

Abstract
The enkephalin signaling pathway regulates various neural functions and can be altered by neurodegenerative disorders. In Alzheimer's disease (AD), elevated enkephalin levels may reflect compensatory processes or contribute to cognitive impairments. To differentiate between these possibilities, we studied transgenic mice that express human amyloid precursor protein (hAPP) and amyloid-β (Aβ) peptides in neurons and exhibit key aspects of AD. Met-enkephalin levels in neuronal projections from the entorhinal cortex and dentate gyrus (brain regions important for memory that are affected in early stages of AD) were increased in hAPP mice, as were preproenkephalin mRNA levels. Genetic manipulations that exacerbate or prevent excitotoxicity also exacerbated or prevented the enkephalin alterations. In human AD brains, enkephalin levels in the dentate gyrus were also increased. In hAPP mice, enkephalin elevations correlated with the extent of Aβ-dependent neuronal and behavioral alterations, and memory deficits were reduced by irreversible blockade of μ-opioid receptors with the antagonist β-funaltrexamine. We conclude that enkephalin elevations may contribute to cognitive impairments in hAPP mice and possibly in humans with AD. The therapeutic potential of reducing enkephalin production or signaling merits further exploration.
Keywords: enkephalin, mu-opioid receptor, amyloid, dentate gyrus, entorhinal cortex, spatial memory
Introduction
Alzheimer's disease (AD) causes progressive memory loss and behavioral abnormalities that may reflect alterations in neurotransmitter and neuromodulatory systems. This notion has led to several treatments (Bales et al., 2006; Lleó et al., 2006). Acetylcholinesterase inhibitors and a glutamate receptor antagonist are widely used, and others are in clinical trials, including inhibitors of calcium channels, GABA receptors, or serotonin receptors (Roberson and Mucke, 2006). Current treatments provide limited benefit, and the prevalence of AD is rising at an alarming pace (Ferri et al., 2005). Additional therapeutic strategies are urgently needed. To address this need, we investigated the role of the enkephalin signaling pathway in the pathogenesis of AD.
Enkephalins are part of the endogenous opioid system, which modulates many functions affected by AD, including learning and memory (Messing et al., 1979; Gallagher, 1982; Gallagher et al., 1983; Bodnar and Klein, 2005), synaptic plasticity (Derrick et al., 1992; Do et al., 2002), and emotional behaviors (Nieto et al., 2005). Preproenkephalin (Penk) is proteolytically cleaved to produce met- and leu-enkephalins in the regulated secretory pathway. Enkephalins are stored in dense-core vesicles and coreleased with fast-acting neurotransmitters, such as glutamate, after high-frequency synaptic stimulation (Caudle et al., 1991).
Enkephalin peptides and Penk mRNA are expressed by diverse cells in the brain and periphery (Mansour et al., 1995). In the hippocampus, enkephalin levels are particularly high in the lateral perforant pathway, which arises in the entorhinal cortex, and in mossy fibers, which arise in the dentate gyrus (Gall et al., 1981; Stengaard-Pedersen, 1983). These brain regions are severely affected by AD (Gomez-Isla et al., 1996; Blennow et al., 2006).
Enkephalins bind and activate μ- and δ-opioid receptors (Duka et al., 1981; Mansour et al., 1995). Enkephalins modulate learning and memory (Messing et al., 1979; Gallagher, 1982; Gallagher et al., 1983) and synaptic plasticity (Derrick et al., 1992; Williams and Johnston, 1996; Do et al., 2002) by activating μ-opioid receptors and emotional behaviors by activating δ-opioid receptors (Nieto et al., 2005). Hippocampal μ-opioid receptors are found on cell bodies, dendrites, and axon terminals of inhibitory interneurons (Kalyuzhny and Wessendorf, 1998; Drake and Milner, 1999; Stumm et al., 2004). Consequently, enkephalins can have complex presynaptic and postsynaptic effects (Arvidsson et al., 1995; Ding et al., 1996). For example, activation of μ-opioid receptors hyperpolarizes GABAergic interneurons (Madison and Nicoll, 1988; Svoboda and Lupica, 1998), disinhibiting principal cells and increasing neuronal excitability (Nicoll et al., 1980; Simmons and Chavkin, 1996).
Most studies of the enkephalin system in AD have focused on changes in opioid receptor binding (Rinne et al., 1993; Mathieu-Kia et al., 2001). AD cases showed significant reductions in μ- and δ-opioid receptor binding in the molecular layer of the dentate gyrus and in the entorhinal cortex (Jansen et al., 1990), possibly reflecting compensatory adjustments to changes in enkephalin levels. Consistent with this possibility, elevated hippocampal enkephalin levels have been identified in aged transgenic mice overexpressing human amyloid precursor protein (hAPP) carrying familial AD (FAD)-linked mutations (Diez et al., 2000, 2003). It was unknown whether alterations in enkephalins and opioid receptors contribute to cognitive and behavioral deficits in AD or in hAPP mice.
To elucidate the role of met-enkephalin (ENK) in Aβ-induced neuronal and behavioral deficits, we studied hAPP mice from lines J20 and J9, which have a range of AD-related synaptic, neuronal, and behavioral deficits (Hsia et al., 1999; Mucke et al., 2000; Palop et al., 2003, 2005, 2007; Chin et al., 2005; Roberson et al., 2007).
Materials and Methods
Animals
hAPP mice and NTG controls were from lines J9 and J20, which express hAPP with the Swedish (K670N, M671L) and Indiana (V717F) FAD mutations, and from line I5, which expresses wild-type hAPP (Rockenstein et al., 1995; Mucke et al., 2000). Mice with neuronal overexpression of Fyn kinase (line N8) were crossed with mice from line J9 to establish FYN/hAPP doubly transgenic mice as described previously (Chin et al., 2004, 2005). hAPP mice from the J20 line were crossed with Tau-deficient mice (Dawson et al., 2001) as described previously (Roberson et al., 2007) to produce offspring with two (Tau+/+), one (Tau+/−), or no (Tau−/−) functional Tau alleles in the presence or absence of hAPP. All lines had been crossed for >10 generations with C57BL/6 mice (The Jackson Laboratory, Bar Harbor, ME). For tissue isolation, mice were deeply anesthetized and flush-perfused transcardially with 0.9% saline. The left hemibrain was snap frozen on dry ice and stored at −70°C. The right hemibrain was drop fixed in 4% paraformaldehyde for 48 h at 4°C, washed in PBS, and placed in a 30% sucrose solution for 24 h at 4°C. All experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Human tissues
Formalin-fixed hippocampal tissues from AD cases (five women and three men; age, 71–90 years; mean ± SD, 83.1 ± 7.2) and nondemented controls (three women and four men; age, 71–102 years; mean ± SD, 84.7 ± 12.8) were obtained from the Alzheimer's Disease Research Center at the University of California at San Diego and processed as described previously (Palop et al., 2003). Formalin-fixed tissue was postfixed in 4% phosphate-buffered paraformaldehyde for 72 h before sectioning.
Aβ ELISAs
Snap-frozen brain tissues were homogenized with guanidine buffer followed by ELISA measurements of human Aβ peptides (Cheng et al., 2007). The ELISA for Aβ1–42 detects this specific peptide. The ELISA for Aβ1–x (approximates total Aβ) recognizes N-terminal fragments of Aβ containing the first 28 aa.
Immunohistochemistry
Vibratome sections (40 μm, humans) and sliding microtome sections (30 μm, mice) were immunostained with the standard avidin–biotin/peroxidase method as described previously (Palop et al., 2003). Briefly, after reducing endogenous peroxidase activity and blocking nonspecific binding, floating sections were incubated for 15–16 h with anti-ENK (1:5000 for mouse tissue; 1:500 for human tissue; Millipore Bioscience Research Reagents, Temecula, CA), anti-calbindin (1:30,000; Swant, Bellinzona, Switzerland), or biotinylated anti-Aβ (3D6; 1:400; Elan Pharmaceuticals, South San Francisco, CA) at 4°C. Primary antibodies (except 3D6) were detected with biotinylated goat anti-rabbit secondary antibodies (1:200; Vector Laboratories, Burlingame, CA). Diaminobenzidine was used as a chromagen. Some sections of human tissue were counterstained with hematoxylin 560 (Surgipath Medical Industries, Richmond, IL) for 30 s. Images were acquired on a BX-60 microscope (Olympus, Melville, NY) with a DEI-470 digital camera (Optronics, Goleta, CA). ENK immunostaining was quantitated in two sections (300 μm apart) per mouse. Integrated optical density (IOD) was determined with a Bioquant Image Analysis (Nashville, TN) system and averaged in two areas (0.04 mm2 each) in each of the following regions: molecular layer of the dentate gyrus, hilar region of the dentate gyrus, stratum lucidum of CA3, stratum lacunosum-moleculare of CA3, and stratum radiatum of CA1. ENK levels were calculated from the IOD in target regions divided by the IOD in stratum radiatum of CA1 of the same section (as an internal control region that did not differ between NTG and hAPPFAD mice). For correlation analyses and when only one region is displayed in a graph, ENK immunoreactivity levels are expressed relative to the average value in NTG mice. Calbindin levels and Aβ plaques were measured as described previously (Palop et al., 2003).
