


Abstract
Many platforms for genome-wide analysis of gene expression contain ‘redundant’ measures for the same gene. For example, the most highly utilized platforms for gene expression microarrays, Affymetrix GeneChip® arrays, have as many as ten or more probe sets for some genes. Occasionally, individual probe sets for the same gene report different trends in expression across experimental conditions, a situation that must be resolved in order to accurately interpret the data. We developed an algorithm, SCOREM, for determining the level of agreement between such probe sets, utilizing a statistical test of concordance, Kendall's W coefficient of concordance, and a graph-searching algorithm for the identification of concordant probe sets. We also present methods for consolidating concordant groups into a single value for its corresponding gene and for post hoc analysis of discordant groups. By combining statistical consolidation with sequence analysis, SCOREM possesses the unique ability to identify biologically meaningful discordant behaviors, including differing behaviors in alternate RNA isoforms and tissue-specific patterns of expression. When consolidating concordant behaviors, SCOREM outperforms other methods in detecting both differential expression and overrepresented functional categories.
INTRODUCTION
The Gene Expression Omnibus (GEO) database at NCBI is an extensive repository of publicly available, genomic-scale data, including gene expression data. The most common platforms in GEO are Affymetrix GeneChip® arrays (http://www.ncbi.nlm.nih.gov/geo). One significant issue with analyzing this type of data is that, for any given gene, a GeneChip®can contain more than one probe set designed to hybridize to the transcript(s) for that gene. In many gene expression studies, a gene is stated to be differentially expressed if any one of its representative probe sets reports differential expression, without regard for the other probe sets. Ideally, a group of probe sets representing the same gene will always behave concordantly, i.e. report similar measures of differential expression. However, this is not always the case. In order to obtain the maximum and most accurate information possible, therefore, redundant probe sets must be addressed.
Various approaches to dealing with redundant measures of gene expression have been proposed, from naïve to elaborate. Naïve approaches include choosing the probe set with the highest variance (1) or the best P-value (2), or simply accepting an average value of the majority of probe sets. More elaborate methods make use of statistical tests to analyze the agreement between probe sets (3,4). Another type of methodology is to reannotate all uniquely assignable probes based on sequence alignment to a given gene (5–7) or specific transcript sequence (8–10) into one gene- or transcript-specific probe set. Each of these approaches has its limitations. In order to overcome those limitations and to take full advantage of all the data available from a given experiment, we have developed the SCOREM algorithm.
The SCOREM algorithm utilizes Kendall's W coefficient of concordance, converts the results to an adjacency matrix and then performs a search for connected components (subgraphs) to identify concordant and discordant groups of probe sets. We then use sequence alignment to form biological interpretations of discordant observations. The SCOREM algorithm improves the interpretation of redundant gene expression measures both by adding statistical confidence to concordant measures, yielding improved detection of differential expression and functional enrichment compared to other methods, and by finding meaningful biological reasons (including the prediction of novel alternate RNA isoforms) for discordant measures, something no statistics-only or sequence-only method can deliver. Although in the remainder of this article we refer only to Affymetrix microarrays, the same issue of redundant probe sets also exists for other gene expression platforms, and the SCOREM algorithm is inherently applicable to those as well.
MATERIALS AND METHODS
Data sources and processing
Eight sets of data including four different Affymetrix GeneChip® arrays were used in this study; five are publicly available through NCBI's GEO website (Table 1). Raw data (where available) were normalized using the R package gcrma (19). Filtering was performed with genefilter (20), and fold change (m) and significance (P) were calculated with eBayes in the limma package (21). Generally, statistical significance is determined using hard cutoffs, e.g. a value of m greater than 2 with P better than 0.01. Here, we have implemented a sliding scale for fold change and significance cutoffs: the critical value for P is α, α = 0.01 when |m| = 1; α increases up to Pmax (e.g. 0.05) when |m| > 1 and decreases when |m| < 1. The critical value for P for any given value of m is given by a sigmoid function, similar to that described in Ref. (22). This sliding scale can be utilized with or without prior P-value adjustments, such as false discovery rate (FDR) correction (23). The analyses of the data sets specified were completed with and without FDR correction. Correction with an FDR of 0.05, however, can be more or less stringent than using a hard cutoff of P < .01, depending on the sample. The use of FDR correction is further discussed in the context of consolidation of concordant probe sets.
Table 1.
Data used in evaluating the SCOREM algorithm
| Experiment | Tissue type | Conditions | Platform | Source (Rawa) | No. of samples |
|---|---|---|---|---|---|
| GSE3678b | Thyroid | Tumor versus normal | Human Genome U133 Plus 2.0 (GPL570) | GEO (yes) | 14 |
| GSE4051 (11) | Retina | Nrl-knockout versus wild-type at 5 time points in development | Mouse Genome 430 2.0 (GPL1261) | GEO (yes) | 39 |
| GSE4799 (12) | Spermatogonial stem cells | Growth factor restoration (3 time points) versus withdrawal and baseline | Mouse Genome 430 2.0 (GPL1261) | GEO (no) | 15 |
| GSE9371 (13) | Aorta | Estrogen versus placebo in wild type, ERα- and ERβ- knockouts | Mouse Genome 430 2.0 (GPL1261) | GEO (yes) | 22 |
| BrCac (14–16) | Breast cancer (MCF7 cell line) | Estrogen versus placebo at 2 time points | Human Genome U133A (GPL96) | GEO (no) | 26 |
| VSMC (17) | Aorta | Estrogen versus placebo at 3 time points | Mouse Genome 430A 2.0 (GPL339) | Authors (yes) | 18 |
| GonFat (18) | Gonadal fat | Female versus male and female versus ovarioectomized female | Mouse Genome 430 2.0 (GPL1261) | Authors (yes) | 21 |
| IngFat (18) | Inguinal fat | Female versus male and female versus ovarioectomized female | Mouse Genome 430 2.0 (GPL1261) | Authors (yes) | 21 |
aWhether or not raw data (.CEL files) were available or only preprocessed data.
bReyes et al. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE3678.
