

Abstract
Nucleobase radicals are the major family of reactive intermediates produced when nucleic acids are exposed to γ-radiolysis. 5,6-Dihydrouridin-5-yl radical (1), the formal product of hydrogen atom addition and a model for hydroxyl radical addition was independently generated from a ketone precursor via Norrish Type I photocleavage in single stranded, and double stranded RNA. Radical 1 produces direct strand breaks at the 5'-adjacent nucleotide and only minor amounts of strand scission are observed at the initial site of radical generation. Strand scission occurs preferentially in double stranded RNA and in the absence of O2. The dependence of strand scission efficiency from 5,6-dihydrouridin-5-yl radical (1) on secondary structure under anaerobic conditions suggests that this reactivity may be useful for extracting additional RNA structural information from hydroxyl radical reactions. Varying the identity of the 5'-adjacent nucleotide has little effect on strand scission. Internucleotidyl strand scission occurs via β-elimination of the 3'-phosphate following C2'-hydrogen atom abstraction by 1. The subsequently formed olefin cation radical yields RNA fragments containing 3'-phosphate or 3'-deoxy-2'-ketonucleotide termini from competing deprotonation pathways. The ketonucleotide end group is favored in the presence of low concentrations of thiol, presumably by reducing the cation radical to the enol. Competition studies with thiol show that strand scission from 5,6-dihydrouridin-5-yl radical (1) is significantly faster than from 5,6-dihydrouridin-6-yl radical (2) and is consistent with computational studies using the G3B3 approach that predict the latter to be more stable than 1 by 2.8 kcal/mol.
Keywords: RNA and DNA damage, reaction mechanism, nucleic acids, free radicals
Introduction
RNA cleavage is an important tool for analyzing structure and folding of this biopolymer.1 Hydroxyl radical (OH•), produced by Fe•EDTA or pulse radiolysis is the most commonly used reagent for this purpose.2, 3 RNA damage induced by reactive oxygen species is also increasingly recognized to play a role in diseases and aging.4–6 Extensive research by radiation scientists has established that OH• and hydrogen atoms preferentially add to the π-bonds of RNA nucleobases.4, 5 The nucleobase radicals also arise from hydration of the cation radicals formed via direct ionization of nucleic acids. Hydrogen atom abstraction by OH• from the ribose rings has been estimated to account for less than 10% of the reactions with polynucleotides. Despite this, RNA cleavage by OH• under aerobic conditions is typically ascribed to hydrogen abstraction by this radical from the C4'- and C5'-positions of the ribose backbone. The high reactivity of OH• suggests that it will exhibit little chemical selectivity. Preferential abstraction of the C4'- and C5'-hydrogen atoms is attributed to their high accessibility to diffusible species.6 The resulting C4'- and C5'-radicals yield direct strand breaks by mechanisms that are partially understood.7, 8 In contrast, strand scission from nucleobase radicals requires subsequent hydrogen atom abstraction from a ribose ring. Herein we describe the reactivity of 5,6-dihydrouridin-5-yl radical (1) and show that it produces direct strand breaks by abstracting a hydrogen atom from the ribose backbone. 5,6-Dihydrouridin-5-yl (1) is the second nucleobase radical shown to form direct strand breaks in RNA by abstracting hydrogen atoms from the ribose backbone.9, 10 However, there are significant differences between the reactivity of 1 and 5,6-dihydrouridin-6-yl (2), which are delineated below.

Hydrogen atoms and OH• preferentially add to the more electron rich C5-carbon of the uracil double bond. However, formation of the regioisomeric C5-radicals (1, 3) can account for up to 40% of the addition reactions.5 The C5-radicals are also preferentially formed upon hydration of cation radicals produced via direct ionization.11 Various proposals concerning the subsequent reactivity of these nucleobase radicals have been put forth. In polynucleotides the nucleobase radicals have been proposed to yield strand breaks by abstracting hydrogen atoms from the C4'- and (in RNA) C2'-positions of the sugar backbone.12–15 The cation radicals have also been proposed to yield strand breaks by abstracting ribose hydrogen atoms from the C4'- and C2'-positions.16, 17 In some instances deciphering the intermediate(s) responsible for strand scission resulting from nucleobase intermediates has been difficult due to hydration of the cation radicals, as well as the reverse process, dehydration of hydroxyl radical adducts.14 Determining the nucleotide from which the nucleobase radical abstracts hydrogen atom(s) was also difficult in these experiments because the initially formed reactive intermediates were generated randomly within the biopolymers. Independent generation of reactive intermediates within nucleic acids greatly simplifies elucidating the chemistry of nucleic acid damage and has provided significant insight into DNA damage and repair.18–22 Recently, we applied this approach to study RNA damage and demonstrated that 5,6-dihydrouridin-6-yl (2) (generated via Norrish Type I photocleavage) produces direct strand breaks by abstracting hydrogen atom(s) from its own ribose ring (intranucleotidyl) and the C2'-hydrogen atom from the 5'-adjacent nucleotide (internucleotidyl) (Scheme 1).9, 10, 23 The C2'-radical undergoes β-elimination to yield a strand break in which the 5'-RNA fragment contains a cation radical at its 3'-terminus.
Scheme 1.