Quantitative fluorogenic RT-PCR
Total RNA from individual hippocampi or dissected entorhinal cortex from 450-μm-thick horizontal mouse brain sections was isolated with RNeasy mini columns (Qiagen, Valencia, CA). Reverse transcription (RT) reactions (Applied Biosystems, Foster City, CA) contained 300 ng of DNase-treated total RNA and random hexamer and oligo-d(T) primers (2.5 μm each). Diluted reactions (6 ng of cDNA) were analyzed with SYBR green PCR reagents in duplicate on an ABI Prism 7700 sequence detector (Applied Biosystems). Penk and hAPP mRNA levels were determined relative to standard curves made with serial dilutions of pooled cDNAs (30, 6, and 1.2 ng) from all samples. Values were normalized to mouse GAPDH gene expression and are reported relative to NTG or wild-type hAPP (hAPPWT) controls. The slopes of standard curves, control reactions without RT, and dissociation curves of products indicated adequate PCR quality. The following primers were used. Penk: forward, 5′-GCCTTGTCAATGATGTTTCTTGTC-3′; reverse, 5′-CAACATAGCCATAAGAGACCAATACTG-3′. mGAPDH: forward, 5′-GGGAAGCCCAT CACCATCTT-3′; reverse, 5′-GCCTTCTCCATGGTGGTGAA-3′. hAPP: forward, 5′-GAGGAGGATGCTCGGATGTCT-3′; reverse 5′-AGCCACTTCTTCCTCCTCTGCTA-3′.
Drug treatments
Mice were treated with the irreversible μ-opioid receptor antagonist β-funaltrexamine (β-FNA) (Takemori et al., 1981) (Tocris Bioscience, Ellisville, MO) or vehicle. β-FNA was dissolved in water to a concentration of 0.5 mg/ml and injected subcutaneously at 5 mg/kg once a week for 3 weeks. To minimize stress effects from the injections, behavioral testing began 24–48 h after the injections. During weeks 2 and 3 of treatment, mice were tested in the water maze. Mice were killed 1 week after the fourth injection.
Behavioral tests
Open field.
Spontaneous locomotor activity was measured with an automated Flex-Field/Open Field Photobeam Activity System (San Diego Instruments, San Diego, CA). Mice were placed individually in a clear plastic chamber (41 × 41 × 30 cm), and their horizontal and vertical movements were monitored for 15 min with two 16 × 16 photobeam arrays. Total ambulations in the periphery of the open field were analyzed as the main measure. The apparatus was thoroughly cleaned with 70% ethanol between trials.
Y-maze.
The Y-maze consisted of three compartments (10 × 10 × 10 cm) connected with 4 × 5 × 10 cm passages. Each mouse was placed in one of the compartments and allowed to explore for 6 min. The total number of arm entries was recorded. The apparatus was thoroughly cleaned with 70% ethanol between trials.
Morris water maze.
The water maze consisted of a pool (122 cm diameter) filled with water (18 ± 1°C) made opaque with nontoxic white tempera paint powder and was placed in a room surrounded by distinct extramaze cues. Two identical pools were used in two different rooms with distinct cues. Pretraining and visible platform training were performed in one room, whereas hidden-platform training was performed in the second room. Mice were first given four pretraining trials in which they had to swim down a channel (15 × 122 cm) and mount a platform hidden 1.5 cm below the water surface at the end of the channel. The following day, mice were trained in the actual water maze. For cued training, the platform (14 × 14 cm) was submerged (1.5 cm) but visibly cued with a black and white striped mast (15 cm in height). On 3 consecutive days, mice received two training sessions per day separated by 3–4 h. Each session consisted of two training trials with an intertrial interval of 10 min. For each session, the platform was moved to a new location, and the drop location was changed semirandomly between trials. Mice were then moved to the second room for hidden-platform training during which a similar platform was submerged (1.5 cm) and not visibly cued. On 5 consecutive days, mice received two training sessions separated by 3–4 h. Each session consisted of three trials (10 min between trials). The platform location remained the same throughout the hidden-platform training, but the drop location was changed semirandomly between trials. Mice had 60 s to locate the platform during training. Mice that did not find the platform were guided to it and placed on it for 10 s. A spatial probe trial was performed 16–18 h after the third day of training (beginning of session 7) and 24 and 72 h after the hidden-platform training. The platform was removed, and mice were allowed to swim in the pool for 60 s. Escape latencies, swim paths, swim speeds, percentage of time spent in each quadrant, and platform crossings were recorded with an EthoVision video tracking system (Noldus, Wageningen, The Netherlands).
Hot plate test
Mice were treated with β-FNA (5 mg/kg, s.c.) or vehicle weekly for 2 weeks. Forty-eight hours after the last β-FNA injection, baseline nociceptive responses were tested on a hot plate analgesia meter (IITC, Woodland Hills, CA) set at 55 ± 0.1°C. Mice were placed in a Plexiglas cylinder (diameter, 4.5 inches) on the hot plate, and the time it took for the mice to lick a hindpaw or jump was recorded. Mice were then immediately removed from the hot plate and injected with morphine (10 or 20 mg/kg, i.p.; Sigma, St. Louis, MO) dissolved in 0.9% saline to a concentration of 1 or 2 mg/ml. Thirty minutes after the morphine injections, mice were placed on the hotplate again and response latencies were recorded. Morphine-induced analgesia was expressed as the percentage of maximum possible effect, calculated as follows: [(test latency − baseline latency)/60 s] × 100. Approximately 1 h after the morphine injection, mice were placed in an open field, and morphine-induced locomotor activity was monitored for 5 min.
Statistical analysis
Statistical analyses were performed with Statview 5.0 (SAS Institute, Cary, NC). Differences between means were assessed by Student's t test and differences among means by one-, two-, or three-way ANOVA (with or without repeated measures), followed by Fisher's PLSD post hoc test or Dunnett's t test. Correlations were assessed by linear regression analysis.
Results
Enkephalin elevations in hAPP mice and in humans with AD
hAPPFAD mice from line J20, which have high levels of Aβ in the hippocampus and neocortex (Mucke et al., 2000), had much higher levels of ENK immunoreactivity in the mossy fiber and lateral perforant pathways than NTG controls (Fig. 1 A–D). At 2–4 months of age, hAPPFAD mice showed increased ENK immunostaining primarily in the hilar region of the dentate gyrus and in mossy fiber terminals in the stratum lucidum of CA3 (Figs. 1A,B, 2A). By 6 months, hAPPFAD mice had increased ENK levels also in the terminal zones of the lateral perforant path: the outer molecular layer of the dentate gyrus and the stratum lacunosum-moleculare of CA1/3 (Figs. 1C,D, 2B). Consistent with these findings, 6-month-old hAPPFAD mice had higher levels of ENK in the entorhinal cortex than NTG mice, particularly in the superficial layer I and the pyramidal cell layers II/III (data not shown), from which the perforant path emanates. In both hAPPFAD mice and NTG controls, ENK expression in the outer molecular layer was confined to the outer two-thirds of this layer and was more prominent in the ventral than the dorsal region of the dentate gyrus (Fig. 1 A–D and data not shown), which is typical for mice (Gall, 1988).
Figure 1.