The analyses discussed below was carried out in R, using Bioconductor annotation packages Mouse4302.db version 2.5 and org.Mm.eg.db version 2.5 in Bioconductor release 2.8.
Measurement of concordance
Kendall's W, or Kendall's coefficient of concordance, is a statistical method used to measure inter-rater reliability (24). Originally devised for subjective human observations in psychological studies, it has been used for the analysis of biological data such as species associations in community ecology (25). Unlike other tests of concordance, Kendall's W does not consider absolute values of ratings, but overall ordering, as it is a rank-sum method based on Spearman's ρ correlation coefficient. When applied to a group of probe sets, a significant value of W (as defined below) indicates a high level of concordance across a group of probe sets. Groups with a value below the critical value are discordant.
Kendall's W is calculated from the mean of all pair-wise Spearman's coefficients as follows (24):
| (1) |
where k is the number of judges (probe sets), kC2 (k choose 2) is the number of pairs of judges, and ρ is the mean Spearman's correlation coefficient. It can further be seen that
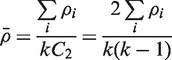 |
(2) |
which when substituted into Equation (1) gives
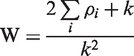 |
(3) |
The significance of an individual value of ρ is determined using the Student's t-test for significance with n − 2 degrees of freedom, where n is the number of observations (arrays) (26).
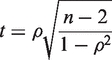 |
(4) |
A critical value (cutoff) for ρ is calculated by rearranging the terms of this equation, using an appropriate critical value for t (e.g. t < 0.01).
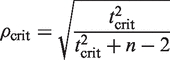 |
(5) |
The critical value for W is then calculated as half the distance between ρcrit and 1 (a perfect correlation), making the W cutoff more stringent than that for an individual ρ.
| (6) |
Detection of concordant subgroups
If the entire group's W does not meet the cutoff Wcrit, a search for concordant subsets within the group (subgroups) is performed. It is useful to note at this point that Equation (3), from the rearrangement of Equation (1), is the same as taking the mean of the symmetric matrix of pairwise coefficients (Table 2). This matrix is then converted into a matrix of true/false values, based on each ρ > ρcrit. This creates an adjacency matrix for an undirected graph, reducing the problem of finding concordant subgroups to that of finding connected components within the graph. If a group is insufficiently concordant (connected under ρ > ρcrit but W < Wcrit), a recursive search for concordant subgroups is performed.
Table 2.
Calculation of W from the mean of the matrix of all pairwise correlation coefficients between k judges
| 1 | 2 | … | k-1 | k | |
|---|---|---|---|---|---|
| 1 | 1.0 | ρ1,2 | … | ρ1,k−1 | ρ1,k |
| 2 | ρ2,1 | 1.0 | … | ρ2,k−1 | ρ2,k |
| … | … | … | … | … | … |
| k-1 | ρk−1,1 | ρk−1,2 | … | 1.0 | ρk−1,k |
| k | ρk,1 | ρk,2 | … | ρk−1,k | 1.0 |
Consolidation of concordant groups
When concordant groups or subgroups are found, a combined P-value can be obtained by performing Fisher's method [Equation (7)] and obtaining the P-value for the resulting χ2 value (27). The individual P-values being combined are those generated by eBayes (21) for each probe set within the group.
| (7) |
Fisher's method is intended for independent measurements (27). Due to the design and experimental protocol under which the gene expression arrays are performed, redundant probe sets can indeed be considered independent measures. The standard Affymetrix sample preparation protocol calls for fragmentation of RNA (resulting fragments are 35–200 bases long), therefore the pool of fragments hybridizing to a given probe set is distinct and independent from that hybridizing to another. For cases in which there are redundant probe sets for the same gene, some represent uniquely processed transcripts; in theses cases, hybridization is clearly independent. However, even when probe sets represent the same transcript, there is minimal overlap between regions covered by those probe sets: the average overlap is 2–3%, with the majority (80–85%) having no overlap at all. Thus, Fisher's method is an appropriate choice.
Finally, each group or subgroup is given a new annotation that indicates which probe sets are included, and the total size of the group, e.g. for the gene with Entrez Gene identifier 99 999, new annotations 99999.1_2_3.5, 99999.4.5, and 99999.5.5 indicate that probe sets 1, 2 and 3 out of 5 have been consolidated, while 4 and 5 have been left as individual values. When there is more than one final value for a given gene, a post hoc analysis is performed, to determine which value(s) makes the most sense, biologically speaking, to use. When necessary, probe sequences were aligned to the most current version of their annotated RefSeq transcripts and gene exon tables downloaded from NCBI (28). When probe sets failed to align to the NCBI transcript sequences, further analysis was performed using the UCSC Blat website (29,30).
R software package SCOREM
All the programs needed to carry out this analysis have been included in an R software package, available for download from the NAR website (Supplementary Data). Requirements are a normalized ExpressionSet object (as, for example, produced by gcrma) and an MArrayLM object with P values, such as produced by eBayes. Appropriate annotation packages must also be available. The SCOREM package includes methods for determination of concordance, consolidation of concordant groups and determination of differential expression, as well as detection of discordant groups remaining after consolidation. Further analysis can be performed using UCSC Blat website or the stand-alone Blat server package available for download. Output of the Blat software (.psl files) can be visualized on the UCSC Genome Browser website, or the Integrated Genome Browser application. Since transcript annotations change on a daily basis, alignment and visualization of probe sets of interest can be repeated as needed.
RESULTS
Redundant probe sets on Affymetrix arrays
The three most common platforms in GEO are the Affymetrix Human Genome U133 Plus 2.0, the Human Genome U133A and the Mouse Genome 430 2.0 GeneChip® arrays. On any of these arrays, a gene may be represented by one or more probe sets. For instance, the U133 Plus 2.0 array averages 2.8 probe sets per gene (54 675 probe sets representing 19 621 genes), while the smaller 133A array averages 1.8 probe sets per gene. Overall, about half of all genes are represented by more than one probe set; a few are represented by ten or more probe sets (Figure 1). Ideally, all the probe sets for a gene would hybridize concordantly, which would provide added confidence to the behavior being observed. However, on occasion some groups of probe sets instead behave discordantly, for a variety of reasons: cross-hybridization to another transcript, misannotation or alternate-transcript-specific binding (which may also be cell type-specific). Such groups of discordant probe sets must be identified and analyzed further, to determine the mostly likely signal for that condition in the biological samples.
