

Abstract
Purpose
Galanin and its three receptors (GALR1-3) are expressed in many normal tissues, but silenced in some tumors. Contradictory roles for galanin and its receptors in various tumors have been reported. To understand their function, investigations of individual GALRs are necessary. In head and neck squamous carcinoma cells (HNSCC) with silenced GALR1 and GALR2, we showed that re-expressed GALR1 suppresses tumor cell proliferation via Erk1/2-mediated effects on cdk inhibitors and cyclin D1. Others showed that GALR2 could induce apoptosis in neuroblastoma cells with wild-type p53, whereas GALR2 stimulated proliferation in small cell lung cancer. In this study, we investigated the role of GALR2 in HNSCC cells that have mutant p53 and do not express GALR1.
Experimental Design and Results
UM-SCC1, a human oral carcinoma cell line with a splice site mutation causing a 46-bp p53 off frame deletion, was stably transfected to express GALR2 (UM-SCC-1-GALR2). Galanin treatment of UM-SCC-1-GALR2 caused morphological changes and a marked decrease in cell number that were not observed in UM-SCC-1-mock cells. Galanin and GALR2 resulted in decreased BrdU incorporation, p27Kip1 and p57Kip2 up-regulation, and decreased cyclin D1 expression. These effects were similar to GALR1 signaling in HNSCC, however, GALR2 also induced caspase-3-dependent apoptosis, which was confirmed by annexin-V staining and DNA fragmentation analysis. These were not observed with GALR1.
Conclusion
This study demonstrates that GALR2 re-expression can inhibit cell proliferation and induce apoptosis in HNSCC cells with mutant p53. GALR2 may be a feasible target for HNSCC therapy.
Keywords: Galanin, G-protein-coupled receptor, apoptosis, p53 independent pathway, head and neck neoplasm
Introduction
Patients with advanced head and neck squamous cell carcinoma (HNSCC) have poor prognosis even when aggressive treatments are used (1). Current progress in molecular biology should make it possible to create new strategies for cancer such as molecular targeting therapy. Approximately 30% of all pharmaceuticals on the market today, exert their therapeutic effect by interacting with a G-protein coupled receptor (GPCR). GPCRs control a wide array of signaling pathways in all tissues of the body and we are only just beginning to understand the full range of their effects (2). Galanin, a 30-amino-acid peptide and its receptors, GALR1, GALR2 and GALR3, are members of the GPCR superfamily and are variably expressed in many normal tissues, including squamous epithelium, and some tumors, such as glioblastoma, neuroblastoma, small cell lung cancer and head and neck cancer (3–6). Reports of the effects of galanin signaling in tumors vary with tumor type. Seufferlein et al. (7) reported that galanin has mitogenic effects in small cell lung carcinoma, whereas, Ishii et al. (8) reported that galanin inhibits pancreatic carcinogenesis. One reason for these conflicting results is lack of knowledge about the effects of individual galanin receptors in cancer cells. To better understand galanin function, studies of individual galanin receptors are needed. Our previous findings indicated galanin and its receptors are likely to act as tumor suppressors in HNSCC because expression of the receptors is frequently silenced by LOH and promoter hypermethylation (9,10, Misawa et al. Clin Can Res. In Press). Using cells that lack endogenous GALR1 expression we demonstrated that in cells expressing exogenous GALR1, galanin stimulation suppressed cell proliferation, consequent to the extracellular signal regulated kinase pathway, suggesting that this receptor is a likely tumor suppressor (6). Berger et al. (4) reported that exogenous expression of GALR2 inhibited cell proliferation and induced apoptosis in neuroblastoma. Furthermore, like GALR1, GALR2 is frequently silenced by methylation and deacetylation in HNSCC (Misawa et al., unpublished data). To further explore how galanin and GALR2 function in HNSCC, we stably transfected HA tagged GALR2 into UM-SCC-1, an HNSCC cell line in which both GALR1 and GALR2 are silenced by methylation (Misawa et al. unpublished data). In the present study, we demonstrated that galanin and GALR2 induce apoptosis and inhibition of cell proliferation and incorporation of BrdU in HNSCC cells with mutant p53. These findings suggest that GALR2 is a potential target gene for molecular therapy in HNSCC.
Materials and Methods
Cell Culture and Proliferation Assay
UM-SCC-1 was established from a human oral carcinoma of the retromolar trigone (Krause, et al. Arch Otolaryngol 107:703–710, 1981). UM-SCC-10B and UM-SCC-74B were established respectively from a lymph node metastasis of a laryngeal cancer and from a recurrent tongue cancer (Bradford et al. Head Neck 25:654–661, 2003). Cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Grand Island, NY) supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin (Irvine Scientific Santa Ana, CA) at 37°C in 5% CO2. The C-terminal HA-tagged GALR2 sequence was obtained from human cDNA library (Invitrogen, Carlsbad, CA), subcloned into the pcDNA3 (Invitrogen) containing the internal ribosomal entry site (Ires) and Green Fluorescent Protein (GFP) sequence. The pCMVIresGFP was used as a negative control. The UM-SCC-1-GALR2 and UM-SCC-1-mock cells were established by transfecting pCMVGALR2IresGFP or pCMVIresGFP into UM-SCC-1 using lipofectamine reagents (Invitrogen). GFP positive cells were selected by Flow Cytometry using FACS Vantage SE (BD Biosciences, San Diego, CA). To examine cell proliferation and morphology, 24 h after the cells were plated, the cells were cultured with serum free media containing with 0.1% BSA (SFM) for 24 h to induce quiescence. Then various concentrations of galanin (ANASPEC, San Jose, CA) were added. A caspase-3 inhibitor, 50 µM Ac-DEVD-CHO (DEVD; Biomol, Plymouth Meeting, PA) was added 2 h prior to galanin treatment. Cell proliferation was determined by counting the cells with a Coulter counter model Z1 (Beckman Coulter Inc., Hialeah, FL). To observe the changes in cell morphology, photographs were taken using the Olympus IX71 inverted microscope (Olympus Corporation, Tokyo, Japan).
p53 Gene Mutation Analysis
The status of the p53 gene in UM-SCC-1 was analyzed by sequencing of cDNA. Total RNA was isolated from UM-SCC-1-GALR2, UM-SCC-74B, and UM-SCC-1-mock cells using the RNAeasy mini kit (Qiagen, Inc., Valencia, CA) according to the manufacturer’s protocol. cDNA was synthesized by the reverse transcription of 2 µg of extracted RNA with 200 units of SuperScript II (Invitrogen) in the presence of random primers and deoxynucleotide triphoshates (Invitrogen). The p53 gene transcripts were amplified using FastStart Taq DNA Polymerase (Roche Diagnostics GmbH). Three overlapping primer sets were used: p53-1 forward 5’-gcgtgctttccacgacg; reverse 5’-ccttccactcggataagatg, p53-2 forward 5’-ttgcattctgggacagccaa; reverse 5’-ggcatccttgagttccaagg, p53-3 forward 5’-caccatcatcacactggaag; reverse 5’-ctgacgcacacctattgcaa. The cycler was programmed with the following conditions: (a) initial denaturation at 94°C for 5 min, followed by (b) 40 cycles of 95°C for 40 s, (c) annealing of the primer template at 58°C for 50 s, (d) extension at 72°C for 50 s, and (e) an additional extension at 72°C for 7 min. The PCR products were separated by electrophoresis through a 1.5% agarose gel containing ethidium bromide and were extracted from the gel with the QIA quick Gel Extraction Kit (Qiagen Inc.). Purified PCR products were sequenced at the University of Michigan DNA Squencing Core on an ABI 3700 (Applied Biosystems, Foster City, CA). p53 sequences and mutations were confirmed using data from the National Center for Biotechnology Information website.