Increased hippocampal ENK immunoreactivity in hAPPFAD mice and humans with AD. A–D, Horizontal brain sections of 2- to 4-month-old (A, B) and 6-month-old (C, D) mice were immunostained for ENK. Compared with NTG controls (A, C), hAPPFAD mice (B, D) had increased ENK immunoreactivity in the outer molecular layer (arrowhead) and hilar region of the dentate gyrus, the stratum lacunosum-moleculare, and the mossy fibers (arrows). E, F, Horizontal sections of human hippocampal tissue depicting differences in ENK immunoreactivity in a 71-year-old nondemented control (E) and a 90-year-old AD patient (F). G–J, Hippocampal sections from different AD cases were immunostained for ENK and counterstained with hematoxylin. Blessed Dementia Scale scores are shown in parentheses. gc, Granular layer; hil, hilus; mf, mossy fibers; oml, outer molecular layer; slm, stratum lacunosum-moleculare; sp, stratum pyramidale; sr, stratum radiatum. Abbreviations are the same in subsequent figures.
Figure 2.

Quantitative comparison of hippocampal ENK and Penk expression in hAPPFAD mice and NTG controls. A, B, Densitometric quantitation of ENK immunoreactivity in specific hippocampal subregions (normalized to values from the stratum radiatum of CA1) from sections of 2- to 4-month-old (A) and 6-month-old (B) mice. n = 10–14 mice per age and genotype. C, Quantitative RT-PCR analysis of Penk mRNA levels in the hippocampus of 2- to 3-month-old mice and in the entorhinal cortex of 4- to 6-month-old mice relative to NTG controls. n = 6 (hippocampus) or 11–15 (entorhinal cortex) mice per age and genotype. D, In 2- to 3-month-old hAPPFAD mice, ENK immunoreactivity levels in the hilus correlated with hippocampal Penk mRNA levels. R2 and p values refer to hAPPFAD mice only. *p < 0.05, **p < 0.01, ***p < 0.001 versus NTG (Student's t test). Values are mean ± SEM.
Compared with nondemented controls, human AD cases also had increased ENK levels in the hilus, outer molecular layer, and stratum lacunosum-moleculare (Fig. 1 E–J), underlining the relevance of the alterations observed in hAPPFAD mice to the human condition. Moreover, the highest levels of ENK were observed in the cases with the highest (worst) scores on the Blessed Dementia Scale (Fig. 1 G–J).
To determine whether the increased ENK immunoreactivity in hAPPFAD mice was caused by changes in gene expression, we measured Penk mRNA levels in the hippocampus and entorhinal cortex by quantitative RT-PCR. In both brain regions, Penk mRNA levels were twofold to threefold higher in hAPPFAD mice than in NTG controls (Fig. 2C). In addition, hippocampal levels of ENK immunoreactivity and Penk mRNA were strongly correlated in hAPPFAD mice (Fig. 2D).
ENK elevations depend on Aβ levels but not on plaque formation
To exclude the possibility that ENK elevations in hAPPFAD mice were caused by hAPP overexpression per se, we measured the levels of Penk mRNA and ENK immunoreactivity in 2- to 3-month-old transgenic mice from line I5. Mice from this line express wild-type hAPP (hAPPWT) at levels comparable with those of hAPPFAD in the J20 line (Fig. 3A). Because of the FAD mutations, total Aβ1–x and Aβ1–42 levels were much higher in the hippocampus of hAPPFAD mice than in hAPPWT mice (Fig. 3B). Hippocampal levels of Penk mRNA were also higher in hAPPFAD mice than in hAPPWT mice (Fig. 3A) and not significantly different between hAPPWT mice and NTG controls (data not shown). Compared with NTG controls, only hAPPFAD mice showed marked increases in ENK immunostaining in the dentate gyrus (Fig. 3C). Densitometric analysis confirmed that there was no significant difference in ENK immunostaining in the hilus, mossy fibers, and stratum lacunosum-moleculare between hAPPWT mice and NTG controls (Fig. 3D), although there was a small increase in ENK immunostaining in the outer molecular layer of the dentate gyrus. These data suggest that increased ENK expression in hAPPFAD mice is dependent on Aβ levels or on another effect of the FAD mutations.
Figure 3.

Comparable expression of wild-type hAPP does not increase hippocampal ENK and Penk expression. A, B, Hippocampal levels of hAPP mRNA (A right), Penk mRNA (A, left), Aβ1–x (B, left), and Aβ1–42 (B, right) in hAPPWT mice from line I5 and hAPPFAD mice from line J20 (n = 7–10 mice per genotype; age, 2–3 months). C, Coronal sections illustrating typical ENK immunostaining in 4- to 6-month-old mice of the indicated genotypes. D, Densitometric quantitation of ENK immunoreactivity revealed no significant differences between hAPPWT mice and age-matched NTG controls in all hippocampal subfields except the oml of the dentate gyrus. E, Levels of ENK immunoreactivity in the hilus did not correlate with hippocampal plaque loads in 6-month-old hAPPFAD mice. *p < 0.05, **p < 0.01, ***p < 0.001 (Student's t test). Values are mean ± SEM.
ENK elevation in the dentate gyrus of aged hAPPFAD mice has previously been attributed to amyloid deposition (Diez et al., 2000, 2003). However, increased ENK expression was detectable in hAPPFAD mice at 2–3 months of age (Figs. 1B, 2A), before Aβ deposits form in line J20 (Mucke et al., 2000). To assess whether Aβ deposition worsens ENK increases in older mice, we examined the relationship between ENK elevation and plaque formation in 4- to 6-month-old hAPPFAD mice (Fig. 3E). We found no correlation between hippocampal ENK expression and Aβ deposition at this age. Thus, the ENK elevations in hAPPFAD mice are not dependent on plaque formation.
ENK elevations correlate with calbindin depletions and behavioral alterations in hAPPFAD mice
hAPPFAD mice with high levels of Aβ have depletions of the calcium-binding protein calbindin-D28K in granule cells of the dentate gyrus that correlate well with other Aβ-dependent alterations, including deficits in the Morris water-maze task (Palop et al., 2003, 2005, 2007; Chin et al., 2004, 2005). Such reductions in calbindin also occur in humans with AD and likely reflect a compensatory response to Aβ-induced alterations in neuronal activity and calcium influx (Palop et al., 2003, 2007). To determine whether the dysregulation of calbindin and ENK expression have related underlying mechanisms, we compared the level of hippocampal immunostaining of calbindin and ENK by densitometry. Calbindin depletions in the outer molecular layer of the dentate gyrus correlated with ENK elevations in the hilus of 2- to 4-month-old and 6-month-old hAPPFAD mice (Fig. 4).
Figure 4.

ENK elevations in hAPPFAD mice correlate with calbindin depletions in the dentate gyrus. A, B, Horizontal brain sections of 2- to 4-month-old (A) and 6-month-old (B) hAPPFAD mice (J20 line) were immunostained for ENK and calbindin, and immunoreactivities were quantitated by densitometry. ENK elevations in the hilus correlated with calbindin depletions in the outer molecular layer in hAPPFAD mice. R2 and p values refer to hAPPFAD mice only.
To test whether ENK elevations also correlate with behavioral alterations, 3- to 5-month-old hAPPFAD mice and NTG controls were trained in a Morris water maze (Fig. 5). Consistent with previous reports (Palop et al., 2003; Kobayashi and Chen, 2005; Cheng et al., 2007; Roberson et al., 2007), hAPPFAD mice showed learning deficits in this test (Fig. 5A). Spatial memory was assessed in a probe trial (hidden platform removed) at the beginning of the fourth day of hidden-platform training (before training session 7). hAPPFAD mice crossed over the target location less often (Fig. 5B) and spent less time searching in the target quadrant (data not shown) than NTG controls. Deficits in water-maze performance correlated with increased ENK levels (Fig. 5C,D) and with calbindin depletion (Palop et al., 2003) (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). hAPPFAD mice also showed hyperactivity in an open field (2258 ± 184 total movements for hAPPFAD vs 1505 ± 53 for NTG; p < 0.001) and a Y-maze (33.8 ± 3.5 arm entries for hAPPFAD vs 23.9 ± 1.3 for NTG; p < 0.01). Hyperactivity is a behavioral phenotype displayed by several hAPP lines (Kobayashi and Chen, 2005) and may relate to impaired hippocampal encoding of contextual information (Godsil et al., 2005). Indeed, hyperactivity in hAPPFAD mice correlated with spatial learning and memory impairments in the water maze (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Notably, hyperactivity also correlated with elevations in hippocampal ENK levels (Fig. 5E,F) and depletion of calbindin (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material) in hAPPFAD mice.
Figure 5.

