


Abstract
DNA methylation is a key regulatory control route in epigenetics, involving gene silencing and chromosome inactivation. It has been recognized that methyl-CpG binding domain (MBD) proteins play an important role in interpreting the genetic information encoded by methylated DNA (mDNA). Although the function of MBD proteins has attracted considerable attention and is well characterized, the mechanism underlying mDNA recognition by MBD proteins is still poorly understood. In this article, we demonstrate that the methyl-CpG dinucleotides are recognized at the MBD–mDNA interface by two MBD arginines through an interplay of hydrogen bonding and cation-π interaction. Through molecular dynamics and quantum-chemistry calculations we investigate the methyl-cytosine recognition process and demonstrate that methylation enhances MBD–mDNA binding by increasing the hydrophobic interfacial area and by strengthening the interaction between mDNA and MBD proteins. Free-energy perturbation calculations also show that methylation yields favorable contribution to the binding free energy for MBD–mDNA complex.
INTRODUCTION
DNA methylation is an epigenetic modification of eukaryotic genomes, including plant genomes, involving the addition of a methyl group at the 5-position of cytosine in CG dinucleotides (1–4). It is now well established that DNA methylation plays an essential role in organismal development, e.g. controlling gene expression, X-chromosome inactivation and gene imprinting (5,6). In the cell, DNA methylation exerts its biological function in at least two ways. First, DNA methylation affects promoter regions to impede binding of transcriptional factors to the gene and, hence, directly interferes with gene activation (7–9). Second, DNA methylation attracts proteins that bind to the methyl-CpG (mCpG) steps and leads to the formation of silenced states of chromatin, which influence gene activity (10–15). A family of proteins that recognize mCpG steps in DNA, known as methyl-CpG binding domain (MBD) proteins, recruit co-repressor complexes that alter chromatin structure, thereby repressing transcription from non-methylated CpG promoters (16–19).
Biological functions of the MBD proteins in epigenetic regulation have been studied over several decades. At present, five proteins with mCpG-binding motifs have been identified in the MBD family, namely MBD1 (20,21), MBD2 (22,23), MBD3 (24), MBD4 (25) and MeCP2 (26–28). In mammals, MBD1, MBD2 and MeCP2 have been observed to bind to mCpG steps and to repress gene transcription. MBD4 binds to T:G mismatches caused by deamination of the methylated cytosine at mCpG sites and is involved in DNA repair (29). Mammalian MBD3 does not bind to the methylated DNA (mDNA) due to the lack of several important amino acids that are conserved in other MBD proteins (16).
All MBD proteins share a similar MBD of about 75 amino acid residues. Sequence alignment of MBD proteins reveals several highly conserved residues on the interface between MBD and methylated DNA (mDNA). Figure 1 shows that the most conserved region in the primary structure of the MBD is located between residues ARG22 and LYS46, including loop L1, strand β3, strand β4 and loop L2. The conserved residues of the MBD form the interfacial surface contacting mDNA and are responsible for recognition of the methyl-CpG steps.
Figure 1.

Sequence alignment of five methyl-CpG binding domain (MBD) proteins from human and mouse. A red-white-blue color scale is used to indicate sequence similarity. The most conserved residues are shown in dark blue, while the less conserved residues are indicated in red.
The NMR structures of MBD1, MBD2 and the X-ray structure of MeCP2 proteins bound to the mDNA provide useful information on how residues in the MBD contact mDNA (21,23,28). Figure 2 illustrates the MBD interaction with the mCpG steps in the major groove of mDNA. The methyl groups of methyl-cytosine are buried in the mDNA–MBD interface. The principal mCpG binding interface composed of hydrophilic and hydrophobic residues involves two types of interaction with mCpG steps (30): the polar atoms in MBD form hydrogen bonds with the polar atoms in the DNA, while the non-polar atoms in MBD form a hydrophobic patch around the methyl groups. However, a detailed picture of how the MBD recognizes mCpG steps over CpG steps, e.g. the influence of methyl groups on the thermodynamics of MBD–DNA binding, is still poorly characterized.
Figure 2.

MBD proteins binding to mDNA. Shown are the structures of (a) MBD1-mDNA, (b) MBD2-mDNA and (c) MeCP2–mDNA complexes. The methyl-cytosines are indicated in red. The DNA sequence for each complex is shown below the corresponding image. The stair motif mCYT ∴ ARG ν GUA is highlighted (cyan rectangle) and the important motifs of the secondary structure, i.e. β-strands (orange), α-helix (purple) and loops (cyan), are labeled. The structures (a)–(c) are compared in Supplementary movies S1–S3.
Binding of a protein to a specific DNA sequence involves complex binding networks of electrostatic and hydrophobic interactions. Earlier investigations suggested that the pyrimidine-guanine dinucleotide step is recognized by an interaction network in which an arginine is featured prominently through a coupled hydrogen bonding/cation-π interaction (31–34). The conformation composed of pyrimidine, guanine and arginine is usually referred to as the PYR ∴ ARG ∨ GUA stair motif (∴ denotes the cation-π interaction and ∨ denotes the hydrogen bonding), which includes three interactions: nucleobase stacking (arising between pyrimidine and guanine), hydrogen bonding (arising between guanine and arginine) and cation-π interaction (arising between pyrimidine and arginine) (35,36). The stair motif was observed in the structure of MBD2–mDNA and MeCP2–mDNA complexes, in which the arginine amino acids form hydrogen bonds with the guanine bases and concomitantly stack with the methyl-cytosines (23,28). Since the two arginines involved in the stair motifs are conserved in all MBD proteins (see Figure 1), it is likely that recognition of the mCpG dinucleotide by MBD proteins is mediated by stair motifs which are stabilized through methyl groups. Although previous studies have reported the two arginine forming hydrogen bonds with the guanines, the stacking interaction between the methyl-cytosines and the arginines has always been neglected.
In the present study, we performed molecular dynamics (MD) simulations (37) of the MBD1–DNA, MBD2–DNA and MeCP2–DNA complexes in order to reveal the general mechanism whereby the MBD recognizes the mCpG steps. The simulations demonstrate formation of the stable stair motif at the MBD–mDNA binding interface. Comparison between MBD–nDNA (nDNA denotes non-methylated DNA) and MBD–mDNA interactions shows that DNA methylation stabilizes the stair motif, thereby facilitating mCpG recognition by the MBD. The strong effect of methylation on reducing the solvent accessible surface at the MBD–mDNA interface is observed through calculation of the contact area between MBD protein and DNA. Quantum-chemistry analysis characterizes quantitatively the influence of methylation on the stability of the MBD–mDNA complex. Free-energy perturbation (FEP) calculations determine the overall methylation-induced free-energy change in MBD–mDNA binding. Finally, MD simulations on mutants of MBD1 bound with mDNA reveal the important role of other residues in MBD–mDNA binding.
