

Abstract
The transcription factor NF-κB is essential for osteoclastogenesis and is considered an immune-modulator of rheumatoid arthritis and inflammatory osteolysis. Activation of NF-κB subunits is regulated by the upstream IκB kinase (IKK) complex which contains IKKα, IKKβ, and IKKγ; the latter also known as NF-κB essential modulator (NEMO). The role of IKKα and IKKβ in the skeletal development and inflammatory osteolysis has been described, whereas little is known regarding the role of NEMO in this setting. Typically, signals induced by RANK ligand (RANKL) or TNF prompt oligomerization of NEMO monomers through the coiled-coil-2 (CC2) and leucine zipper (LZ) motifs. This step facilitates binding to IKKs and further relaying signal transduction. Given the central role of NF-κB in osteoclastogenesis, we asked whether NEMO is essential for osteoclastogenesis and whether interruption of NEMO oligomerization impedes osteoclast differentiation in vitro and in vivo. Using cell-permeable short peptides overlapping the CC2 and LZ motifs we show that these peptides specifically bind to NEMO monomers, prevent trimer formation, and render NEMO monomers susceptible for ubiquitin-mediated degradation. Further, CC2 and LZ peptides attenuate RANKL- and TNF-induced NF-κB signaling in bone marrow-derived osteoclast precursors (OCPs). More importantly, these peptides potently inhibit osteoclastogenesis, in vitro, and arrest RANKL-induced osteolysis, in mice. To further ascertain its role in osteoclastogenesis, we were able to block osteoclastogenesis using NEMO siRNA knockdown approach. Collectively, our data establish that obstruction of NEMO oligomerization destabilizes NEMO monomers, inhibits NF-κB activation, impedes osteoclastogenesis and arrests inflammatory osteolysis. Thus, NEMO presents itself as a promising target for anti-osteolytic intervention.
Keywords: NEMO/IKKγ, OSTEOCLAST, OSTEOLYSIS, NF-κB, COILED-COIL, LEUCINE ZIPPER
The transcription factor NF-κB mediates essential physiological and pathological responses through its regulation of a plethora of genes that control cell differentiation, osteoclastogenesis, oncogenesis, and inflammatory diseases. Activation of NF-κB is under the control of the inhibitory protein IκBα which has been widely investigated [Baldwin, 1996]. Various stimuli trigger a kinase complex which phosphorylates IκBα and marks it for degradation by the proteosome thus allowing nuclear translocation of the transcription factor [Baldwin, 1996; Abu-Amer, 2003]. The predominant kinase complex found in most cells contains two catalytic subunits, IKKα and IKKβ, and a regulatory subunit, IKKγ (also known as NEMO) [DiDonato et al., 1997; Mercurio et al., 1997; Regnier et al., 1997; Stancovski and Baltimore, 1997]. The catalytic serine kinases IKKα and IKKβ were found to catalyze p100 processing into p52 NF-κB and phosphorylate serines 32 and 36 of IκBα, respectively, whereas the role of NEMO was identified as a regulatory subunit. The N-terminal alpha-helical region of NEMO associates with a hexapeptide sequence within the distal carboxyl termini of IKKβ and IKKα termed NEMO-binding domain (NBD) [May et al., 2002].
The requirement of IKKα and IKKβ for normal bone homeostasis has been established [Chaisson et al., 2004; Ruocco et al., 2005]. Canonical NF-κB activation is mediated by IKKβ, the role of which in inflammation, osteoclastogenesis and cancer osteolysis has been widely described [reviewed in Karin et al., 2004; Karin, 2008]. IKKβ is essential for NF-κB induction by pro-osteoclastogenic and pro-inflammatory factors. In this regard, targeted deletion of IKKβ in the myeloid compartment abrogates osteoclastogenesis in vitro and in vivo [Ruocco et al., 2005; Otero et al., 2008]. Similarly, inhibition of IKKβ activation by introduction of NBD peptide abolished osteoclastogenesis, inflammatory osteolysis, and inflammatory arthritis in various animal models of inflammation [Dai et al., 2004; Jimi et al., 2004; Shibata et al., 2007]. These studies established that IKKβ is the link between inflammation and osteolysis. Specifically, IKKβ protects macrophages and osteoclasts from apoptosis and mediates pro-inflammatory cytokine actions, such as in the case of TNF. Deletion of IKKβ renders immune cells, macrophages and osteoclast precursors (OCPs) susceptible for TNF-induced apoptosis through a gain of function in the c-Jun N-terminal kinase (JNK) pathway. This is evident by the findings that inhibition of TNF or JNK restores the osteoclastogenic and inflammatory potential of these cells [Ruocco et al., 2005; Otero et al., 2008].
The alternative NF-κB activation pathway is dominated by NF-κB inducing kinase (NIK) activation of IKKα which in turn induces processing of p100/NF-κB2 subunit to generate p52. p52 then binds to RelB, the dimer translocates to the nucleus and activates target genes. In vitro studies have shown that cells devoid of NIK or IKKα fail to differentiate into osteoclasts, whereas in vivo inactivation of these kinases did not affect the overall skeleton [Novack et al., 2003; Chaisson et al., 2004; Ruocco et al., 2005]. While the precise role of IKKα in baseline osteoclastogenesis remains unclear, an important role for this kinase in regulating and restricting inflammation has been described [Li et al., 2005]. In this regard, Lawrence et al. [2005] have demonstrated that IKKα accelerates the turnover of RelA and c-Rel subunits and their removal from pro-inflammatory gene promoters, resulting with suppression of NF-κB activity.
NEMO was described as the hub for inflammatory diseases [Burns and Martinon, 2004]. In this regard, it has been suggested that lysine 63-linked poly-ubiquitination events of NEMO situate it as a scaffold and signal integrator molecule [Wu et al., 2006]. Mutations specifically targeting the relevant lysine residues responsible for poly-ubiquitination of NEMO identified the role of NEMO as modulator of inflammatory disorders. Using this approach, Ni et al. [2008] have shown that Lys392 modulates TLR signaling and inflammation in vivo.