Immunoblotting
The cells were lysed with 1% Nonidet-P 40 lysis buffer containing protease inhibitors (Calbiochem, La Jolla CA). The supernatant was collected and the protein content was measured using the Bio-Rad protein assay (Bio-Rad, Richmond, CA). Equal amounts of protein were electrophoresed on 10% SDS-PAGE gels and transferred to Hybond-P (Amersham Biosciences Limited, Buckinghamshire, United Kingdom). The membranes were incubated overnight with primary antibody at 4°C, followed by incubation with anti-mouse secondary antibody horseradish peroxidase conjugate (Amersham Biosciences Limited) and detected by chemiluminescence and autoradiography using Hyperfilm obtained from Kodak (Rochester, NY). Densitometric measurements of the bands were carried out using ChemiImager 4400 (Alpha Innotech, San Leandro, CA). To detect exogenous GALR2, the cell lysates were treated with 1000 units of N-Glycosidase F (New England BioLabs, Beverly, MA) and loaded onto the gel without boiling to avoid protein aggregation. The mouse monoclonal anti-HA tag antibody (CONVANCE, Berkeley, CA) was used as the primary antibody. Expression of p27Kip1, p57Kip2, cyclin D1, and p53 was detected using the mouse monoclonal antibodies for p27Kip1, p57Kip2 (Lab Vision, Fremont, CA), cyclin D1 (DakoCytomation Norden A/S, Glostrup, Denmark), and p53 (Calbiochem, La Jolla, CA). UM-SCC-10B cells containing a point mutation were used as a p53 high expression control. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was detected using the mouse monoclonal anti-GAPDH monoclonal antibody (Chemicom International, Temecula, CA) as internal control.
Immunocytochemistry
The cells were seeded on coverslips. After 24 h of incubation, the cells were fixed with 4% paraformaldehyde and then stained with mouse monoclonal anti-HA tag antibody and Hoechst 33342 (Molecular Probes, Leiden, The Netherlands). After incubation with Alexa Fluor 546 goat anti-mouse IgG1 (Molecular Probes), the localization of exogenous GALR1 was determined using the Olympus FV-500 Confocal Microscope (Olympus Corporation).
BrdU Incorporation Assay
The cells were seeded on coverslips. After 24 h of serum-starved incubation, 1 µM galanin was added to the SFM and the cells were incubated for an additional 24 h. After galanin treatment, the cells were incubated with 10 µM BrdU (Caltag Laboratories, Burlingame, CA) for 45 min, fixed with 70% ethanol and the DNA was denatured with 2N HCl. The cells were stained with mouse monoclonal anti-BrdU antibody (Caltag Laboratories), followed by incubation with Alexa Fluor 546 goat anti-mouse IgG1 (Molecular Probes). For each coverslip, 3000 cells were counted and the percentage of BrdU-positive cells was determined.
Apoptosis Analysis
Induction of apoptosis was evaluated by active caspase-3 staining, annexin-V staining, and DNA fragmentation analysis. Serum-starved cells were treated with galanin for 12 or 24 h, and active caspase-3 and annexin-V in UM-SCC-1-GALR2 and UM-SCC-1-mock cells were detected using the Active Caspase-3 PE MAb Apoptosis Kit (BD Biosciences) and the Annexin V-PE Apoptosis Detection Kit I (BD Biosciences) according to the manufacturer’s instructions. Flow cytometric analysis was carried out using FACS Vantage SE and the data analyzed using Cell Quest and ModFit LT software packages (Verity Software House Inc., Topsham, ME). Genomic DNA was isolated using DNAeasy Tissue Kit (Qiagen, Inc.), and then loaded onto a 2 % agarose gel. The DNA ladders stained with ethidium bromide were visualized under UV light.
Statistical Analysis
Results were tested for statistical significance using two-way ANOVA or Student’s t-test.
Results
Exogenous GALR2 expresseion
The extracted protein from UM-SCC-1-GALR2 and UM-SCC-1-mock cells pretreated with PNGase-F was assessed for exogenous GALR2 expression by immunoblotting using the monoclonal antibody for HA-tag. Digestion with PNGase-F, a glycosidase that removes N-linked sugars, and solublization without boiling yielded the appropriate GALR2-HA band in UM-SCC-1-GALR2 but not in UM-SCC-1-mock cells (Fig.1A). To assess the cellular localization of exogenous GALR2, cells grown on coverslips were stained with HA-tag antibody. Immunofluorescence revealed that all the GFP-positive cells show exogenous GALR2 expression localized to the cytoplasmic membrane as expected for a GPCR (Fig. 1B). The expression of GALR2 in the parent UM-SCC-1 cells was undetectable and less than that in normal human tissue (data not shown). Thus, the differences in the results that we obtained with the UM-SCC-1-GALR2 and UM-SCC-1-mock cells following galanin stimulation should reflect mainly the function of GALR2.
Fig. 1.
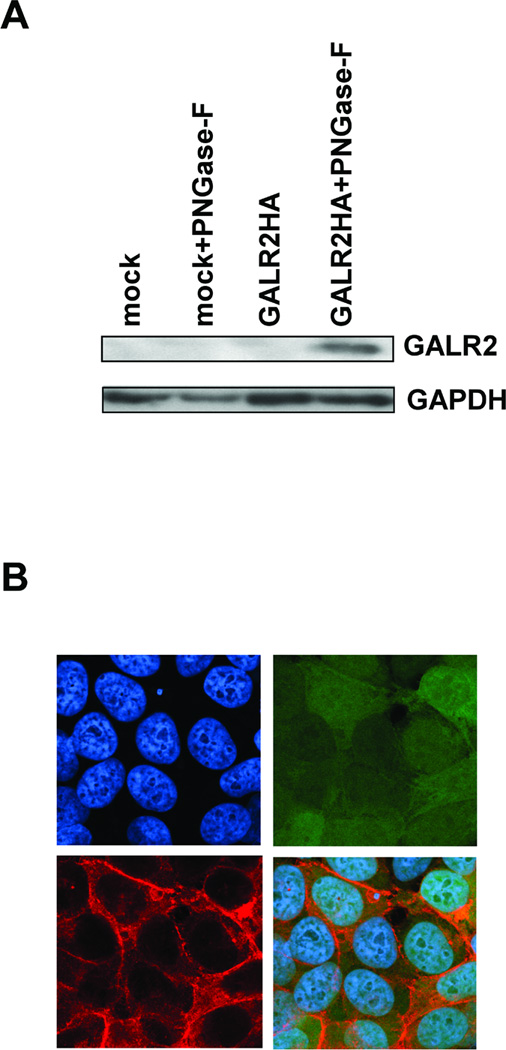
Exogenous GALR2 expression in transfected UM-SCC-1 cells. A, Immunoblotting shows exogenous GALR2 expression in pCMVGALR2IresGFP-transfected cells detected using antibody to HA-tag. Lanes labeled mock and GALR2HA contain protein lysates from mock-transfected and GALR2HA-transfected UM-SCC-1 cells (UM-SCC-1-GALR2). Lanes labeled mock+F and GALR2HA+F contain protein from cell lysates that were digested with N-glycosidase F. B, Exogenous GALR2 localizes to the cell membrane in UM-SCC-1-GALR2 cells. UM-SCC-1-GALR2 cells were stained with mouse monoclonal anti-HA-tag antibody and Hoechst 33342. Photographs show cells stained with Hoechst 33342 (upper left), GFP (upper right), HA-tag (lower left), and the merged image (lower right) (magnification × 400).