Results and Discussion
Precursor synthesis and photochemical generation of 5,6-dihydrouridin-5-yl radical (1)
5,6-Dihydrouridin-5-yl (1) is the formal C6-hydrogen atom adduct of uridine and a model of the respective OH• adduct (3). It was generated in this study from 6 (eqn. 1) via Norrish Type I photocleavage, a photochemical reaction that has been used to generate a number of nucleic acid radicals, including the regioisomeric 5,6-dihydrouridin-6-yl radical (2).23–30 The ketone (8) was synthesized by acylating protected dihydrouridine (7, Scheme 2), as previously described.31 A mixture of diastereomers of 8 was transformed into phosphoramidite 11 by standard methods via the free nucleoside (9). Phosphoramidite 11 was incorporated into oligoribonucleotides via solid phase synthesis. Oligonucleotides containing 6 were deprotected using K2CO3 in methanol, desilylated (Et3N•3HF, pyrrolidinone),32 and purified by denaturing polyacrylamide gel electrophoresis.
 |
(1) |
Scheme 2.a.

The 5'-dimethoxytritylated intermediate (10) was used in monomer photochemical studies to establish the integrity of the radical generation process. High mass balances (Table 1) were obtained when aqueous acetonitrile solutions (1:1 by volume) of 10 (100 µM) were irradiated in the cavity of a Rayonet photoreactor using lamps that emit with maximum intensity at 350 nm. Dimethoxytritylated dihydrouridine (12) and the correspondingly protected 5-hydroxy-5,6-dihydrouridine (13) were the only products observed under anaerobic and aerobic conditions in the presence of β-mercaptoethanol (BME, 5 mM) (Table 1). Photoconversion of 10 is qualitatively less efficient than generation of 2 from the respective t-butyl ketone. This is due at least in part to photoenolization, which was detected by 2H incorporation in recovered 14 that was photolyzed in D2O and analyzed by MALDI-TOF MS.33 Dihydrouridine 12 was also the sole product detected when 10 was photolyzed under anaerobic conditions in the presence of high concentrations of either BME or 2-propanol. Based upon unrecovered starting material, the yield of 12 ranged from 65–98% when the BME concentration was varied between 0.5 and 2.5 M, and from 50–65% when 2-propanol (1.5–10 M) was used as hydrogen atom donor. Mass balances in these reactions ranged from 76–80% in the presence of BME and 75–95% with 2-propanol. Importantly, photoreduction product 15 was not detected in the presence of even the highest concentrations of either hydrogen atom donor. This suggests that the excited state of ketone 6 would not abstract hydrogen atoms from the ribose backbone.
Table 1.
Product yields from the photochemical generation of 5,6-dihydrouridin-5-yl (1) from 10.
| Yield (%)a | ||||
|---|---|---|---|---|
| Conditionsb | 10 | 12 | 13 | Mass Balance |
| Anaerobic | 57.3 ± 2.4 | 27.7 ± 2.6 | - | 85.0 ± 2.3 |
| Aerobic | 62.5 ± 2.9 | - | 29.2 ± 1.4 | 91.7 ± 4.2 |
Yields are the average of 2 independent experiments each carried out in triplicate.
Starting [10] = 100 µM, [BME] = 5 mM.


Direct strand scission from 5,6-dihydrouridin-5-yl radical (1)
Strand scission from 1 was examined in oligonucleotides in which the radical was flanked by uridine (16, 18) or adenosine (17, 19, Table 2). Overall, direct strand scission from 5,6-dihydrouridin-5-yl radical (1) was most efficient in double-stranded substrates under anaerobic conditions. 5,6-Dihydrouridin-6-yl radical (2) also yielded the highest strand scission yields in duplexes under anaerobic conditions.9, 10 Direct strand scission in double stranded RNA (17) was 3.6 ± 0.8 times greater than in single stranded substrate (19) when 1 was flanked by adenosine (Table 2). The average preference for cleavage in duplex RNA (16) was greater when 5,6-dihydrouridin-5-yl radical (1) was flanked by uridine (4.9 ± 1.0) but within experimental error of that observed when 1 was flanked by adenosine. The preference for strand scission in double stranded compared to single stranded RNA by 5,6-dihydrouridin-5-yl radical (1) is approximately 2-fold greater than that of 2.9, 10 The reactivity of the two radicals under aerobic conditions is also very similar. A complex mixture of direct strand scission products is detected in low yield from 1 produced in single and double stranded RNA under aerobic conditions and was not investigated further.
Table 2.
Direct strand scission under anaerobic conditions from 5,6-dihydrouridin-5-yl (1).
| Absolute yield (%)a | ||||
|---|---|---|---|---|
| Substrateb | Inter.c | Intra.d | Total | % Inter.c |
| ds U6U (16) | 31.9 ± 3.0 | 2.0 ± 0.7 | 33.9 ± 3.0 | 94.1 ± 1.9 |
| ss U6U (18) | 3.4 ± 0.6 | 3.2 ± 0.2 | 6.9 ± 1.3 | 52.1 ± 13.5 |
| ds A6A (17) | 26.1 ± 3.8 | 2.4 ± 1.6 | 28.6 ± 3.5 | 91.3 ± 6.0 |
| ss A6A (19) | 5.4 ± 1.4 | 2.4 ± 0.6 | 8.0 ± 1.5 | 68.0 ± 4.5 |
Yields are the average of at least 3 separate experiments each carried out in triplicate.
