

The circadian clock enables an organism to adapt its physiology to daily environmental changes. The master circadian pacemaker in the hypothalamus maintains phase coherence with peripheral cells, and circadian oscillators are able to temperature-compensate, functioning with a stable period over a range of temperatures. Schibler and colleagues find that peripheral clocks are extremely sensitive to even small temperature rhythms and act as systemic cues in synchronizing circadian gene expression throughout the body.
Keywords: circadian gene expression, body temperature rhythms, heat-shock factor 1 (HSF1), heat-shock factor 2 (HSF2)
Abstract
The circadian pacemaker in the suprachiasmatic nuclei (SCN) of the hypothalamus maintains phase coherence in peripheral cells through metabolic, neuronal, and humoral signaling pathways. Here, we investigated the role of daily body temperature fluctuations as possible systemic cues in the resetting of peripheral oscillators. Using precise temperature devices in conjunction with real-time monitoring of the bioluminescence produced by circadian luciferase reporter genes, we showed that simulated body temperature cycles of mice and even humans, with daily temperature differences of only 3°C and 1°C, respectively, could gradually synchronize circadian gene expression in cultured fibroblasts. The time required for establishing the new steady-state phase depended on the reporter gene, but after a few days, the expression of each gene oscillated with a precise phase relative to that of the temperature cycles. Smooth temperature oscillations with a very small amplitude could synchronize fibroblast clocks over a wide temperature range, and such temperature rhythms were also capable of entraining gene expression cycles to periods significantly longer or shorter than 24 h. As revealed by genetic loss-of-function experiments, heat-shock factor 1 (HSF1), but not HSF2, was required for the efficient synchronization of fibroblast oscillators to simulated body temperature cycles.
The circadian clock enables an organism to adapt its physiology and behavior to environmental daily changes in a proactive manner. In mammals, virtually every cell of the body contains a molecular clock. The suprachiasmatic nucleus (SCN) pacemaker in the hypothalamus, which is entrained by daily light/dark cycles, coordinates this multitude of oscillators by controlling various brain regions, which in turn regulate different physiological and behavioral outputs. In the absence of a functional SCN, physiological and behavioral rhythms are quickly lost in constant darkness, and a wide dispersion of phases of circadian gene expression is observed in peripheral cell types (Sakamoto et al. 1998; Akhtar et al. 2002; Yoo et al. 2004; Guo et al. 2006). Under standard laboratory conditions, central and peripheral oscillators are controlled mostly by the imposed light/dark regimen. However, in a more natural setting, organisms have to face additional challenging environmental variations such as daytime-dependent food availability, predator abundance, social interactions, and temperature fluctuations, which may also act as Zeitgebers (German for “time givers,” or synchronization cues).
In homeothermic animals such as mammals, body temperature is kept within a narrow range in most parts of the body in spite of large ambient temperature variations. This necessitates the function of temperature sensors, and such thermoreceptors have indeed been found deep in the body, in the skin, and in the brain (Dhaka et al. 2006; Benarroch 2007; Romanovsky 2007). An average body temperature is maintained owing to the continuous correction of the actual body temperature by thermoregulatory centers located in the preoptic anterior hypothalamus, which instigates behavioral and autonomous body temperature responses. This homeostatic regulation is achieved via feedback mechanisms controlling heat loss and production, resulting in slight variations of the temperature around an average. In addition, the central circadian clock directly acts on thermoregulatory centers in the brain: Body temperature falls during the rest phase and rises during the active phase (Rensing and Ruoff 2002; Refinetti 2010; Weinert 2010). Thus, body temperature exhibits a daily fluctuation resulting from a continuous interplay between rhythmicity and homeostasis. Although the average core body temperature is ∼37°C in most mammals, the amplitude, the wave form, and the range of the oscillations around this value differ in a species-specific fashion (Refinetti 2010).
A landmark feature of circadian oscillators, known as temperature compensation, is their ability to function with a relatively stable period over a wide range of temperatures. One would expect that the period is accelerated and slowed down at higher and lower temperatures, respectively. Yet, the cellular clocks tick at approximately the same rate at different ambient temperatures and, in mammals, even at a slightly higher pace when temperature is decreasing (Izumo et al. 2003; Tsuchiya et al. 2003; Dibner et al. 2009). In spite of the resilience of the circadian period length over physiologically occurring temperature ranges, temperature oscillations have been demonstrated to act as strong Zeitgebers in multiple species from unicellular organisms to mammals (Zimmerman et al. 1968; Francis and Sargent 1979; Underwood and Calaban 1987; Brown et al. 2002; Herzog and Huckfeldt 2003; Boothroyd et al. 2007).
Recently, Kornmann et al. (2007a) have demonstrated that in the mouse liver, many heat-shock protein (Hsp) genes are expressed in a diurnal fashion, reaching zenith and nadir values when body temperature is maximal (∼38°C) and minimal (∼35°C), respectively. The cyclic accumulation of Hsp mRNAs was observed irrespective of whether the hepatocyte circadian clocks were functional or not, suggesting that it was driven by systemic cues, perhaps body temperature oscillations, rather than the core clock circuitry. Heat-dependent Hsp gene transcription has long been known to be regulated by heat-shock transcription factors (HSFs). Of the four HSF family members, HSF1, HSF2, and HSF3 are expressed in many cell types, whereas HSF4 accumulates primarily in lens, brain, and lung tissues (Pirkkala et al. 2001; Fujimoto and Nakai 2010). Using differential display of DNA-binding proteins with liver nuclear extracts, Reinke et al. (2008) have identified HSF1, the dominant HSF, as a rhythmically active transcription factor. Thus, although total HSF1 protein and mRNA levels do not oscillate during the day, a large fraction of HSF1 is translocated into the nucleus during the nocturnal activity phase, when body temperature is elevated. Furthermore, the resetting of Per2 expression by temperature pulses in tissue explants has been shown to be blunted in the presence of KNK437, a pharmacological inhibitor of HSF members (Buhr et al. 2010), or in HSF1-deficient fibroblasts (Tamaru et al. 2011). These observations suggest a possible role of HSF members in the phase entrainment of circadian clocks by temperature.
Here we present evidence that simulated body temperature rhythms with very low amplitudes can act as Zeitgebers for the circadian oscillators operative in NIH3T3 cells and primary tail tip fibroblasts. Surprisingly, smooth temperature oscillations of only a minute amplitude could efficiently synchronize these clocks over a wide temperature range (30°C–40°C). Moreover, we performed genetic loss-of-function experiments with cultured fibroblasts to characterize the possible roles of HSF1 and/or HSF2 in linking circadian gene expression to body temperature. The detailed recording of phase-shifting kinetics in these cells demonstrated that the time span required for the synchronization of circadian gene expression to a large temperature phase change was significantly longer in cells in which HSF1 levels were reduced by RNAi or eliminated by gene disruption. In contrast, a down-regulation of HSF2 by RNAi had little if any effect on the phase entrainment. Hence, HSF1, but probably not HSF2, participated in the synchronization of circadian clocks to body temperature rhythms.
