

Abstract
Objective
Mitochondrial biology appears central to many conditions that progress to death but remains poorly characterized following cardiac arrest. Mitochondrial dysfunction in electron transfer and reactive oxidant species (ROS) leakage during ischemia may lead to downstream events including mitochondrial protein oxidation, tyrosine nitrosylation, cytochrome c loss, and eventual death. We sought to better define early fixed alterations in these mitochondrial functions following whole animal cardiac arrest.
Methods
We used a murine model of 8 minutes of untreated KCl-induced cardiac arrest followed by resuscitation and return of spontaneous circulation (ROSC) to study mitochondrial functions in four groups of animals: (a) after 8 min cardiac arrest (CA8) but no resuscitation, (b) 30 min post-ROSC (R30), (c) 60 min post-ROSC (R60) and in (d) shams. Heart mitochondria were immediately harvested, isolated and stored at −80°C for later spectrophotometric measurements of electron transfer activities and ROS leakage using appropriate substrates and inhibitors. Mitochondrial cytochrome c content and tyrosine nitration were analyzed by western blot and densitometry.
Results
A significant ROS leakage from Complex I was evident after just 8 min of cardiac arrest (CA8 group, P<0.05), which was followed by a progressive reduction in Complex I electron transfer activity (CA8>R30>R60). In contrast, Complex II and II–III activities appeared more resistant to ischemia at the time points evaluated. Early changes in a ~50 kDa and ~25 kDa protein were observed in tyrosine nitration along with a loss of cytochrome c.
Conclusions
A relatively “orderly” process of mitochondrial dysfunction progresses during ischemia and reperfusion. Changes in mitochondrial ROS generation and electron transfer from Complex I occur along with tyrosine nitrosylation and loss of cytochrome c; these may represent important new targets for future human therapies.
Keywords: mitochondria, electron transport, reactive oxygen species, nitric oxide, peroxynitrite, cardiopulmonary resuscitation, heart arrest, sudden death, reperfusion injury
Introduction
Mitochondrial dysfunction following resuscitation from cardiac arrest remains a poorly characterized condition in patients who get return to spontaneous circulation (ROSC), albeit it is a potential therapeutic target for optimal survival (1, 2). In general, mitochondrial dysfunction is characterized by faulty electron transport through the respiratory complexes I–III–IV and II–III–IV (3, 4). Impaired electron transport can be detected in vitro as reduced electron transfer kinetics (i.e. speed) at each electron complex site between specific pairs of reduced electron donors to oxidized electron acceptors. Slow electron transfer is directly related to an abnormal increased generation of reactive oxygen species (ROS) by electron “leakage” at the partially or totally blocked respiratory complex, principally at Complex I (NADH dehydrogenase) or Complex III (3). Electron transfer dysfunction in turn reduces the mitochondrial membrane potential eventually leading to cytosolic release of the pro-apoptotic protein cytochrome c (5, 6). Such mitochondrial defects in diminished electron flow and increased ROS generation are associated with reduced ATP generation, metabolic failure, altered signaling pathways, and induction of apoptosis, which may ultimately lead to cell death.
Prior work supports the general notion that ischemia and reperfusion can result in significant mitochondrial dysfunction. Several groups have identified significant reductions in Complex I activity along with increased ROS generation following ischemia and reperfusion (7, 8). By contrast, Complexes II and III seem more resistant to ischemic modifications while there is conflicting data on alterations in Complex IV (cytochrome oxidase) activity and ROS generation (7, 9, 10, 11). Collectively these prior studies may or may not be relevant to the conditions of human cardiac arrest, as these studies have primarily been conducted in isolated mitochondria, isolated organs, or using focal ischemia models. Despite an enormous impact on human death, there is a paucity of data derived from whole animal studies on mitochondrial dysfunction at the level of specific electron complex activities and ROS generation sites following cardiac arrest. Multiple experts suggest a vital role for understanding mitochondrial dysfunction in the setting of ischemia and reperfusion; they additionally suggest that future therapies may offer significant improvement in life-saving value when modulation of mitochondrial dysfunction becomes a practical therapeutic target (1, 12, 13).