ENK elevations correlate with behavioral alterations in hAPPFAD mice. A, B, hAPPFAD mice from line J20 and NTG controls (n = 10 mice per genotype; age, 3–5 months) were trained in a Morris water maze. A, No differences were found on the first training trial (T1). However, hAPPFAD mice showed learning deficits in the cued-platform (p < 0.05) and hidden-platform (p = 0.001) components of this task (repeated-measures ANOVA). B, During a probe trial (platform removed) at the beginning of the fourth day of training (before session 7), hAPPFAD mice crossed over the target location less often than NTG controls. C–F, ENK elevations in the hilus of hAPPFAD mice correlated with increased average distance required to reach the hidden platform (sessions 1–10) in the water maze (C), decreased target crossings during the probe trial (D), and hyperactivity in the open field (E) and Y-maze (F). R2 and p values refer to hAPPFAD mice only. **p < 0.01 (Student's t test). Values are mean ± SEM.
Together, these results suggest that similar mechanisms may be involved in the Aβ-induced alterations in ENK and calbindin levels, both of which correlated tightly with behavioral abnormalities in hAPPFAD mice.
Genetic manipulations that exacerbate or prevent Aβ-induced neuronal overexcitation also exacerbate or prevent alterations in ENK levels
Increasing neuronal levels of the tyrosine kinase Fyn sensitizes neurons to aberrant overexcitation and to other Aβ-induced abnormalities (Chin et al., 2004, 2005; Palop et al., 2007). Overexpression of Fyn in low-expresser hAPP mice (line J9, hAPPlow) elicits memory deficits as well as prominent calbindin reductions in granule cells of the dentate gyrus (Chin et al., 2005). In contrast, reduction of the microtubule-associated protein Tau prevents cognitive impairments and neuronal depletions of calbindin in hAPPFAD mice with high levels of Aβ (line J20), most likely through a mechanism that protects against aberrant neuronal overexcitation (Palop et al., 2007; Roberson et al., 2007). By experimentally modulating the levels of Fyn and Tau, we tested whether the ENK alterations may be related to the same pathogenic cascade that leads from increased Aβ levels to aberrant neuronal overexcitation and cognitive decline.
First, we compared hippocampal ENK expression in four groups of 7- to 9-month-old mice: FYN transgenic mice with neuronal overexpression of mouse Fyn (Kojima et al., 1997, 1998), hAPP mice from line J9 (hAPPlow) that express hAPPFAD and Aβ at approximately one-half the levels found in hAPP mice from line J20 (Mucke et al., 2000), FYN/hAPPlow doubly transgenic mice, and NTG mice. Compared with NTG mice, only FYN/hAPPlow mice had significant increases in ENK in the dentate gyrus (Fig. 6), demonstrating ENK elevation in an independent hAPPFAD line and implicating Fyn-related pathways in the pathogenesis of this alteration.
Figure 6.

Increased neuronal Fyn expression elicits ENK elevations in low-expresser hAPPFAD mice. hAPPFAD mice from the low-expresser J9 line (hAPPlow) were crossed with mice that overexpress murine Fyn in neurons (FYN). The resulting offspring were analyzed at 7–9 months of age. A, Sagittal sections of the hippocampus immunostained for ENK. B, Densitometric quantitation of ENK immunoreactivity in different subfields of the hippocampus (n = 6–8 mice per genotype). Two-way ANOVA revealed a significant APP × FYN interaction (p < 0.01) for both the hilus and mossy fibers. *p < 0.05, **p < 0.01 versus NTG (Dunnett's test). Values are mean ± SEM.
Next, to determine whether Tau reduction prevents Aβ-dependent increases in ENK levels, we compared hippocampal ENK levels in the following groups of mice at 5–7 months: hAPPFAD mice from line J20 with two (hAPPFAD/Tau+/+), one (hAPPFAD/Tau+/−), or no (hAPPFAD/Tau−/−) functional Tau alleles, and Tau+/+, Tau+/−, or Tau−/− mice without hAPPFAD (Fig. 7). Tau reduction dose-dependently prevented ENK elevations in the hilus (Fig. 7A,B) and mossy fibers (Fig. 7A,C) of hAPPFAD mice, but did not alter ENK expression in the absence of hAPPFAD (Fig. 7).
Figure 7.

Tau reduction prevents ENK elevations in hAPPFAD mice from line J20. Mice were analyzed at 5–7 months of age. A, Coronal sections of the hippocampus immunostained for ENK. B, C, ENK immunoreactivity in the hilus (B) and mossy fibers (C) was quantitated densitometrically in the groups indicated [n = 7–10 mice per genotype, except for hAPPFAD/Tau+/+ mice (n = 3)]. Two-way ANOVA revealed a significant APP effect (p < 0.001), Tau effect (p < 0.05), and APP × Tau interaction (p < 0.01) for both regions. **p < 0.01 versus Tau+/+ mice without hAPP (Dunnett's test); +p < 0.05, ++p < 0.01 versus Tau+/+ mice with hAPP (Fisher's PLSD test). Values are mean ± SEM.
Blocking μ-opioid receptors ameliorates memory deficits in hAPPFAD mice
The association of increased ENK levels with behavioral deficits suggests that ENK either mediates or counteracts Aβ-induced neuronal dysfunction. To differentiate between these possibilities, we assessed whether inhibiting ENK signaling improves or worsens Aβ-induced behavioral alterations. We treated 9- to 10-month-old hAPPFAD and NTG mice with the long-lasting, irreversible μ-opioid receptor antagonist β-FNA or with vehicle once a week for 3 weeks. During the second and third week of treatments, mice were trained in the Morris water maze (Fig. 8).
Figure 8.
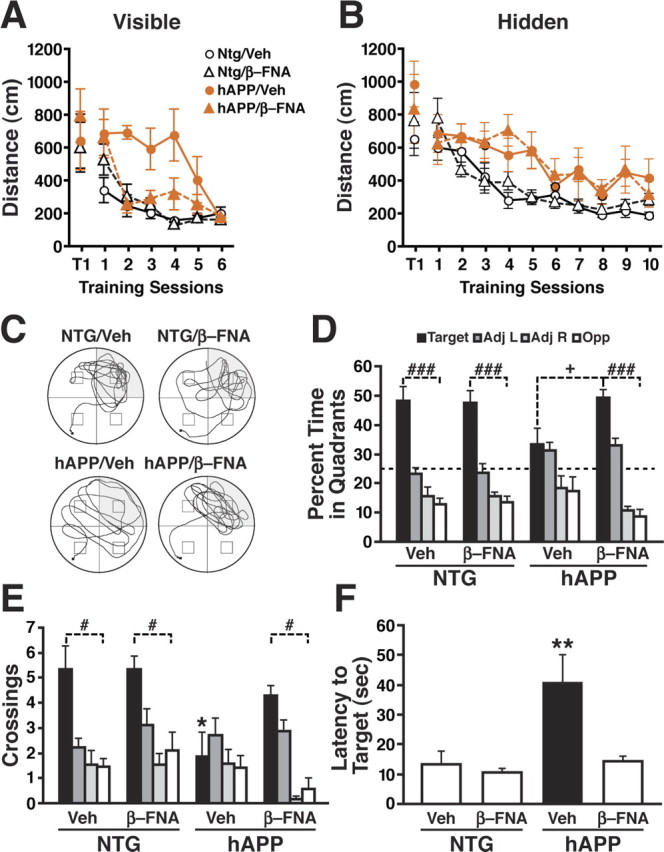
μ-Opioid receptor antagonist ameliorates Aβ-induced behavioral deficits in hAPPFAD mice. hAPPFAD mice and NTG controls were treated with the irreversible μ-opioid receptor antagonist β-FNA or vehicle (Veh) and tested in the Morris water maze (n = 7–9 mice per genotype and treatment; age, 9–10 months). A, B, β-FNA improved the ability of hAPPFAD mice to learn the cued-platform location (p < 0.01 for genotype × treatment interaction by ANOVA; A), but not the hidden-platform location (B). The β-FNA regimen used here had no effect on NTG controls. C–F, Results from a probe trial performed 72 h after the last hidden-platform training trial. C, Representative swim paths show increased preference for the target quadrant in β-FNA-treated hAPPFAD mice. D, Percentage of time spent in different quadrants (p < 0.05 for group × quadrant interaction by ANOVA). E, Number of times mice crossed the platform location or corresponding locations in the other quadrants (p < 0.05 for group × quadrant interaction by ANOVA). F, Latency to reach the platform location (p < 0.05 for genotype × treatment interaction by ANOVA). ###p < 0.001, #p < 0.05 versus all nontarget quadrants; *p < 0.05, **p < 0.01 versus all other groups (Fisher's PLSD test); +p < 0.05 versus β-FNA-treated hAPP mice (Student's t test). Values are mean ± SEM.