Figure 1.

Distribution of the number of probe sets per gene on the three indicated popular Affymetrix GeneChip® arrays.
Several approaches to resolve the issue of different results from supposedly analogous probe sets have been proposed. The simplest approaches for dealing with redundant probe sets do not examine their relative behaviors, but instead employ naïve methods such as choosing the probe set with the highest variance (1), choosing the one with the best P-value (2) or taking a ‘majority rules’ approach. Other approaches use statistical tests to analyze the level of agreement among the data. Jaksik et al. (3) use Dixon's Q test to detect probe sets that are statistical outliers, having significantly higher or lower hybridization levels than the rest, but they do not examine the level of agreement in behavior across conditions. Li et al. (4) use ANOVA to attempt to distinguish probe sets that behave concordantly from those that do not. Their analysis, however, takes an all-or-none approach, ignoring the possibility that some subset of the probe sets might be in agreement. Other groups have implemented a sequence-based approach to redefine the probe sets on the Affymetrix GeneChip® arrays in order to create a one-to-one mapping of probe sets to genes (5–7) or transcripts (8,9). Each of these approaches has its advantages and its limitations, the latter of which the SCOREM algorithm is designed to overcome.
SCOREM: statistical consolidation of redundant expression measures
A typical microarray analysis involves pre-processing the raw data from .CEL files, using linear modeling to fit the samples to conditions, and determining fold change (m-value) between conditions for each probe set (Figure 2a). Optionally, filtering may be performed to remove probe sets reporting extremely low expression or variability. This step is required with the consolidation approach in order to remove probe sets with zero variance, which cannot be included in correlation calculations. Additionally, confidence levels (P-values) are generated for every m-value, and cutoffs may be applied to the m- and P-values to extract a list of genes that are statistically differentially expressed (‘Materials and Methods’ section). In this analysis, we add two additional steps (Figure 2b). First, the processed data are analyzed for concordance of gene groups. Where groups are not in concordance, they are annotated into concordant subgroups (or individually, as warranted). Second, after the m- and P-values are calculated, concordant groups/subgroups are consolidated to one pair of values.
Figure 2.

Flowchart for algorithms for analyzing gene expression data. (a) Typical Affymetrix processing. (b) SCOREM processing with concordance testing and consolidation.
Concordance is measured with Kendall's W test (‘Materials and Methods’ section) (24). When a group is found not to be concordant, the group is searched for subgroups that are concordant. Probe sets are then reannotated with their new group ids—all the same for groups with complete concordance, or distinct ids for two or more subgroups. In the most discordant case, each probe set remains as a separate value. When the analysis is complete, each consolidated (sub)group will have a single m-value (fold change) with a corresponding P-value for each comparison performed in the experiment (e.g. treatment versus control). To determine m for a concordant (sub)group, the individual m-values are averaged. P-values are combined using Fisher's method (27). The resulting values are tested for statistical significance as described in ‘Material and Methods’ section. The final list of differentially expressed genes can then be used for further analysis, such as enrichment of functional categories. In order to understand the biological basis for any probe set groups that were not fully consolidated, they are examined further by sequence alignment to the current version of the transcript/gene.
Application of SCOREM
In all, we applied the SCOREM algorithm to eight data sets, on four different Affymetrix arrays (Table 1). On average, 41% (range 6–56%) of the redundant probe sets were able to be completely consolidated, 19% were partially consolidated, and approximately 40% were not able to be consolidated at all (Table 3). The genes in the latter two categories are examined further, to examine potential reasons for the discordant behaviors of their probe sets.
Table 3.
Degree of probe set consolidation by the SCOREM algorithm in eight data sets
| Experiment | Probe sets (Genes)a |
Consolidation level (%) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| On GeneChip® | After filtering | Unannotated | Single | Redundant | After SCOREM | None | Partial | Complete | |
| GSE3678 | 54 675 (19 798) | 18 725 (10 516) | 2578 | 6802 | 9345 (3714) | 6668 (3714) | 44 | 19 | 37 |
| GSE4051 | 45 101 (20 757) | 16 992 (9963) | 1996 | 6645 | 8351 (3318) | 5019 (3318) | 27 | 19 | 54 |
| GSE4799 | 45 101 (20 757) | 44 062 (20 459) | 6597 | 11 006 | 26 459 (9453) | 24 192 (9453) | 80 | 15 | 5 |
| GSE9371 | 45 101 (20 757) | 20 728 (11 184) | 1937 | 6512 | 12 279 (4672) | 9094 (4672) | 47 | 22 | 30 |
| BrCa | 22 283 (12 718) | 13 397 (8558) | 982 | 5939 | 6476 (2619) | 4031 (2619) | 28 | 19 | 53 |
| VSMC | 45 101 (20 757) | 19 914 (11 717) | 1012 | 7171 | 11 731 (4546) | 8224 (4546) | 44 | 19 | 37 |
| GonFat | 45 101 (20 757) | 24 260 (12 586) | 2472 | 7073 | 14 715 (5513) | 9320 (5513) | 30 | 23 | 48 |
| IngFat | 45 101 (20 757) | 24 364 (12 585) | 2590 | 7080 | 14 694 (5505) | 8966 (5505) | 28 | 20 | 52 |
aNumbers in parentheses indicate the number of distinct genes represented by those probe sets.