Results
Simulated mouse body temperature oscillations can synchronize circadian clocks of cultured fibroblasts
Depending on the species, mammalian body temperature displays daily fluctuations of 1°C–4°C (Brown et al. 2002; Refinetti 2010). Brown et al. (2002) reported that abrupt temperature fluctuations (12 h at 37°C, 12 h at 33°C) can transiently synchronize circadian gene expression in cultured RAT1 fibroblasts and that simulated mouse body temperatures can sustain presynchronized gene expression rhythms for a few days in these cells. More recently, Buhr et al. (2010) showed that PER2::luciferase expression can be entrained by temperature cycles that mimicked body temperature rhythms in mice pituitary and lung explants. Since PER2 functions as both a core clock component and an immediate early regulator in the synchronization of circadian clocks in the SCN (Farre et al. 2005) and in peripheral cell types (Kornmann et al. 2007a), it remains unclear whether the adaptation of its expression to temperature cycles reflects an immediate early function of PER2 or a true phase entrainment of the entire circadian oscillator. Hence, we wished to determine the kinetics of temperature-mediated phase resetting for different circadian genes in NIH3T3 fibroblasts. To this end, we developed an incubator allowing the programming and measurement of the incubation temperatures at high precision and the simultaneous monitoring of bioluminescence in real time. We thus recorded the bioluminescence of NIH3T3 fibroblasts stably expressing firefly luciferase under the control of the Bmal1 promoter (Nagoshi et al. 2004), which were exposed to different temperature regimes. Two parallel cultures were seeded and directly placed for monitoring. The cells were first kept 1 d at constant temperature (37°C). Thereafter, each cell population was subjected to simulated body temperature rhythms (35.5°C–38.5°C) of opposite phases. In parallel, we determined the bioluminescence profile for cells kept at a constant temperature (37°C). As shown in Figure 1A, the simulated body temperature fluctuations efficiently synchronized Bmal1-luciferase expression, and after a few days, the steady-state phases of the bioluminescence cycles were found to be in a fixed relationship with the two respective phases of the applied temperature cycles. While the temperature cycle depicted in blue in Figure 1A appeared to maintain the initial phase of the oscillators, the one depicted in orange did not. Thus, the bioluminescence cycle shown in orange in Figure 1A gradually changed its phase until it was in resonance with the temperature cycles shown in orange. In contrast, cells kept at 37°C progressively lost phase coherence within the examined time window. Conceivably, the increased amplitude of Bmal1 expression observed after successive temperature cycles reflected a rising number of synchronized cells in the population. When released in constant temperature, the oscillators of each cell population free ran from the entrained phases and eventually lost phase coherence.
Figure 1.

Synchronization of cultured fibroblast clocks by simulated body temperature fluctuations. NIH3T3/Bmal1-luc cells (A), NIH3T3/Dbp-luc cells (B), or immortalized tail fibroblasts from PER2::luciferase knock-in mice (C) were exposed to temperature cycles resembling body temperature rhythms measured telemetrically in mice (35.5°C–38.5°C) with opposite phases (blue and orange) and then released into a constant temperature of 37°C. Parallel cell populations were continuously kept at 37°C temperature (gray). All bioluminescence data were filtered by a moving average transformation (see the Materials and Methods). (D) After the synchronization of mouse fibroblasts by simulated body temperature cycles (red), the phase relationship was conserved for the expression of Bmal1, Dbp, and Per2. The differences in phase angles were similar to those observed in cells transiently synchronized by a short treatment with 50% horse serum (green) or dexamethasone (light blue) or those determined in the liver of intact animals (brown).
To analyze the response of other circadian genes to simulated body temperature cycles, we performed similar experiments with two additional reporter cell lines that produce bioluminescence cycles with different phases: NIH3T3 cells stably expressing Dbp-luciferase, and immortalized tail fibroblasts from PER2::luciferase mice (Fig. 1B,C, respectively). Again, the bioluminescence cycles displayed by these reporter cell lines were efficiently synchronized to simulated body temperature rhythms, with phase relationships expected for the expression of the corresponding endogenous genes. These observations were confirmed by the quantification of various circadian transcripts, using quantitative RT–PCR (qRT–PCR) on total RNA from NIH3T3 cells harvested at 4-h intervals during the sixth day of temperature entrainment (Supplemental Fig. S1). As depicted in Figure 1, Bmal1-luciferase and Dbp-luciferase expression adapted to shifted temperature cycles within 3–4 d, while PER2::luciferase expression was phase-entrained almost immediately to the imposed temperature cycles. This indicated that during the first days, the phases of different clock genes were uncoupled and that during this time period, PER2 expression reflected its property as an immediate early gene, rather than that of a core clock gene. However, once the steady-state phases were established for the expression of all three examined luciferase reporter genes, their phase relationships were conserved. This indicated that the molecular oscillator kept its integrity after being synchronized to temperature oscillations and that simulated body temperature cycles are thus valid phase-resetting cues for molecular clocks in cultured fibroblasts. Importantly, the phase relationship between circadian gene expression and simulated body temperature rhythms observed in mouse cultured fibroblasts closely recapitulated the phase relationship observed in vivo between peripheral circadian gene expression and body temperature rhythms (Fig. 1D; Damiola et al. 2000; Brown et al. 2002; Preitner et al. 2002). Moreover, the phase relationship we observed for Per2 is in accordance with another study describing the expression of the PER2::luciferase reporter in response to temperature changes in tissue explants (Buhr et al. 2010).
Circadian gene expression phase shifts in a bimodal manner to simulated body temperature rhythms at the cell population level
Previous reports have shown that circadian gene expression in cultured cells can be transiently synchronized by a puzzling variety of signaling molecules (Balsalobre et al. 1998, 2000a,b; Akashi and Nishida 2000; Hirota et al. 2002; Shirai et al. 2006). These include glucocorticoid hormones, retinoic acid, FGF, forskolin, Ca2+ ionophors, and glucose. Since serum contains many hormones, growth factors, and cytokines, fresh serum-containing culture medium is able to reset circadian gene expression in a large fraction of cultured cells. In our experiments, the medium was changed prior to the implementation of temperature cycles, and this resulted indeed in the partial synchronization of cells before they were exposed to temperature cycles (see Fig. 1, gray curves). We wished to examine how efficiently simulated body temperature rhythms can reset the phases of fully synchronized cells and to what extent the kinetics of this process depends on the phases of the imposed temperature cycles. As shown in Figure 2, cells previously treated with 50% horse serum, which resets the phases of virtually all cells (Balsalobre et al. 1998; Nagoshi et al. 2004), could be synchronized to all phases of simulated body temperature rhythms we examined. The time required for the complete phase resetting depended on the difference between the old and new phase. Interestingly, the bioluminescence profiles (i.e., the occurrence of two daily peaks during the transition from the initial to the new phase) suggested that the phase resetting proceeded in a bimodal manner. Bioluminescence recording of individual cells revealed three different kinetic patterns of phase shifting for cells in a population. A large fraction of cells (49%) were entrained by the initial phase, and this phase entered in conflict with the new phase, giving rise to a “double-peak phenotype.” In the second case, the phase of Bmal1-luciferase expression strictly followed the phase of the temperature cycles and immediately switched to the new phase. In the third case, the cell kept its initial phase without responding to the new phase of the temperature cycle during the period of monitoring (Fig. 3). Since even after synchronization by a serum shock and the subsequent exposure to temperature rhythms not all oscillators in the population have exactly the same phase (Nagoshi et al. 2004), it is conceivable that different cells received the relevant Zeitgeber cues during time windows of different sensitivities to these signals. Nevertheless, since at the population level the large majority of cells eventually adapted to the new phase, the differences observed between individual cells are probably of transient nature and do not necessarily reflect stable differences between cells.