During cardiac arrest there is a variable period of complete global ischemia, followed by whole body reperfusion in those patients who get ROSC. Of the many potentially salvageable patients who gain ROSC temporarily, the majority will die over the next hours during a so called post resuscitation syndrome characterized by progressive failure of hemodynamic function and neurological function. This post resuscitation syndrome has also been associated with alterations of inflammatory mediators (i.e. a “sepsis-like” state), microcirculation, and other organ function (14, 15). Several reports suggest that many of these organ failure conditions are associated with mitochondrial dysfunction (16). A relatively fixed or progressive mitochondrial dysfunction could explain some of the observed deterioration of organ function in patients who despite temporary ROSC, go on to die (17).
We hypothesize that following cardiac arrest a significant mitochondrial dysfunction develops, which can be measured by examining the electron flow activities and ROS generating behavior of complexes I-IV shortly following cardiac arrest. In addition, we sought to identify early changes post cardiac arrest in mitochondrial protein nitrosylation and cytochrome c content. To describe possible alterations, we utilized a well-characterized murine model of potassium-induced cardiac arrest that includes 8 minutes of untreated cardiac arrest followed by resuscitation. The 8 min cardiac arrest results in an 80% rate of successful ROSC and 50% rate of 24 hours survival, a clinically relevant model of cardiopulmonary resuscitation in which to define mitochondrial dysfunction at early time points.
Materials and Methods
Murine model of cardiac arrest and resuscitation
All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Adult female C57BL/6 mice (weight 25 to 35 g; Charles River Production, Wilmington, MA) were freely accessed to food and water before experiments. Mice were anesthetized with 100 mg/kg of ketamine HCl and 10 mg/kg xylazine HCl delivered by intraperitoneal injection. During surgical preparation, body temperature was maintained between 37.0±0.5°C by an incandescent heating lamp and monitored by a rectal thermocouple probe. The trachea was orally intubated with a 20-gauge catheter (Angiocath, Becton Dickinson). Mechanical ventilation (Minivent, Harvard Apparatus) was initiated at rate of 110/min, I:E=1:1, FiO2=1.0. Tidal volume was 8–12 ml/kg regulated by end tidal CO2 (EtCO2) maintained 35–45 mm Hg (Micro-Capnometer, Columbus Instruments, Columbus, OH) and a positive end-expiratory pressure (PEEP) of 2 cm H2O. A heparinized microcatheter (EZ-1101, BioTime Inc) was inserted into the left jugular vein for drug and fluid administration. The other microcatheter was inserted to right carotid artery to monitor the blood pressure. Electrocardiograph (ECG) was recorded by needle probe. After the mice were equilibrated on the ventilator to get target temperature, EtCO2 35–45 mm Hg, heart rate (HR) over 200/min and mean arterial pressure (MAP) >60 mm Hg, the mouse cardiac arrest was quickly induced by 100 μl KCl (80 μg/kg) followed by 200 μl room temperature saline flushing and weaned off ventilator at the same time. The cardiac arrest was confirmed by loss of ECG activity and blood pressure which should sharply decrease below 10 mm Hg. The temperature of rectum of the mice was maintained at 37.0±0.5°C during the whole cardiac arrest period by heating lamp. After 8 minutes of the induction of cardiac arrest, resuscitation via manual cardiopulmonary resuscitation (CPR) (chest compression, about 300–400/min) was begun together with 2 μ/200 μl epinephrine following 200 μl room temperature saline flush via left jugular vein, and resume the ventilation at the rate of 110/min, I:E=1:1, FiO2=1.0, PEEP=0 mm H2O. Chest compression was regulated to provide a uniform rate and to maintain maximal MAP and EtCO2 values. If the mouse does not establish ROSC after 5 minutes, efforts were stopped. ROSC was confirmed by the return of ECG rhythm and significant increase of MAP than that during chest compression. The mouse was maintained on mechanical ventilation after ROSC, during which time 1 μg/0.1 ml epinephrine was used when MAP dropped below 50 mm Hg. Tidal volume was regulated by EtCO2 which was maintained between 35–45mm Hg. Body temperature (measured rectally) was maintained at 37.0±0.5°C after resuscitation by a heating lamp. The sham animals had the same surgical procedures and temperature control as other groups but had no arrest, CPR, or drug administration. Tissues were removed intact and dissected on ice without perfusion at the endpoint of each group for mitochondria isolation.