In comparing any two conditions A and B (e.g. drug treatment versus control, mutant versus wild type, or tumor versus normal tissue), a given probe set or group of probe sets can be (i) higher in A than in B, (ii) lower in A than in B or (iii) the same in A and B (no change). Thus, types of discordant behavior we can examine include when one probe set or subgroup reports differential expression (either higher or lower) in a given condition while another probe set or subgroup is unchanged. Another occurs when two groups report differential expression in opposite directions. A third can be considered to occur when both groups report differential expression, in the same direction, but of very different magnitudes (e.g. two-fold versus eight-fold increases). In an experiment with more than two conditions (e.g. a time course or testing multiple different drugs), discordant subgroups can exhibit one or more of these behaviors. Table 4 summarizes, for each data set, how many instances of the first two types of discordant behaviors exist among the genes reported to have changing expression levels in more than one subgroup of probe sets.
Table 4.
Characterization of differentially expressed groups representing differentially expressed genes from eight gene expression data sets
| Data set | Total number of groups | Number of genes represented by |
|||
|---|---|---|---|---|---|
| A single group | Multiple subgroupsa |
||||
| (a) Yes versus no | (b) Small versus large | (c) Down versus up | |||
| GSE3678 | 1430 | 1383 | 0 | 5 | 0 |
| GSE4051 | 4006 | 3656 | 126 | 24 | 13 |
| GSE4799 | 5122 | 4239 | 348 | 16 | 43 |
| GSE9371 | 1593 | 1509 | 32 | 3 | 2 |
| BrCa | 947 | 915 | 5 | 1 | 0 |
| VSMC | 451 | 447 | 1 | 0 | 0 |
| GonFat | 4559 | 4449 | 14 | 3 | 13 |
| IngFat | 3049 | 2933 | 32 | 2 | 6 |
aColumns refer to discordant groups where (a) one is changing and the other is not; (b) both are changing in the same direction, but with different magnitudes; and (c) one is increasing while the other is decreasing.
Post hoc analysis and the detection of differential processing
Potential reasons for discordant behavior among probe sets representing the same gene include multiple RNA isoforms for the gene, misannotation of probe sets and cross-hybridization of multiple mRNAs to a single probe set. Multiple RNA isoforms can be generated via alternative splice sites, polyadenylation sites and/or promoters. To determine whether probe set inconsistencies in hybridization levels could be due to biologically significant differences, the probes in each probe set or group are mapped to the sequences for all annotated transcript variants of that gene.
Through such mapping we have also validated the relevance of the SCOREM approach, as many of the discordancies can be attributed to known cases of alternative RNA transcripts. In the data sets we analyzed, there were 77 cases in which discordantly expressed subgroups for the same gene were expressed in opposite directions (Table 4, final column). The 76 genes (one is discordant in two data sets) are represented on the arrays by 298 total probe sets (199 after gene filtering). For 26 of these genes, probe sets differentially mapping to distinct, known RNA isoforms can account for the discrepancy (Supplementary Table S1); the alternate RNA isoforms are annotated in RefSeq (28) for 15 of these genes and in the UCSC Genome Browser (29) for an additional 11 cases. These reflect all types of alternative transcript formation: alternative splicing (fifteen genes), alternative polyadenylation (nine genes) and alternative promoters (two genes).
One example where discordant probe sets map to distinct transcripts occurs in Shprh (encoding SNF2 histone linker PHD RING helicase). This gene is represented by four probe sets on the Affymetrix Mouse Genome 430 2.0 array. In the GSE4051 data set, SCOREM measures the correlation coefficient W to be 0.42, not significantly concordant. However, when the discordant subgroups are analyzed, it is clear that two of the probe sets reflect discordant expression values; hybridization to one (1452261_at) increases when that to the other (1457327_at) decreases. There are two known RefSeq transcripts for Shprh, NM_172937 and NM_001077707 (Figure 3a); due to differential splicing these transcripts contain distinct 3′ untranslated regions (UTR). Aligning the probe sequences to the genomic sequence of Shprh reveals that each of the discordant probe sets maps exclusively to only one of the two different isoforms. Thus, it is straightforward to interpret that under conditions of this experiment (in the mouse retina in response to a knockout of the Nrl gene), when expression of the Shprh transcript NM_001077707 increases, expression of transcript NM_172397 decreases. Thus, the SCOREM analysis uncovers novel biological information regarding expression of this gene.
Figure 3.

Examples of post hoc analysis in order to determine causes for discordant expression patterns. Genomic DNA is indicated as a line, with exons indicated as boxes on the line. Nucleotide positions within each genes are given with zero representing the presumed transcription start site for the indicated mRNAs. Mapping of each probe set is color-coded for each gene. Translation start and stop codons are indicated by arrows and asterisks, respectively. Whether each probe set is differentially regulated in the experimental samples, and the direction of regulation, are indicated in parentheses after each probe set id. (a) Detection of different expression patterns in known alternate RNA isoforms of Shprh. The probe sets reporting differential expression correspond to exons unique to each of the known isoforms. Expression changes are for experiment GSE4051 at the post-natal day 10 time point. (b) Identification of a possible novel isoform of the gene Dpys12. The alternate RNA isoform could involve alternate splicing of exons 12–14 or an alternate 3′UTR (polyadenylation site). Expression changes are for experiment GSE4799, deprivation versus untreated. (c) Identification of a potential alternate promoter in Scmh1. Expression changes are for experiment GSE4051 at the embryonic day 16 time point. (d) Identification of potential novel coding exons in Mllt10, in introns 3 and/or 4. Expression changes are for experiment GSE4799, 2 h post-restoration versus untreated.
For an additional 35 genes in which discordantly expressed subgroups were expressed in opposite directions, known isoforms did not distinguish behavior between probe sets. Instead, mapping of the probe sets suggest novel RNA isoforms. The simplest interpretations of various observed scenarios include: (i) when discordant subgroups of probe sets map to distinct coding exons, we propose novel alternatively spliced variant(s); (ii) when a discordant subgroup maps to an intron between coding exons, we propose alternate isoforms containing novel exons; (iii) when discordant subgroups map to distinct regions within the known 3′UTR or when some map downstream of the known 3′UTR, we propose alternative polyadenylation; (iv) when a discordant subgroup maps either to regions upstream of the identified promoter or to an intron prior to the encoded translational initiation codon, we propose an alternative transcription start site (promoter). Other interpretations of novel RNA isoforms for the respective genes are clearly possible. The analysis does not pinpoint the alternate RNA isoform, but it does suggest that more than the known RNAs are being expressed, for subsequent experimental exploration. Finally, it is possible (depending on the source of RNA used in the experiments) that signal from probe sets (especially ones in introns) reflect either partially processed RNA or RNAs unconnected to the gene in question (e.g. non-coding RNAs overlapping the genomic region).