Figure 2.

The resynchronization kinetics of circadian Bmal1-luciferase expression by simulated body temperature cycles depend on the old phase. NIH3T3/Bmal1-luc cells were transiently synchronized with horse serum and then subjected to temperature cycles starting 24, 30, 36, or 42 h after the serum treatment (phases 1, 2, 3, and 4). The top left panel shows bioluminescence from a cell population kept at constant temperature. The next four panels show the responses of cells subjected to temperature phases 1, 2, 3, and 4. All bioluminescence data were filtered by moving average transformation.
Figure 3.

Bioluminescence microscopy of individual cells reveals different sensitivities to temperature cycles. (A) Bioluminescence monitoring of Bmal1-luciferase expression in NIH3T3 fibroblasts subjected to three temperature cycles, followed by a 12-h phase shift (gray curve) at the level of the whole population (dark blue) using photomultiplier tube technology for data acquisition, or at the single-cell level (three panels in C) using a LV200 luminoview microscope (Olympus) equipped with an EM-CCD cooled camera (Hamamatsu Photonics) (see the Materials and Methods). Three different patterns were observed in individual cells. The occurrence of each of them is quantified in B, and a representative example of each of them is shown in C. In the first case (turquoise), the cell was entrained by the initial phase, and this phase entered in conflict with the new phase, giving rise to a “double-peak phenotype.” In the second case (green), the cell kept its initial phase without responding to the new phase of the temperature cycle. In the third case (orange), the cell strictly followed the phase of the temperature cycles and immediately switched to the new phase. All bioluminescence data were filtered by moving average transformation (see the Materials and Methods).
Low-amplitude temperature fluctuations can synchronize circadian gene expression over a wide temperature range
As outlined above, mouse body temperature oscillates by ∼3°C–4°C, with a maximum during the night and a minimum during the day (Brown et al. 2002). Since our previous data suggested that these daily fluctuations could contribute to the synchronization of peripheral cell types, we wished to determine whether temperature cycles of similar amplitude but different magnitude can also entrain circadian gene expression in cultured fibroblasts. Supplemental Figure S2 shows that this was indeed the case. Hence, temperature oscillations with an only 1.1-fold amplitude (when taking 0°C as the baseline) were capable of synchronizing circadian gene expression between 30°C and 40°C. We noticed, however, that the correlation between the phases of circadian gene expression and the imposed temperature cycles varied in a temperature-dependent fashion. Thus, the phase of Bmal1 expression was advanced relative to the temperature cycles in cells entrained in the lower temperature ranges. This trend was probably a direct consequence of the period shortening at reduced temperatures, known as overcompensation (Supplemental Fig. S2, gray curves). Indeed, it is well known that modest changes in period length can provoke large differences in phase under entrained conditions (Duffy et al. 2001).
The phase of circadian gene expression is exquisitely sensitive to temperature oscillations
The experiments described in the previous paragraph revealed that the variation (i.e., amplitude of the temperature cycle), rather than the absolute value of temperature, was relevant for the phase entrainment of cultured fibroblasts. We thus wanted to characterize the relation between the amplitude of the temperature cycles and their synchronization power more closely. To this end, we exposed Bmal1-luc cells to temperature cycles whose nadir and zenith values differed by 1°C, 2°C, 3°C, 4°C, and 8°C and whose average values were 37°C (Fig. 4). As expected, cells synchronized their oscillators more efficiently with increasing temperature cycle amplitudes. Surprisingly, however, a variation as small as 1°C was still able to entrain a significant proportion of the cells. Therefore, body temperature might contribute to the phase entrainment of peripheral cells even in humans, in which it oscillates between ∼36°C and 37°C.
Figure 4.

Circadian clocks can be partially synchronized by temperature fluctuations with very small amplitudes. Circadian gene expression was transiently synchronized by a horse serum shock, and cells were exposed to temperature oscillations with amplitudes differing by 1°C, 2°C, 3°C, 4°C, or 8°C and with a phase opposite to that imposed by the serum shock (corresponding to phase 3 in Fig. 2). Note that all temperature cycles were able to synchronize at least a fraction of NIH3T3/Bmal1-luc cells. All bioluminescence data were filtered by moving average transformation.
Temperature oscillations can synchronize circadian oscillators to widely different period lengths
The molecular mammalian circadian oscillator is thought to be based on interlocked negative feedback loops in gene expression, generating rhythms with a period length of approximately, but not exactly, 24 h. Simulated body temperature cycles are Zeitgebers that can be easily modified in our system and therefore represented a convenient tool to test the period length limits to which circadian oscillators can adapt. Figure 5 shows that simulated body temperature cycles of different period lengths (T-cycles) were capable of synchronizing circadian oscillators of fibroblasts to periods of 6–30 h. However, it appeared that circadian oscillators are not able to resonate with T-cycles of 34 h and longer, at least during the time window examined. Surprisingly, imposed T-cycles with extremely short periods (6–14 h) could still instigate corresponding rhythms in Bmal1-luciferase expression. These bioluminescence fluctuations were not solely caused by temperature-dependent changes in luciferase activity, since the oscillations generated by luciferase expression driven by the noncircadian cytomegalovirus (CMV) promoter had a significantly lower amplitude and a different phase when compared with those regulated by the Bmal1 promoter. Interestingly, the bioluminescence profiles of cells exposed to T-cycles <24 h exhibited a superposition of the imposed period to the circadian period. The circadian component progressively disappeared during the time of monitoring, while only the imposed period persisted.
Figure 5.

Circadian gene expression adapts to widely different T-cycles of simulated body temperature rhythms. NIH3T3 fibroblasts transfected with a Bmal1-luciferase reporter (blue) or a CMV-luciferase reporter (gray; used as a noncircadian control) were kept in constant temperature (first panel) or were exposed to temperature T-cycles of 35.5°C–38.5°C with period lengths of 6, 10, 14, 18, 24, 30, 34, or 40 h (as indicated). All bioluminescence data were filtered by moving average transformation.
Next, we wished to ascertain that the Bmal1-luciferase cycles we observed reflected an adaptation of the entire clockwork circuitry. To this end, we recorded the luminescence cycles of PER2::luciferase immortalized tail fibroblasts and NIH3T3/Dbp-luciferase fibroblasts, which were expected to be nearly anti-phasic to those produced by the Bmal1-luciferase reporter. Similar to the Bmal1-luciferase oscillations, the PER2::luciferase and Dbp-luciferase luminescence cycles exhibited amplitudes and phases significantly different from the CMV-luciferase-generated oscillations. In fibroblasts subjected to T-cycles of 14–30 h, the bioluminescence profiles of these three circadian reporter genes exhibited the expected endogenous phase relationships of Bmal1, Per2, and Dbp expression, indicating that the circadian clocks were synchronized by these T-cycles (Supplemental Figs. S3, S4). Upon imposing shorter or longer T-cycles, however, these phase relationships between bioluminescence cycles produced by different reporter genes were slightly altered. Nevertheless, the quantification of endogenous Bmal1, Rev-erbα, Per2, and Dbp transcripts in whole-cell RNA harvested at 2-h intervals from NIH3T3 cells subjected to 10-h period temperature square cycles did support a transient entrainment of the entire clock to such short 10-h T-cycles (Supplemental Fig. S5). In cells subjected to temperature cycles of extended period lengths (34 and 40 h), the abnormal phase relationships between Bmal1, Dbp, and Per2 expression might reflect that the components of the rhythm-generating feedback loops might have become uncoupled.