METHODS
We performed molecular dynamics simulations for three crystal structures, namely MBD1–mDNA, MBD2–mDNA and MeCP2–mDNA, to examine the mechanism of methyl-CpG recognition by the MBD of the MBD protein family. To characterize the role of the methyl groups in MBD–mDNA binding, we compared the thermodynamics of the binding surface of MBD1–mCpG and MBD1–CpG steps. Five mutants of MBD1 binding with methylated DNA were simulated to characterize the involvement of specific side groups in recognizing mCpG steps. Ab initio quantum-chemistry calculations were employed to account in electronic detail for the influence of methylation on enhancing the interaction between MBD and DNA.
Molecular dynamics simulations
Atomic models
Nine molecular complexes were studied in this work: (i) wild-type MBD1–mDNA, (ii) mutant MBD1–nDNA, (iii) mutant V20A MBD1–mDNA, (iv) mutant R22A MBD1–mDNA, (v) mutant Y34A MBD1–mDNA, (vi) mutant R44A MBD1–mDNA, (vii) mutant S45A MBD1–mDNA, (viii) wild-type MBD2–mDNA and (ix) wild-type MeCP2–mDNA (see Table 1). Atomic coordinates of wild-type MBD1–mDNA, see (i), were taken from the well-defined protein residues 3–69 in the NMR structure of the MBD of human MBD1 complexed with mDNA (Protein Data Bank entry code 1IG4) (21). The mutant structure for MBD1–nDNA, see (ii), was obtained by mutating the methylated cytosines to non-methylated ones. Five mutated structures of the MBD1–mDNA complex, see (iii)–(vii), were obtained by mutating the residues of interest in the wild-type complex using the MUTATOR plug-in in VMD (38). The MUTATOR plug-in removes the structural information of a given residue and generates new coordinates for the desired mutant amino acid based on the internal coordinates provided in standard CHARMM topology files. The atomic coordinates of wild-type MBD2–mDNA, see (viii), and MeCP2–mDNA, see (ix), were taken from the NMR structure of chicken MBD2 bound to mDNA (Protein Data Bank entry code 2KY8) (23) and the X-ray structure of human MeCP2 bound to mDNA (Protein Data Bank entry code 3C2I) (28), respectively.
Table 1.
List of performed simulations
| Index | DNA | Protein | Mutation | Time (ns) |
|---|---|---|---|---|
| (i) | Methylated | MBD1 | – | 100 |
| (ii) | Non-methylated | MBD1 | – | 30 |
| (iii) | Methylated | MBD1 | V20A | 30 |
| (iv) | Methylated | MBD1 | R22A | 30 |
| (v) | Methylated | MBD1 | Y34A | 30 |
| (vi) | Methylated | MBD1 | R44A | 30 |
| (vii) | Methylated | MBD1 | S45A | 30 |
| (viii) | Methylated | MBD2 | – | 30 |
| (ix) | Methylated | MeCP2 | – | 30 |
The topology file of DNA and protein along with the missing hydrogen atoms was generated using the psfgen plug-in of VMD (39). Each complex was placed in a water box with 0.1 mol/l KCl added. The total size of the simulated systems lies in the 50 000–60 000 atom range.
Molecular dynamics
Simulations were performed using the program NAMD 2.7 with the CHARMM27 force field for DNA (40), the CHARMM22 force field for proteins with CMAP corrections (41,42) and the TIP3P water model (43). The parameters for 5-methylcytosine are available in the CHARMM27 force field (40). Periodic boundary conditions were assumed and the particle-mesh Ewald (PME) summation method was employed for evaluating Coulomb forces. The van der Waals (vdW) energy was calculated using a smooth cutoff of 12 Å. The integration time step was 1 fs. The temperature was kept at 310 K by applying Langevin forces with a damping coefficient of 0.1 ps−1 (44) only to the oxygen atoms of water molecules.
Each simulated system was first energy-minimized for 2000 steps and then heated to 310 K in 4 ps. After heating, each simulated system was equilibrated for 500 ps with harmonic restraints applied to all protein and DNA atoms under NPT ensemble conditions using Nosé-Andersen Langevin piston pressure control (44,45), allowing the systems to acquire a constant volume. With restraints turned off each system was then subjected to 2 ns-equilibration under NVT ensemble conditions. Finally, a 30 ns [100 ns in case of simulation (i)] production MD simulation was carried out in the NVT ensemble. Table 1 lists all the simulations carried out in the present study.
Data analysis
Analysis and snapshots of the molecular structures from MD simulations were realized with VMD (38). To evaluate the binding strength of MBD to the mDNA, we analyzed the contact area, σ(t), between protein and DNA through MD trajectories. σ(t) is defined as
| (1) |
where Sprot(t), SDNA(t) and Sprot+DNA(t) denote the time-dependent solvent accessible surface area (SASA) of protein, DNA and protein–DNA complex, respectively. The SASA values of selected atoms were calculated using the built-in SASA function in VMD (38), which uses an assigned radius (van der Waals radius) for each atom and extends the radius to a user-defined value (here we used 1.4 Å) to find the points on a sphere that are exposed to solvent. The SASA function in VMD is based on the Shrake–Rupley algorithm (46).
To characterize the thermodynamics of arginine and guanine pairing, we monitored the time evolution of hydrogen bonding between arginine and guanine. A hydrogen bond is considered formed when the distance between a hydrogen atom and an acceptor atom is <2.5 Å.
Ab initio quantum chemistry calculations
The gas-phase interaction energy of the stair motif in the MBD1–DNA complex was determined using the quantum-chemistry package GAMESS (47), employing second-order Møller–Plesset perturbation theory (MP2) (36,48–50). The electronic wave function was expanded in a sum of Gaussian functions employing the 6-311++G(d, p) Pople-type basis set of triple-ζ accuracy (51). This basis set, which includes polarization d and p functions and double-diffuse functions (51), is important for a proper MP2 estimate of the interaction energy arising in the stair motif of the MBD–DNA interface (36). Tests for accuracy of different basis sets and different quantum-chemistry methods [MP2 (52), coupled-cluster single and double excitation (CCSD) (53) and density functional theory (DFT) (54–57)] are discussed in the Supplementary Data.