The substrate is designated by its secondary structure (ss, ds).
Inter. corresponds to strand scission at the 5'-adjacent nucleotide from 6.
Intra. corresponds to strand scission at the nucleotide where 1 is generated.
Strand scission from 5,6-dihydrouridin-5-yl radical (1) is distributed between the 5'-adjacent nucleotide (internucleotidyl) and nucleotide at which 1 is generated (intranucleotidyl). The preference for internucleotidyl strand scission in duplex RNA is slightly greater from 1 than from 5,6-dihydrouridin-6-yl radical (2).9, 10 More than 90% of the strand breaks in double stranded RNA from 1 (compared to 85% from 2) are formed following reaction with the 5'-adjacent nucleotide regardless of whether the radical is flanked by U or A. Although the mechanism for intranucleotidyl strand scission is unknown for either radical, conformational constraints suggest that 5,6-dihydrouridin-5-yl radical (1) would be unable to abstract hydrogen atoms from the C2'- or C3'-positions. Examination of molecular models suggests that reaction at the C5'-position is more likely but there is no evidence to support this pathway, and it is possible that the small amounts of intranucleotidyl strand scission in photolyzed oligonucleotides containing 6 are an artifact. A greater difference in nucleotide selectivity between the nucleobase radicals is observed in single stranded substrates. Whereas as much as 82% of strand scission from 5,6-dihydrouridin-6-yl radical (2) in single stranded substrates occurs at the nucleotide in which the radical is originally generated, less than 50% of the cleavage from 1 is a result of intranucleotidyl reaction.9, 10 Unlike 2 from which the absolute amount of intranucleotidyl strand scission is greater in single stranded RNA than double stranded, the absolute yield of intranucleotidyl strand scission from 5,6-dihydrouridin-5-yl radical (1) is independent of the secondary structure of the RNA in which it is produced. This too is consistent with the possibility that cleavage at the nucleotide in which 1 is generated may be an artifact.

Characterization of the strand scission products
Products were characterized using enzymatic end group analysis and by MALDI-TOF MS.10, 33 Formation of 5,6-dihydrouridin-5-yl radical (1) under anaerobic conditions in single or double stranded RNA produced differing distributions of the same set of products that were separable by 20% denaturing PAGE. These products were observed regardless of whether the radical precursor was flanked by U or A. Calf alkaline phosphatase treatment of photolyzed 3'-32P-16 confirmed that 5'-phosphates (22, 23) were the exclusive products at the intranucleotidyl and 5'-adjacent nucleotide cleavage sites.33 Polynucleotide kinase treatment of 5'-32P-16 that was photolyzed under anaerobic conditions revealed that two of the products contained 3'-phosphates. The migrations of these products were consistent with 3'-phosphorylated products resulting from inter- (24) and intranucleotidyl cleavage (25a,b). A third product that migrated more slowly than either 3'-phosphate did not react with kinase (26a,b). The migration of this product through a 20% gel was altered by treatment with NaBH4, although the effect of the reducing agent differed depending upon whether 1 was flanked by U or A (Eqn. 2).33 Treatment of the product formed from the 5'-U (5'-32P-16) flanking sequence produced a slower migrating product (28a), whereas photolysis of 5'-32P-17 containing a 5'-A yielded a product that upon reduction moved faster through the gel (28b). Regardless, reaction with NaBH4 suggested that formation of 5,6-dihydrouridin-5-yl radical (1) in either sequence under anaerobic conditions produced a strand scission product containing a carbonyl group at its 3'-terminus. The similarities to the reactivity of 5,6-dihydrouridin-6-yl radical (2) suggested that this product resulting from reaction at the 5'-adjacent nucleotide was the 3'-deoxy-2'-keto product (26a,b). An analogous
 |
(2) |
product resulting from intranucleotidyl cleavage (27a,b) was not observed.
Inferential characterization of 26a was validated by MALDI-TOF MS analysis of 16 photolyzed at pH 3.8 (Figure 1). In addition to 26a, the 3'-phosphorylated product resulting from internucleotidyl strand scission (24) was also observed but not the minor product resulting from intranucleotidyl strand scission (25a). Another significant product detected in the MS spectrum is ascribed to the 3'-fragment formed upon scission at the 5'-adjacent nucleotide (23). The ion of this oligonucleotide corresponds to a molecule containing a 5'-phosphorylated dihydrouridine at its 5'-terminus (23), which is what one would expect if strand scission was due to hydrogen atom abstraction by 1 from the 5'-adjacent nucleotide.
Figure 1.

MALDI-TOF MS analysis of reaction products from 5,6-dihydrouridin-5-yl (1) in duplex RNA (17) at pH 3.8.