UM-SCC-1 cells contain mutant p53
Since previous studies had shown that GALR2 signaling caused apoptosis in neuroblastoma cells with wild-type p53, we examined the p53 status of UM-SCC-1 cells to determine if the effects of galanin and GALR2 required p53-mediated signaling. UM-SCC-1 was previously reported by us to have wild type p532 as assessed by sequencing exons 5–8 (Bradford et al. Head Neck 25:654–661, 2003). However, other experiments indicated that UM-SCC-1 failed to respond to DNA damage with up-regulation of p21. Therefore, we sequenced the p53 transcript (using the method described in Hauser et al. and Mandic et al.3,4) from UM-SCC-1, which revealed a splice site mutation that results in a 46-bp deletion and frame shift mutation beginning in exon 5 that leads to a nonsense sequence after the splice (Fig. 2A). UM-SCC-74B cells have wild-type p53 and were used as a control for transcript analysis (11). UM-SCC-10B contain a point mutation at exon 245 and overexpress p53 (12). These cells were used as a size marker for p53 protein expression. No p53 protein is detectable in UM-SCC-1 cells suggesting that the truncated transcript fails to produce a protein or that it is degraded (Fig. 2B).
Fig. 2.
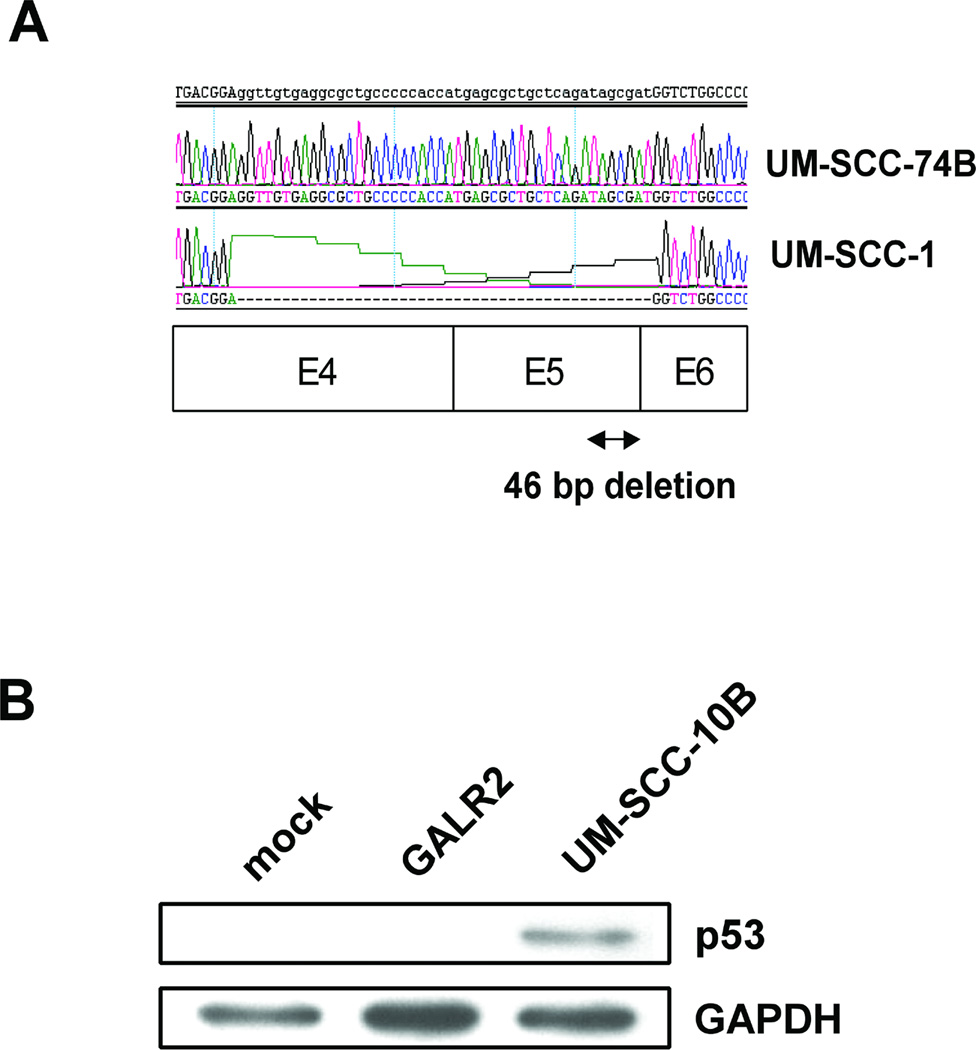
p53 mutation analysis. A, Direct cDNA sequencing of the RT-PCR products of control wild-type p53 expressed by UM-SCC-74B cells and mutant p53 expressed in UM-SCC-1 cells B, p53 protein expressions in UM-SCC-1-mock, UM-SCC-1-GALR2, and UM-SCC-10B cells as positive control.
GALR2-mediated galanin signaling inhibits cell proliferation and induces changes in cell morphology and adhesion
To determine the role of GALR2 in cell proliferation, cells starved for 24 hours were treated with various concentrations of galanin, and counted with a Coulter counter. Fig. 3A shows proliferation of UM-SCC-1-GALR2 and UM-SCC-1-mock cells after galanin treatment in dose-dependent and time-dependent assays. As the concentration of galanin increased, the proliferation UM-SCC-1-GALR2 cells decreased significantly when compared to the mock transfected control cells. Proliferation was reduced by 58% compared to the control UM-SCC-1-mock cells when treated with 1 µM of galanin (P<0.01). Furthermore, UM-SCC-1-GALR2 cells treated with a fixed concentration of galanin (1 µM) and counted at 24, 48 and 72 hours after galanin treatment, also exhibited a progressive decrease in cell number with time. Compared with galanin treated UM-SCC-1-mock cells, galanin treated UM-SCC-1-GALR2 cells showed almost no increase in cell number from control levels at 24 hours after galanin treatment. The inhibitory effect on galanin treated UM-SCC-1-GALR2 cells increased in a time dependent manner, and was significantly different from UM-SCC-1-GALR2 cells without galanin treatment and UM-SCC-1-mock cells (Fig. 3A) (P<0.01). Obvious morphology changes also were seen in UM-SCC-1-GALR2 but not UM-SCC-1-mock cells after treatment with various concentrations of galanin. Within 24 hours after addition of 1 µM of galanin, UM-SCC-1-GALR2 cells started to round up and exhibited decreased adhesion to the culture dishes. By 36 hours after treatment, 60–80% of cells had detached from the culture dishes and were floating in the medium (Fig. 3B). These results indicate that galanin and GALR2 affects cell adhesion in UM-SCC-1 cells.