Examples of three categories of genes containing discordant probe sets that cannot be explained by known RNA isoforms are shown in Figures 3b–d. Dpysl2 is represented by discordant probe sets that both map (partially or completely) in the 3′UTR (Figure 3b); this is an example of potential alternate polyadenylation sites in the terminal exon. For Scmh1, one discordant probe set maps in the intron upstream of the first coding exon (Figure 3c); this is predicted to reflect an alternate promoter, most likely mapping within the annotated intron 4. For Mllt10, there are two probe sets mapping to annotated introns 3 and 4, respectively, where their expression levels change in opposite directions (Figure 3d). This scenario suggests alternative splicing, presumably including novel coding exons within the annotated introns 3 and/or 4.
Finally, 16 of the 77 cases could not be explained by the existence of alternate RNA isoforms. For these, the use of BLAT (30) to align the individual probe sequences against the entire genome can reveal whether some of the probes do not appropriately represent the gene in question (misannotation) or represent more than one gene (cross-hybridization). For eight of these genes, at least one of the probe sets is misannotated (including probe sets that map to the non-coding strand, or to non-coding RNAs overlapping the given gene). Two genes are represented by a probe set that has the potential to cross-hybridize to another gene on a separate chromosome. Finally, six genes do not include annotated exon information, and cannot therefore be assessed. Full details for all 77 cases are provided in Supplementary Table S1.
Overall, the ability to identify and map the locations of probe sets that reflect discordant expression patterns expands and clarifies the information gleaned from analyzing gene expression data. As a validation that SCOREM appropriately separates probe sets into discordant subgroups, over a third of the more severely discordant subgroups can be explained by distinct, known RNA isoforms. For these genes, the discordant expression behavior can add valuable biological insights. In ∼45% of the cases, our analysis suggests novel RNA isoforms that are differentially regulated, thus generating hypotheses to be tested for genes of interest. Finally, by invalidating probe sets in the remaining ∼20% of the discordant cases, incorrect data can be excluded from further consideration.
Detection of tissue-specific behaviors
Since SCOREM performs a new analysis of concordance/discordance in each experiment, the results for any given group of probe sets may differ from experiment to experiment. For instance, this could reflect tissue- or cell type-specific expression of alternate variants. An interesting case in point is that of Rbm39 (Entrez Gene ID 170791). This gene is represented by 10 different probe sets, according to the annotation supplied by Affymetrix. In the analysis of the GSE9371 data sets, the 10 probe sets were not concordant (W = 0.47) (Figure 4a), but two subgroups were highly concordant (Figure 4b and c). If the naïve approach of ‘majority rules’ had been applied, the first group (Figure 4b) with five probe sets would have been taken as the representative value. However, mapping probes to the transcript sequence indicates that only the smaller subgroup of two probe sets (Figure 4c) maps to the annotated (exonic) transcript sequence (Figure 4d). The larger subgroup maps instead to intronic regions of the gene. Whether these represent a separate transcript (e.g. non-coding RNA, overlapping gene) or unannotated alternate exons requires further investigation. Most importantly, Figure 4e–i shows that the clustering of probe sets is different in different experiments. In particular, note that probe sets 2 and 3 do not always behave concordantly, and in one case all 10 probe sets act in concert. Using the standard analysis from Figure 2a, these experiment-dependent differences would not be discerned. Thus, the method described here is sufficiently flexible to provide important additional nuances in understanding the differential RNA processing.
Figure 4.

Graphical analysis of probe sets annotated as representing Rbm39 (encoding RNA binding motif protein 39) from multiple experiments performed on the Mouse 430 2.0 GeneChip®. (a–c) Expression profiles of probe sets across the 22 samples in the GSE9371 data set; W indicates level of concordance. (a) All 10 probe sets; W shows lack of concordance. A dotted line shows a probe set removed by gene filtering (for very low expression or very low variance) and therefore not included in the calculation of W. (b) The largest subgroup includes probe sets 4, 6, 7, 9 and 10; W shows high concordance. (c) The second subgroup includes probe sets 2 and 3; W shows high concordance. (d) Mapping of the 10 probe sets to the genomic sequence of Rbm39 and the exons of its only known transcript. Probes mapped above the line map to the coding (negative) strand, those below the line map to the non-coding (positive) strand. (e–i) Connected subgraphs indicating groups of concordant probe sets in five different experiments. In parentheses are the number of samples in each data set (n) and the critical value for W for a data set of that size. W is given for each group as a whole (bottom) and for each concordant subgroup (below each subgroup). Filled circles indicate statistically significant differential expression in that experiment. A dotted circle indicates a probe set removed during gene filtering in that data set.
We have examined the possibility that some genes would always have discordant probe sets, and have found that not to be the case. In the eight data sets examined, only one gene [WD repeat domain 33 (Wdr33), EntrezGene 74320] was discordant in more than one set (GSE4051 and Inguinal Fat). However, even in this case, the seven probe sets representing this gene were not grouped in the same manner, and so were discordant in different ways (see Supplementary Table S1 for details). This again leads to the conclusion that patterns of concordance and discordance cannot be assumed to be replicable across data sets.
DISCUSSION
The SCOREM algorithm is a novel approach to gene expression analysis, making use of Kendall's coefficient of concordance to determine the level of agreement (concordance) among multiple probe sets representing the same gene in a gene expression experiment. While other methods have been proposed to resolve the issue of so-called redundant probe sets, SCOREM is the only one that also provides information about differential RNA processing, even when no known alternate transcripts exist, and thus can reveal tissue- or cell-specific expression patterns. At the same time, it yields expression measures with increased statistical confidence, leading to further statistical confidence in secondary analyses such as functional enrichment analysis.
