI] [PubMed] [Google Scholar]
- 11.Barter PJ, Caulfield M, Eriksson M, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;357:2109–22. doi: 10.1056/NEJMoa0706628. [DOI] [PubMed] [Google Scholar]
- 12.Rader DJ. Illuminating HDL—is it still a viable therapeutic target? N Engl J Med. 2007;357:2180–3. doi: 10.1056/NEJMe0707210. [DOI] [PubMed] [Google Scholar]
- 13.Kastelein JJ, van Leuven SI, Burgess L, et al. Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007;356:1620–30. doi: 10.1056/NEJMoa071359. [DOI] [PubMed] [Google Scholar]
- 14.Nissen SE, Tardif JC, Nicholls SJ, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–16. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- 15.Krishna R, Anderson MS, Bergman AJ, et al. Effect of the cholesteryl ester transfer protein inhibitor, anacetrapib, on lipoproteins in patients with dyslipidaemia and on 24-h ambulatory blood pressure in healthy individuals: two double-blind, randomised placebo-controlled phase I studies. Lancet. 2007;370:1907–14. doi: 10.1016/S0140-6736(07)61813-3. [DOI] [PubMed] [Google Scholar]
- 16.Mudd JO, Borlaug BA, Johnston PV, et al. Beyond low-density lipoprotein cholesterol: defining the role of low-density lipoprotein heterogeneity in coronary artery disease. J Am Coll Cardiol. 2007;50:1735–41. doi: 10.1016/j.jacc.2007.07.045. [DOI] [PubMed] [Google Scholar]
- 17.Gotto AM, Jr, Whitney E, Stein EA, et al. Relation between baseline and on-treatment lipid parameters and first acute major coronary events in the Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) Circulation. 2000;101:477–84. doi: 10.1161/01.cir.101.5.477. [DOI] [PubMed] [Google Scholar]
- 18.St-Pierre AC, Cantin B, Dagenais GR, et al. Low-density lipoprotein subfractions and the long-term risk of ischemic heart disease in men: 13-year follow-up data from the Quebec Cardiovascular Study. Arterioscler Thromb Vasc Biol. 2005;25:553–9. doi: 10.1161/01.ATV.0000154144.73236.f4. [DOI] [PubMed] [Google Scholar]
- 19.Holvoet P, Harris TB, Tracy RP, et al. Association of high coronary heart disease risk status with circulating oxidized LDL in the well-functioning elderly: findings from the Health, Aging, and Body Composition study. Arterioscler Thromb Vasc Biol. 2003;23:1444–8. doi: 10.1161/01.ATV.0000080379.05071.22. [DOI] [PubMed] [Google Scholar]
- 20.de la Llera Moya M, Atger V, Paul JL, et al. A cell culture system for screening human serum for ability to promote cellular cholesterol efflux. Relations between serum components and efflux, esterification, and transfer. Arterioscler Thromb. 1994;14:1056–65. doi: 10.1161/01.atv.14.7.1056. [DOI] [PubMed] [Google Scholar]
- 21.Fielding PE, Fielding CJ, Havel RJ, Kane JP, Tun P. Cholesterol net transport, esterification, and transfer in human hyperlipidemic plasma. J Clin Invest. 1983;71:449–60. doi: 10.1172/JCI110789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singaraja RR, Fievet C, Castro G, et al. Increased ABCA1 activity protects against atherosclerosis. J Clin Invest. 2002;110:35–42. doi: 10.1172/JCI15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Dam MJ, de Groot E, Clee SM, et al. Association between increased arterial-wall thickness and impairment in ABCA1-driven cholesterol efflux: an observational study. Lancet. 2002;359:37–42. doi: 10.1016/S0140-6736(02)07277-X. [DOI] [PubMed] [Google Scholar]