The ratio of 24 : 26a,b formed from 5,6-dihydrouridin-5-yl radical (1) under anaerobic conditions is independent of flanking sequence, within experimental error. The 3'-terminal phosphate containing product accounts for ~85% from 5'-32P-16 and 5'-32P-17. The ratio of 24 : 26a,b in single stranded RNA resulting from cleavage at the 5'-adjacent nucleotide following generation of 1 flanked by A (5'-32P-19) or U (5'-32P-18) are also within experimental error of one another. However, the absolute yield of 26a,b from the single stranded substrates is <1% and the variation is large. Although this is discussed in more detail below, these observations suggest that the 3'-phosphate (24) and 3'-deoxy-2'-keto (26a,b) products are derived from a common intermediate that differs only with respect to the nucleobase present at the strand scission site (A or U) and this remote difference does not affect its reactivity.
The effect of pH on strand scission and product distribution
The overall strand scission yield resulting from 5,6-dihydrouridin-6-yl radical (2) decreased slightly when the pH was reduced from 7.2 to 3.8. However, the ratio of 5'-fragments containing 3'-deoxy-2'-keto (26a,b) versus 3'-phosphate (24) termini increased significantly. The effect was proposed to result from an influence of the acidity on the reactivity of a cation radical intermediate (5, Scheme 1) formed upon strand scission via β-elimination of the C2'-radical (4) resulting from hydrogen atom abstraction by the nucleobase radical. Consequently, the reactivity of 5,6-dihydrouridin-5-yl radical (1) in duplex RNA at pH 3.8 was compared to that at pH 7.2. When the radical was flanked by either A (5'-32P-17) or U (5'-32P-16) a modest decrease in total strand scission yield was observed when 1 was generated at pH 3.8 (5'-32P-16, 27.7 ± 2.7%; 5'-32P-17, 21.4 ± 1.3%) compared to 7.2 (Table 2). However, in both sequences there was a large increase in the relative amount of 3'-deoxy-2'-keto product (26a,b) compared to the internucleotidyl cleavage product containing a 3'-phosphate (24, Figure 2). The changes in product ratios resulted from an increase in the yield of 26 and a decrease in the amount of 24 formed at pH 3.8. As discussed further below, this is consistent with the partitioning of the cation radical intermediate (5, Scheme 1) formed via β-elimination of the C2'-radical at the 5'-adjacent site.
Figure 2.

Effect of pH on 3'-termini of strand scission products from 5,6-dihydrouridin-5-yl (1) in duplex RNA.
The effects of deuterium substitution on strand scission in duplex RNA resulting from 5,6-dihydrouridin-5-yl radical (1)
Proximity effects, bond dissociation energies, and similarities to products formed by 5,6-dihydrouridin-6-yl radical (2) discussed above, suggested that 1 abstracts the C2'-hydrogen atom en route to strand scission at the 5'-adjacent nucleotide. Indeed, C2'-deuteration of the 5'-adjacent uridine (20) resulted in a significant reduction in strand scission at that site. At pH 7.2 the total strand scission at the 5'-adjacent nucleotide was reduced from slightly less than 32% to less than 9%, giving rise to an approximate KIE = 3.6. Although not rigorously a kinetic isotope effect, the deuterium KIEs reported here (Table 3) are defined by the ratio of cleavage at the 5'-adjacent nucleotide to 1 in 5'-32P-16 to that in 5'-32P-20 (or 5'-32P-21) exposed to the same photolysis conditions. The average KIE at pH 3.8 is larger but statistically within error of the effect at higher pH. Although the KIE at both pH's were significant, the effect of C2'-deuteration on strand scission from 5,6-dihydrouridin-5-yl radical (1) was considerably smaller than from 2.9
Table 3.
Deuterium isotope effect on the reactivity of 5,6-dihydrouridin-5-yl (1) in duplex RNA.
| KIEa | ||
|---|---|---|
| pH | C2'-2H | C3'-2H |
| 7.2 | 3.6 ± 0.7 (5) | 1.1 ± 0.1 (3) |
| 3.8 | 4.7 ± 0.8 (3) | 0.9 ± 0.1 (2) |
KIE determined by measuring cleavage in 5'-32P-16 relative to that in 5'-32P-20 or 5'-32P-21. Each experiment was carried out in triplicate and the number of experiments is in parentheses.
The smaller C2'-2H KIE observed for 5,6-dihydrouridin-5-yl radical (1) compared to 2 may have arisen due to a contribution to strand scission from hydrogen atom abstraction from another ribose position on the 5'-adjacent nucleotide. Hydrogen atom abstraction from the C1'-position would not yield a direct strand break and examination of molecular models suggested that the C4'- and C5'-hydrogen atoms were too far. The C3'-hydrogen atom of the 5'-adjacent nucleotide is in close proximity to 1 in duplex RNA and its C-H bond dissociation energy is calculated with the ONIOM-G3B3 approach to be ~93 kcal/mol, which is comparable to the bond strength of the C1'-carbon-hydrogen bond, albeit considerably higher than the C2'-carbon-hydrogen bond.34 3'-Deuterated uridine was incorporated via its respective phosphoramidite at the 5'-adjacent nucleotide (21), which was synthesized from known ketone 29.33, 35 However, it had no effect on strand scission resulting from 1 at either pH 7.2 or 3.8 (Table 3), indicating that the smaller observed KIE on strand scission emanating from the 5,6-dihydrouridin-5-yl radical (1) than from 2 is not due to abstraction of other hydrogen atoms from the 5'-adjacent nucleotide.

