

Abstract
Transplantation of neural stem cells (NSCs) offers a novel therapeutic strategy for stroke; however, massive grafted cell death following transplantation, possibly due to a hostile host brain environment, lessens the effectiveness of this approach. Here, we have investigated whether reprogramming NSCs with minocycline, a broadly used antibiotic also known to possess cytoprotective properties, enhances survival of grafted cells and promotes neuroprotection in ischemic stroke. NSCs harvested from the subventricular zone of fetal rats were preconditioned with minocycline in vitro and transplanted into rat brains 6 h after transient middle cerebral artery occlusion. Histological and behavioral tests were examined from days 0–28 after stroke. For in vitro experiments, NSCs were subjected to oxygen–glucose deprivation and reoxygenation. Cell viability and antioxidant gene expression were analyzed. Minocycline preconditioning protected the grafted NSCs from ischemic reperfusion injury via upregulation of Nrf2 and Nrf2-regulated antioxidant genes. Additionally, preconditioning with minocycline induced the NSCs to release paracrine factors, including brain-derived neurotrophic factor, nerve growth factor, glial cell-derived neurotrophic factor, and vascular endothelial growth factor. Moreover, transplantation of the minocycline-preconditioned NSCs significantly attenuated infarct size and improved neurological performance, compared with non-preconditioned NSCs. Minocycline-induced neuroprotection was abolished by transfecting the NSCs with Nrf2-small interfering RNA before transplantation. Thus, preconditioning with minocycline, which reprograms NSCs to tolerate oxidative stress after ischemic reperfusion injury and express higher levels of paracrine factors through Nrf2 up-regulation, is a simple and safe approach to enhance the effectiveness of transplantation therapy in ischemic stroke.
Introduction
A growing number of experimental studies highlights the potential of stem cell transplantation as a novel therapeutic approach for stroke (Savitz et al., 2002; Bliss et al., 2007). Moreover, a variety of clinical trials have been performed and others are currently ongoing (Banerjee et al., 2011). Transplantation of neural stem cells (NSCs) in the acute stage of stroke often reduces lesion size and inhibits apoptosis in the penumbra area by providing neuroprotective paracrine factors that enhance host cell survival and function (Bliss et al., 2007; Harms et al., 2010). However, a hostile microenvironment in the ischemic brain offers a significant challenge to survival of transplanted cells. Only a small fraction of grafted cells (1–3%) survived in the ischemic brain 28 d after grafting (Hicks et al., 2009; Nakagomi et al., 2009). The accelerated death of grafted cells might be influenced by production of reactive oxygen species after ischemic reperfusion injury and host inflammatory response mediators (Savitz et al., 2002; Lo et al., 2003). This massive loss of stem cells post-engraftment is an impediment that lessens the effectiveness of cell transplantation therapy.
Considering that cell survival may greatly enhance the effectiveness of transplantation therapy, several remedial approaches have been suggested. Ex vivo gene modification of stem cells for overexpression of pro-survival signaling molecules, such as Bcl-2, reduces grafted cell loss (Wei et al., 2005). An alternative strategy is to genetically modulate them for overexpression of the paracrine factors of interest, such as placental growth factor (Liu et al., 2006). These cells serve as a continuous source of paracrine factors that enhance neuroprotection in the host brain. However, while these methods exhibit a better transplantation outcome, a more beneficial, simpler, and safer approach is needed for future clinical application.
Minocycline, a semisynthetic tetracycline, has been clinically used as an antibiotic and anti-inflammatory drug. Previously, we showed the neuroprotective potential of minocycline in animal models of cerebral ischemia (Yrjänheikki et al., 1999). One of the main biological effects of minocycline is its cytoprotective properties (Zhu et al., 2002). Minocycline selectively manipulates expression of genes, such as Bcl-2 and X chromosome-linked inhibitor-of-apoptosis protein (Keilhoff et al., 2008; Kernt et al., 2010). This finding supports our study rationale that minocycline preconditioning may induce reprogramming of NSCs and promote neuroprotection after transplantation. Therefore, the purpose of the present study was to determine whether preconditioning with minocycline protects grafted cells from ischemic reperfusion injury and enhances the effectiveness of transplantation therapy in ischemic stroke. We also sought to elucidate the underlying mechanisms of minocycline preconditioning in NSCs.
Materials and Methods
Isolation and culturing of fetal NSCs.
All animals were treated in accordance with Stanford University Guidelines and the animal protocols were approved by Stanford University's Administrative Panel on Laboratory Animal Care (Stanford, CA). NSCs were harvested from green fluorescent protein (GFP) transgenic Sprague Dawley rats (SD-Tg(GFP)2BalRrrc) as described previously (Blurton-Jones et al., 2009), with some modification. In brief, bilateral subventricular zones from postnatal day 1 rat brains were dissected in Dulbecco's PBS (14040–182; Invitrogen) and mechanically dissociated. The cells were collected and resuspended in Neurobasal-A medium (catalog no. 10888-022; Invitrogen) containing B-27 supplement (catalog no. 12587-010; Invitrogen), l-glutamine (catalog no. 25030-081; Invitrogen), 20 ng/ml rat fibroblast growth factor-basic (catalog no. 400-29; PeproTech), and 10 ng/ml rat epidermal growth factor (400-25; PeproTech). Cells were grown on a 10 cm plastic dish precoated with poly-l-ornithine hydrobromide (catalog no. P3655-100MG; Sigma-Aldrich) and laminin (L2020–1MG; Sigma-Aldrich) at 37°C and 5% CO2 as adherent monolayers. The medium was changed every 2 d, and cells were passaged once a week. Cells that had been passaged 5–10 times were used for the experiments.
The NSCs were preconditioned with minocycline before the in vitro experiments or transplantation. Minocycline hydrochloride (catalog no. M9511; Sigma-Aldrich) was added to the cell culture medium (final concentration: 0, 1, 3, or 10 μm) for 24 h, followed by drug washout before experiments.
Treatment of the cultures with oxygen–glucose deprivation.
We used oxygen–glucose deprivation (OGD) and reoxygenation, an in vitro model that best mimics in vivo cerebral ischemia–reperfusion. The NSCs were subjected to OGD by replacing the medium with a buffered salt solution without glucose (in mm): 116 NaCl, 1.8 CaCl2, 0.8 MgSO4, 5.4 KCl, 1 NaH2PO4, 14.7 NaHCO3, and 10 HEPES (375368; EMD Chemicals; pH 7.4). The plates were placed in an anaerobic chamber (Plas Labs) at 37°C. After 8 h, the medium was replaced with the regular NSC medium (Neurobasal-A) with glucose, and the plates were returned to a 5% CO2/95% air incubator for various reoxygenation periods.
Assessment of cell death and cell viability in vitro.
Cell death was quantified by a standard measurement of lactate dehydrogenase (LDH) release using a LDH-cytotoxicity assay kit (catalog no. K311-400; BioVision). Cell viability was assessed with a cell proliferation reagent using a WST-1 assay kit (catalog no. 05015944001; Roche Diagnostics). For in situ labeling of DNA fragmentation, an in situ cell death detection kit, TMR red (12156792910; Roche Diagnostics), was used according to the manufacturer's instructions. Slides were covered with VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (catalog no. H-1500; Vector Laboratories) and observed with confocal microscopy (LSM 510; Carl Zeiss).
Immunofluorescent staining.