- 24.Wang X, Collins HL, Ranalletta M, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest. 2007;117:2216–24. doi: 10.1172/JCI32057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yvan-Charvet L, Ranalletta M, Wang N, et al. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest. 2007;117:3900–8. doi: 10.1172/JCI33372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah PK, Kaul S, Nilsson J, Cercek B. Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteins: an idea whose time for testing is coming, part II. Circulation. 2001;104:2498–502. doi: 10.1161/hc4501.098468. [DOI] [PubMed] [Google Scholar]
- 27.Navab M, Anantharamaiah GM, Reddy ST, et al. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. 2004;109:3215–20. doi: 10.1161/01.CIR.0000134275.90823.87. [DOI] [PubMed] [Google Scholar]
- 28.Shah PK, Yano J, Reyes O, et al. High-dose recombinant apolipoprotein A-I(milano) mobilizes tissue cholesterol and rapidly reduces plaque lipid and macrophage content in apolipoprotein e-deficient mice. Potential implications for acute plaque stabilization. Circulation. 2001;103:3047–50. doi: 10.1161/hc2501.092494. [DOI] [PubMed] [Google Scholar]
- 29.Yvan-Charvet L, Matsuura F, Wang N, et al. Inhibition of cholesteryl ester transfer protein by torcetrapib modestly increases macrophage cholesterol efflux to HDL. Arterioscler Thromb Vasc Biol. 2007;27:1132–8. doi: 10.1161/ATVBAHA.106.138347. [DOI] [PubMed] [Google Scholar]
- 30.Chirinos JA, Zambrano JP, Chakko S, et al. Ability of serum to decrease cellular acylCoA:cholesterol acyl transferase activity predicts cardiovascular outcomes. Circulation. 2005;112:2446–53. doi: 10.1161/CIRCULATIONAHA.104.521815. [DOI] [PubMed] [Google Scholar]
- 31.Alam K, Meidell RS, Spady DK. Effect of up-regulating individual steps in the reverse cholesterol transport pathway on reverse cholesterol transport in normolipidemic mice. J Biol Chem. 2001;276:15641–9. doi: 10.1074/jbc.M010230200. [DOI] [PubMed] [Google Scholar]
- 32.Groen AK, Bloks VW, Bandsma RH, Ottenhoff R, Chimini G, Kuipers F. Hepatobiliary cholesterol transport is not impaired in Abca1-null mice lacking HDL. J Clin Invest. 2001;108:843–50. doi: 10.1172/JCI12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Zanotti I, Reilly MP, Glick JM, Rothblat GH, Rader DJ. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–3. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]
- 34.Moore RE, Navab M, Millar JS, et al. Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ Res. 2005;97:763–71. doi: 10.1161/01.RES.0000185320.82962.F7. [DOI] [PubMed] [Google Scholar]
- 35.Naik SU, Wang X, Da Silva JS, et al. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–7. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of SR-BI is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–4. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calpe-Berdiel L, Rotllan N, Palomer X, Ribas V, Blanco-Vaca F, Escola-Gil JC. Direct evidence in vivo of impaired macrophage-specific reverse cholesterol transport in ATP-binding cassette transporter A1-deficient mice. Biochim Biophys Acta. 2005;1738:6–9. doi: 10.1016/j.bbalip.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 38.Wang MD, Franklin V, Marcel YL. In vivo reverse cholesterol transport from macrophages lacking ABCA1 expression is impaired. Arterioscler Thromb Vasc Biol. 2007;27:1837–42. doi: 10.1161/ATVBAHA.107.146068. [DOI] [PubMed] [Google Scholar]