The short T-cycles did not produce a long-lasting “memory,” as the length of the bioluminescence cycles rapidly switched to ∼24 h (albeit with a relatively low amplitude) when the cells were released to constant temperature (data not shown). Nonetheless, when cells were phase-entrained to a T-cycle of 12 h by step gradients (6 h 34°C/6 h 38°C), which synchronize cells more rapidly than smooth temperature cycles and allowed a more precise analysis after shifting to a constant temperature (34°C), we observed a small peak (or shoulder) 12–14 h after the release to 34°C. Thereafter, luminescence cycles displayed ∼24-h periods with a phase determined by the short T-cycle (Supplemental Fig. S4).
HSF1 is required for the efficient synchronization of fibroblast clocks by temperature
In principle, temperature changes could act directly on the molecular oscillator by affecting the activity, synthesis, and degradation rates of core clock components. Alternatively, they could change the activity of temperature sensors, which in turn could act on the expression and/or activity of clock components. Temperature-dependent regulators may include immediate early transcription factors (e.g., HSFs) or temperature-sensitive transient receptor potential cation channels (thermo-TRPs) (Dhaka et al. 2006; Huang et al. 2006). Previous results have shown that the activity of HSF1 oscillates in a robust daily fashion (Reinke et al. 2008), and Buhr et al. (2010) recently reported that KNK437, a pharmacological inhibitor of HSFs, compromised the phase shifting of Per2 expression in tissue explants in response to abrupt temperature pulses. Based on similar experiments with Hsf1-deficient fibroblasts, Tamura et al. (2011) came to the same conclusion. We thus wished to examine by genetic loss-of-function approaches whether synchronization by smooth body temperature cycles still occurred in the absence of HSF1. Therefore, NIH3T3 fibroblasts cotransfected with a Bmal1-luciferase reporter and a shRNA directed against Hsf1 mRNA were synchronized with horse serum and then subjected to simulated body temperature cycles of different phases. Supplemental Figure S6 shows that the Hsf1 shRNA efficiently suppressed Hsf1 mRNA and protein accumulation as well as the heat-shock-induced activation of a luciferase reporter gene carrying four heat-shock response elements (HSEs) (Fig. 6A). As depicted in Figure 6B, cells expressing the Hsf1 shRNA exhibited a delayed adaptation of circadian Bmal1-luciferase expression to the imposed body temperature cycles as compared with cells transfected with an empty vector. Nevertheless, cells with depleted HSF1 levels eventually still adjusted their phase to that of the imposed temperature cycles, suggesting that either the level of HSF1 down-regulation was not sufficient to completely abolish temperature entrainment or HSF1 is not the only regulator implicated in this process. To investigate a possible functional redundancy of HSF family members expressed in fibroblasts, we also performed experiments with an Hsf2 shRNA. We found, however, that the down-regulation of HSF2 accumulation (Supplemental Fig. S6) had little if any effect on the temperature-mediated synchronization of Bmal1-luciferase expression (Fig. 6B). The possible implication of HSF1 in temperature-mediated synchronization of circadian gene expression was also examined in cells exposed to a simulated body temperature rhythm whose phase was then suddenly switched by 12 h. When HSF1 accumulation was down-regulated by RNAi, the kinetics of phase adaptation of Bmal1-luciferase expression was delayed by 1 d, and the amplitude of the bioluminescence cycles following the phase shift in temperature cycles was dampened when compared with control cells (Fig. 6C).
Figure 6.

HSF1 participates in the synchronization of circadian clocks by simulated body temperature cycles. (A) The response of an HSE-luciferase reporter gene to a heat shock (2 h, 42°C) is completely abolished when NIH3T3 cells are cotransfected with a plasmid (pshHSF1) encoding a shRNA targeting Hsf1 mRNA (orange). In contrast, cotransfections with a plasmid (pshHSF2) specifying a shRNA targeting Hsf2 mRNA (light blue) or an empty control plasmid (pSUPER) (blue) had no effect on the heat-induced expression of HSE-luciferase. Bioluminescence intensities were determined before and 5 h after the heat shock and were plotted against the background bioluminescence levels determined for pSUPER before the heat shock. The response of circadian Bmal1-luciferase reporter to simulated body temperature cycles with a phase opposite to the old phase (B) or to a phase inversion in temperature cycles (C) was delayed when NIH3T3 cells were cotransfected with pshHSF1 (orange). In contrast, the cotransfection of cells with pshHSF2 (light blue) or pSUPER (blue) had little effect on the phase-shifting kinetics of circadian Bmal1-luciferase expression. (D) Primary tail fibroblasts from Hsf1 knockout mice that stably express a Bmal1-luciferase reporter (green) were more slowly synchronized by simulated body temperature fluctuations of a contradictory phase when compared with tail fibroblasts from wild-type mice (blue). In B and D, cells were synchronized with horse serum prior to the initiation of bioluminescence monitoring. Note also that the kinetics of adaptation to the new phase depended on the cell type (NIH3T3 vs. primary tail fibroblasts) and on whether the reporter gene was transiently transfected or stably integrated. All bioluminescence data were filtered by moving average transformation.
We also compared primary tail fibroblasts from wild-type mice and Hsf1 knockout mice, transduced with a lentiviral vector harboring a Bmal1-luciferase reporter gene, with regard to their ability to synchronize circadian gene expression to simulated body temperature rhythms. As shown in Figure 6D, fibroblasts from wild-type mice adapted their phase much more rapidly than HSF1-deficient fibroblasts. As expected, the phenotype difference was more pronounced between Hsf1-deficient cells and wild-type cells than between NIH3T3 cells transfected with the Hsf1 shRNA vector or a control vector.
Taken together, our experiments indicated that HSF1, but not HSF2, contributed to the synchronization of molecular clocks by simulated body temperature cycles in cultured cells. However, HSF1-deficient cells were eventually still capable of resetting their oscillators to temperature cycles, suggesting that additional temperature-sensing mechanisms participated in this process.
Discussion
Body temperature rhythms as Zeitgebers for peripheral circadian oscillators
Here we demonstrated that in cultured mouse fibroblasts, the phase of circadian gene expression was exquisitely sensitive to temperature changes. Thus, if applied for several consecutive days, temperature cycles that mimic mouse body temperature oscillations were able to synchronize the phase of circadian gene expression in cultured NIH3T3 cells and primary tail fibroblasts. Surprisingly, even simulated human body temperature rhythms, with an amplitude of only ∼1°C (36°C–37°C), could still synchronize circadian clocks in a measurable fraction of cultured cells. Hence, daily temperature fluctuations controlled directly and indirectly by the SCN through thermoregulatory centers and activity rhythms (Brown and Refinetti 1996; Refinetti 2010; Weinert 2010), respectively, are likely to contribute to the phase coordination of circadian timekeepers in peripheral tissues of mammalian organisms (Fig. 7).