On the first training trial (T1), mice swam a similar distance (Fig. 8A) and required a similar amount of time (data not shown) to find the visible platform, independent of treatment or genotype. Subsequently, it took vehicle-treated hAPPFAD mice longer to learn the visible platform task than vehicle- or β-FNA-treated NTG mice, although by the end of the training vehicle-treated hAPPFAD mice performed as well as the NTG control groups (Fig. 8A). Notably, β-FNA treatment significantly improved the performance of hAPPFAD mice in this task (Fig. 8A).
hAPPFAD mice took significantly longer than NTG mice to learn the hidden-platform component of the water-maze task independent of treatment (Fig. 8B). β-FNA treatment had no effect on the performance of NTG mice (Fig. 8A,B) or on swim speeds in either genotype (data not shown).
Spatial memory was assessed in probe trials performed at the beginning of the fourth day of training (before session 7) and 24 and 72 h after the completion of hidden-platform training (Fig. 8 C–F; supplemental Fig. 3, available at www.jneurosci.org as supplemental material). In the last probe trial, β-FNA treatment significantly increased the amount of time hAPPFAD mice spent searching in the target quadrant (Fig. 8C,D) and the number of times they crossed over the target location (Fig. 8E), compared with vehicle-treated hAPPFAD mice. β-FNA treatment also reduced the time it took hAPPFAD mice to reach the target location (Fig. 8F). In NTG mice, the dose of β-FNA used here increased the percentage of time they spent in the target quadrant on the first probe trial but had no effects on target crossings or time required to reach the target location (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Independent repetition of the above experiment in two additional cohorts of mice with each cohort including 7–11 mice per genotype and treatment yielded similar results (data not shown).
To confirm that the dose of β-FNA used in the behavioral studies was effective in blocking μ-opioid receptor function in hAPP mice and NTG mice, we pretreated mice once a week for 2 weeks with either β-FNA (5 mg/kg, s.c., per week) or vehicle. We then tested whether the β-FNA pretreatment antagonized morphine-induced analgesia and hyperactivity 48 h after the last drug injection (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). β-FNA pretreatment significantly antagonized the antinociceptive actions of morphine in the hot plate test (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). β-FNA pretreatment also antagonized morphine-induced hyperactivity (10 mg/kg morphine) in an open field (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). Because there was no significant interaction between genotype and the effects of β-FNA on morphine-induced analgesia or hyperactivity by ANOVA, the data obtained in different genotypes were combined. Together, these results confirm that β-FNA effectively antagonized μ-opioid receptor activity in our mice, consistent with previous findings (Takemori et al., 1981; Frischknecht et al., 1983; Broadbear et al., 2000).
Discussion
This study shows that high levels of Aβ increase the expression of Penk and ENK in the hippocampus and entorhinal cortex independent of plaque formation. Lower levels of Aβ had similar effects when neurons were sensitized to Aβ by increased expression of Fyn kinase. Lowering endogenous Tau, which increases resistance to aberrant neuronal overexcitation (Palop et al., 2007; Roberson et al., 2007), reduced the Aβ-induced increase in ENK levels. Because ENK levels correlated with behavioral alterations and neuronal calbindin depletion, we hypothesized that overstimulation of μ-opioid receptors by ENK mediates some Aβ-induced neurological impairments. Indeed, treatment of hAPP mice with a μ-opioid receptor antagonist reduced Aβ-dependent water-maze deficits. We also found that ENK elevations in the hippocampus of AD cases increased with the severity of the disease. Because excessive stimulation functionally uncouples opioid receptors from G-proteins and causes internalization (Williams et al., 2001; von Zastrow et al., 2003), reductions in μ-opioid receptor binding in hippocampus and dentate gyrus of AD patients (Jansen et al., 1990; Rinne et al., 1993; Mathieu-Kia et al., 2001) may be compensatory responses to ENK elevation.
Increased ENK levels in the hippocampus and entorhinal cortex of 21- to 27-month-old hAPP mice from two other transgenic lines have been postulated to represent a response to plaque-induced neuronal injury (Diez et al., 2000, 2003). However, ENK elevations were prominent in hAPP mice at 2–4 months of age, when plaques have not yet formed in line J20 mice (Mucke et al., 2000). Moreover, ENK levels did not correlate with the extent of Aβ deposits in older mice. These results are consistent with our recent demonstration that other neuronal alterations and memory deficits in 3- to 4-month-old hAPP mice from line J20 correlate with forebrain levels of the soluble Aβ oligomer Aβ*56, but not with plaque loads (Cheng et al., 2007).
To assess whether ENK elevations in hAPP mice may be caused by hAPP rather than by Aβ, we compared hAPPFAD mice from line J20 with hAPPWT mice from line I5, which have similar hAPP levels but different Aβ levels. Hippocampal levels of Penk mRNA and ENK immunoreactivity were significantly greater in hAPPFAD mice than in hAPPWT mice and NTG controls, which had comparable levels. These results are consistent with the hypothesis that Penk and ENK elevations in hAPP mice are caused by nonfibrillar Aβ, although we cannot exclude contributions by Aβ-independent effects of FAD mutations (De Jonghe et al., 2001; Hecimovic et al., 2004; Wiley et al., 2005).
hAPPFAD mice from the lower-expresser J9 line (hAPPlow) had normal ENK levels, suggesting that relatively high levels of Aβ are required to increase ENK expression. Consistent with reports that Fyn sensitizes neurons to Aβ-induced molecular and functional alterations in hAPPlow mice (Chin et al., 2004, 2005; Palop et al., 2007), increased expression of Fyn elicited prominent increases in ENK expression in hAPPlow/FYN mice. Thus, it is extremely unlikely that ENK elevation in line J20 was caused by a line-specific insertional mutation.
Increased ENK immunoreactivity in mossy fibers and perforant pathway could reflect increased neuronal ENK production, decreased ENK degradation, or accumulation of ENK as a result of reduced synaptic release. Although these possibilities are not mutually exclusive, the significant increases in Penk mRNA levels in the hippocampus and entorhinal cortex of hAPPFAD mice suggest that increased ENK production plays a major role. The tight correlation between increases in ENK immunoreactivity and Penk mRNA across different hAPPFAD mice further supports this conclusion.
Although the precise mechanisms underlying the Aβ-induced increase in Penk and ENK expression remain to be determined, it is likely that aberrant increases in neuronal activity are involved. hAPPFAD mice have increased susceptibility to seizures induced by kainate or pentylenetetrazole (Del Vecchio et al., 2004; Palop et al., 2007; Roberson et al., 2007), which may relate to the increased incidence of seizures in AD patients (Amatniek et al., 2006; Palop et al., 2006). Seizures increase hippocampal ENK levels (Gall, 1988; Morris et al., 1988; Moneta and Höllt, 1990), which may facilitate recurrent limbic seizure activity by disinhibiting neuronal networks via activation of μ-opioid receptors (Kanamatsu et al., 1986; Hong et al., 1988). Intraventricular injection of μ-opioid receptor agonists induced nonconvulsive seizures in rats (Snead and Bearden, 1980; Tortella et al., 1987), whereas blocking μ-opioid receptors with β-FNA attenuated seizure susceptibility in rats treated with ENK (Tortella et al., 1987) or kainate (Liu et al., 2004). Thus, Aβ-induced increases in neuronal excitability may increase neuronal ENK expression, which could promote recurrent seizure activity. Consistent with this possibility, hAPPFAD mice have spontaneous nonconvulsive seizure activity (Palop et al., 2007). Furthermore, ENK elevations in hAPPFAD mice were prevented by Tau reduction and exacerbated by overexpression of Fyn, manipulations that respectively prevent or exacerbate aberrant neuronal overexcitation in hAPPFAD mice (Palop et al., 2007; Roberson et al., 2007).