- 39.Tanigawa H, Billheimer JT, Tohyama J, Zhang Y, Rothblat G, Rader DJ. Expression of cholesteryl ester transfer protein in mice promotes macrophage reverse cholesterol transport. Circulation. 2007;116:1267–73. doi: 10.1161/CIRCULATIONAHA.107.704254. [DOI] [PubMed] [Google Scholar]
- 40.Jolley CD, Woollett LA, Turley SD, Dietschy JM. Centripetal cholesterol flux to the liver is dictated by events in the peripheral organs and not by the plasma high density lipoprotein or apolipoprotein A-I concentration. J Lipid Res. 1998;39:2143–9. [PubMed] [Google Scholar]
- 41.Osono Y, Woollett LA, Marotti KR, Melchior GW, Dietschy JM. Centripetal cholesterol flux from extrahepatic organs to the liver is independent of the concentration of high density lipoprotein-cholesterol in plasma. Proc Natl Acad Sci U S A. 1996;93:4114–9. doi: 10.1073/pnas.93.9.4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spady DK. Reverse cholesterol transport and atherosclerosis regression. Circulation. 1999;100:576–8. doi: 10.1161/01.cir.100.6.576. [DOI] [PubMed] [Google Scholar]
- 43.Czubayko F, Beumers B, Lammsfuss S, Lutjohann D, von Bergmann K. A simplified micro-method for quantification of fecal excretion of neutral and acidic sterols for outpatient studies in humans. J Lipid Res. 1991;32:1861–7. [PubMed] [Google Scholar]
- 44.Nanjee MN, Cooke CJ, Garvin R, et al. Intravenous apoA-I/lecithin discs increase pre-beta-HDL concentration in tissue fluid and stimulate reverse cholesterol transport in humans. J Lipid Res. 2001;42:1586–93. [PubMed] [Google Scholar]
- 45.Eriksson M, Carlson LA, Miettinen TA, Angelin B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I. Potential reverse cholesterol transport in humans. Circulation. 1999;100:594–8. doi: 10.1161/01.cir.100.6.594. [DOI] [PubMed] [Google Scholar]
- 46.Blum CB, Dell RB, Palmer RH, Ramakrishnan R, Seplowitz AH, Goodman DS. Relationship of the parameters of body cholesterol metabolism with plasma levels of HDL cholesterol and the major HDL apoproteins. J Lipid Res. 1985;26:1079–88. [PubMed] [Google Scholar]
- 47.Turner SM, Voogt J, Hellerstein MK. Effect of reconstituted HDL on cholesterol efflux from tissues in vivo. Paper presented at: Arteriosclerosis, Thrombosis, and Vascular Biology Annual Conference; April 19–21, 2007; Chicago, IL. [Google Scholar]
- 48.Voogt J, Killion S, Murphy L, Hellerstein MK, Turner SM. Measurement of cholesterol efflux and global cholesterol transport rates in vivo with stable isotopes. Paper presented at: Arteriosclerosis, Thrombosis, and Vascular Biology Annual Conference; April 19–21, 2007; Chicago, IL. [Google Scholar]
- 49.Barter PJ, Nicholls S, Rye KA, Anantharamaiah GM, Navab M, Fogelman AM. Antiinflammatory properties of HDL. Circ Res. 2004;95:764–72. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- 50.Mineo C, Deguchi H, Griffin JH, Shaul PW. Endothelial and antithrombotic actions of HDL. Circ Res. 2006;98:1352–64. doi: 10.1161/01.RES.0000225982.01988.93. [DOI] [PubMed] [Google Scholar]
- 51.Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell. 1991;65:859–73. doi: 10.1016/0092-8674(91)90393-d. [DOI] [PubMed] [Google Scholar]
- 52.Davies MJ, Gordon JL, Gearing AJ, et al. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol. 1993;171:223–9. doi: 10.1002/path.1711710311. [DOI] [PubMed] [Google Scholar]