A common mechanism for strand scission from 5,6-dihydrouridin-5-yl (1) and 5,6-dihydrouridin-6-yl (2) radicals
Although an explanation for the smaller effect of C2'-deuteration on strand scission from 5,6-dihydrouridin-5-yl radical (1) than from 2 is discussed below, the deuterium isotope effect observed is consistent with the involvement of hydrogen atom abstraction from this position in strand scission. In addition, the products formed and pH effects on strand scission under anaerobic conditions also suggest that 5'-internucleotidyl strand scission from 1 and 5,6-dihydrouridin-6-yl radical (2) occur via a common intermediate, the C2'-radical (4, Schemes 1, 3). The C2'-radical results from hydrogen atom abstraction by 5,6-dihydrouridin-5-yl radical (1) and undergoes β-elimination to produce a strand break and the cation radical (5) concomitantly. Olefin cation radical formation from α-heteroatom stabilized radicals containing leaving groups in the β-position is precedented in nucleic acid chemistry and small molecules.28, 39–42 Based upon the final products and the pH effect on their formation, we previously proposed competing deprotonation pathways leading from 5.10 The data obtained involving the 5,6-dihydrouridin-5-yl radical (1) are consistent with this as well (Scheme 3, b,c).
Scheme 3.

The effect of thiol (BME) on strand scission and product distribution
An alternate possibility for the smaller KIE observed from 1 compared to 5,6-dihydrouridin-6-yl radical (2) is that the former is more reactive and gives rise to an earlier transition state. The ability of 5,6-dihydrouridin-5-yl radical (1) to abstract hydrogen atoms from the ribose backbone was evaluated by measuring the strand break yield produced from the anaerobic photolysis of 5'-32P-16 over a range of BME concentrations (Eqn. 3).
| (3) |
Subtracting the percentage of cleaved RNA in the presence of a given BME concentration from that in the absence of thiol provided the yield of radical trapped by the hydrogen atom donor. Similarly, the amount of strand scission at high BME concentration (5 mM) was subtracted from the corresponding amount of that product formed in the presence of lower thiol concentration (or the absence of thiol) in order to account for any strand scission not due to 1. The ratio of trapped product : cleaved product varied linearly with BME concentration and the slope of this line (3.5 × 103 M−1) corresponded to kTrap/kCleave. A similar study on the reactivity of 5,6-dihydrouridin-6-yl radical (2) (kTrap/kCleave = 8.4 × 104 M−1) produced a rate constant for strand scission, kCleave = 31 ± 10 s−1 by assuming that kTrap = 2.6 ± 0.5 × 106 M−1s−1, which is the approximate rate constant for BME reaction with monomeric 2 that was determined using competitive kinetics.10, 23 Due to the slow rate constant for strand scission from 2, 0.1 mM BME was able to prevent this process. In contrast, millimolar BME concentrations were required to prevent strand scission from 5,6-dihydrouridin-5-yl radical (1). The rate constant for BME trapping of 1 is unknown, and that for 2 is in the lower range for reactions of thiols with alkyl radicals, which are typically ~ 8 × 106 M−1s−1.36 If we assume that 5,6-dihydrouridin-6-yl radical (2) and 1 have equal rate constants for their reactions with BME, then the latter's rate constant for cleavage in double stranded RNA (kCleave) is 24 times greater (744 s−1). This rate constant is a minimum and could be as high as 2.3 × 103 s−1 if the 5,6-dihydrouridin-5-yl radical (1) reacts with BME at rates that are more typical for an alkyl radical.
Although the overall amount of strand scission is inversely proportional to BME concentration, the effect of the thiol on the distribution of end group products is more complicated. The absolute yield of the 3'-deoxy-2'-keto product (26a) increases in the presence of low BME concentrations at the expense of 3'-phosphate containing fragmentation product, reaching a maximum at ~250 – 500 µM (Figure 3). BME has a similar effect on 26a formed from the 5,6-dihydrouridin-6-yl radical (2).33 We propose that the thiol reduces the olefin cation radical (5) produced upon strand scission (Scheme 3, a).37
Figure 3.