Fig. 3.
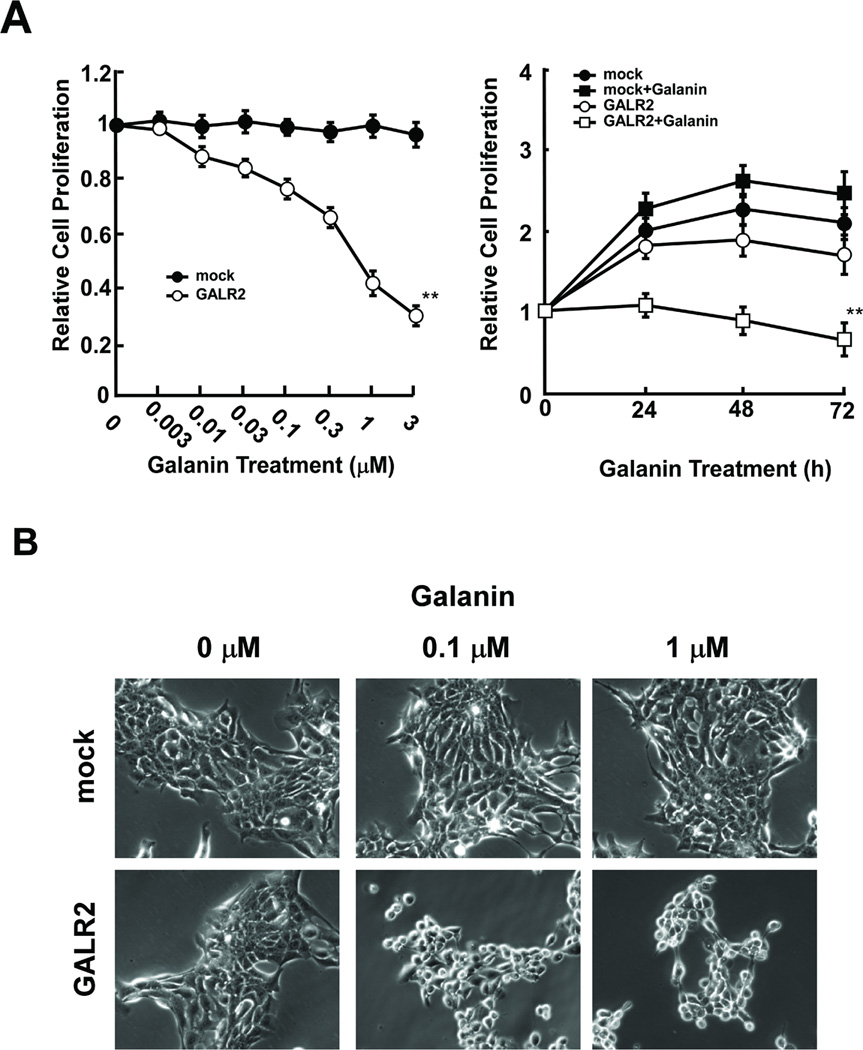
Galanin-induced growth inhibition and cytotoxicity in UMSCC-1-GALR2 cells. A, Relative cell proliferation as a function of galanin concentration. The cells were serum-starved for 24 h and then cultured with various concentrations of galanin for 24 h (left) or with 1 µM galanin for the indicated periods of time (right). Relative cell number is the ratio of cell number after treatment to that before treatment. Data were analyzed by two-way ANOVA (**P<0.01). B, Cell morphology changes induced by galanin in UM-SCC-1-mock and UM-SCC-1-GALR2 cells. The photographs show cells that were cultured with various concentrations of galanin for 24 h (magnification × 200).
GALR2 inhibits BrdU incorporation, induces p27Kip1, p57Kip2 up-regulation and Cyclin D1 down-regulation
To clarify the mechanism of GALR1-induced inhibition of cell proliferation, we examined BrdU incorporation as an index of cell cycle arrest and inhibition of DNA synthesis. As compared with UM-SCC-1-mock cells, UM-SCC-1-GALR2 cells exhibited significantly lower BrdU incorporation after 1 µM of galanin treatment (P<0.01). However, this decrease of BrdU incorporation did not occur in UM-SCC-1-mock regardless of galanin treatment (Fig. 4A). These data indicate that GALR2-mediated inhibition of proliferation is associated with suppression of DNA biosynthesis. Deregulation of the cell cycle is a common feature of human cancers. Tumor-associated alterations in this process frequently affect cyclin-dependent kinases (Cdks) and their regulators such as p27Kip1, p57Kip2 and cyclin D1. To better understand the role of GALR2 in arrest of tumor cell proliferation, starved cells were treated with 1 µM of galanin for 24 hours and the expression of these regulatory proteins was measured by immunoblotting using specific antibodies. As shown in Fig. 4B, p27Kip1 expression in UM-SCC-1-GALR2 was increased by galanin treatment, but this increase was not seen in UM-SCC-1-mock cells. Similarly, p27Kip1, p57Kip2 expression in UM-SCC-1-GALR2 was obviously increased by galanin, but not in UM-SCC-1-mock cells. Densitometric measurements indicate that galanin significantly enhanced p27Kip1 expression at 1.8 fold and p57Kip2 at 1.7 fold (P<0.05). In contrast, cyclin D1 expression decreased in UM-SCC-1-GALR2, but not in UM-SCC-1-mock. Densitometric measurements indicate that galanin significantly suppressed cyclin D1 expression by 0.28 fold (P<0.01). These data suggest that galanin and GALR2 induced arrest of proliferation is associated with regulation of p27Kip1, p57Kip2 and cyclin D1.
Fig. 4.