Calculations were performed for the mCYT106 ∴ ARG22 ∨ GUA107 and mCYT118 ∴ ARG44 ∨ GUA119 stair motifs in the MBD1–mDNA complex. Each stair motif includes a methylated/no-methylated cytosine, a guanine and an arginine, along with hydrogen atoms terminating the side chains of these residues. The mCYT-ARG-GUA stair motif contains 51 atoms; the CYT-ARG-GUA stair motif contains 48 atoms. The geometry of the stair motifs used in the single-point energy calculations was extracted from the structures of the MBD1–DNA complex arising in the molecular dynamic simulations. Optimizations prior to the ab initio calculation of the MBD1–mDNA complex in water was done starting from selected conformations from the 20–30 ns MD simulation interval, and using the conjugate gradient energy minimization method with the CHARMM force field (41) in NAMD (39). Structure optimization is necessary for quantum-chemistry calculations, which should be performed on an equilibrated molecular system to reveal the energy difference in the binding energy of a MBD to methylated and non-methylated DNA, as the difference is relatively small. If non-equilibrium structures were used, the small energy difference may become masked by energy contributions from unfavorable orientation of interacting residues. The optimization was carried out for 50 000 steps. After 50 000 steps of optimization the total energy of the system did not change beyond 0.0001 kcal/mol (∼2 × 10−9 kcal/mol per atom) during one step, implying convergence of the studied structure to a local energy minimum.
In order to examine the influence of the methyl groups on stair motif energetics, the methylated cytosines in the selected snapshots from the MD simulation were substituted by non-methylated cytosine residues and the derived structures were then optimized using NAMD (39) before performing quantum-chemistry single-point energy calculations.
To characterize the MBD1–mDNA and MBD1–nDNA binding we considered the three body interaction energies in the mCYT ∴ ARG ∨ GUA and nCYT ∴ ARG ∨ GUA stair motifs which are defined as
| (2) |
| (3) |
Here, the subscripts m and n denote the methylated and the non-methylated cases of the stair motif, respectively. The superscript x = 1, 2 denotes the CYT106 ∴ ARG22 ∨ GUA107 stair motif (x = 1) and the CYT118 ∴ ARG44 ∨ GUA119 stair motif (x = 2); E(CYT ∴ ARG ∨ GUA) is the total energy of the stair motif, while E(CYT), E(ARG) and E(GUA) are the energies of its individual components, corresponding to the energy of cytosine, guanine and arginine, respectively.
From Equations (2) and (3) the change of the binding energy in the MBD1–DNA stair motifs due to DNA methylation can be calculated as
| (4) |
The ab initio quantum chemistry calculations performed include a correction of the basis set superposition error (BSSE) using the Boys and Bernardi function counterpoise (FCP) method (58); such correction is crucial for the accurate calculation of 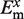 and
and 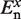 in Equations (2) and (3) (36,59).
in Equations (2) and (3) (36,59).
Alchemical FEP calculations
FEP calculations (60–63) were performed to determine the free-energy change of MBD1–DNA binding upon cytosine methylation using NAMD. According to the thermodynamic cycle shown in Figure 3, the net free-energy change for the MBD–DNA → MBD–mDNA transformation (ΔΔG) can be written
| (5) |
Here, ΔG1 was calculated by mutating non-methylated free DNA into methylated free DNA in water and ΔG2 was calculated by mutating the nDNA–protein complex into mDNA–protein in water. We performed the transformations in both directions and used the Bennett acceptance-ratio (BAR) estimator (63,64) to obtain the maximum-likelihood value of the free energy together with the statistical error.
Figure 3.

Thermodynamic cycle describing DNA methylation in the DNA–MBD complex. The left vertical transition was performed for free DNA (in the absence of the protein) in water; the right vertical transition was performed by mutating DNA to mDNA in the hydrated, native complex. ‘DNA+MBD’ represents the free DNA and the free MBD, while ‘DNA:MBD’ represents the DNA–MBD complex.
The FEP calculations were carried out using 25 intermediate states (Δλ = 0.04), which represent a total simulation time of 40 ns for each unidirectional simulation. λ is a general-extent parameter (65) introduced to transform the system between the reference, non-methylated state, i.e. the state with λ = 0, and the perturbed, methylated state, i.e. the state with λ = 1. The ParseFEP plug-in of VMD (63) was employed to determine the BAR estimator alongside with its variance by combining forward and backward transformations.
RESULTS
In the following section the interaction of methylated and non-methylated DNA with MBD is studied in detail. Particular attention is paid to the structural and energetic analyses of the CYT ∴ ARG ν GUA stair motifs at the MBD–DNA interface. Combination of classical MD simulations and quantum-chemistry calculations is used to explain mDNA recognition by MBD proteins.
Stair motifs at the MBD–mDNA interface
Structures of MBD1–mDNA, MBD2–mDNA and MeCP2–mDNA complexes reveal that the conserved residues ARG22 and ARG44 form hydrogen bonds with guanines in the mCpG steps (21,23,28). The NMR structure of MBD1–mDNA shows that either ARG22 or ARG44 form a single hydrogen bond with guanine, while the X-ray structure of MeCP2–mDNA and the NMR structure of MBD2–mDNA clearly show that two hydrogen bonds are formed between each arginine and guanine. The crystal structures showing that ARG22 and ARG44 interact with mCpG steps in MBD1–mDNA, MBD2–mDNA and MeCP2–mDNA complex are provided in Supplementary Figure S1a, c and e. To investigate the role of ARG22 and ARG44 in recognizing the mCpG steps, we conducted a 100-ns molecular dynamics simulation [simulation (i), see Table 1] of the NMR structure of the human MBD1–mDNA complex (21). Interestingly, within a very short time of simulation (∼1 ns), the side chains of ARG22 and ARG44 rotated, forming two hydrogen bonds with GUA107 and GUA119, respectively, as shown in Figure 4a. The length of the four hydrogen bonds formed between ARG22 and GUA107 as well as between ARG44 and GUA119 is ∼1.7 Å (see Figure 4b). The fluctuation of the hydrogen bond length is small, indicating that the hydrogen bonding between arginine and guanine is stable (see Figure 4b). 30-ns MD simulations were performed for the NMR structure of chicken MBD2–mDNA [simulation (viii), see Table 1] (23) and the X-ray structure of human MeCP2–mDNA complexes [simulation (ix), see Table 1] (28). In both complexes, the four hydrogen bonds between the arginines and the guanines, observed in the crystal structures, remained stable through the entire MD simulations (see Supplementary Figures S1–S3).
Figure 4.