For immunocytochemistry, NSCs cultured on eight-well chamber slides (catalog no. 154941; Thermo Fisher Scientific) were washed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min. They were then washed with PBS and incubated for 1 h in blocking solution (PBS containing 3% bovine serum albumin and 0.3% Triton X-100). For immunohistochemistry, the animals were anesthetized and perfused with PBS followed by 4% paraformaldehyde in PBS, pH 7.4. The brains were postfixed overnight in the same fixative at 4°C. For cryosectioning, fixed tissues were cryoprotected in 10% sucrose in PBS overnight at 4°C, then in 20% sucrose in PBS overnight at 4°C, and embedded in Tissue-Tek O.C.T. compound (catalog no. 4583; Sakura Finetek). Cryostat sections (20 μm) were cut and affixed to glass slides. Cells or tissue sections were subsequently incubated overnight at 4°C in an appropriate mixture of primary antibodies. The following antibodies were used: rabbit anti-GFP (1:100; catalog no. G10362; Invitrogen), goat anti-GFP (1:100; catalog no. LS-C67095; LifeSpan BioSciences), mouse anti-nestin (1:100; catalog no. 556309; BD Biosciences PharMingen), mouse anti-Sox2 (a NSC marker) (1:50; catalog no. 4900; Cell Signaling Technology), rabbit anti-β-tubulin (1:500; catalog no. PRB-435P; Covance), rabbit anti-microtubule-associated protein 2 (MAP2) (1:200; catalog no. AB5622; Millipore), mouse anti-glial fibrillary acidic protein (GFAP) (1:100; catalog no. MAB360; Millipore), rabbit anti-NG2 (an oligodendrocytic marker) (1:100 catalog no. AB5320; Millipore), mouse anti-cytochrome c (1:100; catalog no. 556433; BD Biosciences PharMingen), and rabbit anti-Ki-67 (1:500; catalog no. ab16667; Abcam). After three washes in PBS, cells or tissue sections were incubated for 1 h with a 1:500 dilution of the following secondary antibodies: Alexa Fluor 488-conjugated donkey anti-rabbit IgG (A21206), Alexa Fluor 594-conjugated donkey anti-rabbit IgG (A21207), Alexa Fluor 488-conjugated donkey anti-goat IgG (A11055), Alexa Fluor 594-conjugated donkey anti-mouse IgG (A21203; all from Invitrogen). After three washes with PBS, samples were covered with VECTASHIELD mounting medium with DAPI. The samples were examined by confocal microscopy or fluorescence microscopy (Axioplan 2; Carl Zeiss).
In situ detection of superoxide anion production.
Early production of superoxide anions was investigated with the use of hydroethidine (HEt) as described previously (Murakami et al., 1998). Production of superoxide anions was shown by oxidized HEt as diffuse signals and small particles in the cytosol. For the in vitro study, 5 μm HEt solution (D23107; Invitrogen) was added to the cell culture medium. The cells were incubated for 5 min, followed by fixation with 4% paraformaldehyde in PBS for 15 min. For the in vivo study, HEt solution (1 ml of 1 mg/ml in 1% dimethyl sulfoxide with saline) (D11347; Invitrogen) was administered intravenously immediately after transplantation of the NSCs. Animals were killed 1 h after administration, and tissue sections were prepared. For fluorescent double staining of the HEt signals and GFP, the sections were incubated with anti-GFP (1:100; catalog no. G10362; Invitrogen), followed by Alexa Fluor 488-conjugated donkey anti-rabbit IgG (catalog no. A21206; Invitrogen). Slides were covered with VECTASHIELD mounting medium with DAPI. The sections were observed with a fluorescence microscope, and oxidized HEt fluorescence was examined at an excitation of 510 nm and emission of >580 nm and quantified with ImageJ software (version 1.42q; NIH).
Gene expression study.
After 8 h of OGD and 3 h of reoxygenation, total RNA was isolated from the NSCs using the RNeasy Mini kit (catalog no. 74104; SABiosciences), including DNase digestion. At the end, RNA samples were eluted from the columns using 30 μl of RNase-free water, and their concentrations were determined spectrophotometrically by A260. cDNA was synthesized from 1 μg of total RNA using an RT2 first-strand kit (catalog no. C-03; SABiosciences). Quantitative real-time RT-PCR was performed in triplicate in a 25 μl final volume containing template cDNA, RT2 SYBR Green/ROX qPCR master mix (catalog no. PA-012; SABiosciences) and specific primers (SABiosciences), using a Mx3000p qPCR system (Agilent Technologies). For real-time RT-PCR array, a modified oxidative stress and antioxidant defense PCR array (catalog no. PARN-065; SABiosciences) was used. Data were collected with MxPro qPCR software (Agilent Technologies) and analyzed using the ΔΔ threshold cycle (CT) method. In this study, β-actin was used exclusively as a housekeeping gene. As calibrator samples, we used the non-preconditioned NSCs under normal conditions.
Western blot analysis.
After 8 h of OGD and 3 h of reoxygenation, the NSCs were treated with cell lysis buffer (catalog no. 9803; Cell Signaling Technology) and used as whole-cell lysate samples. Protein concentrations were determined by comparison with a known concentration of bovine serum albumin using a kit (catalog no. 23225; Thermo Fisher Scientific). Equal amounts of the samples (20 μg) were loaded per lane and analyzed by SDS-PAGE on a 10% NuPAGE Bis-Tris gel (catalog no. NP0303; Invitrogen) and then immunoblotted. The primary antibodies were a 1:100 dilution of a rabbit polyclonal anti-Nrf2 antibody (molecular weight, ∼100 kDa) (catalog no. sc-13032; Santa Cruz Biotechnology), a 1:1000 dilution of a rabbit polyclonal anti-heme oxygenase-1 (HO-1) antibody (molecular weight, ∼28 kDa) (catalog no. 5141; Cell Signaling Technology), a 1:1000 dilution of a rabbit polyclonal anti-NAD(P)H:quinone oxidoreductase1 (NQO1) antibody (molecular weight: ∼31 kDa) (catalog no. sc-25591; Santa Cruz Biotechnology), a 1:1000 dilution of a rabbit polyclonal anti-manganese-superoxide dismutase antibody (molecular weight, ∼25 kDa) (catalog no. SOD-110; Enzo Life Sciences), and a 1:100,000 dilution of a mouse monoclonal anti-actin antibody (molecular weight, ∼42 kDa) (catalog no. A5441; Sigma-Aldrich). After incubation with horseradish peroxidase-conjugated anti-mouse IgG (catalog no. 7076; Cell Signaling Technology) or anti-rabbit IgG (catalog no. 7074; Cell Signaling Technology), the antigen was detected by SuperSignal West Pico and/or Femto substrate (catalog nos. 1856135/1856189; Thermo Fisher Scientific). Images were scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories), and the results were quantified using MultiAnalyst software (Bio-Rad).
Transfection of small interfering RNA.
NSCs, which were 60% confluent, were transfected with 20 nm stealth Nrf2-small interfering RNA (siRNA) (Invitrogen) or nonfunctioning negative-control siRNA (catalog no. 45-2001; Invitrogen) using Lipofectamine RNAiMAX (catalog no. 13778; Invitrogen) according to the manufacturer's protocols. The target sequence of the rat-specific Nrf2-siRNA mixture was as follows: UUUAAGUGGCCCAAGUCUUGCUCCA. After 48 h of incubation, the NSCs were used for various experiments and transplantation.
Detection of paracrine factors.
For the in vitro study, culture supernatants were collected for analysis after 4 h of OGD and 48 h of reoxygenation. For the in vivo study, fresh brain tissue was removed 2 d after transplantation. The rectangular cuboid tissue block of the cortex, 2 mm on either side of the NSC-transplanted regions, was dissected (width 4 mm × length 10 mm) and used as a sample. Protein was extracted as described in the Western blot analysis section. Commercial brain-derived neurotrophic factor (BDNF), glial cell-derived neurotrophic factor (GDNF) (catalog nos. G7610 and G7620; Promega), and vascular endothelial growth factor (VEGF) (catalog no. RRV00; R&D Systems) ELISA kits were used to quantify the concentration of BDNF, GDNF, and VEGF in each of the samples.
Focal cerebral ischemia.
Adult male Sprague Dawley rats (260–280 g) were subjected to transient focal cerebral ischemia by intraluminal middle cerebral artery blockade with a suture, as described previously (Fujimura et al., 1998), with some modifications. The rats were anesthetized with 2.0% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Physiological parameters were monitored throughout the surgeries. After a midline skin incision, the right external carotid artery was exposed and its branches were electrocoagulated. A 22 mm 4-0 monofilament nylon suture coated with silicon rubber (catalog no. 4037PK5Re; Doccol) was introduced into the right internal carotid artery through the external carotid artery stump. After 90 min of middle cerebral artery occlusion, blood flow was restored by withdrawal of the suture. The animals were maintained in an air-conditioned room at 20°C with ad libitum access to food and water before and after surgery.