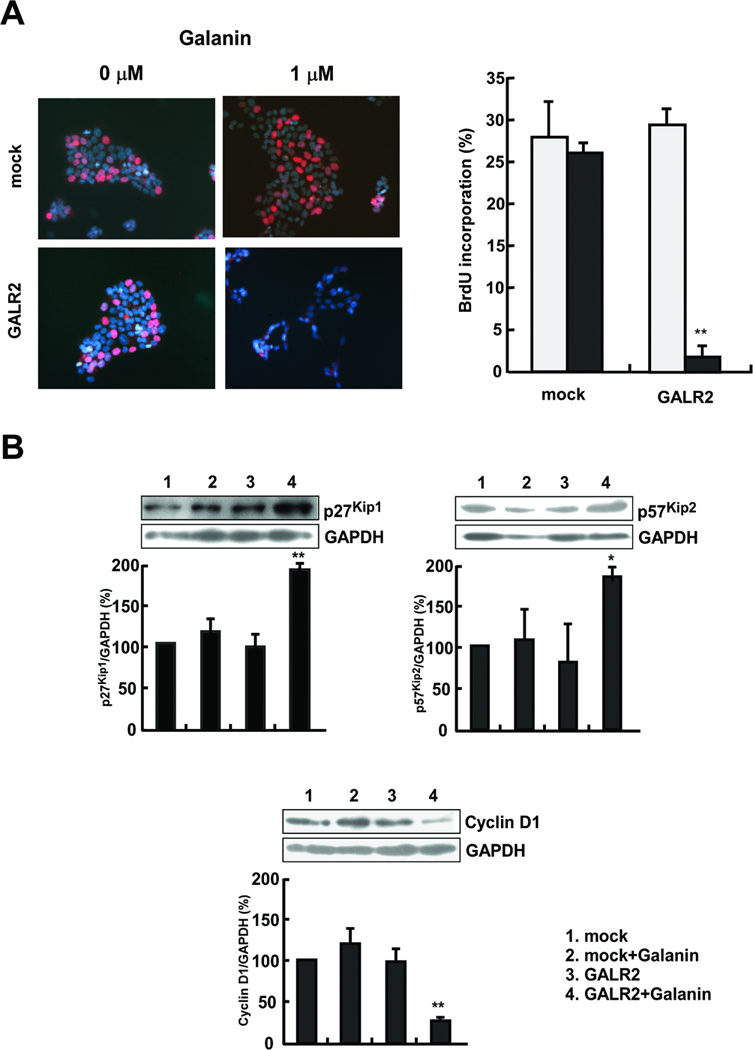
Galanin and GALR2 are associated with arrest of cell proliferation. A, BrdU incorporation after galanin treatment. The BrdU-positive cells (left) were detected as described in “Materials and Methods” (magnification × 200). The BrdU incorporation rate is the ratio of BrdU-positive cell number to the total cell number. Closed bars indicate galanin treatment and open bars indicate no treatment (right) (**P<0.01). B, Galanin and GALR2 regulate cell cycle control genes. Serum-starved cells were treated with 100 nM galanin for 24 h, and the expressions of p27Kip1 (upper left), p57Kip2 (upper right), and cyclin D1 (lower) were assessed by immunoblotting. The band intensities were determined by densitometry and normalized to GAPDH (**P<0.01).
GALR2 activation leads to apoptosis
To investigate whether UM-SCC-1-GALR2 cells die through apoptosis, galanin treated cells were assessed for annexin-V expression by flow cytometry. Annexin-V positive cells increased with galanin concentration in UM-SCC-1-GALR2 but not in UM-SCC-1-mock cells (Fig. 5A) (P<0.01). Furthermore, galanin and GALR2 induced DNA ladder formation, a characteristic of apoptosis. Cells were treated with 100 nM galanin for 24 hours, then genomic DNA was extracted and loaded onto a 2 % agarose gel. As shown in Fig. 5B, galanin treated UM-SCC-1-GALR2 cells exhibited obvious DNA ladder formation. These results provide the evidence that galanin and GALR2 induce apoptosis in UM-SCC-1 cells.
Fig. 5.
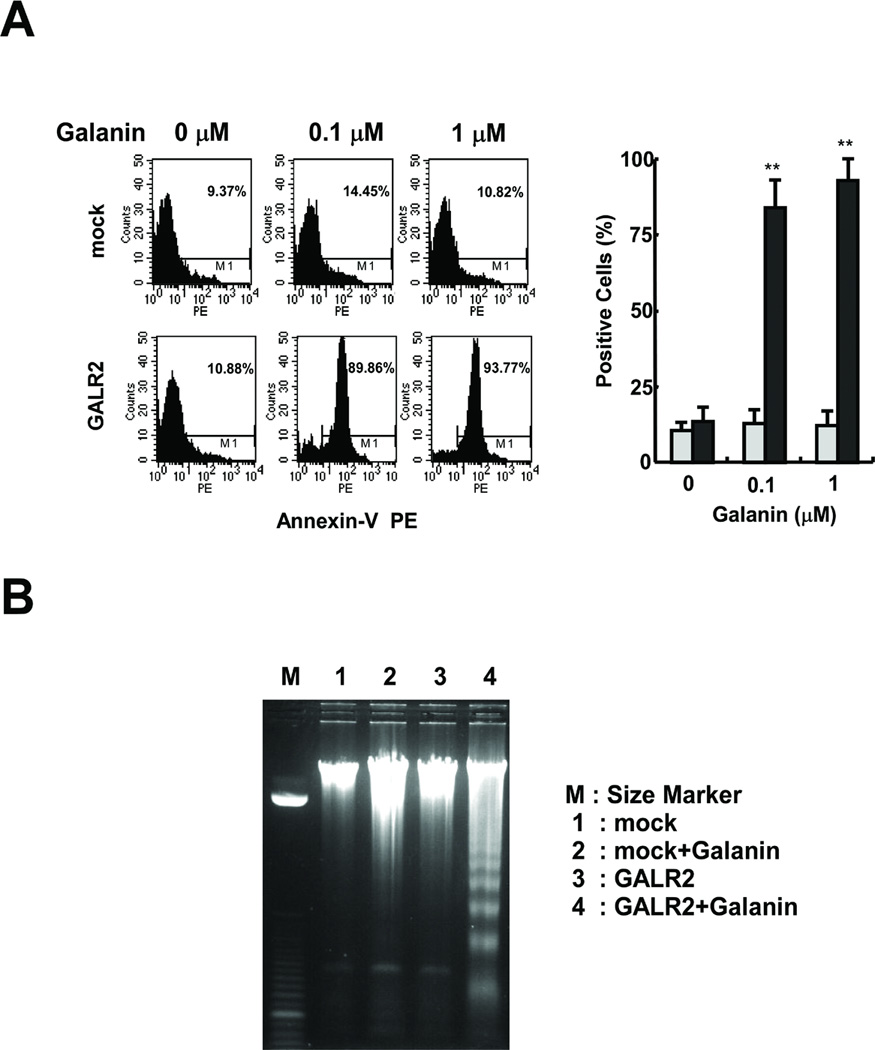
Induction of apoptosis in galanin-treated UM-SCC-1-GALR2 cells. A, Annexin-V staining. The annexin-V-positive cells were stained and counted by flow cytometry (left). Percent positive is the ratio of annexin-V-positive cells to total cell number (right) (**P<0.01). B, DNA fragmentation analysis. Genomic DNA was isolated and electrophoresed on a 2 % agarose gel. The DNA ladders were visualized under UV light with ethidium bromide. Lane M: Size marker. Lane 1: DNA isolated from UM-SCC-1-mock cells without galanin treatment. Lane 2: UM-SCC-1-mock cells with galanin treatment. Lane 3: UM-SCC-1-GALR2 cells without galanin treatment. Lane 4: UM-SCC-1-GALR2 cells with galanin treatment.
Activation of Caspase-3 in GALR2-Mediated Apoptosis
Caspases are a family of cysteine-proteases, which upon activation will cleave cellular protein targets, leading to the biochemical and morphological alterations typical of apoptosis (13). To examine the contribution of caspase-3 to galanin stimulated GALR2-mediated apoptosis, we counted active caspase-3 positive cells by flow cytometry in galanin-treated UM-SCC-1-mock cells and UM-SCC-1-GALR 2 cells. Active-caspase-3 positive cells were significantly increased in a galanin concentration dependent manner in UM-SCC-1-GALR2 (Fig. 6A) (P<0.01). Furthermore, preincubation of cells with 50 µM DEVD prior to 0.1 µM galanin treatment could prevent the apoptotic effect of galanin and GALR2 (Fig. 6B). Thus, these results pointed to the activation of caspase-3 by galanin as a critical step in the process of apoptotic cell death occurring in UM-SCC-1-GALR2 cells.