SCOREM is evaluated here in comparison with a standard (non-consolidating) approach to expression analysis, as well as two other methods for consolidation. Li et al. (4) use ANOVA to analyze agreement between multiple probe sets, while Dai et al. (5) use sequence alignment information to create composite probe sets. We evaluate each method for its ability to detect differentially expressed genes and the statistical confidence of subsequent functional enrichment analyses [measured as the average P-value of the top 20 categories detected as enriched by GOstats (1)].
Comparison with a standard approach
Overall, SCOREM consistently calls more differentially expressed genes than the standard method (Table 5). This includes more genes in each statistically over-represented functional category, resulting in better P-values of the top categories identified by GOstats (1) (Table 5).
Table 5.
Comparison of SCOREM with other methods in terms of detection of differential expression and functional enrichment
| Data Set | Standard (+FDR) | ANOVA | Custom CDF | SCOREM (+FDR) |
|---|---|---|---|---|
| Number of genes called differentially expressed | ||||
| GSE3678 | 1026 (1064) | 402a | 667 | 1405 (821) |
| GSE4051 | 2829 (2400) | 313a | 1678 | 3829 (1930) |
| GSE4799 | 2146 (323) | 832a | NAb | 4660 (298) |
| GSE9371 | 720 (102) | NA | 480 | 1550 (123) |
| BrCa | 73 (1137) | NA | NAb | 931 (592) |
| VSMC | 80 (92) | NA | 126 | 449 (53) |
| GonFat | 1753 (6005) | NA | 1966 | 4502 (3455) |
| IngFat | 777 (3925) | NA | 940 | 2989 (1782) |
| Average P-value of top 20 enriched GO categories | ||||
| GSE3678 | 4.5 × 10−10 (1.8 × 10−7) | 3.5 × 10−2a | 2.5 × 10−6 | 1.3 × 10−10 (5.1 × 10−9) |
| GSE4051 | 6.1 × 10−10 (5.1 × 10−8) | NA | 2.0 × 10−4 | 1.6 × 10−6 (4.9 × 10−8) |
| GSE4799 | 3.6 × 10−10 (5.3 × 10−9) | 1.2 × 10−2a | NAb | 2.0 × 10−13 (2.8 × 10−10) |
| GSE9371 | 6.1 × 10−7 (3.0 × 10−5) | NA | 8.6 × 10−6 | 1.8 × 10−9 (9.5 × 10−6) |
| BrCa | 3.6 × 10−4 (5.0 × 10−5) | NA | NAb | 6.3 × 10−5 (3.0 × 10−5) |
| VSMCc | NA (NA) | NA | 9.8 × 10−4 | 4.5 × 10−4 (NA) |
| GonFat | 3.2 × 10−15 (2.5 × 10−12) | NA | 1.2 × 10−9 | 8.3 × 10−13 (1.7 × 10−13) |
| IngFat | 1.6 × 10−8 (4.4 × 10−9) | NA | 7.9 × 10−6 | 3.5 × 10−8 (1.9 × 10−8) |
aNumber of differentially expressed genes or average P-values of top three or five GO categories as given in Ref. (4).
bNA indicates data sets with no raw data available, where the custom CDF approach could not be applied.
cP-values are for top 5 (Custom CDF) or top 10 (SCOREM) overrepresented GO categories; NA indicates fewer than five GO categories with any overrepresentation.
For example, our previous studies investigated the role of estrogen and estrogen receptors (ER) α and β in regulating gene expression in vascular tissue (GSE9371) (13). This study found that estrogen/ERβ was responsible for downregulation of a number of nuclear-encoded genes in the mitochondrial respiratory chain. Using a standard analysis, 17 genes in the oxidative phosphorylation pathway (KEGG:00190) were identified that had at least one probe set reporting differential expression. Using SCOREM, the list was extended to 26 genes (14 from the original list plus 12 new genes); consequently, the P-value for the statistical significance of this pathway went from 1.9 × 10−04 to 1.8 × 10−7 (Supplementary Table S2).
Since the standard approach makes no attempt to consolidate redundant probe sets, the resulting list of differentially expressed genes typically includes values from redundant probe sets, with no mention of other probe sets corresponding to these genes. There were a total of over 3000 redundant measures in the lists for the eight data sets generated by the standard method, compared to only 334 remaining redundant measures for SCOREM.
Comparison with a statistical approach
The approach developed by Li et al. (4) utilizes ANOVA with the intent of distinguishing between the effects of discordancy on expression measures and true experimental effects. This analysis takes an all-or-none approach, in which all probe sets that are in agreement are consolidated while probe sets not in agreement are all treated as separate measurements, ignoring the possibility that some subset might be in agreement and that this could be the source of valuable information about the gene in question. However, as SCOREM demonstrates, in the majority of cases concordant subgroups can be delineated (Table 3). For example, the ANOVA approach (4) would treat all probe sets as separate signals for Lman1, even though three out of five probe sets are hybridizing concordantly and can be consolidated into one statistically significant increasing value, while a fourth is decreasing (Supplementary Table S1). Finally, when directly compared, SCOREM finds more differentially expressed genes and shows better enrichment of experimentally relevant functional categories than the ANOVA approach (Table 5).
Comparison with a custom CDF approach
Dai et al. (5) use a sequence-based approach to generate a custom Chip Definition File (custom CDF) that redefines the probe sets on the Affymetrix GeneChip® arrays to generate a single probe set per gene. Probes that match more than one transcript (or no annotated transcripts) are removed entirely; the remainder is consolidated into one probe set. As pointed out in Ref. (31), typically more than half of the data from hybridization to an array is eliminated. The number of genes represented by the final probe sets is reduced as well (Table 6). While deLeeuw et al. (31) salvage these lost probe sets by creating hybrid CDFs and annotating ‘less reliable probe-sets’, SCOREM takes the approach of examining probe set behavior in each data set, and annotating the reliability of those whose behavior is inconsistent with the more reliable of its siblings. It is not surprising, given the loss of data in the custom CDF approach, that SCOREM finds more differentially expressed genes and shows greater significance of enriched functional categories (Table 5).