Intracerebral transplantation.
The rats were anesthetized 6 h after stroke, and the NSCs were transplanted using a 10 μl Hamilton syringe with a 26 G needle attached to a stereotaxic apparatus (David Kopf Instruments). The rats were given four 1.0 μl deposits of single cell suspension in Dulbecco's PBS (1 × 105 cells per μl) along the anterior–posterior axis into the cortex at these coordinates: (1) anterior–posterior (A–P), +1.0; medial–lateral (M–L), +3.0; dorsal–ventral (D–V), −3.0; (2) A–P, −1.0; M–L, +3.0; D–V, −3.0; (3) A–P, −3.0; M–L, +3.0; D–V, −2.5; (4) A–P, −5.0; M–L, +3.0; D–V, −2.5. These targets approximated the penumbra area in the cortex. Deposits were delivered at 0.5 μl/min, and the needle was left in situ for 5 min post-injection before being slowly removed.
In situ labeling of DNA fragmentation of the transplanted NSCs.
Every eighth section (160 μm apart) containing the graft region (A–P, −1.0; M–L, +3.0; D–V, −3.0) was chosen for staining using the in situ cell death detection kit, TMR red (Roche Diagnostics). The sections were then incubated with rabbit anti-GFP (1:100; catalog no. G10362; Invitrogen) and mouse anti-nestin (1:100; catalog no. 556309; BD Biosciences PharMingen), followed by Alexa Fluor 488-conjugated donkey anti-rabbit IgG (catalog no. A21206; Invitrogen) and Alexa Fluor 647-conjugated donkey anti-mouse IgG (catalog no. A31571; Invitrogen). Slides were covered with VECTASHIELD mounting medium with DAPI. Terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL)-positive cells, also stained with GFP, were counted under high magnification (×1000) using unbiased computational stereology [fractionator method, using STEREOINVESTIGATOR software (MicroBrightfield)], as described previously (Kelly et al., 2004).
Quantification of survival of the transplanted GFP-positive NSCs.
The transplanted GFP-positive cells were counted using unbiased computational stereology as described above. All the GFP-positive cells were counted on five serial coronal sections per brain (2 mm apart) and stained with GFP and DAPI.
Assessment of NSC proliferation and differentiation profiles.
The proportion of GFP-labeled cells, also stained with a proliferation marker (Ki-67) or lineage-specific phenotype markers (β-tubulin, MAP2, and GFAP), was determined by confocal microscopy. Split panel and z-axis analyses were used for all counting. One hundred or more GFP-positive cells were scored for each marker per animal.
Measurement of infarct size.
The brain sections were stained with H&E. We estimated the infarct size in the cortex/striatum as a percentage of the ipsilateral cortex/striatum using the following: [(area of contralateral cortex/striatum) − (area of remaining ipsilateral cortex/striatum)/(area of contralateral cortex/striatum) × 100]. The area of both sides of the cortex and striatum was measured on nine serial coronal sections per brain (1 mm apart), and the area of the infarct was quantified over these nine levels using Adobe Photoshop (Adobe Systems).
Behavioral analysis.
A rotarod test and beam-balance test were evaluated by two individuals blinded to the rat treatment status on the day of transplantation before the stroke surgery and at 1, 7, 14, 21, and 28 d after transplantation. Beam-balance performance was assessed on a six-point scale: 0, balances with steady posture; 1, grasps side of the beam; 2, hugs the beam and one limb falls from the beam; 3, hugs the beam and two limbs fall from the beam or spins on the beam (>60 s); 4, attempts to balance on the beam but falls off (>40 s); 5, attempts to balance on the beam but falls off (>20 s); and 6, falls off with no attempt to balance or hang onto the beam (<20 s) (Schäbitz et al., 2004).
For the rotarod test, after 3 d of training before the stroke surgery, the rats were placed on the cylinder and the time the animals remained on the rotarod was recorded. The speed was slowly increased from 10 to 40 rpm within a period of 4 min. The trial was ended if the animal fell off the rungs. The maximum duration on the device was recorded with three rotarod measurements before the surgery. Rotarod test data are presented as percentages of the maximal duration, compared with the internal baseline control.
Statistical analysis.
Behavioral data were assessed using repeated-measures ANOVA. We used Scheffé's post hoc analysis of the rotarod test and analyzed the beam-balance test using the Steel–Dwass test. For other experimental data, comparisons among multiple groups were performed with one-way ANOVA, followed by Scheffé's post hoc analysis. Comparisons between two groups were achieved with a Student's unpaired t test. Data are expressed as median for the beam-balance test and mean ± SD for the other experiments. Significance was accepted with p < 0.05.
Results
Minocycline preconditioning conferred cytoprotection on NSCs in vitro
We used self-renewing and multipotent NSCs isolated from fetal GFP transgenic rats (Fig. 1A,B). We first investigated whether minocycline preconditioning could reduce NSC death under ischemic reperfusion injury in vitro. After 8 h of OGD and 24 h of reoxygenation, a LDH assay revealed that minocycline-preconditioned NSCs (PCNSCs) (10 μm) had a significant reduction in death (41%), compared with the non-preconditioned NSCs (non-PCNSCs) (Fig. 1C). This cytoprotective effect was supported by a WST-1 assay (Fig. 1D) and TUNEL staining (Fig. 1E), which showed an increased viability and reduced TUNEL positivity in the PCNSCs compared with the non-PCNSCs. Moreover, fluorescent staining of cytochrome c revealed that a smaller number of PCNSCs showed a release of cytochrome c from mitochondria to the cytoplasm than non-PCNSCs after 8 h of OGD and 3 h of reoxygenation (Fig. 1F). Minocycline also reduced the death of NSCs subjected to H2O2 and diethylenetriamine/nitric oxide, as determined by LDH assay (Fig. 1G,H).
Figure 1.

Reduced NSC death with minocycline preconditioning in vitro. A, The NSCs grown as adherent cultures were examined by immunocytochemistry for GFP (green) and the NSC marker nestin (red). Nearly all the GFP-positive cells colocalized with nestin (yellow). Nuclei were counterstained with DAPI (blue). Scale bar, 20 μm. B, After culturing in differentiation medium containing 1 μm retinoic acid and 1% fetal bovine serum for 5 d, GFP-positive cells (green) expressed the neuronal marker β-tubulin, the astrocytic marker GFAP, and the oligodendrocytic marker NG2 (red) (arrows). Nuclei were counterstained with DAPI (blue). Scale bars, 20 μm. C, LDH assay demonstrated a significant reduction in NSC death with minocycline preconditioning (10 μm) after 8 h of OGD and 24 h of reoxygenation (n = 4). Mino, Minocycline; RO, reoxygenation. D, WST-1 assay showed significantly increased cell viability with 10 μm minocycline after 8 h of OGD and 24 h of reoxygenation (n = 4). E, NSCs analyzed by TUNEL staining (red) and DAPI (blue) after 8 h of OGD and 24 h of reoxygenation. Scale bar, 50 μm. The cell-counting study revealed a significant decrease in TUNEL-positive cells with minocycline preconditioning (10 μm) (n = 4). F, Fluorescent staining of cytochrome c (red) and DAPI (blue) after 8 h of OGD and 3 h of reoxygenation. The NSCs in which cytochrome c was released from mitochondria to the cytoplasm are indicated by arrows. Scale bar, 20 μm. G, H, Preconditioning with 10 μm minocycline significantly reduced the release of LDH from the NSCs under the oxidative stimuli of 200 μm H2O2 (G) and 250 μm diethylenetriamine/nitric oxide (DETA/NO) (H) (n = 4). *p < 0.05; ‡p < 0.005; §p < 0.001.