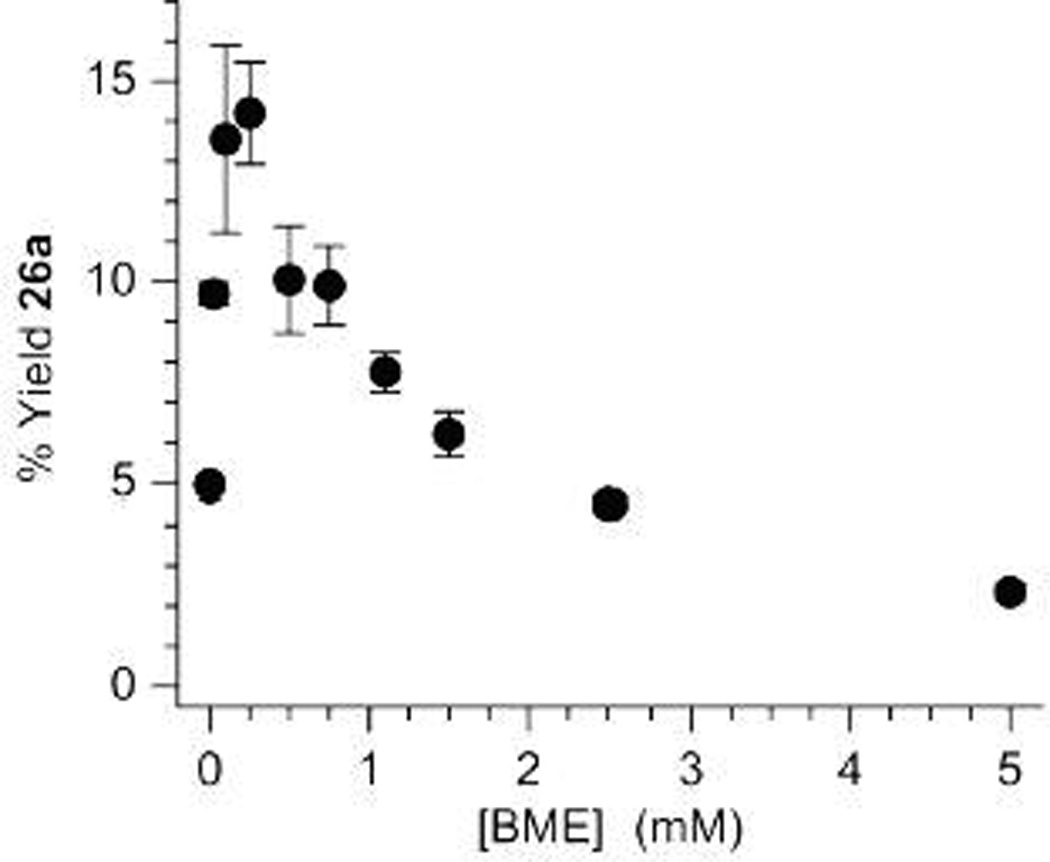
Effect of BME on the 3'-deoxy-2'-keto product (26a) from 5,6-dihydrouridin-5-yl (1) in duplex RNA.
Computational analysis of the reactivity of the 5,6-dihydrouridin-5-yl (2) and 5,6-dihydrouridin-6-yl (1) radicals
The observations that 1 exhibits a smaller KIE than 2 and the need for higher thiol concentrations to quench strand scission from the former suggest that 5,6-dihydrouridin-5-yl (1) is the more reactive of the two radicals. Previous calculations for dihydrothymine at the Hartree Fock (ROHF) level using the isodesmic reaction approach predicted that C6-nucleobase radical was more stable than regioisomeric C5-radical with the C5-H bond (93 kcal/mol) about 4 kcal/mol larger than the C6-H bond (89 kcal/mol).38 We have calculated the bond dissociation enthalpies of the C5- and C6-carbonhydrogen bonds of the more pertinent N1-methyl-5,6-dihydrouracil model system with the thermodynamically predictive G3B3 approach using Gaussian09.39 The G3B3 calculated C5-H bond dissociation enthalpy (95.3 kcal/mol) was 2.8 kcal/mol larger than the respective C6-bond (92.5 kcal/mol). If all of the predicted 2.8 kcal/mol difference in bond dissociation enthalpy were translated to the respective transition states for hydrogen atom abstraction from the C2'-position of the 5'-adjacent nucleotide, 5,6-dihydrouridin-5-yl radical (2) would react more than 125 times faster than the C6-radical. A less than 2 kcal/mol difference in activation energies would account for the minimum (24-fold) difference in kCleave for 1 and 2.
Which step is rate determining in cleavage from 5,6-dihydrouridin-5-yl radical (1)?
The greater rate constant for strand scission via 5,6-dihydrouridin-5-yl radical (1) than from 2 makes the rate determining step uncertain (H•abs or β-Elim in Scheme 3). There is only one study in which the rate constant for a comparable reaction was examined in a nucleic acid. Giese estimated that the C4'-radical in duplex DNA eliminates the 3'-phosphate with a rate constant ~100 s−1 and approximately an order of magnitude faster in single stranded material.40 DNA lesions also cleave faster in single stranded material.41, 42 In contrast, cleavage from 5,6-dihydrouridin-6-yl radical (2) (kCleave) was less than 100 s−1 in either single or double stranded RNA and strand scission was faster in the latter substrate.10 The dependency of kCleave on secondary structure indicated that C2'-radical (4) cleavage could not be the rate limiting step. The rate constant for strand scission from 5,6-dihydrouridin-5-yl radical (1) is considerably faster and from these data alone it is not possible to determine whether the formation of 4 or subsequent elimination from it is the rate determining step. Independent generation of the C2'-radical (4) would be very useful for determining the rate constant for β-elimination to form 5.
Conclusions
5,6-Dihydrouridin-5-yl radical (1) is the second nucleobase radical shown by independent generation from chemical precursors to lead to direct strand scission in RNA. Nucleobase radicals are the major products of the reactions between OH• and pyrimidines in RNA, and radiation chemists have used a variety of tools to study their reactivity.5 The preference for producing direct strand breaks in double stranded compared to single stranded RNA by 1 is even greater than that of 5,6-dihydrouridin-6-yl radical (2) and it is more reactive. It is interesting to note that scientists correctly inferred that C5-radicals would be more reactive than the C6-radicals that are produced in greater amounts from experiments in which the reactive species were generated randomly.14 Computational experiments support the greater reactivity of 5,6-dihydrouridin-5-yl radical (1) than 2, and the approximate rate constants for cleavage (kCleave) and KIEs are also in agreement with this. 5,6-Dihydrouridin-5-yl radical (1) exhibits a higher rate constant for strand scission, and a smaller KIE than 2, which are consistent with an earlier transition state, as would be expected based upon the computational experiments.