Experimental groups
The mice were divided into 4 groups in this study: sham-operated animals (Sham), 8 min of cardiac arrest prior to resuscitation (CA8), 30 min post-ROSC (R30), and 60 min post-ROSC (R60) (Figure 1).
Figure 1.

Experimental surgery protocol including each time point for different groups. K, potassium chloride; CA, cardiac arrest; CPR, cardiopulmonary resuscitation; ROSC, return of spontaneous circulation.
Isolation of heart mitochondria
Fresh-isolated heart from the mouse were put in ice-cold homogenization buffer containing either 0.23 M mannitol, 70 mM sucrose, 10 mM EGTA with 0.1% BSA (pH 7.4), the mitochondrial fraction were isolated by differential centrifugation as described (18). The final pellet enriched in mitochondria was resuspended in homogenization buffer without BSA. Submitochondrial particles were prepared from frozen and thawed mitochondria by sonication and stored at −80°C until use.
Determination of Mitochondrial Complexes I–III and II–III electron flow activities
Mitochondrial Complex I/III electron flow activity was measured in isolated mitochondrial samples as the rotenone-sensitive NADH:cytochrome c reductase activity. Briefly, 50μg/ml submitochondrial particles were thawed and added to a cuvette containing 50 μM oxidized cytochrome c and 1 mM KCN with or without 1 μM rotenone in 100 mM potassium phosphate (pH 7.2) in a thermostatized spectrophotometer (Hitachi U-2810) at 30°C, a baseline of absorbance at λ=550 nm was recorded, and the reaction was initiated with 0.2 mM NADH. Increase in absorbance, corresponding to cytochrome c reduction was continuously recorded.
The electron flow activity of Complex II/III (succinate-cytochrome c reductase) was determined using 50μg/ml submitochondrial particles in 100 mM potassium phosphate buffer containing 50 μM oxidized cytochrome c, 1 mM KCN and 1 μM rotenone and initiated by addition of 6 mM succinate. The activity of Complex I/III and II/III were expressed as nmol reduced cytochrome c min−1 mg−1 (ε550 = 21.1 mM−1 cm−1).
Determination of mitochondrial Complex IV activity
Complex IV electron flow activity was determined by recording the oxidation of 50 oM reduced cytochrome c in a Hitachi U-2810 spectrophotometer at 550 nm at 30°C; ε550 = 21.1 mM−1 cm−1 (18). Cytochrome c was reduced with potassium ascorbate, which was afterward removed by NADH–cytochrome-c24-h dialysis against 10 mM potassium phosphate, pH 7.2.
Measurement of ROS generation leading to hydrogen peroxide production
ROS generation leading to hydrogen peroxide (H2O2) production from Complex III was measured in submitochondrial particles following reaction of H2O2 with p-hydroxyphenyl acetic acid (p-HPA) in the presence of horseradish peroxidase (HRP) at 30°C in an F-2000 HITACHI Fluorescence Spectrophotometer. Briefly, 0.15 mg/ml of the previously frozen submitochondrial particles were thawed and added to a cuvette containing 100 mM potassium phosphate buffer, 12.5 U/mL HRP and 50 μM p-HPA with magnetic stirring for continuous fluorimetric recording. After recording of the baseline, 6 mM succinate with or without 2 μM antimycin A was added to stimulate H2O2 production at Complex III. The slope of fluorescence increase was measured and baseline slope readings subtracted to determine H2O2 production at εex = 315 nm and εem = 425 nm.