Table 6.
Comparison of the number of probes, probe sets used and genes represented in Affymetrix and Brainarray custom CDF files
| Array | CDF | Probes | Probe sets | Genes |
|---|---|---|---|---|
| Human U133 Plus 2.0 | Affymetrix | 604 258 | 54 675 | 19 798 |
| Custom | 277 789 | 19 008 | 18 974 | |
| Human U133A | Affymetrix | 247 965 | 22 283 | 12 718 |
| Custom | 167 345 | 12 078 | 12 065 | |
| Mouse 430 2.0 | Affymetrix | 496 468 | 45 101 | 20 757 |
| Custom | 240 917 | 17 306 | 17 289 |
The custom CDF approach has been favorably reviewed (32) and is currently is use by others, e.g. Carroll et al. (33). However, it does have a number of limitations. First, it can only be applied to raw data (Figure 2a); while gene expression data in GEO are increasingly available in raw format, many data sets are still only available in pre-processed form (34). Second, as a result of eliminating so many probes, the redefined probe sets vary widely in size, from as few as three to over 100 probes, instead of the original 11, thus in any given probe set there may be too few probes left to generate a statistically significant result. For example, analysis of the GSE9371 data set with SCOREM indicates that all three probe sets (33 probes total) for Cox7a2l (Entrez Gene 20463) behave in a highly similar fashion, leading to a highly significant combined expression change (Supplementary Table S2). However, the BrainArray custom CDF file eliminates all but 6 of the 33 probes, and consequently does not consider the results for this gene to be significant. Third, the new probe set definitions are based on the current annotated genomic sequences, which are certain to change, especially as more splice variants are found. As a result, the entire analysis must be repeated each time a new version of the custom CDF is released, requiring periodic downloads of large annotation packages. Fourth, this approach eliminates non-annotated sequences, producing a bias toward well-understood transcripts, and limits the ability of researchers to identify unannotated transcripts that nevertheless may be of critical importance in a particular study. Finally, this approach is based solely on sequence and not at all on behavior, with the same probes being consolidated for each gene expression experiment. However, our method reveals that redundant probe sets can show independent behaviors in different conditions or tissue or cell types.
As an example of the effect of combining probe sets based only on sequence, the gene Shprh is represented by four probe sets on the Affymetrix Mouse 430 2.0 array. The BrainArray custom CDF retains and combines 35 of the 44 probes for these probe sets. SCOREM, however, reveals that two of the probe sets are not hybridizing at all, while the other two uniquely map to (and hybridize with) each of two different known isoforms (Figure 3a). As a result, the BrainArray analysis concludes this gene is not differentially expressed, while SCOREM reveals that both isoforms are, but in different directions.
Further applications
Although in this article we have only presented the results of applying SCOREM to Affymetrix microarray data, SCOREM is platform-independent and can be applied to any type of data containing redundant measures where a single value is desired. All that is needed is an ExpressionSet object containing normalized data and an MArrayLM object with P-values, such as produced by eBayes. In the case of the discordant behaviors where one probe set or subgroup is reporting differentially expression and other subgroup(s) for that gene are not, or where the groups are reporting differential expression of different magnitudes (Table 4, columns a and b), the post hoc analysis described in Results can also be applied. Finally, results of a Blat analysis can be downloaded from the UCSC website, and visualized with a tool such as the Integrated Genome Browser (IGB) (35). Therefore, SCOREM can be applied to any type of data and make sense of any type of discordant behavior present in that data.
CONCLUSIONS
The SCOREM algorithm has several advantages, both technical and biological, over the other methods examined. Its technical advantages include that: (i) it is applicable to raw data (.CEL) or preprocessed data; (ii) its basic analysis does not change, even when sequences are re-annotated, therefore, only the post hoc analysis must be repeated when new information becomes available; (iii) probe sets that do not map to currently known coding sequences are retained for future analysis, not discarded; (iv) it does not require downloading of multiple, large annotation packages from external sources, instead utilizing the latest annotation obtained directly from NCBI; and (v) it is applicable to any type of expression data with redundant expression measures, regardless of platform.
SCOREM also has several key biological advantages over statistical-only or sequencing-only approaches, including that: (i) it identifies differential RNA processing of known isoforms, and it detects potential novel isoforms; (ii) it makes no a priori assumptions that behavior will be the same across experiments, allowing the detection of tissue- or cell-specific differential processing; and (iii) when redundant probe sets behave concordantly, it provides greater statistical confidence in measuring differential expression and in subsequent functional enrichment analyses.
By assessing redundant probe sets based on their behavior, not just their sequence, we achieve an increase in statistical power for concordant sets, and an opportunity for further analysis for discordant sets. Therefore, by utilizing the SCOREM approach, we obtain results with greater statistical confidence in the determination of differential expression and subsequent analysis of functional enrichment, and gain additional information about tissue-and isoform-specific behavior of genes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary R package file.
FUNDING
Evans Center for Interdisciplinary Biomedical Research, Boston University [Affinity Research Collaborative (ARC) Grant: ‘Sex differences in adipose tissue remodeling: mechanisms and role in disease risk associated with obesity’]. Funding for open access charge: Waived by Oxford University Press.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Luis Carvalho and Eric Kolaczyk for helpful technical discussions. We also thank Michael Mendelsohn, Katrin Schnoes and the members of the Society for Women's Health Isis Fund Network on Sex Differences in Metabolism (Susan Fried, Deborah Clegg, Jeffrey Chang, Joel Elmquist, Nori Geary, Andrew Greenberg, Kevin Grove, Jennifer Lovejoy, Philipp Scherer, Randy Seeley, Richard Simerly, Steve Smith and X.Q. Xiao) for making their data available for this study.