Minocycline preconditioning upregulated antioxidant genes and reduced oxidative stress in vitro
We next sought to elucidate the underlying mechanism of minocycline-induced cytoprotection. NSCs subjected to 8 h of OGD and 15 min of reoxygenation exhibited a remarkable increase in HEt signals in the cytosol, which represents superoxide anion production (Fig. 2A) (Murakami et al., 1998). However, this signal increase was reduced by preconditioning with 10 μm minocycline. This finding suggests that resistance to oxidative stress is one of the mechanisms of minocycline-induced cytoprotection. We investigated the expression of antioxidant genes using a RT-PCR array. This analysis identified that with preconditioning, Nrf2, the principal transcription factor that regulates the basal and inducible expression of a battery of antioxidant genes, was up-regulated together with the Nrf2-regulated antioxidant genes, including NQO1 and HO-1 (Fig. 2B). Real-time RT-PCR assays showed that minocycline preconditioning significantly upregulated expression of Nrf2 (2.7-fold), NQO1 (20-fold), and HO-1 (4.0-fold) after 8 h of OGD and 3 h of reoxygenation, whereas it did not change manganese-superoxide dismutase levels (Fig. 2C). This was also supported by Western blot analysis of whole-cell lysate from the NSCs, showing that minocycline induced significantly higher protein expression of Nrf2 (1.5-, 2.0-fold), NQO1 (2.2-, 5.2-fold), and HO-1 (1.9-, 5.9-fold) under normal conditions and after OGD and reoxygenation, respectively (Fig. 2D).
Figure 2.

Upregulation of antioxidant genes in NSCs with minocycline preconditioning in vitro. A, Fluorescent staining of NSCs with HEt (red) and DAPI (blue) in vitro. Production of superoxide anions in the NSCs is shown by oxidized HEt as diffuse signals and small particles in the cytosol. Preconditioning with 10 μm minocycline reduced the increase in HEt signals after 8 h of OGD and 15 min of reoxygenation (n = 4). Scale bar, 20 μm. B, Expression of antioxidant genes in non-PCNSCs and PCNSCs under normal conditions or after OGD and reoxygenation analyzed by a real-time RT-PCR array (n = 3). Data are expressed as mean fold induction (FI), compared with the non-PCNSCs under normal conditions (−/−). C, D, Real-time RT-PCR assays (C) (n = 3) and Western blot analyses of whole-cell lysate from the NSCs (D) (n = 4) revealed that preconditioning with 10 μm minocycline induced higher expression of Nrf2 and the Nrf2-regulated antioxidant genes NQO1 and HO-1. β-Actin was used as an internal control. Mino, Minocycline; RO, reoxygenation; SOD2, manganese-superoxide dismutase; OD, optical density. *p < 0.05; †p < 0.01; ‡p < 0.005; §p < 0.001.
Upregulation of Nrf2 was essential for minocycline-induced cytoprotection in vitro
To identify the role of Nrf2 in minocycline preconditioning, we pretreated non-PCNSCs and PCNSCs (10 μm) with control- or Nrf2-siRNA before the experiments. Nrf2-siRNA significantly downregulated minocycline-mediated gene expression of Nrf2, NQO1, and HO-1 (Fig. 3A-C). Minocycline-induced reduction in HEt signals after OGD and reoxygenation was reversed by Nrf2-siRNA (Fig. 3D). LDH and WST-1 assays revealed that Nrf2-siRNA abolished minocycline-induced cytoprotection (Fig. 3E,F).
Figure 3.

Inhibition of Nrf2 abolished minocycline-induced cytoprotection in vitro. non-PCNSCs and PCNSCs (10 μm) were pretreated with control- or Nrf2-siRNA before OGD and reoxygenation. A–C, Real-time RT-PCR assays of the NSCs. Expression of Nrf2 (A), together with NQO1 (B) and HO-1 (C), was downregulated by transfection with Nrf2-siRNA (n = 3). D, Reduced HEt signals with minocycline preconditioning after 8 h of OGD and 15 min of reoxygenation were reversed by Nrf2-siRNA (n = 4). E, F, Death and viability of the NSCs analyzed by LDH assay (E) and WST-1 assay (F) after 8 h of OGD and 24 h of reoxygenation. Nrf2-siRNA abolished cytoprotection offered by minocycline preconditioning (n = 4). Mino, Minocycline; c, control-siRNA. *p < 0.05; †p < 0.01; ‡p < 0.005; §p < 0.001.
Minocycline preconditioning promoted paracrine factor expression in vitro
Because Nrf2 and HO-1 regulate secretion of paracrine factors in other types of cells (Dulak et al., 2008; Afonyushkin et al., 2010), we tested whether minocycline changed their expression in NSCs in vitro. Significantly higher gene expression of BDNF, GDNF, nerve growth factor, and VEGF was detected in the PCNSCs (10 μm) after OGD and reoxygenation, compared with the non-PCNSCs (Fig. 4A). This upregulation was partially suppressed by Nrf2-siRNA transfection. The levels of BDNF and GDNF in the conditioned medium 48 h after OGD were measured by sandwich ELISA, showing that both factors were significantly increased by preconditioning with 10 μm minocycline (Fig. 4B). However, Nrf2-siRNA partially inhibited their increase.
Figure 4.

Induction of paracrine factors with minocycline preconditioning in vitro. A, Real-time RT-PCR assays of NSCs. mRNA expression of BDNF, GDNF, nerve growth factor (NGF), and VEGF was significantly upregulated in the PCNSCs (10 μm) after 8 h of OGD and 3 h of reoxygenation, but this was partially blocked by transfection with Nrf2-siRNA (n = 3). Mino, Minocycline; c, control-siRNA. B, In vitro ELISA of the culture supernatant 48 h after OGD. Minocycline preconditioning (10 μm) increased the levels of BDNF and GDNF, which were partially suppressed by Nrf2-siRNA (n = 4). *p < 0.05; †p < 0.01; ‡p < 0.005.
Minocycline preconditioning reduced grafted-cell death
We transplanted NSCs into the cortical ischemic penumbra 6 h after stroke (Fig. 5A). We asked whether minocycline preconditioning could protect the grafted cells from ischemic reperfusion injury in vivo. When non-PCNSCs were transplanted into the ischemic penumbra, HEt signals in these cells increased remarkably 1 h after transplantation, compared with those in the grafted cells in the intact brain (Fig. 5B). However, this signal increase was reduced with minocycline preconditioning (10 μm).
Figure 5.
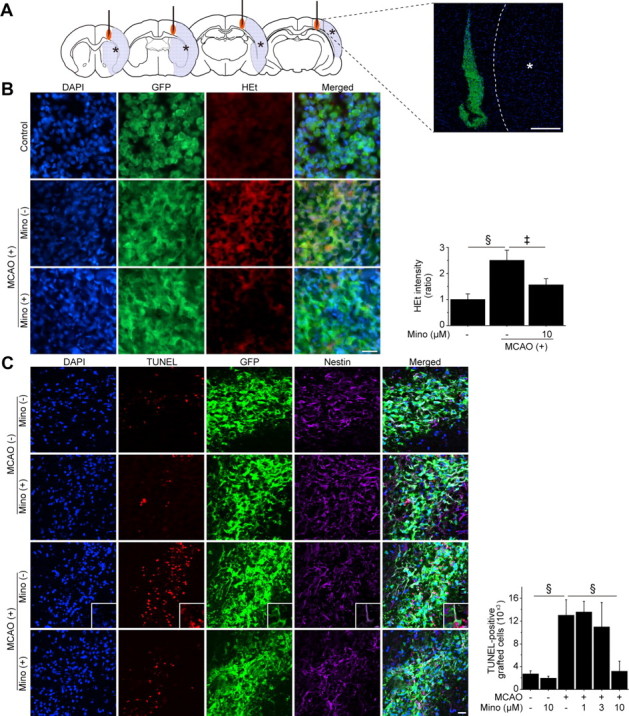
Reduced grafted-cell death with minocycline preconditioning in vivo. A, A schematic diagram and fluorescent staining with DAPI (blue) and GFP (green) 1 h after transplantation show the location of the graft and the ischemic lesion (*). The NSCs, with or without minocycline preconditioning, were transplanted 6 h after stroke. Scale bar, 200 μm. B, Fluorescent staining with DAPI (blue), GFP (green), and HEt (red) in brain sections 1 h after transplantation. HEt signals increased in the non-PCNSCs under ischemic reperfusion injury, but this signal increase was significantly reduced in the PCNSCs (10 μm) (n = 4). Scale bar, 20 μm. MCAO, Middle cerebral artery occlusion; Mino, minocycline. C, Fluorescent staining with DAPI (blue), TUNEL (red), GFP (green), and nestin (magenta) 2 d after transplantation. The number of TUNEL-positive grafted cells was similar in the non-PCNSC and PCNSC groups in the intact brain. When the non-PCNSCs were transplanted into the ischemic brain, the number of TUNEL-positive grafted cells increased remarkably. Minocycline preconditioning (10 μm) significantly reduced the number of TUNEL-positive grafted cells. The insets represent high magnification images showing the colocalization of TUNEL with nestin- and GFP-positive grafted cells (n = 4). Scale bar, 20 μm. ‡p < 0.005; §p < 0.001.