Increased ENK expression may contribute to neural network dysfunction in hAPPFAD mice and AD. ENK elevations correlated tightly with calbindin depletions in the dentate gyrus, which correlate with reductions in other synaptic activity-dependent proteins and with learning and memory deficits (Palop et al., 2003, 2005). In NTG mice and rats, peripheral administration of ENK, leu-enkephalin, or morphine caused dose-dependent impairments in acquisition and retention of active and passive avoidance conditioning (Izquierdo et al., 1980; Izquierdo and Dias, 1981; Martinez et al., 1985). In rats, morphine also impaired spatial learning and memory in a water maze, an effect that was blocked with the opioid receptor antagonist naloxone (McNamara and Skelton, 1991). Even in untreated mice and rats, peripheral injection of naloxone or selective μ-opioid receptor antagonists after training enhanced retention of active and passive avoidance conditioning (Izquierdo, 1979; Messing et al., 1979; Izquierdo and Graudenz, 1980; Gallagher, 1982, 1985). Opioid antagonists also improved spatial memory in a water maze (Decker et al., 1989) and working memory in a radial arm maze (Gallagher et al., 1983; Canli et al., 1990). These results suggest that activation of μ-opioid receptors by exogenous or endogenous opioids inhibits memory formation or retention. Indeed, water-maze deficits and hyperactivity in an open field and a Y-maze correlated with hippocampal ENK elevations in hAPPFAD mice, suggesting a strong association between Aβ-induced increases in ENK expression and behavioral abnormalities.
To test more directly whether ENK elevations play a causal role in Aβ-induced behavioral deficits, we treated hAPPFAD mice with β-FNA. In hAPPFAD mice, this irreversible μ-opioid receptor antagonist improved cued-platform learning but not hidden-platform learning, possibly because the former task is easier. Although the cued-platform task can be learned in the absence of distal spatial cues, NTG mice and rats do use such cues to advantage in solving this task, a strategy that requires normal hippocampal function (Whishaw et al., 1995; Hamilton et al., 2004). Partial improvement in hippocampal function may have helped hAPPFAD mice to solve the cued-platform task, but may have been insufficient to solve the hidden-platform task, which depends more heavily on spatial learning.
β-FNA may also have affected other brain regions and behavioral domains. Although hAPPFAD mice have normal visual acuity, they have difficulty associating a visual cue with escape from water (deIpolyi et al., 2008), an impairment that might relate to visual processing deficits (O'Brien et al., 2001; Kergoat et al., 2002; Mapstone et al., 2003) or topographic agnosia (Kiyosawa et al., 1989; Mendez and Cherrier, 2003) in AD. Although the precise neural substrates of these behavioral deficits are unknown, aberrant activation of μ-opioid receptors might be involved. β-FNA might also improve motivation, because morphine appears to impair water-maze acquisition in part by reducing the impetus to escape onto the platform, an effect that can be blocked with naloxone (McNamara and Skelton, 1991).
β-FNA did not affect hidden-platform learning but improved spatial memory retention in hAPPFAD mice, as revealed by their performance in the probe trial. We speculate that β-FNA improves memory retention in hAPPFAD mice by counteracting Aβ-induced increases in ENK signaling through μ-opioid receptor stimulation in the hippocampus and entorhinal cortex.
In a double-blind, placebo-controlled clinical study, naloxone improved cognitive and behavioral performance of AD patients (Reisberg et al., 1983). Although subsequent studies found no significant improvements (Pomara et al., 1985; Serby et al., 1986; Tariot et al., 1986), the opioid receptor antagonist naltrexone markedly improved cognitive performance in specific tests in 1 of 9 (Serby et al., 1986) and 1 of 10 (Pomara et al., 1985) AD patients. Overstimulation of μ-opioid receptors might contribute more critically to cognitive failure in some AD patients than others. It is also possible that a more specific μ-opioid receptor antagonist might enhance cognitive performance in a larger proportion of AD patients. Additional studies are warranted to further explore the therapeutic potential of reducing ENK production or, specifically, μ-opioid receptor activity in the context of AD-related cognitive impairments.
Footnotes
This work was supported by National Institutes of Health Grants AG022074, AG023501, and NS041787 to L.M. and RR018928 to the J. David Gladstone Institutes. We thank E. Masliah for human tissue samples; H. Solanoy, X. Wang, and D. Kim for technical support; J. Harris and M. Cisse for comments on this manuscript; G. Howard and S. Ordway for editorial review; J. Carroll for preparation of graphics; and D. McPherson and T. Hamto for administrative support.
References
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, Albert M, Brandt J, Stern Y. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–872. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Arvidsson U, Riedl M, Chakrabarti S, Lee JH, Nakano AH, Dado RJ, Loh HH, Law PY, Wessendorf MW, Elde R. Distribution and targeting of a μ-opioid receptor (MOR1) in brain and spinal cord. J Neurosci. 1995;15:3328–3341. doi: 10.1523/JNEUROSCI.15-05-03328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales KR, Tzavara ET, Wu S, Wade MR, Bymaster FP, Paul SM, Nomikos GG. Cholinergic dysfunction in a mouse model of Alzheimer disease is reversed by an anti-Aβ antibody. J Clin Invest. 2006;116:825–832. doi: 10.1172/JCI27120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Bodnar RJ, Klein GE. Endogenous opiates and behavior: 2004. Peptides. 2005;26:2629–2711. doi: 10.1016/j.peptides.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Broadbear JH, Sumpter TL, Burke TF, Husbands SM, Lewis JW, Woods JH, Traynor JR. Methocinnamox is a potent, long-lasting, and selective antagonist of morphine-mediated antinociception in the mouse: Comparison with clocinnamox, β-funaltrexamine, and β-chlornaltrexamine. J Pharmacol Exp Ther. 2000;294:933–940. [PubMed] [Google Scholar]
- Canli T, Cook RG, Miczek KA. Opiate antagonists enhance the working memory of rats in the radial maze. Pharmacol Biochem Behav. 1990;36:521–525. doi: 10.1016/0091-3057(90)90250-l. [DOI] [PubMed] [Google Scholar]
- Caudle RM, Wagner JJ, Chavkin C. Endogenous opioids released from perforant path modulate norepinephrine actions and inhibitory postsynaptic potentials in guinea pig CA3 pyramidal cells. J Pharmacol Exp Ther. 1991;258:18–26. [PubMed] [Google Scholar]
- Cheng I, Scearce-Levie K, Legleiter J, Palop J, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe K, Muchowski P, Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Yu G-Q, Kojima N, Masliah E, Mucke L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J Neurosci. 2004;24:4692–4697. doi: 10.1523/JNEUROSCI.0277-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, Palop JJ, Puoliväli J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci. 2001;114:1179–1187. doi: 10.1242/jcs.114.6.1179. [DOI] [PubMed] [Google Scholar]
- Decker MW, Introini-Collison IB, McGaugh JL. Effects of naloxone on Morris water maze learning in the rat: Enhanced acquisition with pretraining but not posttraining administration. Psychobiology. 1989;17:270–275. [Google Scholar]
- deIpolyi A, Fang S, Wang F, Mucke L. Altered navigational strategy use and visuospatial deficits in hAPP transgenic mice. Neurobiol Aging. 2008;29:253–266. doi: 10.1016/j.neurobiolaging.2006.10.021. [DOI] [PubMed] [Google Scholar]
- De Jonghe C, Esselens C, Kumar-Singh S, Craessaerts K, Serneels S, Checler F, Annaert W, Van Broeckhoven C, De Strooper B. Pathogenic APP mutations near the γ-secretase cleavage site differentially affect Aβ secretion and APP C-terminal fragment stability. Hum Mol Genet. 2001;10:1665–1671. doi: 10.1093/hmg/10.16.1665. [DOI] [PubMed] [Google Scholar]
- Del Vecchio RA, Gold LH, Novick SJ, Wong G, Hyde LA. Increased seizure threshold and severity in young transgenic CRND8 mice. Neurosci Lett. 2004;367:164–167. doi: 10.1016/j.neulet.2004.05.107. [DOI] [PubMed] [Google Scholar]