Strand scission via nucleobase radicals in RNA is driven by the relatively weak C2'-carbonhydrogen bond dissociation energy, which provides a viable pathway for transferring spin to the carbohydrate backbone.34 This pathway is absent in DNA and it is reflected in the inability of DNA nucleobase radicals to produce direct strand breaks efficiently.19, 20, 28, 43, 44 The reactivity of RNA nucleobase radicals is contrary with respect to previous observations that indicated that it was more stable to oxidative stress than DNA.45 However, these studies focused on direct oxidation of the carbohydrate backbone. For instance, C4'-radicals lead to strand scission more efficiently in DNA than in RNA.46 Our studies on pyrimidine radicals suggest that RNA is more susceptible to strand scission when nucleobase radicals are formed.
Upon completing studies on the reactivity of 5,6-dihydrouridin-6-yl radical (2) we proposed that the preference for strand scission in double stranded versus single stranded RNA under anaerobic conditions may be useful for distinguishing between single and double stranded RNA by comparing cleavage in hydroxyl radical reactions under aerobic and anaerobic conditions.10 We also suggested that enhanced cleavage under anaerobic conditions by 2 was contrary to the generally accepted oxygen enhancement by ionizing radiation, and that nucleobase radicals are not major contributors to direct strand scission under aerobic conditions.4 The results described herein reinforce these notions.
Supplementary Material
Acknowledgment
M.M.G. and M.J.E.R. are grateful for support of this research by the National Institute of General Medical Sciences (GM-054996). M.J.E.R. thanks the NIGMS for a Research Supplement to Promote Diversity in Health-Related Research. M.D.S. and V.B. thank the National Cancer Institute for support (CA-045424). MALDI-TOF MS data were collected on an instrument purchased with support from the NSF-0840463.
Footnotes
Supporting Information Available. Experimental procedures, thiol effects on products, mass spectral data, and sample autoradiograms. Complete Reference 39. These materials are available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Adilakshmi T, Bellur DL, Woodson SA. Nature. 2008;455:1268–1272. doi: 10.1038/nature07298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ralston CY, Sclavi B, Sullivan M, Deras ML, Woodson SA, Chance MR, Brenowitz M. Methods Enzymol. 2000;317:353–368. doi: 10.1016/s0076-6879(00)17024-7. [DOI] [PubMed] [Google Scholar]
- 3.Tullius TD, Greenbaum JA. Curr. Opin. Chem. Biol. 2005;9:127–134. doi: 10.1016/j.cbpa.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 4.von Sonntag C. The Chemical Basis of Radiation Biology. London: Taylor & Francis; 1987. [Google Scholar]
- 5.von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair. Berlin: Springer-Verlag; 2006. [Google Scholar]
- 6.Balasubramanian B, Pogozelski WK, Tullius TD. Proc. Nat. Acad. Sci. USA. 1998;95:9738–9743. doi: 10.1073/pnas.95.17.9738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dussy A, Meggers E, Giese B. J. Am. Chem. Soc. 1998;120:7399–7403. [Google Scholar]
- 8.Pitié M, Pratviel G. Chem. Rev. 2010;110:1018–1059. doi: 10.1021/cr900247m. [DOI] [PubMed] [Google Scholar]
- 9.Jacobs AC, Resendiz MJE, Greenberg MM. J. Am. Chem. Soc. 2010;132:3668–3669. doi: 10.1021/ja100281x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs AC, Resendiz MJE, Greenberg MM. J. Am. Chem. Soc. 2011;133:5152–5159. doi: 10.1021/ja200317w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melvin T, Butchway SW, Parker AW, O'Neill P. J. Am. Chem. Soc. 1996;118:10031–10036. [Google Scholar]
- 12.Deeble DJ, Schulz D, Von Sonntag C. Int. J. Radiat. Biol. 1986;49:915–926. doi: 10.1080/09553008514553151. [DOI] [PubMed] [Google Scholar]
- 13.Lemaire DGE, Bothe E, Schulte-Frohlinde D. Int. J. Radiat. Biol. 1987;51:319–330. doi: 10.1080/09553008714550791. [DOI] [PubMed] [Google Scholar]