The essential role of NEMO for NF-κB activation has been demonstrated in various studies. At the molecular level, recent studies have identified the coil-zipper domain of NEMO as crucial for NF-κB activation [Bloor et al., 2008] and that oligomerization of NEMO monomers through their coil-zipper domains is essential for signalsome assembly and activation of IKKs [Agou et al., 2002, 2004a,b].
In osteoclasts and bone, the role of NEMO has been described indirectly using NBD inhibitor [Dai et al., 2004; Jimi et al., 2004]. Importantly, short cell-permeable peptide spanning the IKKβ NBD disrupts the association of NEMO with IKKβ, blocks NF-κB activation, and ameliorates responses in animal models of inflammation [May et al., 2000, 2002]. In addition, sporadic mutations in the ikbkg/nemo gene were manifested by bone abnormalities [Courtois et al., 2001; Ku et al., 2005] suggesting that this gene plays a key role in bone homeostasis.
Given these observations and the indirect evidence that NEMO regulates osteoclastogenesis, we surmised that inhibition of NEMO trimerization may provide direct evidence regarding its role in osteoclast differentiation and osteolysis. In this study, we show that administration of decoy peptides, designed to bind to exposed coiled-coil and leucine zipper (LZ) domains of NEMO and prevent its oligomerization, inhibit NF-κB activation, abolish basal and inflammatory osteoclastogenesis, and hinder inflammatory osteolysis in mice.
EXPERIMENTAL PROCEDURES
REAGENTS
All cytokines were purchased from R&D Systems (Minneapolis, MN). All antibodies were purchased from Santa Cruz Biotech (Santa Cruz, CA) and Cell Signaling Technologies, Inc. (Danvers, MA). All other chemicals are from Sigma (St. Louis, MO) unless otherwise indicated.
CELL ISOLATION AND PURIFICATION
OCPs in the form of marrow macrophages were isolated from whole bone marrow of 4- to 6-week mice and incubated in tissue culture plates, at 37°C in 5% CO2, in the presence of 10 ng/ml M-CSF. After 24 h in culture, the non-adherent cells are collected and layered on a Ficoll–Hypaque gradient. Cells at the gradient interface are collected and plated in α-MEM, supplemented with 10% heat-inactivated fetal bovine serum, at 37°C in 5% CO2 in the presence of 10 ng/ml M-CSF, and plated according to each experimental conditions.
NEMO PEPTIDE SEQUENCES USED IN THIS STUDY
TAT-CC2: MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSKLGYGRKKRRQRRRGGSTMSGYPYDVPDYAGSMAGTSKGMQLEDLRQQLQQAEEALVAKQELIDKLKEEAEQHKIVFEA.
TAT-LZ: MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSKLGYGRKKRRQRRRGGSTMSGYPYDVPDYAGSMAGTLKAQADIYKADFQAERHAREKLVEKKEYLQEQLEQLQREFNKLFEA.
TAT-CC2-LZ (CNLZ): MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSKLGYGRKKRRQRRRGGSTMSGYPYDVPDYAGSMAGTSKGMQLEDLRQQLQQAEEALVAKQELIDKLKEEAEQHKIVMETVPVLKAQADIYKADFQAERHAREKLVEKKEYLQEQLEQLQREFNKLFEA.
TAT-CC2-mutant: MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSKLGYGRKKRRQRRRGGSTMSGYPYDVPDYAGSMAGTSKGMQLEDLRQQgQQAEEAgVAKQELgDKLKEEAEQHKIVFEA.
TAT-LZ-mutant: MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSKLGYGRKKRRQRRRGGSTMSGYPYDVPDYAGSMAGTLKAQADIYKADFQAERHAREKLVEKKEYsQEQLEQsQREFNKLFEA.
TAT-CC2-LZ-mutant: MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDRWGSKLGYGRKKRRQRRRGGSTMSGYPYDVPDYAGSMAGTSKGMQLEDLRQQgQQAEEAgVAKQELgDKLKEEAEQHKIVMETVPVLKAQADIYKADFQAERHAREKLVEKKEYsQEQLEQsQREFNKLFEA.
TAT-NBD PEPTIDES
We synthesized and HPLC-purified (using Washington University facility) three peptides. (1) Functional wild-type TAT-NBD (YGRKKRRQRRR-G-TTLDWSWLQME). (2) Negative control mutant TAT-NBD (YGRKKRRQRRR-G-TTLDASALQME). (3) GFP-conjugated TAT-NBD to trace distribution (GFP-YGRKKRRQRRR-G-TTLDWSWLQME).
pTAT CONSTRUCT AND PROTEIN COUPLING
Construction of pTAT-fusion proteins, expression and purification was previously described [Abu-Amer et al., 1997, 2001]. Briefly, we have cloned the TAT transduction domain followed by a multiple cloning site into the pREST (Invitrogen) bacterial expression vector. The plasmid expresses a six histidine tag for purification, HA tag for detection followed by the TAT transduction domain (RKKRRQRRRPP). The various IKK forms were cloned in the MCS using standard procedures. The plasmids containing CC2 or LZ inserts are expressed in the BL21 (DE3) strain of E. coli followed by purification as described previously [Abu-Amer et al., 1997, 2001].