- 53.Kume N, Cybulsky MI, Gimbrone MA., Jr Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J Clin Invest. 1992;90:1138–44. doi: 10.1172/JCI115932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krejcy K, Schwarzacher S, Ferber W, Plesch C, Cybulsky MI, Weidinger FF. Expression of VCAM-1 in rabbit iliac arteries is associated with vasodilator dysfunction of regenerated endothelium following balloon injury. Atherosclerosis. 1996;122:59–67. doi: 10.1016/0021-9150(95)05747-1. [DOI] [PubMed] [Google Scholar]
- 55.Iiyama K, Hajra L, Iiyama M, et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999;85:199–207. doi: 10.1161/01.res.85.2.199. [DOI] [PubMed] [Google Scholar]
- 56.Rohde LE, Lee RT, Rivero J, et al. Circulating cell adhesion molecules are correlated with ultrasound-based assessment of carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 1998;18:1765–70. doi: 10.1161/01.atv.18.11.1765. [DOI] [PubMed] [Google Scholar]
- 57.Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet. 1998;351:88–92. doi: 10.1016/S0140-6736(97)09032-6. [DOI] [PubMed] [Google Scholar]
- 58.Barter PJ, Baker PW, Rye KA. Effect of HDL on the expression of adhesion molecules in endothelial cells. Curr Opin Lipidol. 2002;13:285–8. doi: 10.1097/00041433-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 59.Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol. 1995;15:1987–94. doi: 10.1161/01.atv.15.11.1987. [DOI] [PubMed] [Google Scholar]
- 60.Ashby DT, Rye KA, Clay MA, Vadas MA, Gamble JR, Barter PJ. Factors influencing the ability of HDL to inhibit expression of vascular cell adhesion molecule-1 in endothelial cells. Arterioscler Thromb Vasc Biol. 1998;18:1450–5. doi: 10.1161/01.atv.18.9.1450. [DOI] [PubMed] [Google Scholar]
- 61.Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Phospholipid composition of reconstituted HDL influences their ability to inhibit endothelial cell adhesion molecule expression. J Lipid Res. 2000;41:1261–7. [PubMed] [Google Scholar]
- 62.Baker PW, Rye KA, Gamble JR, Vadas MA, Barter PJ. Ability of reconstituted HDL to inhibit cytokine-induced expression of vascular cell adhesion molecule-1 in human umbilical vein endothelial cells. J Lipid Res. 1999;40:345–53. [PubMed] [Google Scholar]
- 63.Stannard AK, Khan S, Graham A, Owen JS, Allen SP. Inability of plasma high-density lipoproteins to inhibit cell adhesion molecule expression in human coronary artery endothelial cells. Atherosclerosis. 2001;154:31–8. doi: 10.1016/s0021-9150(00)00444-5. [DOI] [PubMed] [Google Scholar]
- 64.Nicholls SJ, Dusting GJ, Cutri B, et al. Reconstituted HDL inhibits the acute pro-oxidant and proinflammatory vascular changes induced by a periarterial collar in normocholesterolemic rabbits. Circulation. 2005;111:1543–50. doi: 10.1161/01.CIR.0000159351.95399.50. [DOI] [PubMed] [Google Scholar]
- 65.Dimayuga P, Zhu J, Oguchi S, et al. Reconstituted HDL containing human apolipoprotein A-1 reduces VCAM-1 expression and neointima formation following periadventitial cuff-induced carotid injury in apoE null mice. Biochem Biophys Res Commun. 1999;264:465–8. doi: 10.1006/bbrc.1999.1278. [DOI] [PubMed] [Google Scholar]
- 66.Dansky HM, Barlow CB, Lominska C, et al. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. 2001;21:1662–7. doi: 10.1161/hq1001.096625. [DOI] [PubMed] [Google Scholar]
- 67.Oguchi S, Dimayuga P, Zhu J, et al. Monoclonal antibody against vascular cell adhesion molecule-1 inhibits neointimal formation after periadventitial carotid artery injury in genetically hypercholesterolemic mice. Arterioscler Thromb Vasc Biol. 2000;20:1729–36. doi: 10.1161/01.atv.20.7.1729. [DOI] [PubMed] [Google Scholar]