Since the p-hydroxyphenyl acetic method utilizes wavelengths in the range of interference of NADH, we determined relative H2O2 production by Complex I using another fluorescent probe, Amplex red (Molecular Probes, Eugene, OR, USA) as previously described (19, 20). Similar recordings were performed but with 0.25 mg/ml submitochondrial particles, 5 U HRP, 50 μM Amplex red, 20 U/ml Cu-Zn SOD, and the use of 0.2 mM NADH as electron donor with or without 1 μM rotenone. H2O2 production was measured using εex = 585 nm and εem = 550 nm.
Cytochrome c and tyrosine nitration analysis by western blotting
Mitochondrial protein tyrosine nitration and cytochrome c content were determined by western blot. Briefly 50μg of mitochondrial protein were loaded and separated in a 4–12% SDS-PAGE gel (Invitrogen), transferred to a nitrocellulose membrane (Bio-Rad). Cytochrome c was then assessed using an anti-cyt c antibody from Cell Signaling at 1:1000 dilution, and normalized by detection of a Complex IV subunit (anti Cox IV 1:1000 from Cell Signaling). After stripping the membrane, tyrosine nitration was detected with a monoclonal anti-nitrotyrosine antibody at 1:1000 dilution (a generous gift of Dr. Alvaro Estevez, Weil Cornell University). Secondary antibodies were peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (Amersham) used at 1:10,000 dilution. Signals were developed by enhanced chemiluminescence (Amersham). Densitometric quantification of the immunoblots was performed by using Image J software.
Statistical Analysis
Data are presented as mean ± standard deviation unless otherwise stated. For animal’s physiological parameters, one-way ANOVA was performed, followed by Tukey post hoc tests to identify differences in all groups at baseline and unpaired t-test was used to compare the differences between R30 and R60 groups after ROSC. For mitochondrial measurements, one-way ANOVA followed by unpaired t-test was used. Statistical software SPSS for Windows and Microsoft Excel were used. A p value <.05 was considered statistically significant.
Results
There are no difference of body weight, HR, MAP and temperature among each group at baseline as shown in Table 1. The time of CPR, HR and MAP at ROSC are also no difference between R30 and R60 groups.
Table 1.
The characteristics of each group at baseline and return of spontaneous circulation
| Sham | CA8 | R30 | R60 | |
|---|---|---|---|---|
| Weight (g) | 24.5 ± 0.6 | 26.2 ± 1.2 | 27.8 ± 2.1 | 27.9 ± 2.6 |
| HR at BL (bpm) | 255.0 ± 18.0 | 221.3 ± 10.0 | 256.7 ± 15.3 | 218.0 ± 19.3 |
| MAP at BL (mm Hg) | 76.7 ± 4.6 | 69.0 ± 5.6 | 74.7 ± 9.1 | 66.7 ± 9.1 |
| Temperature at BL (°C) | 36.8 ± 0.1 | 36.7 ± 0.3 | 36.8 ± 0.1 | 36.9 ± 0.2 |
| CPR Time to ROSC (s) | NA | NA | 70.0 ± 8.7 | 93.7 ± 57.6 |
| HR at ROSC (bpm) | NA | NA | 436.7 ± 86.2 | 431.7 ± 50.1 |
| MAP at ROSC (mm Hg) | NA | NA | 97.0 ± 19.7 | 108.7 ± 9.7 |
NA indicates not applicable. BL, baseline; ROSC, return of spontaneous circulation; CPR, cardiopulmonary resuscitation; CA8, 8 min of cardiac arrest prior to resuscitation; R30, 30 min post-ROSC; R60, 60 min post-ROSC.