ELECTROPHORETIC MOBILITY SHIFT ASSAY (EMSA)
Nuclear fractions were prepared as previously described [Dai et al., 2004]. In brief, cells were resuspended in hypotonic lysis buffer A (10 mM HEPES [pH 7.8] 10 mM KCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol 0.5 mM 4-(2-aminoethyl) benzenesulfonyl fluoride (AEBSF), 5 mg/ml leupeptin) and incubated on ice for 15 min, and NP-40 was added to a final concentration of 0.64%. Nuclei were pelleted and the cytosolic fraction was carefully removed. The nuclei were then resuspended in nuclear extraction buffer B (20 mM HEPES [pH 7.8] 420 mM NaCl, 1.2 mM MgCl2, 0.2 mM EDTA, 25% glycerol, 0.5 mM dithiothreitol, 0.5 mM AEBSF, 5 μg/ml Pepstatin A, 5 μg/ml leupeptin), vortexed for 30 s and rotated for 30 min in 4°C. The samples were then centrifuged and the nuclear proteins in the supernatant were transferred to fresh tubes and protein content was measured using standard BCA kit (Pierce, Rockford, IL). Nuclear extracts (10 μg) were incubated with an end-labeled double stranded oligonucleotide probe containing the sequence 5′-AAA CAG GGG GCT TTC CCT CCT C-3′ derived from the κB3 site of the TNF promoter. The reaction was performed in a total of 20 μl of binding buffer (20 mM HEPES [pH 7.8], 100 mM NaCl, 0.5 mM dithiothreitol, 1 μg poly dI – dC, and 10% glycerol) for 30 min at room temperature. Samples were then fractionated on a 4% polyacrylamide gel and visualized by exposing dried gel to film.
KINASE ASSAY
Extracts prepared from the various cells under use were suspended in lysis buffer containing 40 mM Tris–HCl, pH 8.0, 500 mM NaCl, 0.1% Nonidet P-40, 6 mM EDTA, 6 mM EGTA, 5 mM β-glycerophosphate, 5 mM NaF, 1 mM NaVO4, pH 10.0, and protease inhibitor (Roche Molecular Biochemicals). The cellular lysates (500 μg) were subjected to immunoprecipitation with the relevant antibody and gamma-bind Sepharose (25 μl). After washing of the immunoprecipitates, kinase assays were performed at 30°C for 30 min with buffer containing 50 mM Tris–HCl, pH 8.0, 100 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol, 10 μM ATP, 5 mM β-glycerophosphate, 5 mM NaF, 1 mM NaVO4, pH 10.0, 5 μCi of [γ-32P]ATP, 5 μg of the GST-IκBα (aa 1–54). The kinase reaction mixtures were then resolved by SDS–polyacrylamide gel electrophoresis, and phosphorylation of IκBα was detected by autoradiography.
siRNA KNOCKDOWN
The target sequence for NEMO was: (5′-AGGATCGGCAAGCTT-TAGA-UU). Scrambled sequence was used as a negative control (5′-AGTACTGCTTACGATACGG-UU). Sequences were introduced into OCPs using retroviral approach. Cells were infected with siRNAs using Fugene6 reagent. Cells were then stimulated with RANK ligand (RANKL) and M-CSF and maintained in culture for 4 days after which they were either lysed to measure protein expression or fixed and stained with tartrate-resistant acid phosphatase (TRAP) to detect osteoclasts.
HISTOLOGY
Calvaria and long bones were preserved in 10% buffered formalin (24 h) and subjected to a decalcification process using 10% EDTA, pH 7.0 for 7 days with gentle rocking and daily replacement of solution. Decalcified bones were then dehydrated in graded alcohol, cleared through xylene and embedded in paraffin. Paraffin blocks were then sectioned longitudinally. Five-micron sections were stained with either H&E or histochemically for TRAP to visualize osteoclasts.
OSTEOCLAST FORMATION ASSAY
OCPs were plated in triplicate at a density of 3.0 × 104 cells in 200 μl whole media supplemented with 10 ng/ml m-CSF and RANKL in 96-well tissue culture plates. Mature osteoclasts form between day 4 and day 6 of culture, at which point, the cells are fixed and stained for TRAP to visualize osteoclasts (Leukocyte Acid Phosphatase Kit, Sigma). TRAP-positive multinucleated cells with three or more nuclei were scored as osteoclasts.
WESTERN BLOT ASSAY
Total cell lysates were boiled in the presence of an equal volume of 2× SDS sample buffer consisting of (0.5 M Tris–HCl, pH 6.8, 10% (w/v) SDS, 10% glycerol, 0.05% (w/v) bromophenol blue, 3% β-mercaptoethanol, and distilled water) for 5 min and subjected to electrophoresis on 8–10% SDS–PAGE. The proteins were transferred to nitrocellulose membranes using a semi-dry blotter (Bio-Rad, Hercules, CA) and incubated in blocking solution (10% skim milk prepared in phosphate-buffered saline containing 0.05% Tween-20) to reduce non-specific binding. The membranes were washed with phosphate-buffered saline/Tween buffer and exposed to primary antibodies (16 h at 4°C), washed again four times, and incubated with the respective secondary horseradish peroxidase-conjugated antibodies (1 h at room temperature). The membranes were washed extensively (4 × 15 min), and an ECL detection assay was performed following the manufacturer’s directions.
RESULTS
CC2-PEPTIDE BINDS TO ENDOGENOUS NEMO IN OCPs AND BLOCKS TRIMERIZATION
The proposed mechanism underlying inhibition of NEMO oligomerization entails binding of CC2 and LZ peptides to their corresponding regions in NEMO monomers and hindrance of NEMO oligomer formation. To test this initial step, OCPs were treated with cell-permeable TAT-CC2 or TAT-LZ peptides or their mutated forms (mut-CC2) in the absence or presence of RANKL or TNF for 30 min. Immunoprecipitations with anti HA antibody (HA tag is fused to TAT-peptides) of cell lysates showed that endogenous NEMO bind to HA-CC2 and HA-LZ peptides whereas no appreciable binding was detected when mutant forms of the peptides were utilized (Fig. 1A and data not shown). The functional significance of this peptide binding was further evident by inhibition of dimer and trimer formation of NEMO molecules after incubation with CC2 peptide for 20–30 min (Fig. 1B).
Fig. 1.