- 68.Cockerill GW, Huehns TY, Weerasinghe A, et al. Elevation of plasma high-density lipoprotein concentration reduces interleukin-1-induced expression of E-selectin in an in vivo model of acute inflammation. Circulation. 2001;103:108–12. doi: 10.1161/01.cir.103.1.108. [DOI] [PubMed] [Google Scholar]
- 69.Garber DW, Datta G, Chaddha M, et al. A new synthetic class A amphipathic peptide analogue protects mice from diet-induced atherosclerosis. J Lipid Res. 2001;42:545–52. [PubMed] [Google Scholar]
- 70.Gupta H, Dai L, Datta G, et al. Inhibition of lipopolysaccharide-induced inflammatory responses by an apolipoprotein AI mimetic peptide. Circ Res. 2005;97:236–43. doi: 10.1161/01.RES.0000176530.66400.48. [DOI] [PubMed] [Google Scholar]
- 71.Kushi LH, Lew RA, Stare FJ, et al. Diet and 20-year mortality from coronary heart disease. The Ireland-Boston Diet-Heart Study. N Engl J Med. 1985;312:811–8. doi: 10.1056/NEJM198503283121302. [DOI] [PubMed] [Google Scholar]
- 72.Nicholls SJ, Lundman P, Harmer JA, et al. Consumption of saturated fat impairs the anti-inflammatory properties of HDL and endothelial function. J Am Coll Cardiol. 2006;48:715–20. doi: 10.1016/j.jacc.2006.04.080. [DOI] [PubMed] [Google Scholar]
- 73.Rudel LL, Parks JS, Sawyer JK. Compared with dietary monounsaturated and saturated fat, polyunsaturated fat protects African green monkeys from coronary artery atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:2101–10. doi: 10.1161/01.atv.15.12.2101. [DOI] [PubMed] [Google Scholar]
- 74.Navab M, Hough GP, Stevenson LW, Drinkwater DC, Laks H, Fogelman AM. Monocyte migration into the subendothelial space of a coculture of adult human aortic endothelial and smooth muscle cells. J Clin Invest. 1988;82:1853–63. doi: 10.1172/JCI113802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ansell BJ, Navab M, Hama S, et al. Inflammatory/antiinflammatory properties of high-density lipoprotein distinguish patients from control subjects better than high-density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation. 2003;108:2751–6. doi: 10.1161/01.CIR.0000103624.14436.4B. [DOI] [PubMed] [Google Scholar]
- 76.Navab M, Anantharamaiah GM, Hama S, et al. Oral administration of an Apo A-I mimetic peptide synthesized from D-amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation. 2002;105:290–2. doi: 10.1161/hc0302.103711. [DOI] [PubMed] [Google Scholar]
- 77.Navab M, Anantharamaiah GM, Hama S, et al. D-4F and statins synergize to render HDL antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1426–32. doi: 10.1161/01.ATV.0000167412.98221.1a. [DOI] [PubMed] [Google Scholar]
- 78.Navab M, Anantharamaiah GM, Reddy ST, et al. An oral apoJ peptide renders HDL antiinflammatory in mice and monkeys and dramatically reduces atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2005;25:1932–7. doi: 10.1161/01.ATV.0000174589.70190.e2. [DOI] [PubMed] [Google Scholar]
- 79.Yan D, Navab M, Bruce C, Fogelman AM, Jiang XC. PLTP deficiency improves the anti-inflammatory properties of HDL and reduces the ability of LDL to induce monocyte chemotactic activity. J Lipid Res. 2004;45:1852–8. doi: 10.1194/jlr.M400053-JLR200. [DOI] [PubMed] [Google Scholar]