Stair motifs in the interface of MBD1–mDNA. (a) Orientation of stair motifs mCYT106 ∴ ARG22 ∨ GUA107 (left) and mCYT118 ∴ ARG44 ∨ GUA119 (right) in the MBD1–mDNA complex. (b) Time evolution of hydrogen bonding between the H- and N-atoms in the ARG22-GUA107 pair (black line), H- and O-atoms in the ARG22-GUA107 pair (red line), H- and N-atoms in the ARG44-GUA119 pair (green line), H- and O-atoms in the ARG44-GUA119 pair (blue line). (c) Time evolution of the distance between the center of mass of mCYT106 residue in the DNA and ARG22 residue in the MBD1 protein (blue line), and the distance between the center of mass of mCYT118 residue in the DNA and ARG44 residue in the MBD1 protein (red line). mCYT indicates the methylated cytosine residues.
Protein binding often induces structural changes in the contacted DNA (66,67). In the case of the MBD–mDNA complex, ARG22 and ARG44 insert into the major groove of the DNA, forming hydrogen bonds with the guanines and shifting them into the minor groove (see Supplementary Figure S4). This shift decreases the stacking interaction between guanine and cytosine in mCpG steps, but allows the positively charged ARG22 and ARG44 to form cation-π interactions with cytosine bases, in addition to forming hydrogen bonds to guanines. Figure 4c shows that the distance between the center of mass of the arginine side chains and the center of mass of the methyl-cytosine base is well preserved during the 100-ns simulation. Two snapshots in Figure 4a illustrate a coupled hydrogen bonding/cation-π interaction between the arginine and the mCpG-steps in the MBD1–mDNA complex. The spatial configurations of mCYT106 ∴ ARG22 ∨ GUA107 and mCYT118 ∴ ARG44 ∨ GUA119 adopt a so-called stair motif. Similar stair motifs are formed and remain stable at the MBD2–mDNA and MeCP2–mDNA interface, as shown in Supplementary Figures S2–S3.
Methylation stabilizes stair motif
The results presented above show that arginine residues recognize mCpG steps through simultaneous cation-π and hydrogen bonding interactions. Arginine residues are expected to interact with CpG steps at the MBD–nDNA interface as well (33). However, mutational studies in which mCYT106 and mCYT118 were substituted by nCYT106 and nCYT118 (nCYT denotes non-methylated cytosine) revealed that MBD1 does not bind to non-methylated DNA (21), indicating that the 5-methyl groups on the methyl-cytosines are critical for the formation of the stair motif in MBD–mDNA binding interfaces. The influence of methylation on the stability of the stair motif was investigated through a 30-ns MD simulation [simulation (ii), see Table 1] for the MBD1–nDNA complex, where the mCYT106 and mCYT118 nucleotides were mutated into two non-methylated cytosines. The analysis of the MD simulation trajectories showed that the stair motifs in the MBD1–nDNA complex arising at mCYT106 and mCYT118 remained stable in both places for 30 ns. However, the average root-mean-square deviation (RMSD) of the distance between ARG22 and nCYT106 is 1.8 Å, while for ARG22 and mCYT106 it is 1.0 Å; the RMSD between ARG44 and nCYT118 is 1.5 Å and for ARG22 and mCYT118 it is 0.8 Å, indicating that the interactions between ARG and mCYT are more stable than those between ARG and nCYT (see Figure 5). Snapshots in Figure 5 demonstrate the spatial configurations of ARG-nCYT106, ARG-mCYT106, ARG-nCYT118 and ARG-mCYT118. The performed analysis of the MBD1–DNA complex suggests two contributions to MBD–mDNA recognition as discussed below.
Figure 5.

Stabilization of the MBD1–mDNA complex due to CYT106 and CYT118 methylation. (a) Comparison of the RMSD for ARG22-nCYT106 (blue line) and ARG22-mCYT106 (red line). (b) Comparison of the RMSD for ARG44-nCYT118 (blue line) and ARG22-mCYT118 (red line). The structure of the relevant part of the methylated and non-methylated MBD1–DNA complexes is shown on the right. The van der Waals surface of residues is shown in wireframe representation. Two movies are provided in Supplementary Data showing the dynamics of mCYT106 ∴ ARG22 ∨ GUA107 (Suplementary Movie S4) and CYT106 ∴ ARG22 ∨ GUA107 (Supplementary Movie S5).
The first contribution stems from the contact area between mDNA and the MBD, which increases upon cytosine methylation. The contact area, σ, defined in Equation (1), is the surface buried at an interface between DNA and protein, which affects the binding free energy (68) and, therefore, contributes to the stability of the DNA–protein complex. In the case of the MBD–mDNA binding, the methyl groups lead to an increase of the contact area between cytosine and arginine in the stair motif, as illustrated in the snapshots in Figure 5 and Figure 6. Moreover, since several other amino acids in the MBD have direct contact with the mCpG steps, e.g. ASP32 and TYR34, replacing a hydrogen atom with a methyl group likely increases the total interfacial area between DNA and MBD. Indeed, the contact area between mCYT106 and MBD1 is ∼200 Å2, the contact area between mCYT118 and MBD1 is ∼150 Å2 and the total contact area between mDNA and MBD1 is ∼804 Å2 (see Figure 6). After altering mCYT to nCYT, the values of the contact area decrease significantly: the contact area between nCYT106 and MBD1 drops to ∼170 Å2, the contact area between nCYT118 and MBD1 decreases to ∼130 Å2 and the total contact area between DNA and MBD1 decreases to ∼700 Å2.
Figure 6.

Effect of CYT106 and CYT118 methylation on the MBD1–DNA contact area. Time evolution of the surface contact area calculated, using Equation (1), between (a) CYT106 and MBD1, (b) CYT118 and MBD1 and (c) DNA and MBD1. Red and blue lines correspond to the case of methylated and the non-methylated DNA, respectively.
The second contribution to MBD–mDNA recognition stems from the formation of the mCYT ∴ ARG ∨ GUA stair motif that arises twice in the MBD–mDNA interface and is stabilized by cytosine methylation. The motifs exhibit a favorable contact potential energy between MBD and mDNA. In fact, the stair motif induces a cation-π interaction between the aromatic cytosine and the positively charged arginine, and two hydrogen bonds between arginine and guanine. The classical force field used in our MD simulations does not accurately account for polarizability and charge-transfer effects, especially those arising through cation-π interaction (69–72). The quantum many-body effects, i.e. when the interactions between each monomer in the stair motif are non-additive, are also not included in MD simulations (36).