REFERENCES
- 1.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 2.Mieczkowski J, Tyburczy ME, Dabrowski M, Pokarowski P. Probe set filtering increases correlation between Affymetrix GeneChip®and qRT-PCR expression measurements. BMC Bioinformatics. 2010;11:104. doi: 10.1186/1471-2105-11-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaksik R, Polańska J, Herok R, Rzeszowska-Wolny J. Calculation of reliable transcript levels of annotated genes on the basis of multiple probe-sets in Affymetrix microarrays. Acta Biochim Pol. 2009;56:271–277. [PubMed] [Google Scholar]
- 4.Li H, Zhu D, Cook M. A statistical framework for consolidating “sibling” probe sets for Affymetrix GeneChip®data. BMC Genomics. 2008;9:188. doi: 10.1186/1471-2164-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip®data. Nucleic Acids Res. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrari F, Bortoluzzi S, Coppe A, Sirota A, Safran M, Shmoish M, Ferrari S, Lancet D, Danieli GA, Bicciato S. Novel definition files for human GeneChip®arrays based on GeneAnnot. BMC Bioinformatics. 2007;8:446. doi: 10.1186/1471-2105-8-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harbig J, Sprinkle R, Enkemann SA. A sequence-based identification of the genes detected by probesets on the Affymetrix U133 plus 2.0 array. Nucleic Acids Res. 2005;33:e31. doi: 10.1093/nar/gni027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui X, Loraine AE. Consistency analysis of redundant probe sets on Affymetrix three-prime expression arrays and applications to differential mRNA processing. PLoS One. 2009;4:e4229. doi: 10.1371/journal.pone.0004229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu J, Lee JC, Salit ML, Cam MC. Transcript-based redefinition of grouped oligonucleotide probe sets using AceView: High-resolution annotation for microarrays. BMC Bioinformatics. 2007;8:108. doi: 10.1186/1471-2105-8-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H, Wang F, Tu K, Xie L, Li YY, Li YX. Transcript-level annotation of Affymetrix probesets improves the interpretation of gene expression data. BMC Bioinformatics. 2007;8:194. doi: 10.1186/1471-2105-8-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, et al. Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc. Natl Acad. Sci. USA. 2006;103:3890–3895. doi: 10.1073/pnas.0508214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc. Natl Acad. Sci. USA. 2006;103:9524–9529. doi: 10.1073/pnas.0603332103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Lone R, Knorr K, Jaffe IZ, Schaffer ME, Martini PG, Karas RH, Bienkowska J, Mendelsohn ME, Hansen U. Estrogen receptors alpha and beta mediate distinct pathways of vascular gene expression, including genes involved in mitochondrial electron transport and generation of reactive oxygen species. Mol. Endocrinol. 2007;21:1281–1296. doi: 10.1210/me.2006-0497. [DOI] [PubMed] [Google Scholar]
- 14.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–4842. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 15.Frasor J, Chang EC, Komm B, Lin CY, Vega VB, Liu ET, Miller LD, Smeds J, Bergh J, Katzenellenbogen BS. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006;66:7334–7340. doi: 10.1158/0008-5472.CAN-05-4269. [DOI] [PubMed] [Google Scholar]
- 16.Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA, Katzenellenbogen BS. Estrogen receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol. Endocrinol. 2008;22:1032–1043. doi: 10.1210/me.2007-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schnoes KK, Jaffe IZ, Iyer L, Dabreo A, Aronovitz M, Newfell B, Hansen U, Rosano G, Mendelsohn ME. Rapid recruitment of temporally distinct vascular gene sets by estrogen. Mol. Endocrinol. 2008;22:2544–2556. doi: 10.1210/me.2008-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grove KL, Fried SK, Greenberg AS, Xiao XQ, Clegg DJ. A microarray analysis of sexual dimorphism of adipose tissues in high-fat-diet-induced obese mice. Int. J. Obes. 2010;34:989–1000. doi: 10.1038/ijo.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu J, Irizarry R, Gentleman R, Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J.A.S.A. 2004;99:909–917. [Google Scholar]
- 20.Gentleman R, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smyth GK. Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 22.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005;4:17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 23.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 24.Kendall MG, Babington-Smith B. The problem of m rankings. Ann. Math. Stat. 1939;10:275–287. [Google Scholar]
- 25.Legendre P. Species associations: the Kendall coefficient of concordance revisited. J. Agri. Biol. Environ. Stat. 2005;10:226–245. [Google Scholar]
- 26.Press WH, Vettering WT, Teukolsky SA, Flannery BP. Numerical Recipes in C: The Art of Scientific Computing. 2nd edn. Cambridge, UK: Cambridge University Press; 1992. p. 640. [Google Scholar]
- 27.Fisher RA. Statistical Methods for Research Workers. 14th edn. Edinburgh: Oliver and Boyd; 1970. pp. 99–100. [Google Scholar]
- 28.Pruitt KD, Tatusova T, Maglott DR. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu F, Kent WJ, Clawson H, Kuhn RM, Diekhans M, Haussler D. The UCSC Known Genes. Bioinformatics. 2006;22:1036–1046. doi: 10.1093/bioinformatics/btl048. [DOI] [PubMed] [Google Scholar]
- 30.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Leeuw WC, Rauwerda H, Jonker MJ, Breit TM. Salvaging Affymetrix probes after probe-level re-annotation. BMC Res Notes. 2008;1:66. doi: 10.1186/1756-0500-1-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandberg R, Larsson O. Improved precision and accuracy for microarrays using updated probe set definitions. BMC Bioinformatics. 2007;8:48. doi: 10.1186/1471-2105-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- 34.Larsson O, Sandberg R. Lack of correct data format and comparability limits future integrative microarray research. Nat. Biotech. 2006;24:1322–1323. doi: 10.1038/nbt1106-1322. [DOI] [PubMed] [Google Scholar]
- 35.Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE. The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009;25:2730–2731. doi: 10.1093/bioinformatics/btp472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.