Fig. 6.
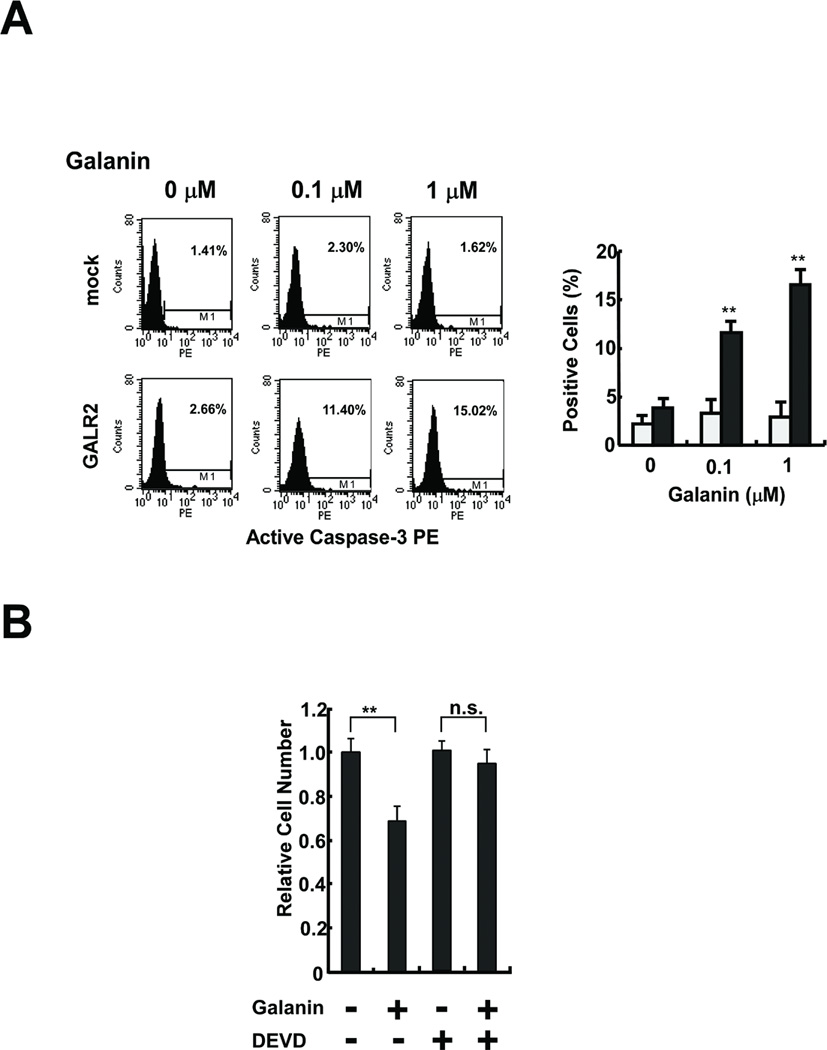
GALR2 induced apoptosis through caspase-3 activation. A, Active caspase-3 staining. The active-caspase-3-positive cells were stained and counted by flow cytometry (left). Percent positive is the ratio of active-caspase-3-positive cells to total cell number (right) (**P<0.01). B, Effects of galanin and caspase-3 inhibition on GALR2-induced cell death. Serum-starved UM-SCC-1-GALR2 cells were pretreated with 50 µM DEVD before galanin treatment. Relative cell number is the ratio of the cell number after treatment to that before treatment (**P<0.01).
Discussion
In this study, we demonstrated that re-introduction of GALR2 into HNSCC cells makes the tumor cells susceptible to galanin-induced growth inhibition and apoptosis. These results suggest that the galanin-GALR2 pathway has significant growth regulatory activity and that loss of this pathway could be an important factor in tumor development and progression. Furthermore, since GALR2 can induce apoptosis in p53 mutant HNSCC cells, the loss or inactivation of the galanin-GALR2 signaling cascade provides tumor cells with another mechanism of avoiding cell death. Our results also suggest the possibility that reactivation of the galanin receptor(s) pathway could be a therapeutic strategy in HNSCC. In fact, our recent data demonstrate that the galanin receptors are frequently silenced by hypermethylation in HNSCC (14), and that galanin and GALR1 can inhibit cell proliferation (6).
It was reported earlier that GALR2 can initiate a complex signaling cascade by coupling to Gq, Gi and G12 proteins (5), but that in small cell lung cancer, galanin activated MAP kinase and had mitogenic effects (7). Recently, it was reported that in neuroblastoma cells, exogenous expression of GALR2 inhibited cell proliferation by inducing apoptosis (4). It is known that some kinds of GPCRs, including the N-formyl peptide receptor (FPR), and the V2 vasopressin, angiotensin II, and CXCR2 receptors are capable of initiating apoptosis upon stimulation (15). Although the mechanism by which the GALR2 pathway induces antiproliferative effects is not completely understood, a few studies about other GPCRs have been reported. Angiotensin II-induced apoptosis is mediated via its receptors through the induction of transforming growth factor-β production, which is followed by increased transcription of cell death genes such as Fas, FasL, Bax and caspase-3 (16). The FPR receptor also induces apoptosis by the activation of several signaling proteins, such as p38, MEK and caspases (17). Recently, To fighi et al. demonstrated exogenous GALR2-transfected pheochromocytoma cells induced apoptosis, by decrease in the phosphorylated Bad level followed by suppression of the PI3K/Akt pathway (18). In our preliminary data, galanin induced activation of the extracellular signal regulated kinase (Erk) pathway activation in UM-SCC-1-GALR2 cells (data not shown), and Erk pathway and PI3K/Akt pathway could cross-talk in cancer cells (19), but their roles for GALR2 induced apoptosis in HNSCC cells are unclear.
In our study, GALR2 up-regulated p27Kip1, p57Kip2 while down-regulating cyclin D1. p27Kip1, p57Kip2 and p21Cip1 belong to the Cip/Kip family of inhibitors of cyclin-dependent kinases. Their roles are not fully understood but many studies have been reported. One recent finding has shown that p21Cip1 is implicated in the antiapoptotic response to chemotherapy (20). As a negative regulator of apoptosis, p21Cip1 interacts with several caspases, such as caspase-3, caspase-8 and caspase-10 (20). We could not detect p21Cip1 expression in either UM-SCC-1-GALR2 or UM-SCC-1-mock cells (data not shown), but its absence is consistent with the fact that UM-SCC-1 cells have mutated p53. Meanwhile, it has been reported that p27Kip1 positively regulates the apoptotic response (21, 22). Actually, galanin and GALR2 mediated p27Kip1 up-regulation and caspase-3 dependent apoptosis in our experimental models, but the role of p27Kip1 for GALR2-mediated apoptosis is unclear. Clinically, p27Kip1 and p57Kip2 are known to be tumor suppressor genes, and their expression levels are related to prognosis in many different tumors, including non-small cell lung carcinoma, gastric carcinoma, and HNSCC (23–26). Overexpression of cyclin D1 occurs at a high frequency in a variety of carcinomas, including those arising in the breast, colorectum, and head and neck, and is associated with poor prognosis (27–29). The fact that galanin and GALR2 can regulate these genes suggests GALR2 is a tumor suppressor in head and neck cancer cells.