We next counted the number of TUNEL-positive cells (which were also stained with GFP) 2 d after transplantation (Fig. 5C). When transplanted in the intact brain, the number of TUNEL-positive grafted cells was similar between the non-PCNSC and PCNSC groups. When transplanted into the ischemic brain, the number of TUNEL-positive grafted cells increased approximately five times as much as those in the intact brain. Minocycline preconditioning (10 μm) reduced by 76% the number of TUNEL-positive grafted cells in the ischemic brain. This protective effect was also observed when the NSCs were transplanted 7 d after the induction of stroke (data not shown). Together, minocycline preconditioning protected the grafted NSCs from a hostile host–brain environment.
Minocycline preconditioning increased proliferation capacity of NSCs
To assess the proliferation capacity of the NSCs, the number of Ki-67-positive cells, also stained with GFP, was counted 2 d after stroke and transplantation. The percentage of Ki-67-positive grafted cells significantly increased in the PCNSC group, compared with the non-PCNSC group (8.50 ± 2.89 vs 14.75 ± 3.3; p < 0.05) (Fig. 6A). In contrast, there were no detectable Ki-67-positive grafted cells in either group 28 d after stroke and transplantation (data not shown). Increased proliferation with minocycline preconditioning was also confirmed in vitro (14.5 ± 2.53 vs 19.97 ± 3.82; p < 0.05) (Fig. 6B).
Figure 6.

Increased proliferation of NSCs with minocycline preconditioning. A, Immunostaining images of the NSCs 2 d after stroke and transplantation. The sections were stained with GFP (green) and Ki-67 (red). Minocycline preconditioning (10 μm) enhanced proliferation of the grafted cells. Scale bar, 20 μm. Mino, Minocycline. B, Fluorescent staining of the NSCs with Ki-67 (red) and DAPI (blue) after culturing in differentiation medium for 2 d in vitro. The percentage of Ki-67-positive cells was increased with minocycline preconditioning (10 μm). Scale bar, 50 μm.
Minocycline preconditioning increased survival of grafted cells
Twenty-eight days after transplantation, fluorescent staining with GFP revealed an extensive migration of the grafted cells toward the ischemic lesion border in both the non-PCNSC and PCNSC groups (Fig. 7A). No signs of tumor formation caused by the transplanted NSCs were detected in any of the 14 rats. Importantly, a significantly greater number of PCNSCs than non-PCNSCs survived in the ischemic brains (Fig. 7B). In addition, we checked whether transplantation of the non-PCNSCs (n = 4) or PCNSCs (n = 4) caused tumor formation at a later time point. No rats had tumors or teratomas in their brains 3 months after stroke and transplantation.
Figure 7.
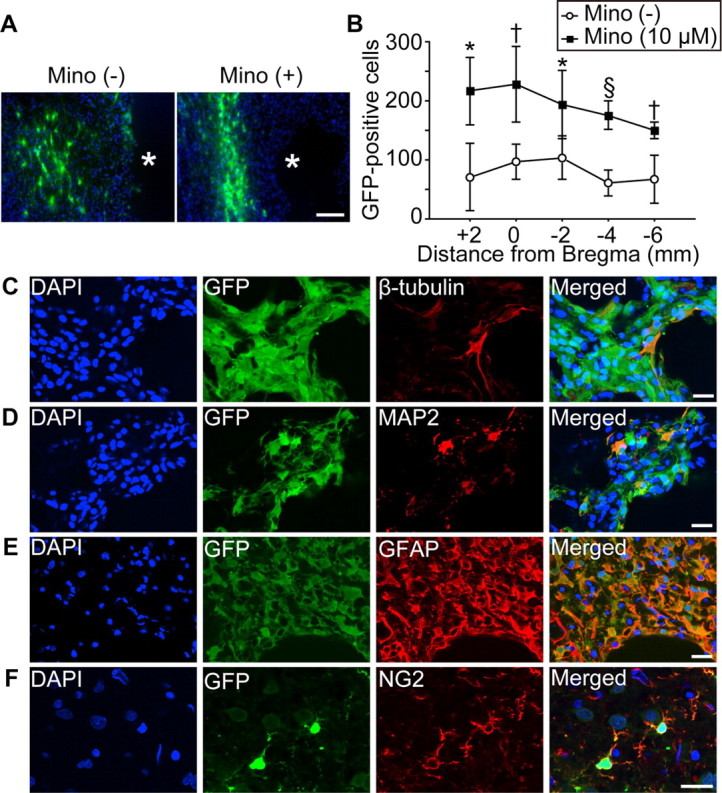
Enhanced survival of the grafted cells with minocycline preconditioning in vivo. A, Fluorescent staining with GFP (green) and DAPI (blue) revealed the migration of the grafted cells toward the lesion border zone 28 d after stroke and transplantation. Asterisk (*) indicates ischemic lesion. Scale bar, 100 μm. Mino, Minocycline. B, Quantification of the number of surviving grafted cells 28 d after stroke and transplantation. Specimens were picked up every 2 mm, and the number of GFP-positive cells was counted. Minocycline preconditioning (10 μm) significantly increased survival of the grafted cells (n = 7). C–F, Fluorescent staining with GFP (green) and β-tubulin (C), MAP2 (D), GFAP (E), or NG2 (red) (F), revealed that the grafted NSCs differentiated into neurons (β-tubulin+, MAP2+), astrocytes (GFAP+), and oligodendrocytes (NG2+) 28 d after transplantation. Nuclei were counterstained with DAPI. Scale bars, 20 μm. *p < 0.05; †p < 0.01; §p < 0.001.
We also analyzed fluorescent double staining of lineage-specific phenotype markers and GFP, which demonstrated that the grafted NSCs differentiated into neurons, astrocytes, or oligodendrocytes 28 d after transplantation (Fig. 7C-F). The percentage of neurons (12% vs 13%) and astrocytes (41% vs 42%) differentiated from the grafted cells was similar between the non-PCNSCs and PCNSCs.
Transplantation of PCNSCs enhanced neuroprotection in ischemic stroke
We investigated whether PCNSCs could facilitate neuroprotection in ischemic stroke. First, we measured BDNF, GDNF, and VEGF levels in the cortex using a sandwich ELISA 2 d after stroke and transplantation. These paracrine factors significantly increased in the PCNSC group (10 μm) compared with the non-transplanted control and non-PCNSC groups (Fig. 8A–C). The increased level of VEGF with minocycline preconditioning was significantly inhibited by transfecting the PCNSCs with Nrf2-siRNA before transplantation. In addition, the levels of BDNF and GDNF were decreased in the PCNSC group with Nrf2-siRNA transfection (15 and 19%) compared to the PCNSC group with control-siRNA transfection, although they did not reach a statistical significance. Twenty-eight days after transplantation, the cortical infarct significantly decreased by 22 and 14% in the PCNSC group (10 μm) compared with the non-transplanted control and non-PCNSC groups, respectively, as determined by H&E staining (Fig. 8D,E). Infarct size in the striatum was similar among the five groups.
Figure 8.