For this reason we performed quantum chemistry calculations of the interaction energy in the mCYT ∴ ARG ∨ GUA and nCYT ∴ ARG ∨ GUA stair motifs in order to determine the impact of the methyl group on the stair motif stabilization. The interaction energy differences ΔEx [see Equation (4)] between methylated and non-methylated stair motif at x = 1 (CYT106 ∴ ARG22 ∨ GUA107) and x = 2 (CYT118 ∴ ARG44 ∨ GUA119) are presented in Figure 7. The average value of ΔE1 is −0.81 ± 0.33 kcal/mol; the fluctuations arise mainly due to the fact that, after optimizing the structure of the MBD1–DNA complex in the solvent, the structures of the stair motif in the complex are at different local minima, with slightly different potential energies. The average value of ΔE2 is −0.80 ± 0.44 kcal/mol, i.e. very close to ΔE1. Thus, the total change of the interaction energy in the two stair motifs is −1.61 ± 0.77 kcal/mol. The corresponding three-body interaction energies are given in Supplementary Table S1.
Figure 7.

Influence of cytosine methylation on MBD1 binding energy. The energy difference ΔE is obtained from quantum chemistry calculations for the two stair motifs arising in the MBD1–DNA interface (see Figure 4). The total energy ΔE = ΔE1 + ΔE2 [see Equation (4)] shows the three-body interaction energy difference of the mCYT ∴ ARG ∨ GUA stair motifs on the MBD1–DNA interface calculated for methylated (Em) and non-methylated (En) cytosine. The blue line (squares) shows the interaction energy calculated for the mCYT106 ∴ ARG22 ∨ GUA107 stair motif, the red line (triangles) shows the interaction energy calculated for the mCYT118 ∴ ARG44 ∨ GUA119 stair motif, while the green line (dots) shows the combined energy difference.
In order to test whether the MP2 method properly describes the interaction between CYT106 and ARG22, two alternative quantum-chemistry methods (CCSD and DFT) were used for the calculation of the energy differences between the methylated and the non-methylated states of the CYT ∴ ARG ∨ GUA stair motif. As shown in Supplementary results (see Supplementary Table S2 and Figure S7), the results obtained using the CCSD method (53) are consistent with those at the MP2 level, while the density functional theory does not describe appropriately the long-range dispersion interactions (73,74), i.e. a part of van der Waals interactions. This result is expected; the discrepancy between CCSD and DFT methods suggests that the dispersion interaction contributes strongly to stair motif stabilization. The three-body (mCYT ∴ ARG ∨ GUA) and two-body (mCYT106 ∴ ARG22) interaction energy differences upon methylation were also calculated using the CHARMM force field (see Supplementary Figures S5 and S8). Consistent with QM calculations, the results show a stabilization effect of cytosine methylation on the studied system.
Free-energy contributions of DNA methylation to MBD1–DNA complex stabilization
The protein–DNA binding affinity is affected by different interactions. In order to account for the total effect of methylation on the stability of the MBD–DNA complex, a thermodynamic analysis of the changes in binding free-energy is needed. For this purpose, FEP calculations were performed to characterize the free-energy contribution of the methyl group to the MBD–DNA complex. The free-energy changes for the MBD1–DNA complex and free DNA are shown in Supplementary Figure S6. The BAR (see ‘Methods’ section) free-energy difference for the free DNA was determined to be ΔG1 = 54.9 ± 0.0 kcal/mol, with −0.12 kcal/mol hysteresis between forward and backward transition; for the MBD–DNA complex, the free-energy difference was determined to be ΔG2 = 53.7 ± 0.1 kcal/mol, with 0.2 kcal/mol hysteresis between forward and backward transition. The hysteresis can be interpreted as an estimate of the systematic error of the FEP calculation, restricted to finite-length errors, i.e. not accounting for force-field inaccuracies or inadequate algorithms (63). Following Equation (5), one obtains ΔΔG = −1.2 ± 0.1 kcal/mol, hence, indicating that methylation makes a favorable contribution to the binding free energy.
Binding affinity of mutated MBD for mDNA
Previous mutagenesis studies have shown that several residues of the MBD play key roles in binding to mDNA. Ohki et al. (21) suggested that the recognition of mCpG steps is due to combined hydrophobic and polar contacts made by five residues of MBD, namely VAL20, ARG22, TYR34, ARG44 and SER45. The hydrophobic region of each residue forms a hydrophobic patch around the methylation sites, while the polar moieties form hydrogen bonds with the polar atoms of DNA. Mutation of these residues can significantly reduce the binding affinity for mDNA (21). Consistent with mutagenesis studies, MD simulations revealed that mutants of MBD1 cause a decrease of the contact interfacial mDNA binding area, reducing the mCpG-binding affinity. Figure 8a shows that the average contact area between mDNA and wild MBD1 is 804 Å2, while for the V20A mutant the average contact area is 752 Å2, for the R22A mutant 678 Å2, for the D32A mutant 767 Å2, for the Y34A mutant 730 Å2, for the R44A mutant 625 Å2 and for the S45A mutant 723 Å2. Remarkably, mutations of ARG22 and ARG44 to alanines decrease the average contact area significantly, namely by 150 Å2, which again emphasizes the important role of ARG22 and ARG44 in mCpG recognition. We also monitored hydrogen bonding between mDNA and MBD. Figure 8b shows the average number of interfacial hydrogen bonds for wild-type MBD1 MBD (13 bonds on average), V20A mutant (9 bds.), R22A mutant (9 bds.), D32A mutant (10 bds.), Y34A mutant (10 bds.), R44A mutant (9 bds.) and S45A (10 bds.). MBD mutation is found to reduce the number of hydrogen bonds between MBD1 and mDNA. The R22A and R44A mutants are seen to have the largest impact on reducing hydrogen bonding with mDNA. The last column in the histogram in Figure 8b depicts the average number of hydrogen bonds in non-methylated DNA bound to wild-type MBD1. In the case of the MBD–DNA complex, although methyl groups are not involved in any hydrogen bonding, removing two methyl groups leads to a decrease of the MBD–DNA contact area (Figure 6), reducing the number of interfacial hydrogen bonds. We note that prior studies have also shown that mutation causes structural rearrangement at the protein–DNA interface, changing DNA-binding affinity (75,76).
Figure 8.

Mutation effects on characteristics of the MBD1–DNA interface. (a) Comparison of the contact area between DNA and mutants of MBD1. The dashed line shows the average contact area for wild-type MBD1–mDNA. (b) Comparison of the number of hydrogen bonds between DNA and mutants of MBD1. The dashed line shows the average number of interfacial hydrogen bonds for wild-type MBD1–mDNA.