- 80.Navab M, Imes SS, Hama SY, et al. Monocyte transmigration induced by modification of low density lipoprotein in cocultures of human aortic wall cells is due to induction of monocyte chemotactic protein 1 synthesis and is abolished by high density lipoprotein. J Clin Invest. 1991;88:2039–46. doi: 10.1172/JCI115532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kunitake ST, Jarvis MR, Hamilton RL, Kane JP. Binding of transition metals by apolipoprotein A-I-containing plasma lipoproteins: inhibition of oxidation of low density lipoproteins. Proc Natl Acad Sci U S A. 1992;89:6993–7. doi: 10.1073/pnas.89.15.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parthasarathy S, Barnett J, Fong LG. High-density lipoprotein inhibits the oxidative modification of low-density lipoprotein. Biochim Biophys Acta. 1990;1044:275–83. doi: 10.1016/0005-2760(90)90314-n. [DOI] [PubMed] [Google Scholar]
- 83.Navab M, Hama SY, Anantharamaiah GM, et al. Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: steps 2 and 3. J Lipid Res. 2000;41:1495–508. [PubMed] [Google Scholar]
- 84.Van Lenten BJ, Wagner AC, Anantharamaiah GM, et al. Influenza infection promotes macrophage traffic into arteries of mice that is prevented by D-4F, an apolipoprotein A-I mimetic peptide. Circulation. 2002;106:1127–32. doi: 10.1161/01.cir.0000030182.35880.3e. [DOI] [PubMed] [Google Scholar]
- 85.Navab M, Hama SY, Hough GP, Subbanagounder G, Reddy ST, Fogelman AM. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J Lipid Res. 2001;42:1308–17. [PubMed] [Google Scholar]
- 86.Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–95. doi: 10.1161/CIRCULATIONAHA.106.652859. [DOI] [PubMed] [Google Scholar]
- 87.Yuhanna IS, Zhu Y, Cox BE, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001;7:853–7. doi: 10.1038/89986. [DOI] [PubMed] [Google Scholar]
- 88.Uittenbogaard A, Shaul PW, Yuhanna IS, Blair A, Smart EJ. High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J Biol Chem. 2000;275:11278–83. doi: 10.1074/jbc.275.15.11278. [DOI] [PubMed] [Google Scholar]
- 89.Braman RS, Hendrix SA. Nanogram nitrite and nitrate determination in environmental and biological materials by vanadium (III) reduction with chemiluminescence detection. Anal Chem. 1989;61:2715–8. doi: 10.1021/ac00199a007. [DOI] [PubMed] [Google Scholar]
- 90.Ou Z, Ou J, Ackerman AW, Oldham KT, Pritchard KA., Jr L-4F, an apolipoprotein A-1 mimetic, restores nitric oxide and superoxide anion balance in low-density lipoprotein-treated endothelial cells. Circulation. 2003;107:1520–4. doi: 10.1161/01.cir.0000061949.17174.b6. [DOI] [PubMed] [Google Scholar]
- 91.Ou J, Wang J, Xu H, et al. Effects of D-4F on vasodilation and vessel wall thickness in hypercholesterolemic LDL receptor-null and LDL receptor/apolipoprotein A-I double-knockout mice on Western diet. Circ Res. 2005;97:1190–7. doi: 10.1161/01.RES.0000190634.60042.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kuvin JT, Ramet ME, Patel AR, Pandian NG, Mendelsohn ME, Karas RH. A novel mechanism for the beneficial vascular effects of high-density lipoprotein cholesterol: enhanced vasorelaxation and increased endothelial nitric oxide synthase expression. Am Heart J. 2002;144:165–72. doi: 10.1067/mhj.2002.123145. [DOI] [PubMed] [Google Scholar]
- 93.Li XP, Zhao SP, Zhang XY, Liu L, Gao M, Zhou QC. Protective effect of high density lipoprotein on endothelium-dependent vasodilatation. Int J Cardiol. 2000;73:231–6. doi: 10.1016/s0167-5273(00)00221-7. [DOI] [PubMed] [Google Scholar]