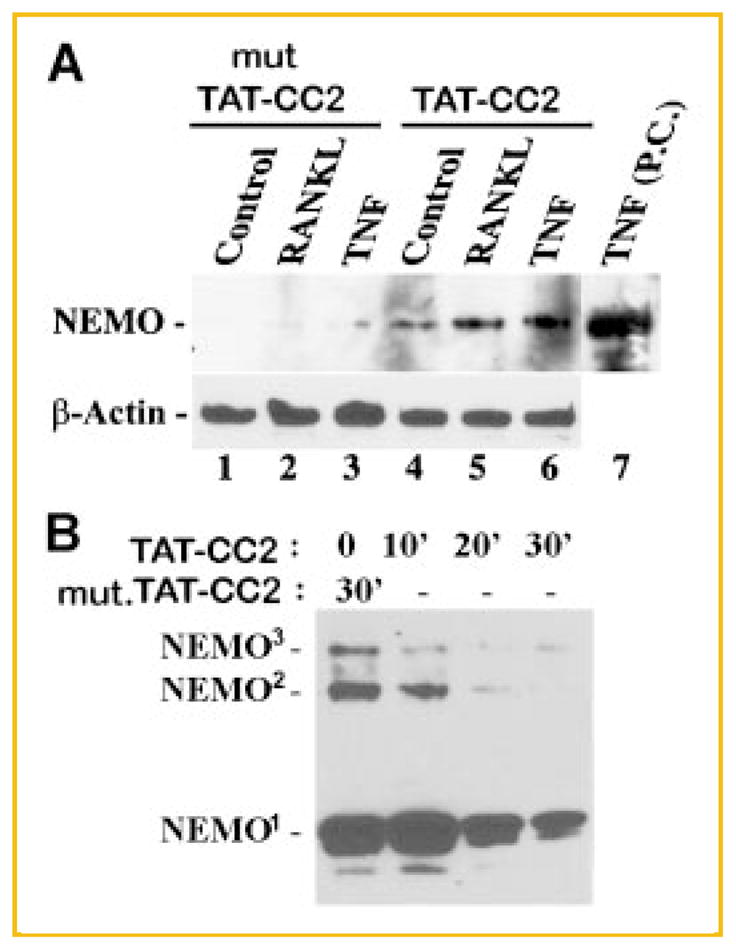
TAT-CC2 peptide binds to endogenous NEMO in OCPs and inhibits NEMO oligomerization. Wild-type (WT) and mutated (mut) CC2 peptides (50 μM) were added to cells and (A) stimulated with vehicle, RANKL, or TNF for 30 min. Lysates (lanes 1–6) were pre-cleared with IgG, immunoprecipitated with anti HA antibody and then blotted with anti NEMO antibody. Lane 7 represents a positive control (PC) for NEMO expressed in TNF-stimulated cells (crude lysate). Lower panel represents β-actin expression in equal concentrations of total protein from crude lysates collected from the respective samples before immunoprecipitation, used as loading control. B: Cells were treated with either WT-CC2 or mut-CC2 for the time points indicated. Cell lysates were processed for immunoblots in non-reducing conditions. Superscript numbers 1, 2, and 3 indicate NEMO monomer, dimer, or trimer, respectively.
EXPRESSION LEVELS OF NEMO IN OCP CELLS TREATED WITH RANKL ARE REDUCED UNDER CONTINUOUS PRESENCE OF CC2 OR LZ PEPTIDES
In the course of conducting immunoprecipitation studies, we observed that NEMO binding with CC2 or LZ peptides is short lived (approximately 30 min). To further examine the kinetics of this phenomenon, we performed a time course study and discovered that expression levels of NEMO are abolished following exposure of cells to CC2 or LZ in the presence of RANKL for time points longer than 30 min (Fig. 2A,C). These findings suggested that CC2 and LZ-bound NEMO monomers are unstable. Indeed, we found that NEMO is ubiquitinated (Fig. 2B) and undergoes proteosome-mediated degradation evident by restoration of NEMO levels in the presence of the proteosome inhibitor MG132 (Fig. 2C, NEMO levels after 1 h in Fig. 2A,C).
Fig. 2.

Expression levels of NEMO in OCPs are reduced in continuous presence of TAT-CC2 peptide. OCPs were left untreated or pre-stimulated with RANKL (30 min) in the absence or presence of TAT-CC2 peptide (50 μM), for the time points indicated. All conditions except for lane 1 were treated with RANKL for a total of 4.5 h. Cells were then lysed, immunoprecipitated, and blotted with NEMO antibody (A). Crude cell lysates from identical cultures were probed for β-actin expression (A). B: OCPs were treated with RANKL for 30 min followed by TAT-CC2 as indicated. Cells were then lysed, the lysate was pre-cleared and immunoprecipitated with NEMO followed by immunoblot with anti-ubiquitin and anti-NEMO antibodies. C: RANKL-stimulated OCPs (30 min) were treated with TAT-CC2 for the time point shown (similar to panel A) in the continuous presence of the proteosome inhibitor MG-132 (10 μM). Expression levels of NEMO (and actin as a loading control) were measured by Western blots.