DISCUSSION
In the present study, we investigated the role of the stair motif that combines arginine–guanine hydrogen bonding and arginine–cytosine cation-π stacking in the recognition of mCpG steps by MBD. Sequence alignment of human and mouse MBD family proteins shows that two arginine amino acids (ARG22 and ARG44) involved in stair motifs formation are strictly conserved, suggesting that these arginines play a crucial role in MBD–mDNA binding. Indeed, MD simulations and quantum-chemistry calculations, the latter restricted to the stair motif formed by arginine and mCpG steps at mCYT106 and mCYT118, show that methylated cytosine renders the stair motif more stable than does non-methylated cytosine. FEP calculations take into account the energy costs of methylation-induced deformations at the MBD–DNA binding interface, e.g. the displacement of mCpG step guanine to the minor groove, and the potential energy change in the stair motif, e.g. the change in cation-π interaction energy between arginine and cytosine upon methylation, quantifying the overall effect of DNA methylation on MBD–mDNA complex stability.
Our results quantified also that methylation increases the buried hydrophobic surface between MBD and DNA by ∼100 Å2. Previous theoretical and experimental studies suggested that decreasing the hydrophobic surface by 1 Å2 reduces the interface stabilization free energy by 15 ± 1.2 cal/mol (68,77). Therefore, in the case of the MBD–mDNA complex, methylation contributes about 100 × 15 cal/mol = 1.5 kcal/mol to the stability of MBD–mDNA binding, which is close to the value (−1.2 ± 0.1 kcal/mol) obtained through FEP calculations.
The recognition of the 5′-pyrimidine-purine-3′ step via stair motifs through arginine residues has been observed in many protein–DNA complexes (33,36). An example is the sporulation-specific transcription factor Ndt80. Lamoureux et al. (32–34) reported that two 5′-TpG-3′ steps are recognized by Ndt80 via two arginines; the non-polar region of the arginines forms a hydrophobic half-pocket for the methyl group of a thymine, while the polar region contacts the guanine bases. Interestingly, the methyl group in the thymine was found to be crucial for the cation-π interaction between the thymine and the arginine, as substitution of the thymine with uracil, which lacks a methyl group, effectively results in a significant loss of Ndt80 binding affinity (32–34). Similarly, the 5-methyl group on methyl-cytosine is crucial for MBD proteins to recognize mCpG steps. However, previous studies did not address how the methyl groups in DNA lead to such a significant protein binding specificity, leaving a gap for speculation. Filling this gap, we have demonstrated in the present study that the effects of the 5-methyl group arise from a change of binding affinity due to an increased contact area and cation-π interaction.
Binding of a protein to a particular site in DNA involves guidance through protein tertiary structure, DNA local conformation and DNA sequence (78,79). It is worth noting that the binding affinities of MBD proteins for mDNA are sequence-dependent as they target different methylated promoters (17,80). For example, MeCP2 binds more efficiently to mCpG steps with adjacent A/T bases (81), while MBD1 strongly prefers an mCpG step in the context of TmCGCA and TGmCGCA in vitro and in vivo (82). A recent NMR study of MBD2 bound to mDNA revealed that MBD2 has high affinity for an mCGG sequence (23). Earlier studies suggested that the binding affinities of MBD proteins for different mDNA sequences can be attributed to specific residues within the MBD. Future investigations on specific binding properties of MBD proteins at atomic resolution are needed to explain the observed sequence specificities.
In this article we have addressed an important question in epigenetics, namely how MBD proteins are capable of recognizing methylated DNA. Although we answered some questions, others remain open, such as those regarding the mDNA interaction with other mDNA-binding proteins, e.g. Kaiso protein, the influence of the solvent on the MBD–mDNA interaction, temperature and pressure effects on mDNA binding. Epigenetics is still a long way from a thorough understanding of how methylated DNA conducts its functions in the cell and further studies should be initiated to learn more about the crucial, although subtle mDNA recognition process.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–2, Supplementary Figures 1–8, Supplementary Movies 1–5 and Supplementary Results.
FUNDING
Funding for open access charge: National Institutes of Health (P41-RR005969, R01 GM073655); National Science Foundation (PHY0822613).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors gladly acknowledge supercomputer time provided by the Texas Advanced Computing Center and the National Center for Supercomputing Applications via TeraGrid Resource Allocation Committee grant MCA93S028. I.S. acknowledges support as a Beckman Fellow. C.C. acknowledges the Centre National de la Recherche Scientifique for travel support through its Programme International de Coopération Scientifique.
REFERENCES
- 1.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 2.Razin A, Riggs AD. DNA methylation and gene function. Science. 1980;210:604–610. doi: 10.1126/science.6254144. [DOI] [PubMed] [Google Scholar]
- 3.Rottach A, Leonhardt H, Spada F. DNA methylation-mediated epigenetic control. J. Cell. Biochem. 2009;108:43–51. doi: 10.1002/jcb.22253. [DOI] [PubMed] [Google Scholar]
- 4.Raj S, Bräutigam K, Hamanishi ET, Wilkins O, Thomas BR, Schroeder W, Mansfield SD, Plant AL, Campbell MM. Clone history shapes Populus drought responses. Proc. Natl. Acad. Sci. USA. 2011 doi: 10.1073/pnas.1103341108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bird AP. The essentials of DNA methylation. Cell. 1992;70:5–8. doi: 10.1016/0092-8674(92)90526-i. [DOI] [PubMed] [Google Scholar]
- 6.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 7.Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription? Trends Gen. 1997;13:444–449. doi: 10.1016/s0168-9525(97)01268-7. [DOI] [PubMed] [Google Scholar]
- 8.Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006;31:89–97. doi: 10.1016/j.tibs.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keshet I, Lieman-Hurwitz J, Cedar H. DNA methylation affects the formation of active chromatin. Cell. 1986;44:535–543. doi: 10.1016/0092-8674(86)90263-1. [DOI] [PubMed] [Google Scholar]