Selective impairment of electron transfer Complexes I and IV in heart mitochondria following cardiac arrest and resuscitation
Since the respiratory complexes localize to the inner membrane, and since their activities are dynamically modulated by changes in permeability, very likely to occur in our model, we decided to study the existence of permanent defects in electron transfer in submitochondrial particles (SMPs). SMPs consist of a mixed population of right-side out and inside-out vesicles of inner mitochondrial membrane and are devoid of the outer membrane and matrix components. Following mitochondrial isolation from the whole heart of the four experimental groups, and SMPs preparation, we measured the enzymatic activities of the respiratory complexes. The activity of Complex I–III was relatively unaffected by 8 min cardiac arrest and remained at control levels even after 30 minutes of reperfusion. However, a ~ 50% reduction of electron flow occurred between 30 and 60 minutes of reperfusion injury (Figure 2, A and C). In contrast, Complex II–III activity was not significantly affected in any of the groups (Figure 2B). This pattern of preserved electron flow from complex II–III but decreased electron flow within complex I–III would indicate that mitochondrial Complex I is the more vulnerable site of electron flow dysfunction after 30–60 minutes of ischemia and reperfusion.
Figure 2.

Mitochondrial respiratory complexes activities. Submitochondrial particles prepared from the four animal groups were analyzed for Complex I–III activity (A), Complex II–III (B), or Complex IV (D). The ratio of Complex I–III to Complex II–III activities is shown as a surrogate of isolated Complex I activity (C). CA8, 8 min of cardiac arrest prior to resuscitation; R30, 30 min post-ROSC; R60, 60 min post-ROSC; *p <.05, n = 3 in each group.
In contrast to Complex I–III, the activity of Complex IV showed a biphasic early and late pattern of dysfunction. A strong trend towards reduced electron flow was seen immediately at the end of the 8 min cardiac arrest, this dysfunction recovered to near normal electron flow levels by 30 minutes after reperfusion. After 60 minutes of reperfusion, Complex IV displayed a significant reduction of specific activity (Figure 2D). Note that this dysfunction can not be attributed to simple nitric oxide (NO) overproduction and binding to the complex since the process of isolation and preparation of SMPs washes the reversible gas away. This change, instead, suggest a more-permanent or relatively fixed modification of protein complex itself or of the surrounding lipid environment.
Generation of reactive oxygen and nitrogen species during cardiac arrest and reperfusion
Since blockade of the electron transfer chain is usually associated with, and can also be the result of, increased reactive oxygen and nitrogen species, we sought to estimate ROS generation leading to production of H2O2 by SMPs. Using the Amplex Red probe for fluorometric H2O2 determination, we found that heart mitochondria produce increasing levels of H2O2 at Complex I at the end of 8 min cardiac arrest, this H2O2 production peaks at 30 minutes post-reperfusion and was observed to decrease later (Table 2), possibly decreasing due to the parallel impairment of electron flow within the enzymatic complex. This increase is evident even in the absence of rotenone, which is usually added to promote a detectable H2O2 production, suggesting an early and significant generation of ROS from Complex I. The production of ROS at Complex III is not altered in SMPs, as seen in Table 2.
Table 2.
Hydrogen peroxide production by mitochondrial Complex I and Complex III
| Group/Substrate | NADH | NADH + Rotenone | Succinate | Succinate + Antimycin A |
|---|---|---|---|---|
| Sham | 401 ± 15 | 704 ± 12 | 10.1 ± 501 | 44 ± 13 |
| CA8 | 621 ± 39a | 1064 ±142a | 18.3 ± 8.8 | 64.3 ± 13 |
| R30 | 884 ± 39a | 1353 ± 18a | 14 ± 5.8 | 65 ± 17 |
| R60 | 689 ± 117a | 1023 ± 208a | 16 ± 4 | 36 ± 5 |
Results are express as relative arbitrary units/min/mg mitochondrial protein.
p <.05, compared with sham; CA8, 8 min of cardiac arrest prior to resuscitation; R30, 30 min post-ROSC; R60, 60 min post-ROSC.