NEMO PEPTIDES CC2 AND LZ INHIBIT NF-κB ACTIVATION
NEMO trimer assembly and association with the IKK complex mediate signal-induced NF-κB activation. Varieties of factors, including RANKL, induce the NEMO-containing IKK complex and require this signaling complex to transmit their biological effect. To test the significance of NEMO-derived peptides on RANKL-induced activation of the NF-κB pathway, several parameters including expression levels of endogenous IκBα, phosphorylation of endogenous IκBα, activation of IKKβ, and transcriptional activity of NF-κB were analyzed. To this end, OCPs were incubated with mutated or wild-type TAT-CC2 and/or TAT-LZ peptides (50 μM) and stimulated with RANKL. First, IκBα protein expression and phosphorylation were examined (Fig. 3A). The results show that whereas RANKL-induced rapid phosphorylation of IκBα which was paralleled by degradation of the protein, TAT-CC2 peptide inhibited phosphorylation and stabilized expression of IκBα Next, we sought to determine if reduced phosphorylation of IκBα correlates with diminished IKK activity. Given the hypothesis that NEMO oligomerization is essential for IKK complex activation through association with IKKβ and IKKα, we asked if TAT-CC2 peptide modulates the kinase expression and/or activity. To address this latter point, we first show that TAT-CC2 does not alter IKKβ protein expression below control levels (compare lanes 5–7 with lane 1; Fig. 3A), although it reduces RANKL-induced accumulation of IKKβ, suggesting that signals normally transmitted through NEMO are abrogated by these peptides. Next, OCPs treated with CC2 and stimulated with RANKL for 30 min were subjected to immunoprecipitations of NEMO followed by assessing associated kinase activity. The results of this exercise show that CC2 significantly inhibits kinase-mediated phosphorylation of GST-IκB substrate compared with normal phosphorylation of this substrate when mutated (inactive) CC2 peptide was used (Fig. 3B). This result suggests that CC2 disrupts NEMO binding to IKKβ and that NEMO precipitates do not include IKK activity. Having established that TAT-CC2 abolished activity of the IKK complex, we proceeded to determine if this outcome altered signal-induced NF-κB transcriptional activity. Thus, nuclear extracts were isolated and subjected to NF-κB activity assay, namely EMSA. Nuclear extract data show that whereas NF-κB activity was increased in the presence of RANKL, this activity was significantly reduced in the presence of TAT-CC2 and TAT-LZ peptides (Fig. 3C). AP-1 activity was not affected by either TAT-CC2 or TAT-LZ peptides (Fig. 3C, lower panel). Finally, using luciferase assay, we show that a peptide encompassing CC2-LZ (CNLZ) inhibits activation of NF-κB as efficient as do IκBα and NBD (Fig. 3D). Altogether, the data suggest that these NEMO-derived peptides obstruct activation of NF-κB by interrupting RANKL induction of NEMO-IKK complex formation and activity.
Fig. 3.

NEMO peptides CC2 and LZ inhibit NF-κB activation. A: TAT-CC2 inhibits RANKL-induced IκBα phosphorylation and degradation. OCPs were treated with RANKL for the time points indicated in the presence of mutant (mut) or wild-type TAT-CC2. Cells were then lysed and equal amounts of total protein were probed with IKKβ, β-actin, IκBα and phospho-IκBα antibodies. B: Kinase activity: OCPs were co-treated with RANKL for the time points shown in the presence of mutant or wild-type TAT-CC2 peptide. Cells were then lysed; a fraction of the crude lysate was used to determine NEMO expression, while the remainder of the lysate was pre-cleared and immunoprecipitated with NEMO followed by a kinase assay using GST-IκB as a substrate. PC in lane #1 of upper panel denotes positive control for in vitro phosphorylation of GST-IκB with recombinant IKKβ. C: OCPs were left untreated or stimulated with RANKL (15, 30 min) in the absence or presence of TAT-CC2 or TAT-LZ peptides (50 μM each). DNA binding activity was measured using EMSA which was performed with κB3 or AP-1 oligonucleotides. D: The direct effect of CC2-LZ peptides (CNLZ) on NF-κB promoter–reporter activity was determined using HEK cells. Cells were transfected with NF-κB promoter–reporter construct and treated with TNF, CNLZ or both. Positive controls include TNF, constitutively active IKKβ (IKKβ-CA), whereas negative (inhibitor) controls include IκBα and NBD peptide.
NEMO PEPTIDES INHIBIT OSTEOCLASTOGENESIS
The critical requirement of NF-κB for osteoclast formation and activity has been established (double NF-κB1/2-KO) [Franzoso et al., 1997]. Furthermore, we and others have shown that NEMO binding to IKKs is also essential for osteoclastogenesis evident by blunted osteoclast formation in vitro and reduced osteolysis in vivo, in the presence of the NEMO decoy peptide, NBD [Dai et al., 2004; Jimi et al., 2004]. To further examine if NF-κB inhibition by CC2 and LZ impacts osteoclastogenesis, we tested the effect of these peptides on RANKL-induced differentiation of OCPs. Our findings show that CC2 and LZ inhibit basal (RANKL-induced) osteoclastogenesis when added at the first day (day 0) or 3 days after stimulation with RANKL (Fig. 4A and Table I). More importantly, CC2 and LZ hinder TNF-stimulated osteoclastogenesis when added simultaneously to RANKL-primed pre-osteoclasts. These results imply that NEMO is essential for basal and inflammatory osteoclastogenesis. To further support this reputed role of NEMO as crucial for osteoclastogenesis, we took a direct approach. To this end, we knocked down expression of NEMO in OCPs using a retroviral siRNA approach and subjected these cells to osteoclastogenic conditions. The results of this experiment clearly show that whereas a scrambled oligonucleotide (SC) did not affect expression levels of NEMO protein (Fig. 4B) or basal levels of RANKL-induced osteoclasts (Fig. 4C: 126 ± 16 in pSi-SCR compared with 143 ± 18 osteoclasts in control), a specific siRNA nucleotide which successfully and significantly reduced protein expression of NEMO (Fig. 4B; N1), also blunted osteoclastogenesis (83% inhibition compared with control) (Fig. 4C; pSi-N1). We also noted that higher level expression of wild-type NEMO in OCPs elevates RANKL-induced osteoclastogenesis by 37% compared with control (pMx-NEMO; Fig. 4C). These results provide direct evidence regarding the necessity of NEMO for osteoclastogenesis and lend support for our proposition that CC2 and LZ-mediated inhibition of NEMO signaling abrogates osteoclastogenesis.
Fig. 4.

NEMO peptides inhibit osteoclastogenesis. A: OCPs were stimulated with RANKL in the absence or presence of TAT-CC2 (50 μM). The peptide was added at the beginning of culture (d0) or 3 days after stimulation with RANKL (d3). One-half of cultures was further stimulated with TNF on day 3 (panels B, D, F). NBD peptide was utilized as a positive control for NEMO inhibition (panel G) whereas a mutant peptide served as a negative control (panel H). B: siRNA knockdown of NEMO arrests osteoclastogenesis. OCPs were transduced with NEMO siRNA oligonucleotides (N1, N2) or with a scrambled oligo (SC) using retrovirus delivery approach (pSilencer). Cells were then incubated with RANKL and M-CSF for 6 days. Western blot was used to detect NEMO expression (B). C: Cell cultures treated as described in panel B were maintained in with RANKL and M-CSF for 5 days after which cells were fixed and TRAP-stained to detect osteoclasts. pMx-NEMO denotes cells infected with virally expressed wild-type NEMO as a positive control. pSi—pSilencer. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
TABLE I.