- 11.Hashimshony T, Zhang J, Keshet I, Bustin M, Cedar H. The role of DNA methylation in setting up chromatin structure during development. Nat. Genet. 2003;34:187–192. doi: 10.1038/ng1158. [DOI] [PubMed] [Google Scholar]
- 12.Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat. Struct. Biol. 2004;11:1068–1075. doi: 10.1038/nsmb840. [DOI] [PubMed] [Google Scholar]
- 13.Lopez-Serra L, Esteller M. Proteins that bind methylated DNA and human cancer: reading the wrong words. Br. J. Cancer. 2008;98:1881–1885. doi: 10.1038/sj.bjc.6604374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choy JS, Wei S, Lee JY, Tan S, Chu S, Lee T-H. DNA methylation increases nucleosome compaction and rigidity. J. Am. Chem. Soc. 2010;132:1782–1783. doi: 10.1021/ja910264z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rottach A, Frauer C, Pichler G, Bonapace IM, Spada F, Leonhardt H. The multi-domain protein Np95 connects DNA methylation and histone modification. Nucleic Acids Res. 2010;38:1796–804. doi: 10.1093/nar/gkp1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31:1765–1774. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rauch C, Trieb M, Wibowo FR, Wellenzohn B, Mayer E, Liedl KR. Towards an understanding of DNA recognition by the methyl-CpG binding domain 1. J. Biomol. Struct. Dyn. 2005;22:695–705. doi: 10.1080/07391102.2005.10507036. [DOI] [PubMed] [Google Scholar]
- 19.Inomata K, Ohki I, Tochio H, Fujiwara K, Hiroaki H, Shirakawa M. Kinetic and thermodynamic evidence for flipping of a methyl-CpG binding domain on methylated DNA. Biochemistry. 2008;47:3266–3271. doi: 10.1021/bi7019029. [DOI] [PubMed] [Google Scholar]
- 20.Ohki I, Shimotake N, Fujita N, Nakao M, Shirakawa M. Solution structure of the methyl-CpG-binding domain of the methylation-dependent transcriptional repressor MBD1. EMBO J. 1999;18:6653–6661. doi: 10.1093/emboj/18.23.6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohki I, Shimotake N, Fujita N, Jee J-G, Ikegami T, Nakao M, Shirakawa M. Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell. 2001;105:487–497. doi: 10.1016/s0092-8674(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 22.Rupon JW, Wang SZ, Gaensler K, Lloyd J, Ginder GD. Methyl binding domain protein 2 mediates γ-globin gene silencing in adult human βYAC transgenic mice. Proc. Natl Acad. Sci. USA. 2006;103:6617–6622. doi: 10.1073/pnas.0509322103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scarsdale JN, Webb HD, Ginder GD, Williams DCJ. Solution structure and dynamic analysis of chicken MBD2 methyl binding domain bound to a target-methylated DNA sequence. Nucleic Acids Res. 2011;39:6741–6752. doi: 10.1093/nar/gkr262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown SE, Suderman MJ, Hallett M, Szyf M. DNA demethylation induced by the methyl-CpG-binding domain protein MBD3. Gene. 2008;420:99–106. doi: 10.1016/j.gene.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Screaton RA, Kiessling S, Sansom OJ, Millar CB, Maddison K, Bird A, Clarke AR, Frisch SM. Fas-associated death domain protein interacts with methyl-CpG binding domain protein 4: a potential link between genome surveillance and apoptosis. Proc. Natl Acad. Sci. USA. 2003;100:5211–5216. doi: 10.1073/pnas.0431215100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell. 1989;58:499–507. doi: 10.1016/0092-8674(89)90430-3. [DOI] [PubMed] [Google Scholar]
- 27.Wakefield RID, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, Barlow PN. The solution structure of the domain from MeCP2 that binds to methylated DNA. J. Mol. Biol. 1999;291:1055–1065. doi: 10.1006/jmbi.1999.3023. [DOI] [PubMed] [Google Scholar]
- 28.Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell. 2008;29:525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 29.Bird AP, Wolffe AP. Methylation-induced repression–belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 30.Clouaire T, Stancheva I. Methyl-CpG binding proteins: specialized transcriptional repressors or structural components of chromatin? Cell. Mol. Life Sci. 2008;65:1509–1522. doi: 10.1007/s00018-008-7324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luscombe NM, Laskowski RA, Thornton JM. Amino acidbase interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lamoureux JS, Stuart D, Tsang R, Wu C, Glover JNM. Structure of the sporulation-specific transcription factor Ndt80 bound to DNA. EMBO J. 2002;21:5721–5732. doi: 10.1093/emboj/cdf572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamoureux JS, Maynes JT, Glover JNM. Recognition of 5′-YpG-3′ sequences by coupled stacking/hydrogen bonding interactions with amino acid residues. J. Mol. Biol. 2004;335:399–408. doi: 10.1016/j.jmb.2003.10.071. [DOI] [PubMed] [Google Scholar]
- 34.Lamoureux JS, Glover JNM. Principles of protein-DNA recognition revealed in the structural analysis of Ndt80-MSE DNA complexes. Structure. 2006;14:555–565. doi: 10.1016/j.str.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 35.Rooman M, Liévin J, Buisine E, Wintjens R. Cation-π/H-bond stair motifs at protein-DNA interfaces. J. Mol. Biol. 2002;319:67–76. doi: 10.1016/s0022-2836(02)00263-2. [DOI] [PubMed] [Google Scholar]
- 36.Biot C, Wintjens R, Rooman M. Stair motifs at protein-DNA interfaces: nonadditivity of H-Bond, stacking, and cation-π Interactions. J. Am. Chem. Soc. 2004;126:6220–6221. doi: 10.1021/ja049620g. [DOI] [PubMed] [Google Scholar]
- 37.MacKerell AD, Jr, Nilsson L. Molecular dynamics simulations of nucleic acid-protein complexes. Curr. Opin. Struct. Biol. 2008;18:194–199. doi: 10.1016/j.sbi.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Humphrey W, Dalke A, Schulten K. VMD—visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comp. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Foloppe N, MacKerell AD., Jr All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comp. Chem. 2000;21:86–104. [Google Scholar]
- 41.MacKerell A, Jr, Bashford D, Bellott M, Dunbrack RL, Jr, Evanseck J, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 42.MacKerell AD, Jr, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comp. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 43.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 44.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994;101:4177–4189. [Google Scholar]
- 45.Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation—the Langevin piston method. J. Chem. Phys. 1995;103:4613–4621. [Google Scholar]
- 46.Shrake A, Rupley JA. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973;79:351–364. doi: 10.1016/0022-2836(73)90011-9. [DOI] [PubMed] [Google Scholar]
- 47.Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, et al. The general atomic and molecular electronic structure system. J. Comp. Chem. 1993;14:1347–1363. [Google Scholar]