It is significant that GALR2 can mediate this potent anti-proliferative effect in the absence of p53 function. Most studies have focused on the involvement of p53 in regulating apoptosis, because the therapeutic action of many anti-neoplastic agents depends on p53-induced apoptosis. However, p53 is frequently mutated in HNSCC, and thus it would be valuable to find a molecule that can kill tumor cells in a p53-independent manner. Although the p53-independent mechanism activated by GALR2 has not yet been clearly defined, it is known that some kinds of receptors and signaling proteins, such as adenovirus E1A, E2F1 and p73, induce apoptosis independent of p53 (30–33). Conflicting results have been reported regarding GPCR- induced apoptosis and p53 status. Wang et al. reported a potential role for GPCR signaling in the modulation of p53-mediated apoptosis and indicated that β-arrestin 2 may serve as a signaling linker between the GPCR and p53 pathways via sequestration of MDM2 (17). On the other hand, the somatostatin analogue AN-238 and its GPCR receptor induce apoptosis though a p53-independent pathway (34). The orexin receptor was also reported to induce p53-transcription-independent apoptosis though p38 activation (35). Until now, it had not been determined whether GALR2 needs p53 function to induce apoptosis, because GALR2-mediated apoptosis was demonstrated in the neuroblastoma cell line SH-SY5Y (4). In general, p53 is wild-type in human neuroblastoma cell lines (36). SH-SY5Y cells are known to have wild-type p53 and were used as a typical model to study the mechanism of p53-induction of apoptosis (37). In the present study, GALR2 induced-anti-proliferative and apoptotic effects in UM-SCC-1, a cell line that expresses only a truncated p53 message that lacks the C-terminal sequence encoding the nuclear localization signal and lacks detectable p53 protein. Our results demonstrated that GALR2 can induce apoptosis and cell cycle arrest though a p53-independent but caspase-3 dependent pathway. These results suggest that GALR2 might act through a novel apoptotic pathway, that is not yet well-defined.
In conclusion, we have shown that galanin and GALR2 induce apoptosis and cell cycle arrest independent of p53. These results suggest that GALR2 probably controls normal cell proliferation and survival in the epithelium and that its loss may facilitate tumor development and progression. Furthermore, this pathway is a possible target in head and neck cancer.
Translational Relevance.
In this study, we investigated the role of GALR2 in HNSCC, a tumor type that frequently contains mutated p53. HA-tagged GALR2 was stably expressed in a human oral carcinoma cell line UM-SCC-1 that contains mutant p53 and does not express GALR1 (Misawa, et al. Clin Can Res, In Press). Galanin treatment of GALR2 transfected UM-SCC-1 cells caused distinct morphological changes and a marked decrease in cell number. Galanin and GALR2 inhibited BrdU incorporation and induced p27Kip1 and p57Kip2 up-regulation and cyclin D1 down-regulation. These effects were similar to GALR1 signaling,1 however, GALR2 also induced caspase-3-dependent apoptosis, which was not observed with GALR1. The results demonstrate that exogenous GALR2 expression can inhibit cell proliferation and induce apoptosis in HNSCC cells with mutant p53, and thus GALR2 may be a feasible target for HNSCC therapy.
Acknowledgement
This research is supported in part by the NIH NCI through the University of Michigan’s Cancer Center Support Grant (5P30 CA46592), Head and Neck SPORE grant (1 P50 CA97248), and a Grant-in-Aid for Scientific Research (No. 20592027) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Kanazawa T, Iwashita T, Kommareddi Pet al. Galanin and galanin receptor type 1 suppress proliferation in squamous carcinoma cells: activation of the extracellular signal regulated kinase pathway and induction of cyclin-dependent kinase inhibitors. Oncogene 2007;26:5762–5771.
Bradford CR, Zhu S, Ogawa H et al. P53 mutation correlates with cisplatin sensitivity in head and neck squamous cell carcinoma lines. Head Neck 2003;25:654–661.
Hauser U, Balz V, Carey TE et al. Reliable detection of p53 aberrations in squamous cell carcinomas of the head and neck requires transcript analysis of the entire coding region. Head Neck 2002;24:868–873.
Mandic R, Schamberger CJ, Muller JF et al. Reduced cisplatin sensitivity of head and neck squamous cell carcinoma cell lines correlates with mutations affecting the COOH-terminal nuclear localization signal of p53. Clin Cancer Res 2005;11:6845–6852.
References
- 1.Nishino H, Miyata M, Morita M, Ishikawa K, Kanazawa T, Ichimura K. Combined therapy with conservative surgery, radiotherapy, and regional chemotherapy for maxillary sinus carcinoma. Cancer. 2000;89:1925–1932. doi: 10.1002/1097-0142(20001101)89:9<1925::aid-cncr8>3.3.co;2-8. [DOI] [PubMed] [Google Scholar]
- 2.Hill SJ. G-protein-coupled receptors: past, present and future. Br J Pharmacol. 2006;147:S27–S37. doi: 10.1038/sj.bjp.0706455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berger A, Santic R, Almer D, et al. Galanin and galanin receptors in human gliomas. Acta Neuropathol (Berl) 2003;105:555–560. doi: 10.1007/s00401-003-0680-7. [DOI] [PubMed] [Google Scholar]
- 4.Berger A, Lang R, Moritz K, et al. Galanin receptor subtype GalR2 mediates apoptosis in SH-SY5Y neuroblastoma cells. Endocrinology. 2004;145:500–507. doi: 10.1210/en.2003-0649. [DOI] [PubMed] [Google Scholar]
- 5.Wittau N, Grosse R, Kalkbrenner F, Gohla A, Schultz G, Gudermann T. The galanin receptor type 2 initiates multiple signaling pathways in small cell lung cancer cells by coupling to G(q), G(i) and G(12) proteins. Oncogene. 2000;19:4199–4209. doi: 10.1038/sj.onc.1203777. [DOI] [PubMed] [Google Scholar]
- 6.Kanazawa T, Iwashita T, Kommareddi P, et al. Galanin and galanin receptor type 1 suppress proliferation in squamous carcinoma cells: activation of the extracellular signal regulated kinase pathway and induction of cyclin-dependent kinase inhibitors. Oncogene. 2007;26:5762–5771. doi: 10.1038/sj.onc.1210384. [DOI] [PubMed] [Google Scholar]
- 7.Seufferlein T, Rozengurt E. Galanin, neurotensin, and phorbol esters rapidly stimulate activation of mitogen-activated protein kinase in small cell lung cancer cells. Cancer Res. 1996;56:5758–5764. [PubMed] [Google Scholar]