- Derrick BE, Rodriguez SB, Lieberman DN, Martinez JL., Jr Mu opioid receptors are associated with the induction of hippocampal mossy fiber long-term potentiation. J Pharmacol Exp Ther. 1992;263:725–733. [PubMed] [Google Scholar]
- Diez M, Koistinaho J, Kahn K, Games D, Hökfelt T. Neuropeptides in hippocampus and cortex in transgenic mice overexpressing V717F β-amyloid precursor protein—initial observations. Neuroscience. 2000;100:259–286. doi: 10.1016/s0306-4522(00)00261-x. [DOI] [PubMed] [Google Scholar]
- Diez M, Danner S, Frey P, Sommer B, Staufenbiel M, Wiederhold KH, Hokfelt T. Neuropeptide alterations in the hippocampal formation and cortex of transgenic mice overexpressing β-amyloid precursor protein (APP) with the Swedish double mutation (APP23) Neurobiol Dis. 2003;14:579–594. doi: 10.1016/j.nbd.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Ding YQ, Kaneko T, Nomura S, Mizuno N. Immunohistochemical localization of μ-opioid receptors in the central nervous system of the rat. J Comp Neurol. 1996;367:375–402. doi: 10.1002/(SICI)1096-9861(19960408)367:3<375::AID-CNE5>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Do VH, Martinez CO, Martinez JL, Jr, Derrick BE. Long-term potentiation in direct perforant path projections to the hippocampal CA3 region in vivo. J Neurophysiol. 2002;87:669–678. doi: 10.1152/jn.00938.2000. [DOI] [PubMed] [Google Scholar]
- Drake CT, Milner TA. μ opioid receptors are in somatodendritic and axonal compartments of GABAergic neurons in rat hippocampal formation. Brain Res. 1999;849:203–215. doi: 10.1016/s0006-8993(99)01910-1. [DOI] [PubMed] [Google Scholar]
- Duka T, Wuster M, Schubert P, Stoiber R, Herz A. Selective localization of different types of opiate receptors in hippocampus as revealed by in vitro autoradiography. Brain Res. 1981;205:181–186. doi: 10.1016/0006-8993(81)90730-7. [DOI] [PubMed] [Google Scholar]
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y, Jorm A, Mathers C, Menezes PR, Rimmer E, Scazufca M. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–2117. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht HR, Siegfried B, Riggio G, Waser PG. Inhibition of morphine-induced analgesia and locomotor activity in strains of mice: a comparison of long-acting opiate antagonists. Pharmacol Biochem Behav. 1983;19:939–944. doi: 10.1016/0091-3057(83)90395-7. [DOI] [PubMed] [Google Scholar]
- Gall C. Seizures induce dramatic and distinctly different changes in enkephalin, dynorphin, and CCK immunoreactivities in mouse hippocampal mossy fibers. J Neurosci. 1988;8:1852–1862. doi: 10.1523/JNEUROSCI.08-06-01852.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall C, Brecha N, Karten HJ, Chang KJ. Localization of enkephalin-like immunoreactivity to identified axonal and neuronal populations of the rat hippocampus. J Comp Neurol. 1981;198:335–350. doi: 10.1002/cne.901980211. [DOI] [PubMed] [Google Scholar]
- Gallagher M. Naloxone enhancement of memory processes: effects of other opiate antagonists. Behav Neural Biol. 1982;35:375–382. doi: 10.1016/s0163-1047(82)91020-2. [DOI] [PubMed] [Google Scholar]
- Gallagher M. Effect of β-funaltrexamine on retention of passive-avoidance conditioning. Behav Neural Biol. 1985;44:499–502. doi: 10.1016/s0163-1047(85)90976-8. [DOI] [PubMed] [Google Scholar]
- Gallagher M, King RA, Young NB. Opiate antagonists improve spatial memory. Science. 1983;221:975–976. doi: 10.1126/science.6879198. [DOI] [PubMed] [Google Scholar]
- Godsil BP, Stefanacci L, Fanselow MS. Bright light suppresses hyperactivity induced by excitotoxic dorsal hippocampus lesions in the rat. Behav Neurosci. 2005;119:1339–1352. doi: 10.1037/0735-7044.119.5.1339. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton DA, Rosenfelt CS, Whishaw IQ. Sequential control of navigation by locale and taxon cues in the Morris water task. Behav Brain Res. 2004;154:385–397. doi: 10.1016/j.bbr.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Hecimovic S, Wang J, Dolios G, Martinez M, Wang R, Goate AM. Mutations in APP have independent effects on Aβ and CTFγ generation. Neurobiol Dis. 2004;17:205–218. doi: 10.1016/j.nbd.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Hong JS, McGinty JF, Grimes L, Kanamatsu T, Obie J, Mitchell CL. Seizure-induced alterations in the metabolism of hippocampal opioid peptides suggest opioid modulation of seizure-related behaviors. NIDA Res Monogr. 1988;82:48–66. [PubMed] [Google Scholar]
- Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izquierdo I. Effect of naloxone and morphine on various forms of memory in the rat: possible role of endogenous opiate mechanisms in memory consolidation. Psychopharmacology (Berl) 1979;66:199–203. doi: 10.1007/BF00427631. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Dias RD. Retrograde amnesia caused by Met-, Leu- and des-Try-Met-enkephalin in the rat and its reversal by naloxone. Neurosci Lett. 1981;22:189–193. doi: 10.1016/0304-3940(81)90086-0. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Graudenz M. Memory facilitation by naloxone is due to release of dopaminergic and β-adrenergic systems from tonic inhibition. Psychopharmacology. 1980;67:265–268. doi: 10.1007/BF00431268. [DOI] [PubMed] [Google Scholar]
- Izquierdo I, Paiva AC, Elisabetsky E. Post-training intraperitoneal administration of leu-enkephalin and β-endorphin causes retrograde amnesia for two different tasks in rats. Behav Neural Biol. 1980;28:246–250. doi: 10.1016/s0163-1047(80)91663-5. [DOI] [PubMed] [Google Scholar]
- Jansen KL, Faull RL, Dragunow M, Synek BL. Alzheimer's disease: changes in hippocampal N-methyl-D-aspartate, quisqualate, neurotensin, adenosine, benzodiazepine, serotonin and opioid receptors—an autoradiographic study. Neuroscience. 1990;39:613–627. doi: 10.1016/0306-4522(90)90246-z. [DOI] [PubMed] [Google Scholar]
- Kalyuzhny AE, Wessendorf MW. Relationship of μ- and δ-opioid receptors to GABAergic neurons in the central nervous system, including antinociceptive brainstem circuits. J Comp Neurol. 1998;392:528–547. [PubMed] [Google Scholar]
- Kanamatsu T, Obie J, Grimes L, McGinty JF, Yoshikawa K, Sabol S, Hong JS. Kainic acid alters the metabolism of Met5-enkephalin and the level of dynorphin A in the rat hippocampus. J Neurosci. 1986;6:3094–3102. doi: 10.1523/JNEUROSCI.06-10-03094.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kergoat H, Kergoat MJ, Justino L, Chertkow H, Robillard A, Bergman H. Visual retinocortical function in dementia of the Alzheimer type. Gerontology. 2002;48:197–203. doi: 10.1159/000058350. [DOI] [PubMed] [Google Scholar]
- Kiyosawa M, Bosley TM, Chawluk J, Jamieson D, Schatz NJ, Savino PJ, Sergott RC, Reivich M, Alavi A. Alzheimer's disease with prominent visual symptoms. Clinical and metabolic evaluation. Ophthalmology. 1989;96:1077–1085. doi: 10.1016/s0161-6420(89)32769-2. discussion 1085–1086. [DOI] [PubMed] [Google Scholar]
- Kobayashi DT, Chen KS. Behavioral phenotypes of amyloid-based genetically modified mouse models of Alzheimer's disease. Genes Brain Behav. 2005;4:173–196. doi: 10.1111/j.1601-183X.2005.00124.x. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SGN, Mayford M, Kandel ER. Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci USA. 1997;94:4761–4765. doi: 10.1073/pnas.94.9.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Ishibashi H, Obata K, Kandel ER. Higher seizure susceptibility and enhanced tyrosine phosphorylation on N-methyl-d-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem. 1998;5:429–445. [PMC free article] [PubMed] [Google Scholar]
- Liu H, Gao HM, Zhang WQ, Tang YY, Song HS. Effects of chronic administration of PL017 and β-funaltrexamine hydrochloride on susceptibility of kainic acid-induced seizures in rats. Sheng Li Xue Bao. 2004;56:101–106. [PubMed] [Google Scholar]