- 48.Møller C, Plesset MS. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934;46:618–622. [Google Scholar]
- 49.Foresman JB, Frisch A. Exploring Chemistry with Electronic Structure Methods. Pittsburgh, PA: Gaussian Inc.; 1996. [Google Scholar]
- 50.Wintjens R, Biot C, Rooman M, Liévin J. Basis set and electron correlation effects on ab Initio calculations of cation-π/H-Bond stair motifs. J. Phys. Chem. A. 2003;107:6249–6258. [Google Scholar]
- 51.Frish MJ, Pople JA, Binkley JS. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984;80:3265–3269. [Google Scholar]
- 52.Dunning TH. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989;90:1007–1023. [Google Scholar]
- 53.Piecuch P, Kucharski SA, Kowalski K, Musial M. Efficient computer implementation of the renormalized coupled-cluster methods: the R-CCSD[T], R-CCSD(T), CR-CCSD[T], and CR-CCSD(T) approaches. Comput. Phys. Commun. 2002;149:71–96. [Google Scholar]
- 54.Parr RG, Yang W. New York: Oxford University Press; 1989. Density-functional theory of atoms and molecules. [Google Scholar]
- 55.Yakubovich A, Solov'yov I, Solov'yov A, Greiner W. Conformational changes in glycine tri- and hexapeptide. Eur. Phys. J. D. 2006;39:23–34. [Google Scholar]
- 56.Solov'yov I, Yakubovich A, Solov'yov A, Greiner W. Ab initio study of alanine polypeptide chain twisting. Phys. Rev. E. 2006;73:021916. doi: 10.1103/PhysRevE.73.021916. [DOI] [PubMed] [Google Scholar]
- 57.Solov'yov I, Yakubovich A, Solov'yov A, Greiner W. On the fragmentation of biomolecules: fragmentation of alanine dipeptide along the polypeptide chain. J. Exp. Theor. Phys. 2006;103:463–471. [Google Scholar]
- 58.Boys SF, Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970;19:553–566. [Google Scholar]
- 59.Wintjens R, Liévin J, Rooman M, Buisine E. Contribution of cation-π interactions to the stability of protein-DNA complexes. J. Mol. Biol. 2000;302:393–408. doi: 10.1006/jmbi.2000.4040. [DOI] [PubMed] [Google Scholar]
- 60.Zwanzig RW. High-temperature equation of state by a perturbation method. I. Nonpolar gases. J. Chem. Phys. 1954;22:1420–1426. [Google Scholar]
- 61.Chipot C, Pearlman DA. Free energy calculations. The long and winding gilded road. Mol. Sim. 2002;28:1–12. [Google Scholar]
- 62.Chipot C, Pohorille A. Free Energy Calculations: Theory and Applications in Chemistry and Biology. Berlin Heidelberg: Springer; 2007. [Google Scholar]
- 63.Pohorille A, Jarzynski C, Chipot C. Good practices in free-energy calculations. J. Phys. Chem. B. 2010;114:10235–10253. doi: 10.1021/jp102971x. [DOI] [PubMed] [Google Scholar]
- 64.Bennett CH. Efficient estimation of free energy differences from Monte Carlo data. J. Chem. Phys. 1976;22:245–268. [Google Scholar]
- 65.Kirkwood J. Statistical mechanics of fluid mixtures. J. Chem. Phys. 1935;3:300–313. [Google Scholar]
- 66.Erikssona M, Härda T, Nilsson L. Molecular dynamics simulations of the glucocorticoid receptor DNA-binding domain in complex with DNA and free in solution. Biophys. J. 1995;68:402–426. doi: 10.1016/S0006-3495(95)80203-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prévost C, Takahashi M, Lavery R. Deforming DNA: from physics to biology. Chem. Phys. Chem. 2009;10:1399–1404. doi: 10.1002/cphc.200900253. [DOI] [PubMed] [Google Scholar]
- 68.Vallone B, Miele AE, Vecchini P, Chiancone E, Brunori M. Free energy of burying hydrophobic residues in the interface between protein subunits. Proc. Natl Acad. Sci. USA. 1998;95:6103–6107. doi: 10.1073/pnas.95.11.6103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caldwell JW, Kollman PA. Cation-π interactions: nonadditive effects are critical in their accurate representation. J. Am. Chem. Soc. 1995;117:4177–4178. [Google Scholar]
- 70.Cubero E, Luque FJ, Orozco M. Is polarization important in cation-π interactions? Proc. Natl Acad. Sci. USA. 1998;95:5976–5980. doi: 10.1073/pnas.95.11.5976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Minoux H, Chipot C. Cation-π interactions in proteins: can simple models provide an accurate description? J. Am. Chem. Soc. 1999;121:10366–10372. [Google Scholar]
- 72.Dehez F, Angyan JG, Gutierrez IS, Luque FJ, Schulten K, Chipot C. Modeling induction phenomena in intermolecular interactions with an ab initio force field. J. Chem. Theor. Comp. 2007;3:1914–1926. doi: 10.1021/ct700156a. [DOI] [PubMed] [Google Scholar]
- 73.van Mourik T, Gdanitz RJ. A critical note on density functional theory studies on rare-gas dimers. J. Chem. Phys. 2002;116:9620–9623. [Google Scholar]
- 74.Šponer J, Jurečka P, Hobza P. Base stacking and base pairing. In: Šponer J, Lankaš F, editors. Computational Studies of RNA and DNA. Netherlands: Springer; 2006. pp. 343–388. [Google Scholar]
- 75.Anderson PC, Daggett V. The R46Q, R131Q and R154H polymorphs of human DNA glycosylase/β-Lyase hOgg1 severely distort the active site and DNA recognition site but do not cause unfolding. J. Am. Chem. Soc. 2009;131:9506–9515. doi: 10.1021/ja809726e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Rutherford K, Daggett V. Polymorphisms and disease: hotspots of inactivation in methyltransferases. Trends Biochem. Sci. 2010;35:531–538. doi: 10.1016/j.tibs.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Juffer AH, Eisenhaber F, Hubbard SJ, Walther D, Argos P. Comparison of atomic solvation parametric sets: applicability and limitations in protein folding and binding. Prot. Sci. 1995;4:2499–2509. doi: 10.1002/pro.5560041206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Travers AA. DNA conformation and protein binding. Annu. Rev. Biochem. 1989;58:427–452. doi: 10.1146/annurev.bi.58.070189.002235. [DOI] [PubMed] [Google Scholar]
- 79.Lavery R, Zakrzewska K, Beveridge D, Bishop TC, Case DA, Cheatham T, III, Dixit S, Jayaram B, Lankas F, Laughton C, et al. A systematic molecular dynamics study of nearest-neighbor effects on base pair and base pair step conformations and fluctuations in B-DNA. Nucleic Acids Res. 2010;38:299–313. doi: 10.1093/nar/gkp834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. Eur. J. Biochem. 2001;268:1–6. doi: 10.1046/j.1432-1327.2001.01869.x. [DOI] [PubMed] [Google Scholar]
- 81.Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol. Cell. 2005;19:667–678. doi: 10.1016/j.molcel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 82.Clouaire T, de las Heras JI, Merusi C, Stancheva I. Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res. 2010;38:4620–4634. doi: 10.1093/nar/gkq228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.