- 94.Zhang X, Zhao SP, Li XP, Gao M, Zhou QC. Endothelium-dependent and -independent functions are impaired in patients with coronary heart disease. Atherosclerosis. 2000;149:19–24. doi: 10.1016/s0021-9150(99)00288-9. [DOI] [PubMed] [Google Scholar]
- 95.Kuvin JT, Patel AR, Sidhu M, et al. Relation between high-density lipoprotein cholesterol and peripheral vasomotor function. Am J Cardiol. 2003;92:275–9. doi: 10.1016/s0002-9149(03)00623-4. [DOI] [PubMed] [Google Scholar]
- 96.Lupattelli G, Marchesi S, Roscini AR, et al. Direct association between high-density lipoprotein cholesterol and endothelial function in hyperlipemia. Am J Cardiol. 2002;90:648–50. doi: 10.1016/s0002-9149(02)02575-4. [DOI] [PubMed] [Google Scholar]
- 97.Toikka JO, Ahotupa M, Viikari JS, et al. Constantly low HDL-cholesterol concentration relates to endothelial dysfunction and increased in vivo LDL-oxidation in healthy young men. Atherosclerosis. 1999;147:133–8. doi: 10.1016/s0021-9150(99)00186-0. [DOI] [PubMed] [Google Scholar]
- 98.Wang TD, Chen WJ, Lin JW, Cheng CC, Chen MF, Lee YT. Efficacy of fenofibrate and simvastatin on endothelial function and inflammatory markers in patients with combined hyperlipidemia: relations with baseline lipid profiles. Atherosclerosis. 2003;170:315–23. doi: 10.1016/s0021-9150(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 99.Spieker LE, Sudano I, Hurlimann D, et al. HDL restores endothelial function in hypercholesterolemic men. Circulation. 2002;105:1399–402. doi: 10.1161/01.cir.0000013424.28206.8f. [DOI] [PubMed] [Google Scholar]
- 100.Benjo AM, Maranhao RC, Coimbra SR, et al. Accumulation of chylomicron remnants and impaired vascular reactivity occur in subjects with isolated low HDL cholesterol: effects of niacin treatment. Atherosclerosis. 2006;187:116–22. doi: 10.1016/j.atherosclerosis.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 101.Fathi R, Haluska B, Isbel N, Short L, Marwick TH. The relative importance of vascular structure and function in predicting cardiovascular events. J Am Coll Cardiol. 2004;43:616–23. doi: 10.1016/j.jacc.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 102.Shaul PW. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J Physiol. 2003;547:21–33. doi: 10.1113/jphysiol.2002.031534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kimura T, Sato K, Malchinkhuu E, et al. High-density lipoprotein stimulates endothelial cell migration and survival through sphingosine 1-phosphate and its receptors. Arterioscler Thromb Vasc Biol. 2003;23:1283–8. doi: 10.1161/01.ATV.0000079011.67194.5A. [DOI] [PubMed] [Google Scholar]
- 104.Nofer JR, Levkau B, Wolinska I, et al. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem. 2001;276:34480–5. doi: 10.1074/jbc.M103782200. [DOI] [PubMed] [Google Scholar]
- 105.Desai K, Bruckdorfer KR, Hutton RA, Owen JS. Binding of apoE-rich high density lipoprotein particles by saturable sites on human blood platelets inhibits agonist-induced platelet aggregation. J Lipid Res. 1989;30:831–40. [PubMed] [Google Scholar]
- 106.Lerch PG, Spycher MO, Doran JE. Reconstituted high density lipoprotein (rHDL) modulates platelet activity in vitro and ex vivo. Thromb Haemost. 1998;80:316–20. [PubMed] [Google Scholar]
- 107.Pajkrt D, Lerch PG, van der Poll T, et al. Differential effects of reconstituted high-density lipoprotein on coagulation, fibrinolysis and platelet activation during human endotoxemia. Thromb Haemost. 1997;77:303–7. [PubMed] [Google Scholar]