Effects of the PCNSCs on infarct size and behavioral performance following focal cerebral ischemia. A–C, In vivo ELISA of the cortex revealed a significant elevation of BDNF (A), GDNF (B), and VEGF (C) in the PCNSC group 2 d after stroke and transplantation. Transfection with Nrf2-siRNA significantly abolished the VEGF increase and partially blocked the BDNF and GDNF increases (n = 4). Mino, Minocycline; c, control siRNA; MCAO, middle cerebral artery occlusion. D, E, Measurement of the infarct size by H&E staining 28 d after stroke and transplantation. Cortical infarct size was significantly attenuated in the PCNSC group (10 μm), compared with the non-transplanted control and non-PCNSC groups (n = 7). Lesion size in the striatum was similar among the five groups. F, Indices of behavioral performance using the rotarod test and beam-balance test. Transplantation of PCNSCs (10 μm) resulted in the greatest functional recovery 28 d after transplantation as shown by both tests (n = 7). Black bars denote non-transplanted control group; red bars denote non-PCNSC group; blue bars denote PCNSC group (10 μm). The labels show p value compared with the non-transplanted control group. *p < 0.05; †p < 0.01; §p < 0.001.
We next monitored neurological performance using the rotarod test and the beam-balance test. The PCNSC group (10 μm) showed a significant functional recovery from days 1 through 28 using the rotarod test and on day 28 using the beam-balance test, compared with the non-transplanted control group (Fig. 8F). Significant behavioral improvement was not observed in the non-PCNSC group at any time point compared with the non-transplanted control group.
Minocycline preconditioning enhanced neuroprotection via Nrf2 upregulation
We transplanted PCNSCs (10 μm) or non-PCNSCs (pretreated with control- or Nrf2-siRNA) into the ischemic penumbra. The decreased number of TUNEL-positive grafted cells by minocycline was reversed by Nrf2-siRNA (Fig. 9A). The number of Ki-67-positive grafted cells that was increased by minocycline preconditioning was decreased by Nrf2-siRNA (Fig. 9B). The PCNSCs, also transfected with Nrf2-siRNA, failed to attenuate cortical infarct size (Fig. 9C). Furthermore, enhanced behavioral improvement offered by PCNSCs was blocked by Nrf2-siRNA from days 1 through 28 as assessed by both the rotarod test and the beam-balance test (Fig. 9D).
Figure 9.
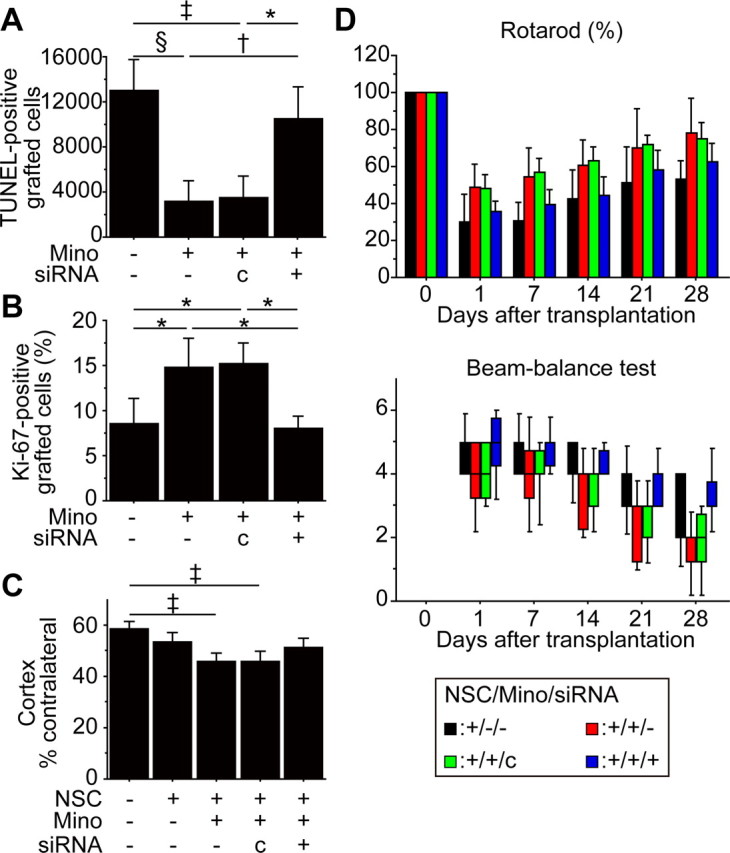
Inhibition of Nrf2 abolished minocycline-induced neuroprotection in vivo. The PCNSCs (10 μm) or non-PCNSCs, transfected with control- or Nrf2-siRNA, were transplanted following focal cerebral ischemia. A, B, Stereological counting of TUNEL-positive (A) and Ki-67-positive (B) grafted cells 2 d after stroke and transplantation. Nrf2-siRNA suppressed minocycline-induced cytoprotection and proliferation of the grafted cells (n = 4). Mino, Minocycline; c, control-siRNA. C, Cortical infarct size evaluated by H&E staining 28 d after transplantation. The PCNSCs, along with Nrf2-siRNA pretreatment, failed to attenuate lesion size (n = 7). D, Behavioral performance analyzed by the rotarod test and beam-balance test. Nrf2-siRNA diminished the enhanced behavioral improvement observed in the PCNSC group from 1 d through 28 d after transplantation (n = 7). Black bars denote non-PCNSC group; red bars denote PCNSC group (10 μm); green bars denote PCNSC group (10 μm) with control-siRNA transfection; blue bars denote PCNSC group (10 μm) with Nrf2-siRNA transfection. *p < 0.05; †p < 0.01; ‡p < 0.005; §p < 0.001.
Discussion
We have shown in this study that PCNSCs not only have a better survival rate than non-PCNSCs, but also express higher levels of paracrine factors, resulting in stronger therapeutic effects in ischemic stroke. The major findings are these: (1) minocycline protected NSCs from ischemic reperfusion injury via upregulation of Nrf2 and Nrf2-regulated antioxidant genes; (2) minocycline increased the proliferation capacity of the NSCs; (3) minocycline preconditioning induced the NSCs to release paracrine factors at least in part by upregulated Nrf2; and (4) transplantation of the PCNSCs resulted in attenuated infarct size and improved neurological recovery, compared with non-PCNSCs. Because minocycline, whose safety profile, clinical features, and potential adverse effects are well characterized, is not necessarily required to be carried by stem cells, we believe that our chemical preconditioning approach is simple and safe to follow from a clinical perspective.
PCNSCs attenuated infarct size and improved the outcome of the rotarod test from 1 d after stroke and transplantation onward, indicating that neuroprotection is one of the main mechanisms of stem cell action in the present study. Minocycline preconditioning enhanced survival of the grafted cells and induced paracrine factors from individual NSCs. These effects might synergistically lead to augmentation of the total amount of neuroprotective paracrine factors in the ischemic brain, resulting in enhanced neuroprotection. Since the effects of PCNSCs on behavioral performance are relatively large compared to the effects on infarct size, transplantation of PCNSCs might also facilitate other mechanisms of stem cell action, such as immunomodulation, angiogenesis, and cell replacement.
In our study, a substantial number of non-PCNSCs died in the ischemic brain. Oxidative stress is induced by ischemia–reperfusion and inflammation in ischemic stroke (Chen et al., 2011). Although not much information is available on the vulnerability and response of NSCs to oxidative stress, they are generally less susceptible to damage from environmental stresses than differentiated cells (Ramalho-Santos et al., 2002). NSCs resist oxidative stress better than neurons because of their higher expression of antioxidant enzymes at a steady-state and faster upregulation following oxidative stress stimuli (Madhavan et al., 2006). Contrarily, studies other than the present one have shown a rapid and massive loss of NSCs under oxidative stress (Wei et al., 2005; Sahlgren et al., 2006; Theus et al., 2008; Acharya et al., 2010). These opposite findings may indicate that oxidative stress induced by pathophysiological conditions exceeds the limitation of the antioxidant capacity of NSCs.
Nrf2, a basic leucine zipper redox-sensitive transcription factor, is a pleiotropic protein that regulates the basal and inducible expression of a battery of antioxidants and other cytoprotective genes (Kaspar et al., 2009). In the present study, we found the vital link between minocycline and Nrf2 as follows. First, minocycline preconditioning induced overexpression of Nrf2 at both mRNA and protein levels. Second, minocycline upregulated expression of Nrf2-regulated antioxidant genes, including NQO1 and HO-1. Third, minocycline-induced expression of Nrf2-regulated antioxidant genes was attenuated by inhibition of Nrf2 by Nrf2-siRNA. These findings provide new evidence that minocycline preconditioning has potential to promote expression of antioxidant genes in NSCs via Nrf2 upregulation. Interestingly, expression of Nrf2 is synergistically upregulated by minocycline preconditioning and OGD stimulation, although the underlying mechanism of this synergistic effect is unknown. Further study is needed to clarify this important issue.