- 8.Iishi H, Tatsuta M, Baba M, et al. Inhibition by galanin of experimental carcinogenesis induced by azaserine in rat pancreas. Int J Cancer. 1998;75:396–399. doi: 10.1002/(sici)1097-0215(19980130)75:3<396::aid-ijc12>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 9.Takebayashi S, Hickson A, Ogawa T, et al. Loss of chromosome arm 18q with tumor progression in head and neck squamous cancer. Genes Chromosomes Cancer. 2004;41:145–154. doi: 10.1002/gcc.20066. [DOI] [PubMed] [Google Scholar]
- 10.Takebayashi S, Ogawa T, Jung KY, et al. Identification of new minimally lost regions on 18q in head and neck squamous cell carcinoma. Cancer Res. 2000;60:3397–3403. [PubMed] [Google Scholar]
- 11.Bauer JA, Trask DK, Kumar B, et al. Reversal of cisplatin resistance with a BH3 mimetic, (−)-gossypol, in head and neck cancer cells: role of wild-type p53 and Bcl-xL. Mol Cancer Ther. 2005;4:1096–1104. doi: 10.1158/1535-7163.MCT-05-0081. [DOI] [PubMed] [Google Scholar]
- 12.Oliver CL, Bauer JA, Wolter KG, et al. In vitro effects of the BH3 mimetic, (−)-gossypol, on head and neck squamous cell carcinoma cells. Clin Cancer Res. 2004;10:7757–7763. doi: 10.1158/1078-0432.CCR-04-0551. [DOI] [PubMed] [Google Scholar]
- 13.Gorman AM, Orrenius S, Ceccatelli S. Apoptosis in neuronal cells: role of caspases. Neuroreport. 1998;9:R49–R55. doi: 10.1097/00001756-199807130-00001. [DOI] [PubMed] [Google Scholar]
- 14.Misawa K, Ueda Y, Kanazawa T, et al. Epigenetic Inactivation of GALR1 in Head and Neck Cancer. Clin Cancer Res. doi: 10.1158/1078-0432.CCR-07-4673. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins block G protein-coupled receptor-mediated apoptosis. J Biol Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- 16.Bhaskaran M, Reddy K, Radhakrishanan N, Franki N, Ding G, Singhal PC. Angiotensin II induces apoptosis in renal proximal tubular cells. Am J Physiol Renal Physiol. 2003;284:F955–F965. doi: 10.1152/ajprenal.00246.2002. [DOI] [PubMed] [Google Scholar]
- 17.Wang P, Gao H, Ni Y, et al. Beta-arrestin 2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J Biol Chem. 2003;278:6363–6370. doi: 10.1074/jbc.M210350200. [DOI] [PubMed] [Google Scholar]
- 18.Tofighi R, Joseph B, Xia S, et al. Galanin decreases proliferation of PC12 cells and induces apoptosis via its subtype 2 receptor (GalR2) Proc Natl Acad Sci U S A. 2008;105:2717–2722. doi: 10.1073/pnas.0712300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moelling K, Schad K, Bosse M, Zimmermann S, Schweneker M. Regulation of Raf-Akt Cross-talk. J Biol Chem. 2002;277:31099–31106. doi: 10.1074/jbc.M111974200. [DOI] [PubMed] [Google Scholar]
- 20.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- 21.Wang X, Gorospe M, Huang Y, Holbrook NJ. p27Kip1 overexpression causes apoptotic death of mammalian cells. Oncogene. 1997;15:2991–2997. doi: 10.1038/sj.onc.1201450. [DOI] [PubMed] [Google Scholar]
- 22.Otsubo T, Akiyama Y, Yanagihara K, Yuasa Y. SOX2 is frequently downregulated in gastric cancers and inhibits cell growth through cell-cycle arrest and apoptosis. Br J Cancer. 2008;98:824–831. doi: 10.1038/sj.bjc.6604193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hommura F, Dosaka-Akita H, Mishina T, et al. Prognostic significance of p27KIP1 protein and ki-67 growth fraction in non-small cell lung cancers. Clin Cancer Res. 2000;6:4073–4081. [PubMed] [Google Scholar]
- 24.Sgambato A, Migaldi M, Leocata P, et al. Loss of p27Kip1 expression is a strong independent prognostic factor of reduced survival in N0 gastric carcinomas. Cancer. 2000;89:2247–2257. doi: 10.1002/1097-0142(20001201)89:11<2247::aid-cncr13>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 25.Mineta H, Miura K, Suzuki I, et al. Low p27 expression correlates with poor prognosis for patients with oral tongue squamous cell carcinoma. Cancer. 1999;85:1011–1017. doi: 10.1002/(sici)1097-0142(19990301)85:5<1011::aid-cncr1>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 26.Fan GK, Xu F, Yang B, Fujieda S. p57(kip2) expression is related to carcinogenesis and tumor progression in laryngeal tissues. Acta Otolaryngol. 2006;126:301–305. doi: 10.1080/00016480500388851. [DOI] [PubMed] [Google Scholar]
- 27.Kenny FS, Hui R, Musgrove EA, et al. Overexpression of cyclin D1 messenger RNA predicts for poor prognosis in estrogen receptor-positive breast cancer. Clin Cancer Res. 1999;5:2069–2076. [PubMed] [Google Scholar]
- 28.Kong S, Amos CI, Luthra R, Lynch PM, Levin B, Frazier ML. Effects of cyclin D1 polymorphism on age of onset of hereditary nonpolyposis colorectal cancer. Cancer Res. 2000;60:249–252. [PubMed] [Google Scholar]
- 29.Akervall J, Bockmuhl U, Petersen I, Yang K, Carey TE, Kurnit DM. The gene ratios c-MYC:cyclin-dependent kinase (CDK)N2A and CCND1:CDKN2A correlate with poor prognosis in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2003;9:1750–1755. [PubMed] [Google Scholar]
- 30.Clarke AR, Purdie CA, Harrison DJ, et al. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 31.Holmberg C, Helin K, Sehested M, Karlstrom O. E2F-1-induced p53-independent apoptosis in transgenic mice. Oncogene. 1998;17:143–155. doi: 10.1038/sj.onc.1201915. [DOI] [PubMed] [Google Scholar]
- 32.Putzer BM, Stiewe T, Parssanedjad K, Rega S, Esche H. E1A is sufficient by itself to induce apoptosis independent of p53 and other adenoviral gene products. Cell Death Differ. 2000;7:177–188. doi: 10.1038/sj.cdd.4400618. [DOI] [PubMed] [Google Scholar]
- 33.Urist M, Tanaka T, Poyurovsky MV, Prives C. p73 induction after DNA damage is regulated by checkpoint kinases Chk1 and Chk2. Genes Dev. 2004;18:3041–3054. doi: 10.1101/gad.1221004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lasfer M, Vadrot N, Schally AV, et al. Potent induction of apoptosis in human hepatoma cell lines by targeted cytotoxic somatostatin analogue AN-238. J Hepatol. 2005;42:230–237. doi: 10.1016/j.jhep.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 35.Ammoun S, Lindholm D, Wootz H, Akerman KE, Kukkonen JP. G-protein-coupled OX1 orexin/hcrtr-1 hypocretin receptors induce caspase-dependent and -independent cell death through p38 mitogen-/stress-activated protein kinase. J Biol Chem. 2006;281:834–842. doi: 10.1074/jbc.M508603200. [DOI] [PubMed] [Google Scholar]
- 36.Moll UM, LaQuaglia M, Benard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci U S A. 1995;92:4407–4411. doi: 10.1073/pnas.92.10.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shibata T, Iio K, Kawai Y, et al. Identification of a lipid peroxidation product as a potential trigger of the p53 pathway. J Biol Chem. 2006;281:1196–1204. doi: 10.1074/jbc.M509065200. [DOI] [PubMed] [Google Scholar]