Quantification of the Number of Osteoclasts Per Well
| Mutant TAT-CC2 | TAT-CC2 (d0) | TAT-CC2 (d3) | |
|---|---|---|---|
| RANKL (#oc/well) | 152 ± 17* | 21 ± 12 | 26 ± 14 |
| RANKL + TNF (#oc/well) | 261 ± 27* | 18 ± 10 | 31 ± 20 |
OCPs were plated and treated as described for Figure 4A. TRAP-positive osteoclasts (3+nuclei) were counted and shown as the mean average of three wells from three independent experiments.
P <0.05.
NEMO-DECOY PEPTIDES ARREST RANKL-INDUCED OSTEOLYSIS, IN VIVO
Having established that inhibition of NEMO assembly by CC2 and LZ decoy peptides halts in vitro osteoclastogenesis, we turned to investigate the functional significance of this observation, in vivo. Mice were injected over their calvaria and in the knee joint space with RANKL (50 μg/20 μl injected locally) in the presence of PBS or CC2 and LZ peptides (100 μg/injection). Calvaria and knee joint images taken from the various experimental conditions demonstrate severe osteolysis in RANKL-injected animals as evident by elevated levels of TRAP-reactive osteoclasts (arrows) and increased resorbed space (asterisk), whereas CC2 and LZ peptides obliterated this response when administered simultaneously with RANKL (Fig. 5A,B). Thus, NEMO decoy peptides impede RANKL-induced osteolysis, in vivo.
Fig. 5.

TAT-CC2 and TAT-LZ peptides inhibit RANKL-induced osteolysis, in vivo. Mice were injected over the calvaria and in the knee joint space with RANKL (50 μg/20 μl) in the absence or presence of the peptides TAT-CC and TAT-LZ (100 μg injected locally). Peptides were injected every 48 h as we have established previously [Clohisy et al., 2003; Dai et al., 2004; Hirayama et al., 2005]. Mice were sacrificed after 7 days and (A) calvarial and (B) knee joint osteolysis was analyzed by histochemistry. Arrows point to osteoclasts. Asterisks denote resorbed bone areas.
DISCUSSION
The NF-κB activation pathway plays a crucial role in bone homeostasis through its established and essential role in osteoclast differentiation and survival. The individual role of a number of NF-κB proteins in osteoclastogenesis has been described. Using genetic animal models, it has been shown that downstream NF-κB molecules, such as p50, p52, p65, and RelB, as well as upstream activators of the pathway including IKKα and IKKβ are all required for normal bone homeostasis [Franzoso et al., 1997; Chaisson et al., 2004; Ruocco et al., 2005; Otero et al., 2008; Vaira et al., 2008a,b]. Although, several studies using inhibitory approaches suggest that NEMO is also essential for osteoclasts and osteolysis, the precise direct role of NEMO in this process remains enigmatic. Based on the fact that NEMO oligomerization is required for IKK complex assembly and NF-κB activation, we surmised that inhibiting NEMO oligomerization provides a direct approach to assess its role in osteoclastogenesis. Indeed, administration of cell-permeable decoy peptides targeting cognate motifs within the CC2 and LZ regions of NEMO efficiently bind to NEMO monomers. These monomers are short lived and undergo proteosome-mediated degradation. As a result of NEMO depletion, activation of NF-κB signaling is hindered as evident by reduced IκBα phosphorylation and diminished NF-κB activity. Perhaps, the most significant aspect of inhibiting NEMO assembly is arresting osteoclastogenesis and osteolysis.
The rationale for targeting NEMO is based on accumulating evidence suggesting that this scaffold protein is a crucial modulator of inflammatory responses and that certain mutations in the nemo gene in human subjects cause severe infections and bone defects such as hypodontia, conical incisors, and osteopetrosis [Courtois et al., 2001; Ku et al., 2005].
The first indirect evidence regarding the role of NEMO in bone homeostasis was established by using the decoy peptide NBD which was designed to obstruct NEMO binding to the carboxyl domain of IKKα and IKKβ [Dai et al., 2004; Jimi et al., 2004]. In this regard, we have shown that NBD “short-circuits” NF-κB activation leading to attenuation of osteoclast formation and impediment of osteolysis [Dai et al., 2004]. However, assessing the role of NEMO directly in this process was never described. Although the utility of NBD appears so far promising, this compound only impedes binding of NEMO to IKKs, but not other potential functions of NEMO that may not require IKKs. Therefore, direct and focused approaches targeting NEMO elements with potential functional significance appeared plausible. In this regard, the utility of CC2 and LZ peptides as inhibitors of NEMO oligomerization and subsequent NF-κB activity which has been established by Agou et al. [2002, 2004a,b] lends itself as a feasible tool. According to this body of work, it has been established that the carboxyl terminal coiled-coil and LZ domains compose the trimerization domain of NEMO and that inhibition of NEMO oligomerization using overlapping short cognate peptides that obviate mutual NEMO monomer binding inhibits activation of NF-κB [Tegethoff et al., 2003; Agou et al., 2004b]. Importantly, it was further established that these short peptides prevent rearrangement of natively disordered NEMO into organized hexamer bundles [Agou et al., 2004b]. Consistent with these observations, we provide evidence that TAT-fused CC2 and LZ peptides bind avidly to NEMO monomers. Interestingly, however, peptide-bound NEMO monomers undergo rapid degradation following cellular stimulation with RANKL. Preliminary evidence suggests that this event is mediated by ubiquitination implying that conformational changes in folded NEMO molecules or in complex-associated NEMO limit lysine 48 ubiquitination-mediated NEMO destruction, and that native structures of NEMO monomers unable to oligomerize are more susceptible for such signal-induced ubiquitination-targeted degradation.