Figure 7.

Model for the integration of body temperature rhythms in the synchronization of peripheral oscillators. (A) In mammals, the SCN regulates virtually all clock outputs, including rest/activity cycles, feeding/fasting cycles, and temperature rhythms. In peripheral cell types, elevated temperature induces HSF1 activity, which, in cooperation with other temperature-sensitive regulators, promotes the expression of immediate early genes (IEGs) such as Per2. In turn, the IEGs phase-reset other clock genes such as Bmal1 or Dbp. A direct action of temperature on the activity and stability of clock components may also occur. (B) Representative expression kinetics of Per2 versus Bmal1 in response to an altered body temperature rhythm. While Per2 expression immediately adapts to the temperature stimulus, Bmal1 expression requires few days to oscillate in resonance with the newly imposed phase.
Circadian clocks in cultured cells can be transiently synchronized by a short treatment with a variety of signals, including serum, glucocorticoid hormones, retinoic acid, Ca2+ ionophors, tumor promoters, and growth factors (for review, see Stratmann and Schibler 2006). These pathways immediately reset all oscillators in the population to the same new phase. However, under natural conditions, the phase resetting is probably accomplished by gradual differences in Zeitgeber strength. For example, the SCN master pacemaker is synchronized to the photoperiod by increasing and decreasing light intensity during dawn and dusk, respectively. Likewise, oscillators in peripheral cell types are phase-entrained by gradual fluctuations of metabolites, blood-borne signals, and, probably, body temperature rhythms. The latter can be simulated in vitro (Brown et al. 2002; Reinke et al. 2008; Buhr et al. 2010; this study), and the response of circadian gene expression to such smooth daily fluctuations can be conveniently recorded. In our experiments, we observed complex Bmal1-luciferase expression profiles during the transition period from one phase to an opposite phase (i.e., a phase differing by 12 h). Two bioluminescence peaks, one corresponding to the old and the other to the new phase, could regularly be discerned after the application of conflictual temperature cycles (e.g., Figs. 1, 2, 6). In primary tail fibroblasts, double peaks (or a peak with a shoulder) could be detected during multiple consecutive days. Bioluminescence microscopy revealed that individual cells could express either the old phase, the new phase, or both phases simultaneously (Supplemental Fig. S2). Asher et al. (2010) and Abraham et al. (2011) have previously presented similar bimodal phase-shifting profiles for the adaptation of circadian gene expression in intact mice and lung explants, respectively.
In contrast to the experiments presented here, Brown et al. (2002) have previously reported that simulated body temperature rhythms can sustain presynchronized cycles of circadian gene expression in cultured fibroblasts, but not entrain a new phase. However, these investigators used mRNA quantification to measure circadian gene expression and followed gene expression during shorter time spans than those used in the experiments presented here. We therefore believe that the different conclusions just reflect differences in the recording techniques and the duration of the experiments. Indeed, the phase inversion elicited by simulated body temperature rhythms may be difficult to assess by measuring moderate differences of mRNA accumulation, in particular during the phase transition period. In contrast, even small differences in amplitude can be recorded by determining bioluminescence in real time.
During the process of synchronization of circadian oscillators by temperature cycles, we noticed that PER2::luciferase expression was synchronized more rapidly than the expression of Bmal1-luciferase or Dbp-luciferase. Asher et al. (2010) observed the same trend during the synchronization of circadian oscillators by feeding cycles in the livers of mice. The rapid synchronization of Per2 expression is perhaps not surprising, given that Per2 can serve as both a core clock gene and an immediate early gene in phase shifting (Albrecht et al. 1997; Spoelstra et al. 2004; Kornmann et al. 2007a). Nevertheless, our results demonstrated that the phase relationship between the expressions of different core clock genes can be transiently disrupted during the phase transition period.
Buhr et al. (2010) recently reported that the phase of the SCN, unlike that of peripheral organs, is resilient to temperature changes when studied in cultured tissue sections. According to these investigators, the logic underlying this difference is that the SCN should not react to the timing cues it emits to synchronize peripheral oscillators. A similar scenario holds true for glucocorticoid signaling, shown to participate in the phase resetting of clocks in peripheral organs but not in the SCN (Balsalobre et al. 2000a; Le Minh et al. 2001). Nonetheless, it should be noted that the resilience of the SCN to temperature changes is somewhat controversial. Herzog and Huckfeldt (2003) reported that in vitro cultured SCN preparations from rats synchronized readily to 24-h temperature cycles. Ruby (2011) suggested that different explant sizes and culture conditions may account for the somewhat incoherent observations made in studies with SCN tissue.
The flexibility of circadian oscillators to short and long temperature cycles
Depending on the system under investigation, circadian oscillators produce oscillations with a period length longer or shorter than 24 h. Therefore, these fluctuations must be expanded or contracted in order to be in resonance with geophysical time. We applied temperature oscillations of various durations (T-cycles) to determine the flexibility of circadian oscillators. Surprisingly, the Bmal1-luciferase expression in cultured NIH3T3 fibroblasts produced very short bioluminescence cycles of 6–14 h when exposed to equivalent T-cycles. The amplitudes of these oscillations were small, probably due to a relatively high ratio of half-life time for luciferase mRNA and protein over T-cycle time. To investigate whether these ultradian bioluminescence oscillations indeed reflected a complete synchronization of the oscillator, we also recorded the expression of a Dbp-luciferase reporter gene and PER2::luciferase immortalized tail fibroblasts, which should produce bioluminescence cycles nearly anti-phasic to those driven by Bmal1-luciferase. Our results showed that, similar to Bmal1-luciferase expression rhythms, PER2::luciferase and Dbp-luciferase expression cycles were able to resonate with short T-cycles of 6–14 h. Moreover, RT–PCR quantification revealed that temperature square cycles of 10 h produced expected anti-phasic patterns for endogenous Bmal1, Rev-erbα, Per2, and Dbp expressions. Hence, even very short T-cycles may indeed have synchronized the entire clockwork circuitry. In contrast, the expression of neither Bmal1-luciferase nor PER2- and Dbp-luciferase was able to resonate with very long T-cycles (>30 h). Thus, in cultured fibroblasts, the entrainable range of period lengths spanned from probably <14 h to ∼30 h. The range of free-running period lengths observed for different tissue explants (Yoo et al. 2004) is considerably narrower than this, and the clocks of peripheral organs can therefore readily be synchronized by rhythmic Zeitgebers, such as body temperature cycles. Irrespective of the length of the T-cycles we imposed on cells, circadian gene expression eventually assumed their normal intrinsic period length (∼24 h) when the cells were released into constant temperature (data not shown). Hence, excessively long or short T-cycles did not generate a long-lasting memory with regard to the period length of fibroblast circadian oscillators.
Abraham et al. (2011), using square wave temperature T-cycles, also observed a large entrainment flexibility of lung explants from PER2::luc mice, albeit with narrower limits than the ones shown here for cultured fibroblasts. Importantly, they found that SCN explants displayed a more restricted entrainment range, indicating that the SCN has more rigid oscillators than lung tissue. The additional oscillator strength is probably conferred to SCN cells through intercellular coupling (Liu et al. 2007).