- Lleó A, Greenberg SM, Growdon JH. Current pharmacotherapy for Alzheimer's disease. Annu Rev Med. 2006;57:513–533. doi: 10.1146/annurev.med.57.121304.131442. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Enkephalin hyperpolarizes interneurones in the rat hippocampus. J Physiol (Lond) 1988;398:123–130. doi: 10.1113/jphysiol.1988.sp017033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A, Fox CA, Akil H, Watson SJ. Opioid-receptor mRNA expression in the rat CNS: Anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- Mapstone M, Steffenella TM, Duffy CJ. A visuospatial variant of mild cognitive impairment: getting lost between aging and AD. Neurology. 2003;60:802–808. doi: 10.1212/01.wnl.0000049471.76799.de. [DOI] [PubMed] [Google Scholar]
- Martinez JL, Jr, Conner P, Dana RC. Central versus peripheral actions of Leu-enkephalin on acquisition of a one-way active avoidance response in rats. Brain Res. 1985;327:37–43. doi: 10.1016/0006-8993(85)91496-9. [DOI] [PubMed] [Google Scholar]
- Mathieu-Kia AM, Fan LQ, Kreek MJ, Simon EJ, Hiller JM. μ-, δ- and κ-opioid receptor populations are differentially altered in distinct areas of postmortem brains of Alzheimer's disease patients. Brain Res. 2001;893:121–134. doi: 10.1016/s0006-8993(00)03302-3. [DOI] [PubMed] [Google Scholar]
- McNamara RK, Skelton RW. Pretraining morphine impairs acquisition and performance in the Morris water maze: motivation reduction rather than amnesia. Psychobiology. 1991;19:313–322. [Google Scholar]
- Mendez MF, Cherrier MM. Agnosia for scenes in topographagnosia. Neuropsychologia. 2003;41:1387–1395. doi: 10.1016/s0028-3932(03)00041-1. [DOI] [PubMed] [Google Scholar]
- Messing RB, Jensen RA, Martinez JL, Jr, Spiehler VR, Vasquez BJ, Soumireu-Mourat B, Liang KC, McGaugh JL. Naloxone enhancement of memory. Behav Neural Biol. 1979;27:266–275. doi: 10.1016/s0163-1047(79)92328-8. [DOI] [PubMed] [Google Scholar]
- Moneta ME, Höllt V. Perforant path kindling induces differential alterations in the mRNA levels coding for prodynorphin and proenkephalin in the rat hippocampus. Neurosci Lett. 1990;110:273–278. doi: 10.1016/0304-3940(90)90859-8. [DOI] [PubMed] [Google Scholar]
- Morris BJ, Feasey KJ, Ten Bruggencate G, Herz A, Höllt V. Electrical stimulation in vivo increases the expression of proenkephalin mRNA and decreases the expression of prodynorphin mRNA in rat hippocampal granule cells. Proc Natl Acad Sci USA. 1988;85:3226–3230. doi: 10.1073/pnas.85.9.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Alger BE, Jahr CE. Enkephalin blocks inhibitory pathways in the vertebrate CNS. Nature. 1980;287:22–25. doi: 10.1038/287022a0. [DOI] [PubMed] [Google Scholar]
- Nieto MM, Guen SL, Kieffer BL, Roques BP, Noble F. Physiological control of emotion-related behaviors by endogenous enkephalins involves essentially the Δ opioid receptors. Neuroscience. 2005;135:305–313. doi: 10.1016/j.neuroscience.2005.06.025. [DOI] [PubMed] [Google Scholar]
- O'Brien HL, Tetewsky SJ, Avery LM, Cushman LA, Makous W, Duffy CJ. Visual mechanisms of spatial disorientation in Alzheimer's disease. Cereb Cortex. 2001;11:1083–1092. doi: 10.1093/cercor/11.11.1083. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci USA. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu G-Q, Mucke L. Vulnerability of dentate granule cells to disruption of Arc expression in human amyloid precursor protein transgenic mice. J Neurosci. 2005;25:9686–9693. doi: 10.1523/JNEUROSCI.2829-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Mucke L. A network dysfunction perspective on neurodegenerative diseases. Nature. 2006;443:768–773. doi: 10.1038/nature05289. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu G-Q, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomara N, Roberts R, Rhiew HB, Stanley M, Gershon S. Multiple, single-dose naltrexone administrations fail to effect overall cognitive functioning and plasma cortisol in individuals with probable Alzheimer's disease. Neurobiol Aging. 1985;6:233–236. doi: 10.1016/0197-4580(85)90055-7. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Ferris SH, Anand R, Mir P, Geibel V, De Leon MJ, Roberts E. Effects of naloxone in senile dementia: a double-blind trial. N Engl J Med. 1983;308:721–722. doi: 10.1056/NEJM198303243081213. [DOI] [PubMed] [Google Scholar]
- Rinne JO, Lönnberg P, Marjamäki P, Mölsä P, Säkö E, Paljärvi L. Brain methionine- and leucine-enkephalin receptors in patients with dementia. Neurosci Lett. 1993;161:77–80. doi: 10.1016/0304-3940(93)90144-a. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, Mucke L. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science. 2007;316:750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- Rockenstein EM, McConlogue L, Tan H, Gordon M, Power M, Masliah E, Mucke L. Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of transgenic mice and humans with Alzheimer's disease. J Biol Chem. 1995;270:28257–28267. doi: 10.1074/jbc.270.47.28257. [DOI] [PubMed] [Google Scholar]
- Serby M, Resnick R, Jordan B, Adler J, Corwin J, Rotrosen JP. Naltrexone and Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10:587–590. doi: 10.1016/0278-5846(86)90028-x. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Chavkin C. Endogenous opioid regulation of hippocampal function. Int Rev Neurobiol. 1996;39:145–196. doi: 10.1016/s0074-7742(08)60666-2. [DOI] [PubMed] [Google Scholar]
- Snead OC, III, Bearden LJ. Anticonvulsants specific for petit mal antagonize epileptogenic effect of leucine enkephalin. Science. 1980;210:1031–1033. doi: 10.1126/science.6254150. [DOI] [PubMed] [Google Scholar]
- Stengaard-Pedersen K. Comparative mapping of opioid receptors and enkephalin immunoreactive nerve terminals in the rat hippocampus. A radiohistochemical and immunocytochemical study. Histochemistry. 1983;79:311–333. doi: 10.1007/BF00491768. [DOI] [PubMed] [Google Scholar]
- Stumm RK, Zhou C, Schulz S, Hollt V. Neuronal types expressing μ and δ-opioid receptor mRNA in the rat hippocampal formation. J Comp Neurol. 2004;469:107–118. doi: 10.1002/cne.10997. [DOI] [PubMed] [Google Scholar]
- Svoboda KR, Lupica CR. Opioid inhibition of hippocampal interneurons via modulation of potassium and hyperpolarization-activated cation (Ih) currents. J Neurosci. 1998;18:7084–7098. doi: 10.1523/JNEUROSCI.18-18-07084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori AE, Larson DL, Portoghese PS. The irreversible narcotic antagonistic and reversible agonistic properties of the fumaramate methyl ester derivative of naltrexone. Eur J Pharmacol. 1981;70:445–451. doi: 10.1016/0014-2999(81)90355-1. [DOI] [PubMed] [Google Scholar]
- Tariot PN, Sunderland T, Murphy DL, Cohen MR, Welkowitz JA, Weingartner H, Newhouse PA, Cohen RM. Design and interpretation of opiate antagonist trials in dementia. Prog Neuropsychopharmacol Biol Psychiatry. 1986;10:611–626. doi: 10.1016/0278-5846(86)90031-x. [DOI] [PubMed] [Google Scholar]
- Tortella FC, Robles L, Mosberg HI. Evidence for μ opioid receptor mediation of enkephalin-induced electroencephalographic seizures. J Pharmacol Exp Ther. 1987;240:571–577. [PubMed] [Google Scholar]
- von Zastrow M, Svingos A, Haberstock-Debic H, Evans C. Regulated endocytosis of opioid receptors: cellular mechanisms and proposed roles in physiological adaptation to opiate drugs. Curr Opin Neurobiol. 2003;13:348–353. doi: 10.1016/s0959-4388(03)00069-2. [DOI] [PubMed] [Google Scholar]
- Whishaw IQ, Cassel JC, Jarrad LE. Rats with fimbria-fornix lesions display a place response in a swimming pool: a dissociation between getting there and knowing where. J Neurosci. 1995;15:5779–5788. doi: 10.1523/JNEUROSCI.15-08-05779.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley JC, Hudson M, Kanning KC, Schecterson LC, Bothwell M. Familial Alzheimer's disease mutations inhibit γ-secretase-mediated liberation of β-amyloid precursor protein carboxy-terminal fragment. J Neurochem. 2005;94:1189–1201. doi: 10.1111/j.1471-4159.2005.03266.x. [DOI] [PubMed] [Google Scholar]
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev. 2001;81:299–343. doi: 10.1152/physrev.2001.81.1.299. [DOI] [PubMed] [Google Scholar]
- Williams SH, Johnston D. Actions of endogenous opioids on NMDA receptor-independent long-term potentiation in area CA3 of the hippocampus. J Neurosci. 1996;16:3652–3660. doi: 10.1523/JNEUROSCI.16-11-03652.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]