The choice of CC2 and LZ domain for modulation of NEMO-mediated signaling is rather fundamental in light of their vital role relative to NEMO polyubiquitination-mediated signaling. In this regard, recent studies found that mutations in the CC2 and LZ domains of NEMO, such as D304N, cause signaling defects, whereas another mutation in K270A causes constitutive activation owing to high affinity of CC2-LZ (K270A) domain for homophilic binding without the need for ubiquitination [Bloor et al., 2008]. Another study demonstrated that the LUBAC ligase complex (composed of two RING finger proteins HOIL-1L and HOIP) binds to NEMO and conjugates linear polyubiquitin chains onto specific lysine residues in the CC2 and LZ domains of NEMO [Tokunaga et al., 2009]. These findings buttress the fact that intact CC2–LZ NEMO domains are essential for signal-induced NF-κB activation.
To further bolster the functional significance of inhibiting NEMO oligomerization, we provide direct evidence that cell permeable TAT-CC2 and TAT-LZ peptides are efficient inhibitors of osteoclast differentiation, block inflammatory (TNF-induced) osteoclastogenesis, and abrogate osteolysis, in vivo. These findings are significant owing to the dual role of NEMO as a modulator of inflammatory responses and osteoclastogenesis. Although, specificity and selectivity of these peptides remains to be elucidated, nevertheless, our observations position CC2 and LZ peptides as potential targets for future therapeutic approaches.
Acknowledgments
Grant sponsor: National Institutes of Health Grants; Grant numbers: AR049192, AR054326; Grant sponsor: Shriners Hospital for Children Grants; Grant numbers: 8570, 8510.
This work was funded by National Institutes of Health Grants: AR049192, AR054326 (to Y.A.-A.) and by grants #8570, #8510 from the Shriners Hospital for Children (to Y.A.-A.).
Abbreviations used
- OCPs
osteoclast precursors
- RANK
receptor activator of NF-κB
- RANKL
RANK ligand
- TRAP
tartrate-resistant acid phosphatase
- NBD
NEMO-binding domain
- CC2
coiled-coil-2
- LZ
leucine zipper
References
- Abu-Amer Y. Mechanisms of inflammatory mediators in bone loss diseases. In: Rosier RN, Evans CH, editors. Molecular biology in orthopedics. Rosemont, Illinois: AAOS; 2003. pp. 229–239. [Google Scholar]
- Abu-Amer Y, Ross FP, Edwards J, Teitelbaum SL. Lipopolysaccharide-stimulated osteoclastogenesis is mediated by tumor necrosis factor via its p55 receptor. J Clin Invest. 1997;100:1557–1565. doi: 10.1172/JCI119679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Amer Y, Dowdy S, Ross F, Clohisy J, Teitelbaum S. TAT fusion proteins containing tyrosine 42-deleted IkBa arrest osteoclastogenesis. J Biol Chem. 2001;276:30499–30503. doi: 10.1074/jbc.M104725200. [DOI] [PubMed] [Google Scholar]
- Agou F, Ye F, Goffinont S, Courtois G, Yamaoka S, Israel A, Veron M. NEMO trimerizes through its coiled-coil C-terminal domain. J Biol Chem. 2002;277:17464–17475. doi: 10.1074/jbc.M201964200. [DOI] [PubMed] [Google Scholar]
- Agou F, Courtois G, Chiaravalli J, Baleux F, Coic Y-M, Traincard F, Israel A, Veron M. Inhibition of NF-{kappa}B activation by peptides targeting NF-{kappa}B essential modulator (NEMO) oligomerization. J Biol Chem. 2004a;279:54248–54257. doi: 10.1074/jbc.M406423200. [DOI] [PubMed] [Google Scholar]
- Agou F, Traincard F, Vinolo E, Courtois G, Yamaoka S, Israel A, Veron M. The trimerization domain of NEMO is composed of the interacting C-terminal CC2 and LZ coiled-coil subdomains. J Biol Chem. 2004b;279:27861–27869. doi: 10.1074/jbc.M314278200. [DOI] [PubMed] [Google Scholar]
- Baldwin AS., Jr The NF-kB and IkB proteins: New discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [Review] [180 refs] [DOI] [PubMed] [Google Scholar]
- Bloor S, Ryzhakov G, Wagner S, Butler PJG, Smith DL, Krumbach R, Dikic I, Randow F. Signal processing by its coil zipper domain activates IKK{gamma} Proc Natl Acad Sci. 2008;105:1279–1284. doi: 10.1073/pnas.0706552105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, Martinon F. Inflammatory diseases: Is ubiquitinated NEMO at the hub? Curr Biol. 2004;14:R1040–R1042. doi: 10.1016/j.cub.2004.11.040. [DOI] [PubMed] [Google Scholar]
- Chaisson ML, Branstetter DG, Derry JM, Armstrong AP, Tometsko ME, Takeda K, Akira S, Dougall WC. Osteoclast differentiation is impaired in the absence of IkB kinase-alpha. J Biol Chem. 2004;279:54841–54848. doi: 10.1074/jbc.M406392200. [DOI] [PubMed] [Google Scholar]
- Clohisy JC, Roy BC, Biondo C, Frazier E, Willis D, Teitelbaum SL, Abu-Amer Y. Direct inhibition of NF-kappa B blocks bone erosion associated with inflammatory arthritis. J Immunol. 2003;171:5547–5553. doi: 10.4049/jimmunol.171.10.5547. [DOI] [PubMed] [Google Scholar]
- Courtois G, Smahi A, Israel A. NEMO/IKK-gamma: Linking NF-kB to human disease. Trends Mol Med. 2001;7:427–430. doi: 10.1016/s1471-4914(01)02154-2. [DOI] [PubMed] [Google Scholar]
- Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IkappaB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J Biol Chem. 2004;279:37219–37222. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- DiDonato J, Hayakawa M, Rothwarf D, Zandi E, Karin M. A cytokine-responsive I-κB kinase that activates the transcription factor Nf-kB. Nature. 1997;388:548–554. doi: 10.1038/41493. [DOI] [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Poljak L, Shores E, Brown K, Leonardi A, Tran T, Boyce B, Siebenlist U. Requirment for NF-kB in osteoclast and B-cell development. Genes Dev. 1997;11:3482–3496. doi: 10.1101/gad.11.24.3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama T, Dai S, Abbas S, Yamanaka Y, Abu-Amer Y. Inhibition of inflammatory bone erosion by constitutively active STAT-6 through blockade of JNK and NF-kB activation. Arthritis Rheum. 2005;52:2719–2729. doi: 10.1002/art.21286. [DOI] [PubMed] [Google Scholar]