HSF1 participates in the resetting of circadian gene expression by temperature
Kornmann et al. (2007a) established a transgenic mouse strain with conditionally active hepatocyte clocks. By using this system in conjunction with genome-wide transcriptome profiling, they identified ∼50 genes whose cyclic expression was driven by systemic cues supposedly emanating from the SCN rather than driven by local hepatocyte oscillators (Kornmann et al. 2007b). The list of these genes contains Per2 and several genes whose expression is known to be modulated by temperature, such as Hsp genes and genes encoding cold-inducible RNA-binding proteins. Using a completely different approach, differential display of DNA-binding proteins, Reinke et al. (2008) discovered that the nuclear translocation of HSF1 and its occupancy of HSEs surrounding Hsp genes were robustly rhythmic. This observation, together with the demonstration that simulated body temperature rhythms act as Zeitgebers, opened the possibility that HSF1 might serve as an immediate early transcription factor in the temperature-dependent entrainment of circadian clocks. Recent reports by Buhr et al. (2010) and Tamaru et al. (2011) lent support to this hypothesis. However, these investigators limited their analysis to the expression of the Per2 gene, whose expression in hepatocytes is system-driven and does not require functional local oscillators (Kornmann et al. 2007a). Therefore, we scrutinized the conjecture that HSF1 and, perhaps, HSF2 might participate in the synchronization of circadian clocks by genetic loss-of-function studies. Our experiments with cultured cells clearly established a role for HSF1 in the phase entrainment of circadian oscillators by simulated body temperature rhythms. In all experiments, we observed that the down-regulation or ablation of Hsf1 gene expression delayed the synchronization of circadian clocks to a new phase. Yet, even HSF1-deficient cells eventually assumed the new phase, suggesting that additional players participate in this process. As shown by this study, HSF2 had little effect on the kinetics of phase entrainment. Moreover, HSF3 and HSF4, the other two HSF family members (Fujimoto and Nakai 2010), are not expressed at detectable levels in NIH3T3 fibroblasts (J Morf and U Schibler, unpubl.). Thus, temperature sensors other than HSF members (for example, thermo-TRPs) (Vay et al. 2012) are likely to contribute to the residual synchronization of HSF1-deficient cells to simulated body temperature rhythms. Genome-wide siRNA screens may be capable of identifying the genes encoding these regulators.
Materials and methods
Cell culture, generation of temperature cycles, and real-time bioluminescence recording
NIH3T3 or immortalized tail fibroblasts were cultured in DMEM medium (GIBCO) containing 1% PSG (penicillin, streptomycin, and glutamine) and 10% or 20% FBS (fetal bovine serum), respectively.
Confluent NIH3T3 were trypsinized and seeded at either a 1:3 dilution 2 d before or a 1:2 dilution directly before they were used for bioluminescence monitoring by photomultiplier tube (PMT) technology (Nagoshi et al. 2004). Depending on the experiment, cells were transfected with the luciferase reporter genes Bmal1-luciferase (Nagoshi et al. 2004) or HSE-luc (Reinke et al. 2008) or with shRNA expression vectors (see below). For transfections, FuGENE 6 transfection reagent (Roche) was used according to the manufacturer's instructions. Before the bioluminescence recording was initiated, the medium was replaced with phenol red-free DMEM (21063, Invitrogen) supplemented with 0.1 mM luciferin and 10% FBS. Depending on the experiment, cells were synchronized by a 2-h treatment with 50% horse serum (Balsalobre et al. 1998) before bioluminescence was recorded.
Immortalized tail fibroblasts either contained a PER2::luciferase knock-in transgene (Yoo et al. 2004) or were transduced with a Bmal1-luc lentiviral vector (Liu et al. 2008) and were selected for stable expression with 2.5 μg/mL blasticidin (Invitrogen).
The Dbp-luciferase cell line was constructed by cloning a 9.6-kb DNA fragment encompassing the full-length Dbp gene, including 0.5 kb and 0.6 kb of 5′- and 3′-flanking regions, respectively, and firefly luciferase translated from an internal ribosome entry site (IRES) (Brown et al. 2005) into the BamHI and NruI sites of pSFVdhfr (Shimizu et al. 2001). NIH3T3 mouse fibroblasts were transfected with 20 μg of pSFVDbp(luc) using Lipofectamine 2000 according to the manufacturer's instructions. Stable transformants were selected in 5 μg/mL blasticidine (Sigma), and a colony harboring ∼250 Dbp-luciferase copies and yielding bioluminescence cycles of high amplitude and magnitude was kept for further analysis.
Bioluminescence and temperature were monitored in a custom-built device designed and constructed by André Liani, Yves-Alain Poget, and Georges Severi (members of the Mechanical Workshop of the Department of Molecular Biology, University of Geneva). Briefly, this incubator consists of a light-tight incubator allowing the programming and recording of temperature profiles in real time with an accuracy of 0.1°C. Simultaneously, bioluminescence is monitored continuously using Hamamatsu PMTs. Photon and temperature counts were integrated over 1-min intervals.
Single-cell bioluminescence microscopy was recorded in 3.5-cm plastic dishes (Falcon) using a LV200 luminoview microscope (Olympus) equipped with an EM-CCD cooled camera (Hamamatsu Photonics, EM-CCD C9100-13), as previously described (Suter et al. 2011). Briefly, before the bioluminescence recording was initiated, the medium was replaced with phenol red-free DMEM (21063, Invitrogen) supplemented with 0.5 mM luciferin and 10% FBS. The microscope environment was maintained at 5% of CO2, and the cells were monitored for 144–168 h using a 20-fold magnification objective. Photon counts were acquired over 15-min intervals. Temperature coprogramming and comonitoring were performed using a custom-built device designed and constructed by André Liani, Yves-Alain Poget, and Georges Severi (Mechanical Workshop of the Department of Molecular Biology, University of Geneva). Temperature counts were integrated over 1-min intervals.
Preparation and immortalization of mouse primary fibroblasts
Primary fibroblasts from Hsf1 knockout and wild-type mice were prepared from tail tips. These were minced and digested overnight with 1 mg/mL collagenase type IA (Sigma) at 37°C in medium containing 20% FBS, 1% PSG (Sigma), and amphotericin B (1/100; Sigma). The next day, disaggregated tissue pieces were washed and plated into fresh medium, and the fibroblasts emigrating from these pieces were allowed to grow to 80% confluence. Cells were then trypsinized, plated in a threefold dilution into new plates, and left in the incubator until spontaneously immortalized cell clones were observed.
Construction of HSF1 and HSF2 shRNA expression plasmids and determination of knockdown efficiencies
shRNA sequences were selected from sequences already tested in studies done in human cells and that are identical in the mouse and human orthologs. The oligonucleotides 5′-GCTCATTCAGTTCCTGATC-3′ (Ostling et al. 2007) and 5′-GCACCTTTTGCTTTTCTCA-3′ (Xing et al. 2005) were chosen for HSF1 and HSF2 shRNAs, respectively. Construction and expression of shRNA plasmids were carried out according to the pSUPER RNAi system's instructions (OligoEngine).