Minocycline preconditioning reduced oxidative stress in the NSCs and conferred cytoprotection in this study. Moreover, Nrf2-siRNA abolished the antioxidant capacity and cytoprotection offered by minocycline, indicating that Nrf2, together with Nrf2-regulated antioxidant genes, plays a critical role in these minocycline effects. In accordance with our findings, others have shown that increased Nrf2 activity is highly neuroprotective by reducing oxidative stress in mixed neuronal/glial cultures and in animal models of ischemic stroke (Kraft et al., 2004; Shih et al., 2005). Curiously, minocycline itself possesses antioxidant properties by directly scavenging free radicals and inhibiting enzymes, such as nitric oxide synthase and lipoxygenases (Kraus et al., 2005). This raises the possibility that minocycline directly reduces the level of reactive oxygen species and protects the NSCs. In addition, minocycline exerts its cytoprotective effect through various mechanisms, including direct inhibition of cytochrome c release from mitochondria and selective upregulation of the anti-apoptotic protein Bcl-2 (Kim and Suh, 2009). Although we cannot exclude the possibility of the involvement of these minocycline-mediated mechanisms in cytoprotection, our findings from the Nrf2-siRNA study indicate that overexpression of Nrf2 is at least the main factor responsible for the minocycline-induced antioxidant capacity and cytoprotection.
Human fetal NSCs are less tumorigenic than embryonic stem cells, and in a clinical trial using human fetal NSCs in Batten disease, no tumors were detected in five patients 2 years after transplantation (Lindvall and Kokaia, 2011). However, since the unfavorable microenvironment after ischemic stroke might influence tumorigenesis and differentiation profiles of grafted NSCs (Seminatore et al., 2010), a thorough characterization of the cells is vital. In the present study, many of the grafted NSCs differentiated into astrocytes (41–42%) 28 d after stroke and transplantation, and some of them seemed to have formed glial scarring, which might hamper recovery from stroke. Nonetheless, NSCs have glial scar-inhibitory effects through downregulation of genes promoting astrogliosis in the brain (Bacigaluppi et al., 2009). Importantly, minocycline preconditioning did not change the differentiation profiles of the grafted NSCs. In contrast, proliferation of the NSCs was transiently increased with minocycline preconditioning, although minocycline is known to reduce the proliferation of resting microglia (Tikka et al., 2001). Interestingly, this enhanced proliferation capacity of the NSCs was abolished by Nrf2-siRNA transfection. Recent studies have shown the association of Nrf2 with cell proliferation (Homma et al., 2009; Wakabayashi et al., 2010). Nrf2 recognizes a functional antioxidant response element in the promoter of Notch1 (Wakabayashi et al., 2010), which regulates processes of NSC proliferation (Aguirre et al., 2010). These studies suggest that minocycline enhances NSC proliferation through cross-talk between the Nrf2 and Notch1 signaling pathways. Since neither Ki-67-positive grafted cells nor tumor formation were observed in the brains transplanted with PCNSCs 28 d and 3 months after stroke and transplantation, minocycline-induced Nrf2 overexpression temporarily and securely enhances the proliferation capacity.
Another interesting finding of this study was the induction of paracrine factors with minocycline preconditioning. These paracrine factors have a neuroprotective potential in animal ischemic stroke models (Wang et al., 1997; Schäbitz et al., 2000; Zhang et al., 2000; Saito et al., 2004). The partial reduction in paracrine factor expression observed in NSCs transfected with Nrf2-siRNA indicates, at least in part, the contribution of Nrf2 to the induction of these factors. Activation of Nrf2 stimulates transcription of ATF4, which in turn directly transactivates the VEGF gene in endothelial cells (Afonyushkin et al., 2010, 2011). In addition, HO-1, whose expression level was regulated by Nrf2 in the present study, mediates the synthesis of VEGF in various cell types (Dulak et al., 2008), as well as BDNF and GDNF in astrocytes and neurons (Morita et al., 2009; Hung et al., 2010). These studies suggest that minocycline-induced overexpression of Nrf2 and subsequent upregulation of HO-1 lead to increased paracrine factor expression. However, since the increased expression of these paracrine factors with minocycline preconditioning was not completely inhibited by Nrf2-siRNA transfection, other minocycline-mediated pathways might also be involved in their upregulation. A future study using NSCs would address this important issue. In light of earlier study results showing that ex vivo gene modification of stem cells for overexpression of individual paracrine factors is neuroprotective (Liu et al., 2006), we anticipated that delivery of stem cells capable of releasing a multitude of pro-survival paracrine factors would be more appealing.
In the present study, we administered an intracerebral injection of NSCs in the acute setting of stroke, which might not be a clinically relevant scenario. Evaluating the effects of minocycline preconditioning using other routes/timing of stem cell delivery is warranted in future studies. In addition, we expect that this chemical preconditioning approach might be applied to other types of cells, such as mesenchymal stem cells.
In conclusion, we have demonstrated that minocycline preconditioning reprograms NSCs to tolerate oxidative stress and to express higher levels of paracrine factors, resulting in enhanced effectiveness of transplantation therapy in ischemic stroke. The beneficial effects of minocycline preconditioning, as well as the simplicity, easy adoptability, and lack of safety issues, make this approach highly appealing for future clinical applications.
Footnotes
This work was supported by National Institutes of Health Grants PO1 NS014543, RO1 NS025372, and RO1 NS038653, and by the James R. Doty Endowment (P.H.C.). We thank Liza Reola and Bernard Calagui for technical assistance, Cheryl Christensen for editorial assistance, and Elizabeth Hoyte for assistance with figure preparation. We also thank Dr. Carlos Lois (California Institute of Technology) for supplying SD-Tg(GFP)2BalRrrc rats.
The authors declare no competing financial interests.