- 108.Rosenson RS, Lowe GD. Effects of lipids and lipoproteins on thrombosis and rheology. Atherosclerosis. 1998;140:271–80. doi: 10.1016/s0021-9150(98)00144-0. [DOI] [PubMed] [Google Scholar]
- 109.Knetsch ML, Aldenhoff YB, Koole LH. The effect of high-density-lipoprotein on thrombus formation on and endothelial cell attachment to biomaterial surfaces. Biomaterials. 2006;27:2813–9. doi: 10.1016/j.biomaterials.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 110.Li D, Weng S, Yang B, et al. Inhibition of arterial thrombus formation by ApoA1 Milano. Arterioscler Thromb Vasc Biol. 1999;19:378–83. doi: 10.1161/01.atv.19.2.378. [DOI] [PubMed] [Google Scholar]
- 111.Naqvi TZ, Shah PK, Ivey PA, et al. Evidence that high-density lipoprotein cholesterol is an independent predictor of acute platelet-dependent thrombus formation. Am J Cardiol. 1999;84:1011–7. doi: 10.1016/s0002-9149(99)00489-0. [DOI] [PubMed] [Google Scholar]
- 112.Eichinger S, Pecheniuk NM, Hron G, et al. High-density lipoprotein and the risk of recurrent venous thromboembolism. Circulation. 2007;115:1609–14. doi: 10.1161/CIRCULATIONAHA.106.649954. [DOI] [PubMed] [Google Scholar]
- 113.Eitzman DT, Westrick RJ, Shen Y, et al. Homozygosity for factor V Leiden leads to enhanced thrombosis and atherosclerosis in mice. Circulation. 2005;111:1822–5. doi: 10.1161/01.CIR.0000160854.75779.E8. [DOI] [PubMed] [Google Scholar]
- 114.Griffin JH, Fernandez JA, Deguchi H. Plasma lipoproteins, hemostasis and thrombosis. Thromb Haemost. 2001;86:386–94. [PubMed] [Google Scholar]
- 115.Griffin JH, Kojima K, Banka CL, Curtiss LK, Fernandez JA. High-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein C. J Clin Invest. 1999;103:219–27. doi: 10.1172/JCI5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nicholls SJ, Cutri B, Worthley SG, et al. Impact of short-term administration of HDL and atorvastatin on atherosclerosis in rabbits. Arterioscler Thromb Vasc Biol. 2005;25:2416–21. doi: 10.1161/01.ATV.0000184760.95957.d6. [DOI] [PubMed] [Google Scholar]
- 117.Carson SD. Plasma high density lipoproteins inhibit the activation of coagulation factor X by factor VIIa and tissue factor. FEBS Lett. 1981;132:37–40. doi: 10.1016/0014-5793(81)80422-x. [DOI] [PubMed] [Google Scholar]
- 118.Levin EG, Miles LA, Fless GM, et al. Lipoproteins inhibit the secretion of tissue plasminogen activator from human endothelial cells. Arterioscler Thromb. 1994;14:438–42. doi: 10.1161/01.atv.14.3.438. [DOI] [PubMed] [Google Scholar]
- 119.Viswambharan H, Ming XF, Zhu S, et al. Reconstituted high-density lipoprotein inhibits thrombin-induced endothelial tissue factor expression through inhibition of RhoA and stimulation of phosphatidylinositol 3-kinase but not Akt/endothelial nitric oxide synthase. Circ Res. 2004;94:918–25. doi: 10.1161/01.RES.0000124302.20396.B7. [DOI] [PubMed] [Google Scholar]
- 120.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–94. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 121.Chesterman CN, Berndt MC. Platelet and vessel wall interaction and the genesis of atherosclerosis. Clin Haematol. 1986;15:323–53. [PubMed] [Google Scholar]
- 122.Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;9:61–7. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 123.Massberg S, Brand K, Gruner S, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. 2002;196:887–96. doi: 10.1084/jem.20012044. [DOI] [PMC free article] [PubMed] [Google Scholar]