Knockdown efficiencies in shRNA transfection experiments were measured by real-time PCR quantification of Hsf1 and Hsf2 mRNAs, and Western blot analysis of the corresponding proteins (Supplemental Fig. S6) and monitoring of the bioluminescence produced by HSE-luc reporter genes upon a 2-h heat shock at 42°C were conducted as previously described (Fig. 6A; Reinke et al. 2008). The cells transfected with shRNA plasmids also expressed green fluorescent protein (GFP) from a phosphoglycerate kinase (PGK) gene promoter and could thus be FACS-sorted before RNA and protein extracts were prepared. This ensured that only shRNA-expressing cells were included for the Western blot and RT-qPCR experiments.
RNA analysis by real-time qPCR
RNAs were prepared from cultured cells by phenol/CHCl3 extraction and reverse-transcribed using random hexamers and SuperScriptTM RNase H-Free reverse transcriptase (Invitrogen). The resulting cDNAs were analyzed by real-time qPCR using TaqMan Universal PCR Master Mix No AmpErase UNG (Roche) supplemented with specific primers and probes in an ABI Prism 7700 sequence detection system from PE-Applied Biosystems. The primers and TaqMan probes used in these assays are listed in Supplemental Table 1. Other sets of cDNAs were analyzed by real-time qPCR using SYBR Green detection. The primers used in these reactions are also listed in Supplemental Table 1.
Western blot experiments
Western blot experiments were performed on whole-cell protein extracts following standard protocols and using antibodies against HSF1 and HSF2 (Stressgen Bioreagents SPA-950 and SPA-960, respectively), and U2AF65 (Sigma-Aldrich). The signals obtained for the latter protein served as loading controls.
Filtering of bioluminescence data by moving average transformation
To compare bioluminescence values with regard to phase and amplitude, we eliminated variations in the magnitude of the signals resulting from different transfection efficiencies or from different confluency states of cells. To this end, raw data were filtered by applying a moving average transformation on 24-h intervals. Briefly, the ratio of each value on the average of values from the 12 h before and the 12 h after this value was calculated as follows:
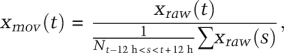 |
where xraw(t) is the original time series, xmov(t) is the transformed time series, and N is the number of time points between t − 12 and t + 12. For enlarged graphs in Supplemental Figure S3, the moving average transformation was also calculated according to each imposed period length.
Acknowledgments
We thank André Liani, Yves-Alain Poget, and Georges Severi for designing and constructing the bioluminescence and temperature recording system; Ivor J. Benjamin for his generous gift of the Hsf1 knockout mice; Joseph S. Takahashi for the PER2::luciferase knock-in mice; Steve Kay for the Bmal1-luc lentiviral vector; David Suter and Hans Reinke for their helpful advice and comments; Guillaume Rey (EPFL, Lausanne) for his help in bioluminescence data filtering; Christophe Bauer and Jerome Bosset for their help at the bioimaging platform; Nicolas Roggli for the artwork; and all Schibler laboratory members for their continuous support. This research was supported by the Swiss National Science Foundation (individual grants SNF 31-113565 and SNF 31-128656/1, and the NCCR program grant Frontiers in Genetics), the European Research Council (ERC-2009-AdG 20090506), the State of Geneva, the Louis Jeantet Foundation of Medicine, and the Sixth European Framework Project EUCLOCK.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.183251.111.
References
- Abraham U, Granada AE, Westermark PO, Heine M, Kramer A, Herzel H 2011. Coupling governs entrainment range of circadian clocks. Mol Syst Biol 6: 438 doi: 10.1038/msb.2010.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi M, Nishida E 2000. Involvement of the MAP kinase cascade in resetting of the mammalian circadian clock. Genes Dev 14: 645–649 [PMC free article] [PubMed] [Google Scholar]
- Akhtar RA, Reddy AB, Maywood ES, Clayton JD, King VM, Smith AG, Gant TW, Hastings MH, Kyriacou CP 2002. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Curr Biol 12: 540–550 [DOI] [PubMed] [Google Scholar]
- Albrecht U, Sun ZS, Eichele G, Lee CC 1997. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell 91: 1055–1064 [DOI] [PubMed] [Google Scholar]
- Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U 2010. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell 142: 943–953 [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Damiola F, Schibler U 1998. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 93: 929–937 [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Brown SA, Marcacci L, Tronche F, Kellendonk C, Reichardt HM, Schutz G, Schibler U 2000a. Resetting of circadian time in peripheral tissues by glucocorticoid signaling. Science 289: 2344–2347 [DOI] [PubMed] [Google Scholar]
- Balsalobre A, Marcacci L, Schibler U 2000b. Multiple signaling pathways elicit circadian gene expression in cultured Rat-1 fibroblasts. Curr Biol 10: 1291–1294 [DOI] [PubMed] [Google Scholar]
- Benarroch EE 2007. Thermoregulation: Recent concepts and remaining questions. Neurology 69: 1293–1297 [DOI] [PubMed] [Google Scholar]
- Boothroyd CE, Wijnen H, Naef F, Saez L, Young MW 2007. Integration of light and temperature in the regulation of circadian gene expression in Drosophila. PLoS Genet 3: e54 doi: 10.1371/journal.pgen.0030054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CM, Refinetti R 1996. Daily rhythms of metabolic heat production, body temperature, and locomotor activity in golden hamsters. J Therm Biol 21: 227–230 [Google Scholar]
- Brown SA, Zumbrunn G, Fleury-Olela F, Preitner N, Schibler U 2002. Rhythms of mammalian body temperature can sustain peripheral circadian clocks. Curr Biol 12: 1574–1583 [DOI] [PubMed] [Google Scholar]
- Brown SA, Ripperger J, Kadener S, Fleury-Olela F, Vilbois F, Rosbash M, Schibler U 2005. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 308: 693–696 [DOI] [PubMed] [Google Scholar]
- Buhr ED, Yoo SH, Takahashi JS 2010. Temperature as a universal resetting cue for mammalian circadian oscillators. Science 330: 379–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, Schibler U 2000. Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev 14: 2950–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaka A, Viswanath V, Patapoutian A 2006. Trp ion channels and temperature sensation. Annu Rev Neurosci 29: 135–161 [DOI] [PubMed] [Google Scholar]
- Dibner C, Sage D, Unser M, Bauer C, d'Eysmond T, Naef F, Schibler U 2009. Circadian gene expression is resilient to large fluctuations in overall transcription rates. EMBO J 28: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy JF, Rimmer DW, Czeisler CA 2001. Association of intrinsic circadian period with morningness-eveningness, usual wake time, and circadian phase. Behav Neurosci 115: 895–899 [DOI] [PubMed] [Google Scholar]
- Farre EM, Harmer SL, Harmon FG, Yanovsky MJ, Kay SA 2005. Overlapping and distinct roles of PRR7 and PRR9 in the Arabidopsis circadian clock. Curr Biol 15: 47–54 [DOI] [PubMed] [Google Scholar]