References
- Acharya MM, Lan ML, Kan VH, Patel NH, Giedzinski E, Tseng BP, Limoli CL. Consequences of ionizing radiation-induced damage in human neural stem cells. Free Radic Biol Med. 2010;49:1846–1855. doi: 10.1016/j.freeradbiomed.2010.08.021. [DOI] [PubMed] [Google Scholar]
- Afonyushkin T, Oskolkova OV, Philippova M, Resink TJ, Erne P, Binder BR, Bochkov VN. Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol. 2010;30:1007–1013. doi: 10.1161/ATVBAHA.110.204354. [DOI] [PubMed] [Google Scholar]
- Afonyushkin T, Oskolkova OV, Binder BR, Bochkov VN. Involvement of CK2 in activation of electrophilic genes in endothelial cells by oxidized phospholipids. J Lipid Res. 2011;52:98–103. doi: 10.1194/jlr.M009480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre A, Rubio ME, Gallo V. Notch and EGFR pathway interaction regulates neural stem cell number and self-renewal [Letter] Nature. 2010;467:323–327. doi: 10.1038/nature09347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacigaluppi M, Pluchino S, Peruzzotti-Jametti L, Kilic E, Kilic U, Salani G, Brambilla E, West MJ, Comi G, Martino G, Hermann DM. Delayed post-ischaemic neuroprotection following systemic neural stem cell transplantation involves multiple mechanisms. Brain. 2009;132:2239–2251. doi: 10.1093/brain/awp174. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Williamson D, Habib N, Gordon M, Chataway J. Human stem cell therapy in ischaemic stroke: a review. Age Ageing. 2011;40:7–13. doi: 10.1093/ageing/afq133. [DOI] [PubMed] [Google Scholar]
- Bliss T, Guzman R, Daadi M, Steinberg GK. Cell transplantation therapy for stroke. Stroke. 2007;38:817–826. doi: 10.1161/01.STR.0000247888.25985.62. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Yoshioka H, Kim GS, Jung JE, Okami N, Sakata H, Maier CM, Narasimhan P, Goeders CE, Chan PH. Oxidative stress in ischemic brain damage: mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid Redox Signal. 2011;14:1505–1517. doi: 10.1089/ars.2010.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathobiology. Focus on angiogenesis. Circulation. 2008;117:231–241. doi: 10.1161/CIRCULATIONAHA.107.698316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18:1239–1247. doi: 10.1097/00004647-199811000-00010. [DOI] [PubMed] [Google Scholar]
- Harms KM, Li L, Cunningham LA. Murine neural stem/progenitor cells protect neurons against ischemia by HIF-1α–regulated VEGF signaling. PLoS One. 2010;5:e9767. doi: 10.1371/journal.pone.0009767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks AU, Lappalainen RS, Narkilahti S, Suuronen R, Corbett D, Sivenius J, Hovatta O, Jolkkonen J. Transplantation of human embryonic stem cell-derived neural precursor cells and enriched environment after cortical stroke in rats: cell survival and functional recovery. Eur J Neurosci. 2009;29:562–574. doi: 10.1111/j.1460-9568.2008.06599.x. [DOI] [PubMed] [Google Scholar]
- Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N, Itoh K, Yamamoto M. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res. 2009;15:3423–3432. doi: 10.1158/1078-0432.CCR-08-2822. [DOI] [PubMed] [Google Scholar]
- Hung SY, Liou HC, Fu WM. The mechanism of heme oxygenase-1 action involved in the enhancement of neurotrophic factor expression. Neuropharmacology. 2010;58:321–329. doi: 10.1016/j.neuropharm.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilhoff G, Schild L, Fansa H. Minocycline protects Schwann cells from ischemia-like injury and promotes axonal outgrowth in bioartificial nerve grafts lacking Wallerian degeneration. Exp Neurol. 2008;212:189–200. doi: 10.1016/j.expneurol.2008.03.028. [DOI] [PubMed] [Google Scholar]
- Kelly S, Bliss TM, Shah AK, Sun GH, Ma M, Foo WC, Masel J, Yenari MA, Weissman IL, Uchida N, Palmer T, Steinberg GK. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc Natl Acad Sci U S A. 2004;101:11839–11844. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernt M, Hirneiss C, Neubauer AS, Kampik A. Minocycline is cytoprotective in human corneal endothelial cells and induces anti-apoptotic B-cell CLL/lymphoma 2 (Bcl-2) and X-linked inhibitor of apoptosis (XIAP) Br J Ophthalmol. 2010;94:940–946. doi: 10.1136/bjo.2009.165092. [DOI] [PubMed] [Google Scholar]
- Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav. 2009 doi: 10.1016/j.bbr.2008.09.040. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus RL, Pasieczny R, Lariosa-Willingham K, Turner MS, Jiang A, Trauger JW. Antioxidant properties of minocycline: neuroprotection in an oxidative stress assay and direct radical-scavenging activity. J Neurochem. 2005;94:819–827. doi: 10.1111/j.1471-4159.2005.03219.x. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Kokaia Z. Stem cell research in stroke. How far from the clinic? Stroke. 2011;42:2369–2375. doi: 10.1161/STROKEAHA.110.599654. [DOI] [PubMed] [Google Scholar]
- Liu H, Honmou O, Harada K, Nakamura K, Houkin K, Hamada H, Kocsis JD. Neuroprotection by PIGF gene-modified human mesenchymal stem cells after cerebral ischaemia. Brain. 2006;129:2734–2745. doi: 10.1093/brain/awl207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Madhavan L, Ourednik V, Ourednik J. Increased “vigilance” of antioxidant mechanisms in neural stem cells potentiates their capability to resist oxidative stress. Stem Cells. 2006;24:2110–2119. doi: 10.1634/stemcells.2006-0018. [DOI] [PubMed] [Google Scholar]
- Morita K, Lee MS, Her S. Possible relation of hemin-induced HO-1 expression to the upregulation of VEGF and BDNF mRNA levels in rat C6 glioma cells. J Mol Neurosci. 2009;38:31–40. doi: 10.1007/s12031-008-9156-5. [DOI] [PubMed] [Google Scholar]
- Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi N, Nakagomi T, Kubo S, Nakano-Doi A, Saino O, Takata M, Yoshikawa H, Stern DM, Matsuyama T, Taguchi A. Endothelial cells support survival, proliferation, and neuronal differentiation of transplanted adult ischemia-induced neural stem/progenitor cells after cerebral infarction. Stem Cells. 2009;27:2185–2195. doi: 10.1002/stem.161. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- Sahlgren CM, Pallari HM, He T, Chou YH, Goldman RD, Eriksson JE. A nestin scaffold links Cdk5/p35 signaling to oxidant-induced cell death. EMBO J. 2006;25:4808–4819. doi: 10.1038/sj.emboj.7601366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Narasimhan P, Hayashi T, Okuno S, Ferrand-Drake M, Chan PH. Neuroprotective role of a proline-rich Akt substrate in apoptotic neuronal cell death after stroke: relationships with nerve growth factor. J Neurosci. 2004;24:1584–1593. doi: 10.1523/JNEUROSCI.5209-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz SI, Rosenbaum DM, Dinsmore JH, Wechsler LR, Caplan LR. Cell transplantation for stroke. Ann Neurol. 2002;52:266–275. doi: 10.1002/ana.60000. [DOI] [PubMed] [Google Scholar]
- Schäbitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S. Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke. 2000;31:2212–2217. doi: 10.1161/01.str.31.9.2212. [DOI] [PubMed] [Google Scholar]
- Schäbitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, Schwab S, Sommer C. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35:992–997. doi: 10.1161/01.STR.0000119754.85848.0D. [DOI] [PubMed] [Google Scholar]
- Seminatore C, Polentes J, Ellman D, Kozubenko N, Itier V, Tine S, Tritschler L, Brenot M, Guidou E, Blondeau J, Lhuillier M, Bugi A, Aubry L, Jendelova P, Sykova E, Perrier AL, Finsen B, Onteniente B. The postischemic environment differentially impacts teratoma or tumor formation after transplantation of human embryonic stem cell-derived neural progenitors. Stroke. 2010;41:153–159. doi: 10.1161/STROKEAHA.109.563015. [DOI] [PubMed] [Google Scholar]
- Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theus MH, Wei L, Cui L, Francis K, Hu X, Keogh C, Yu SP. In vitro hypoxic preconditioning of embryonic stem cells as a strategy of promoting cell survival and functional benefits after transplantation into the ischemic rat brain. Exp Neurol. 2008;210:656–670. doi: 10.1016/j.expneurol.2007.12.020. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich BL, Goldsteins G, Keinänen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi N, Shin S, Slocum SL, Agoston ES, Wakabayashi J, Kwak MK, Misra V, Biswal S, Yamamoto M, Kensler TW. Regulation of Notch1 signaling by Nrf2: implications for tissue regeneration. Sci Signal. 2010;3:ra52. doi: 10.1126/scisignal.2000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lin SZ, Chiou A-L, Williams LR, Hoffer BJ. Glial cell line-derived neurotrophic factor protects against ischemia-induced injury in the cerebral cortex. J Neurosci. 1997;17:4341–4348. doi: 10.1523/JNEUROSCI.17-11-04341.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Cui L, Snider BJ, Rivkin M, Yu SS, Lee CS, Adams LD, Gottlieb DI, Johnson EM, Jr, Yu SP, Choi DW. Transplantation of embryonic stem cells overexpressing Bcl-2 promotes functional recovery after transient cerebral ischemia. Neurobiol Dis. 2005;19:183–193. doi: 10.1016/j.nbd.2004.12.016. [DOI] [PubMed] [Google Scholar]
- Yrjänheikki J, Tikka T, Keinänen R, Goldsteins G, Chan PH, Koistinaho J. A tetracycline derivative, minocycline, reduces inflammation and protects against focal cerebral ischemia with a wide therapeutic window. Proc Natl Acad Sci U S A. 1999;96:13496–13500. doi: 10.1073/pnas.96.23.13496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZG, Zhang L, Jiang Q, Zhang R, Davies K, Powers C, van Bruggen N, Chopp M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J Clin Invest. 2000;106:829–838. doi: 10.1172/JCI9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice [Letter] Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]