- Jimi E, Aoki K, Saito H, D’Acquisto F, May MJ, Nakamura I, Sudo T, Kojima T, Okamoto F, Fukushima H, Okabe K, Ohya K, Ghosh S. Selective inhibition of NF-kappa B blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat Med. 2004;10:617–624. doi: 10.1038/nm1054. [DOI] [PubMed] [Google Scholar]
- Karin M. The IkappaB kinase—A bridge between inflammation and cancer. Cell Res. 2008;18:334–342. doi: 10.1038/cr.2008.30. [DOI] [PubMed] [Google Scholar]
- Karin M, Yamamoto Y, Wang M. The IKK NF-kB system: A treasure trove for drug development. Nat Rev. 2004;3:17–26. doi: 10.1038/nrd1279. [DOI] [PubMed] [Google Scholar]
- Ku CL, Dupuis-Girod S, Dittrich AM, Bustamante J, Santos OF, Schulze I, Bertrand Y, Couly G, Bodemer C, Bossuyt X, Picard C, Casanova JL. NEMO mutations in 2 unrelated boys with severe infections and conical teeth. Pediatrics. 2005;115:e615–e619. doi: 10.1542/peds.2004-1754. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKK-alpha-limits macrophage NF-kB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- Li Q, Lu Q, Bottero V, Estepa G, Morrison L, Mercurio F, Verma IM. Enhanced NF-kB activation and cellular function in macrophages lacking IkB kinase 1 (IKK1) Proc Natl Acad Sci. 2005;102(35):12425–12430. doi: 10.1073/pnas.0505997102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-kappa B activation by a peptide that blocks the interaction of NEMO with the Ikappa B kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- May MJ, Marienfeld RB, Ghosh S. Characterization of the Ikappa B-kinase NEMO binding domain. J Biol Chem. 2002;277:45992–46000. doi: 10.1074/jbc.M206494200. [DOI] [PubMed] [Google Scholar]
- Mercurio F, Zhu H, Murray BW, Shevchenko A, Bennett BL, Li J, Young DB, Barbosa M, Mann M, Manning A, Rao A. IKK-1 and IKK-2: Cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science (New York, NY) 1997;278:860–866. doi: 10.1126/science.278.5339.860. [DOI] [PubMed] [Google Scholar]
- Ni C-Y, Wu Z-H, Florence WC, Parekh VV, Arrate MP, Pierce S, Schweitzer B, Van Kaer L, Joyce S, Miyamoto S, Ballard DW, Oltz EM. Cutting edge: K63-linked polyubiquitination of NEMO modulates TLR signaling and inflammation in vivo. J Immunol. 2008;180:7107–7111. doi: 10.4049/jimmunol.180.11.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, Teitelbaum SL. The I-kappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003;198:771–781. doi: 10.1084/jem.20030116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero JE, Dai S, Foglia D, Alhawagri M, Vacher J, Pasparakis M, Abu-Amer Y. Defective osteoclastogenesis by IKKbeta-null precursors is a result of receptor activator of NF-kappaB ligand (RANKL)-induced JNK-dependent apoptosis and impaired differentiation. J Biol Chem. 2008;283:24546–24553. doi: 10.1074/jbc.M800434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier C, Song H, Gao X, Goeddel D, Cao Z, Rothe M. Identification and characterization of an I-κB kinase. Cell. 1997;90:373–383. doi: 10.1016/s0092-8674(00)80344-x. [DOI] [PubMed] [Google Scholar]
- Ruocco MG, Maeda S, Park JM, Lawrence T, Hsu L-C, Cao Y, Schett G, Wagner EF, Karin M. IkB kinase-beta, but not IKK-alpha, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J Exp Med. 2005;201:1677–1687. doi: 10.1084/jem.20042081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata W, Maeda S, Hikiba Y, Yanai A, Ohmae T, Sakamoto K, Nakagawa H, Ogura K, Omata M. Cutting edge: The I{kappa}B kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks inflammatory injury in murine colitis. J Immunol. 2007;179:2681–2685. doi: 10.4049/jimmunol.179.5.2681. [DOI] [PubMed] [Google Scholar]
- Stancovski I, Baltimore D. NF-kB activation: The IkB kinase revealed? Cell. 1997;91:299–302. doi: 10.1016/s0092-8674(00)80413-4. [DOI] [PubMed] [Google Scholar]
- Tegethoff S, Behlke J, Scheidereit C. Tetrameric oligomerization of IkappaB kinase gamma (IKKgamma) is obligatory for IKK complex activity and NF-kappaB activation. Mol Cell Biol. 2003;23:2029–2041. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S-I, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, Iwai K. Involvement of linear polyubiquitylation of NEMO in NF-[kappa]B activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- Vaira S, Alhawagri M, Anwisye I, Kitaura H, Faccio R, Novack DV. RelA/p65 promotes osteoclast differentiation by blocking a RANKL-induced apoptotic JNK pathway in mice. J Clin Invest. 2008a;118:2088–2097. doi: 10.1172/JCI33392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaira S, Johnson T, Hirbe AC, Alhawagri M, Anwisye I, Sammut B, O’Neal J, Zou W, Weilbaecher KN, Faccio R, Novack DV. RelB is the NF-kappaB subunit downstream of NIK responsible for osteoclast differentiation. Proc Natl Acad Sci USA. 2008b;105:3897–3902. doi: 10.1073/pnas.0708576105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation. Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [corrected] [DOI] [PubMed] [Google Scholar]