- Francis CD, Sargent ML 1979. Effects of temperature perturbations on circadian conidiation in neurospora. Plant Physiol 64: 1000–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Nakai A 2010. The heat shock factor family and adaptation to proteotoxic stress. FEBS J 277: 4112–4125 [DOI] [PubMed] [Google Scholar]
- Guo H, Brewer JM, Lehman MN, Bittman EL 2006. Suprachiasmatic regulation of circadian rhythms of gene expression in hamster peripheral organs: Effects of transplanting the pacemaker. J Neurosci 26: 6406–6412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog ED, Huckfeldt RM 2003. Circadian entrainment to temperature, but not light, in the isolated suprachiasmatic nucleus. J Neurophysiol 90: 763–770 [DOI] [PubMed] [Google Scholar]
- Hirota T, Okano T, Kokame K, Shirotani-Ikejima H, Miyata T, Fukada Y 2002. Glucose down-regulates Per1 and Per2 mRNA levels and induces circadian gene expression in cultured Rat-1 fibroblasts. J Biol Chem 277: 44244–44251 [DOI] [PubMed] [Google Scholar]
- Huang J, Zhang X, McNaughton PA 2006. Modulation of temperature-sensitive TRP channels. Semin Cell Dev Biol 17: 638–645 [DOI] [PubMed] [Google Scholar]
- Izumo M, Johnson CH, Yamazaki S 2003. Circadian gene expression in mammalian fibroblasts revealed by real-time luminescence reporting: Temperature compensation and damping. Proc Natl Acad Sci 100: 16089–16094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Schaad O, Bujard H, Takahashi JS, Schibler U 2007a. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biol 5: e34 doi: 10.1371/journal.pbio.0050034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornmann B, Schaad O, Reinke H, Saini C, Schibler U 2007b. Regulation of circadian gene expression in liver by systemic signals and hepatocyte oscillators. Cold Spring Harb Symp Quant Biol 72: 319–330 [DOI] [PubMed] [Google Scholar]
- Le Minh N, Damiola F, Tronche F, Schutz G, Schibler U 2001. Glucocorticoid hormones inhibit food-induced phase-shifting of peripheral circadian oscillators. EMBO J 20: 7128–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, Buhr ED, Singer O, Meeker K, Verma IM, et al. 2007. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell 129: 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu AC, Tran HG, Zhang EE, Priest AA, Welsh DK, Kay SA 2008. Redundant function of REV-ERBα and β and non-essential role for Bmal1 cycling in transcriptional regulation of intracellular circadian rhythms. PLoS Genet 4: e1000023 doi: 10.1371/journal.pgen.1000023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagoshi E, Saini C, Bauer C, Laroche T, Naef F, Schibler U 2004. Circadian gene expression in individual fibroblasts: Cell-autonomous and self-sustained oscillators pass time to daughter cells. Cell 119: 693–705 [DOI] [PubMed] [Google Scholar]
- Ostling P, Bjork JK, Roos-Mattjus P, Mezger V, Sistonen L 2007. Heat shock factor 2 (HSF2) contributes to inducible expression of hsp genes through interplay with HSF1. J Biol Chem 282: 7077–7086 [DOI] [PubMed] [Google Scholar]
- Pirkkala L, Nykanen P, Sistonen L 2001. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J 15: 1118–1131 [DOI] [PubMed] [Google Scholar]
- Preitner N, Damiola F, Lopez-Molina L, Zakany J, Duboule D, Albrecht U, Schibler U 2002. The orphan nuclear receptor REV-ERBα controls circadian transcription within the positive limb of the mammalian circadian oscillator. Cell 110: 251–260 [DOI] [PubMed] [Google Scholar]
- Refinetti R 2010. The circadian rhythm of body temperature. Front Biosci 15: 564–594 [DOI] [PubMed] [Google Scholar]
- Reinke H, Saini C, Fleury-Olela F, Dibner C, Benjamin IJ, Schibler U 2008. Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes Dev 22: 331–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensing L, Ruoff P 2002. Temperature effect on entrainment, phase shifting, and amplitude of circadian clocks and its molecular bases. Chronobiol Int 19: 807–864 [DOI] [PubMed] [Google Scholar]
- Romanovsky AA 2007. Thermoregulation: Some concepts have changed. Functional architecture of the thermoregulatory system. Am J Physiol Regul Integr Comp Physiol 292: R37–R46 doi: 10.1152/ajpregu.00668.2006 [DOI] [PubMed] [Google Scholar]
- Ruby NF 2011. Rethinking temperature sensitivity of the suprachiasmatic nucleus. J Biol Rhythms 26: 368–370 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Nagase T, Fukui H, Horikawa K, Okada T, Tanaka H, Sato K, Miyake Y, Ohara O, Kako K, et al. 1998. Multitissue circadian expression of rat period homolog (rPer2) mRNA is governed by the mammalian circadian clock, the suprachiasmatic nucleus in the brain. J Biol Chem 273: 27039–27042 [DOI] [PubMed] [Google Scholar]
- Shimizu N, Miura Y, Sakamoto Y, Tsutsui K 2001. Plasmids with a mammalian replication origin and a matrix attachment region initiate the event similar to gene amplification. Cancer Res 61: 6987–6990 [PubMed] [Google Scholar]
- Shirai H, Oishi K, Ishida N 2006. Bidirectional CLOCK/BMAL1-dependent circadian gene regulation by retinoic acid in vitro. Biochem Biophys Res Commun 351: 387–391 [DOI] [PubMed] [Google Scholar]
- Spoelstra K, Albrecht U, van der Horst GT, Brauer V, Daan S 2004. Phase responses to light pulses in mice lacking functional per or cry genes. J Biol Rhythms 19: 518–529 [DOI] [PubMed] [Google Scholar]
- Stratmann M, Schibler U 2006. Properties, entrainment, and physiological functions of mammalian peripheral oscillators. J Biol Rhythms 21: 494–506 [DOI] [PubMed] [Google Scholar]
- Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F 2011. Mammalian genes are transcribed with widely different bursting kinetics. Science 332: 472–474 [DOI] [PubMed] [Google Scholar]
- Tamaru T, Hattori M, Honda K, Benjamin I, Ozawa T, Takamatsu K 2011. Synchronization of circadian Per2 rhythms and HSF1–BMAL1:CLOCK interaction in mouse fibroblasts after short-term heat shock pulse. PLoS ONE 6: e24521 doi: 10.1371/journal.pone.0024521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya Y, Akashi M, Nishida E 2003. Temperature compensation and temperature resetting of circadian rhythms in mammalian cultured fibroblasts. Genes Cells 8: 713–720 [DOI] [PubMed] [Google Scholar]
- Underwood H, Calaban M 1987. Pineal melatonin rhythms in the lizard Anolis carolinensis: I. Response to light and temperature cycles. J Biol Rhythms 2: 179–193 [DOI] [PubMed] [Google Scholar]
- Vay L, Gu C, McNaughton PA 2012. The thermo-TRP ion channel family: Properties and therapeutic implications. Br J Pharmacol 165: 787–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert D 2010. Circadian temperature variation and ageing. Ageing Res Rev 9: 51–60 [DOI] [PubMed] [Google Scholar]
- Xing H, Wilkerson DC, Mayhew CN, Lubert EJ, Skaggs HS, Goodson ML, Hong Y, Park-Sarge OK, Sarge KD 2005. Mechanism of hsp70i gene bookmarking. Science 307: 421–423 [DOI] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. 2004. PERIOD2:LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci 101: 5339–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman WF, Pittendrigh CS, Pavlidis T 1968. Temperature compensation of the circadian oscillation in Drosophila pseudoobscura and its entrainment by temperature cycles. J Insect Physiol 14: 669–684 [DOI] [PubMed] [Google Scholar]