- 14.Hildenbrand K, Schulte-Frohlinde D. Int. J. Radiat. Biol. 1989;55:725–738. doi: 10.1080/09553008914550781. [DOI] [PubMed] [Google Scholar]
- 15.Jones GDD, O'Neill P. Int. J. Radiat. Biol. 1991;59:1127–1145. doi: 10.1080/09553009114551031. [DOI] [PubMed] [Google Scholar]
- 16.Schulte-Frohlinde D, Opitz J, Görner H, Bothe E. Int. J. Radiat. Biol. 1985;48:397–408. doi: 10.1080/09553008514551401. [DOI] [PubMed] [Google Scholar]
- 17.Catterall H, Davies MJ, Gilbert BC. J. Chem. Soc. Perkin Trans. II. 1992:1379–1385. [Google Scholar]
- 18.Giese B. Annu. Rev. Biochem. 2002;71:51–70. doi: 10.1146/annurev.biochem.71.083101.134037. [DOI] [PubMed] [Google Scholar]
- 19.Greenberg MM. In: Wiley Ser. React. Intermed. Chem. Biol. Greenberg MM, editor. Vol. 2. 2009. pp. 135–162. [Google Scholar]
- 20.Greenberg MM. Org. Biomol. Chem. 2007;5:18–30. doi: 10.1039/b612729k. [DOI] [PubMed] [Google Scholar]
- 21.Bales BC, Kodama T, Weledji YN, Pitie M, Meunier B, Greenberg MM. Nucleic Acids Res. 2005;33:5371–5379. doi: 10.1093/nar/gki856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kroeger KM, Hashimoto M, Kow YW, Greenberg MM. Biochemistry. 2003;42:2449–2455. doi: 10.1021/bi027168c. [DOI] [PubMed] [Google Scholar]
- 23.Newman CA, Resendiz MJE, Sczepanski JT, Greenberg MM. J. Org. Chem. 2009;74:7007–7012. doi: 10.1021/jo9012805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koerner S, Bryant-Friedrich A, Giese B. J. Org. Chem. 1999;64:1559–1564. doi: 10.1021/jo982022y. [DOI] [PubMed] [Google Scholar]
- 25.Giese B, Beyrich-Graf X, Erdmann P, Petretta M, Schwitter U. Chem. & Biol. 1995;2:367–375. doi: 10.1016/1074-5521(95)90217-1. [DOI] [PubMed] [Google Scholar]
- 26.Tronche C, Goodman BK, Greenberg MM. Chem. Biol. 1998;5:263–271. [PubMed] [Google Scholar]
- 27.Barvian MR, Greenberg MM. J. Org. Chem. 1995;60:1916–1917. [Google Scholar]
- 28.Carter KN, Greenberg MM. J. Am. Chem. Soc. 2003;125:13376–13378. doi: 10.1021/ja036629u. [DOI] [PubMed] [Google Scholar]
- 29.Carter KN, Greenberg MM. J. Org. Chem. 2003;68:4275–4280. doi: 10.1021/jo034038g. [DOI] [PubMed] [Google Scholar]
- 30.Lahoud G, Fancher J, Grosu S, Cavanaugh B, Bryant-Friedrich A. Bioorg. Med. Chem. 2006;14:2581–2588. doi: 10.1016/j.bmc.2005.11.038. [DOI] [PubMed] [Google Scholar]
- 31.Hayakawa H, Tanaka H, Miyasaka T. Tetrahedron. 1985;41:1675–1683. [Google Scholar]
- 32.Wincott F, DiRenzo A, Shaffer C, Grimm S, Tracz D, Workman C, Sweedler D, Gonzalez C, Scaringe S, Usman N. Nucleic Acids Res. 1995;23:2677–2684. doi: 10.1093/nar/23.14.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.See Supporting Information.
- 34.Li M-J, Liu L, Wei K, Fu Y, Guo QX. J. Phys. Chem. B. 2006;110:13582–13589. doi: 10.1021/jp060331j. [DOI] [PubMed] [Google Scholar]
- 35.Serafinowski PJ, Barnes CL. Tetrahedron. 1996;52:7929–7938. [Google Scholar]
- 36.Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 37.Burke M, Edge R, Land EJ, McGarvey DJ, Truscott TG. FEBS Lett. 2001;500:132–136. doi: 10.1016/s0014-5793(01)02601-1. [DOI] [PubMed] [Google Scholar]
- 38.Colson A-O, Becker D, Eliezer I, Sevilla MD. J. Phys. Chem. A. 1997;101:8935–8941. [Google Scholar]
- 39.Frisch MJ, et al. Gaussian 09. Wallingford CT: Gaussian, Inc.; 2009. [Google Scholar]
- 40.Giese B, Dussy A, Meggers E, Petretta M, Schwitter U. J. Am. Chem. Soc. 1997;119:11130–11131. [Google Scholar]
- 41.Kim J, Gil JM, Greenberg MM. Angew. Chem. Int. Ed. 2003;42:5882–5885. doi: 10.1002/anie.200352102. [DOI] [PubMed] [Google Scholar]
- 42.Chen J, Stubbe J. Biochemistry. 2004;43:5278–5286. doi: 10.1021/bi0495376. [DOI] [PubMed] [Google Scholar]
- 43.Hong IS, Carter KN, Greenberg MM. J. Org. Chem. 2004;69:6974–6978. doi: 10.1021/jo0492158. [DOI] [PubMed] [Google Scholar]
- 44.Hong IS, Carter KN, Sato K, Greenberg MM. J. Am. Chem. Soc. 2007;129:4089–4098. doi: 10.1021/ja0692276. [DOI] [PubMed] [Google Scholar]
- 45.Thorp HH. Chem. & Biol. 2000;7:R33–R36. doi: 10.1016/s1074-5521(00)00080-6. [DOI] [PubMed] [Google Scholar]
- 46.Crich D, Mo XS. J. Am. Chem. Soc. 1997;119:249–250. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.