Given that mitochondrial Complex I is exquisitely sensitive to both reversible inhibition by S-nitrosylation by NO (21) and to irreversible inactivation by peroxynitrite (ONOO−), the product of NO and superoxide (O2−) anion reaction (22), we studied the levels of tyrosine nitration in heart mitochondria. Tyrosine nitration is a footprint of the rapid reaction between ONOO− and tyrosine residues in proteins. Western blot of mitochondrial proteins using a monoclonal anti-nitrotyrosine antibody revealed a very early increase in tyrosine nitration of a ~50 kDa protein, which peaks during cardiac arrest, and a slower accumulation in nitration of a ~25 kDa protein with reperfusion (Figure 3). These data indicate that during cardiac arrest and early reperfusion there is an elevated generation of NO and O2−, which undergo reactions to produce both ONOO− and H2O2.
Figure 3.

Protein tyrosine nitration in mitochondria isolated from the four experimental groups. Arrowheads indicate three predominant nitrated proteins, of apparent MW 50, 25 and 20 kDa (A). Densitometric analysis of tyrosine nitration of the 20–25 kDa proteins (B) and the ~50 kDa protein (C). Data were normalized to COX subunit densitometry. CA8, 8 min of cardiac arrest prior to resuscitation; R30, 30 min post-ROSC; R60, 60 min post-ROSC.
Evaluation of cytochrome c content in heart mitochondria following cardiac arrest
We then determined the endogenous levels of cytochrome c in heart mitochondria by western blot. Densitometric analysis was performed and normalized to the content of a subunit of the cytochrome oxidase complex (an integral protein, marker of mitochondrial inner membrane). The results, presented in Figure 4, show a marginally-significant 10–20% loss of mitochondrial cytochrome c (p <.1). The biological significance of this reduction is unclear, but it does not seem to be the basis of Complexes I and IV defects, since we supplement SMPs with excess cytochrome c for those measurements. Additional studies are necessary to determine whether the amount of released cytochrome c is related to the induction of caspase-mediated apoptosis.
Figure 4.
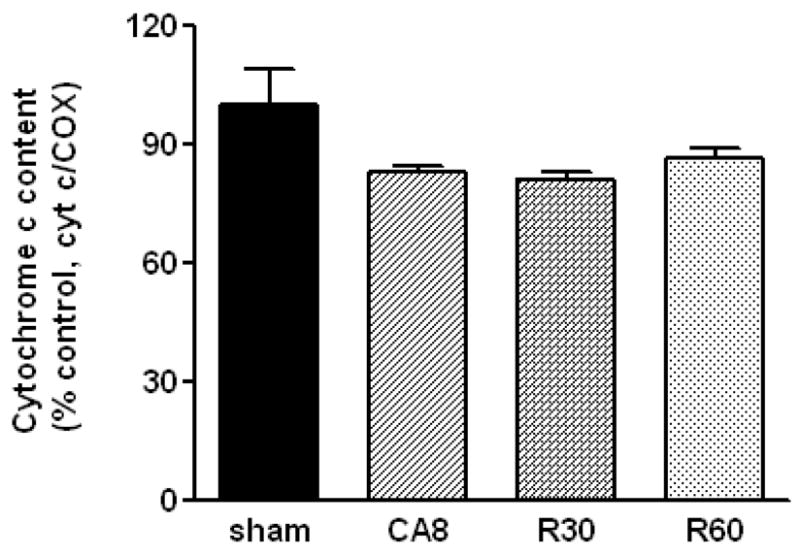
Densitometric analysis of cytochrome c content in mitochondria prior, during and after cardiac arrest. Data represent n = 3 samples/group and are normalized to COX content. CA8, 8 min of cardiac arrest prior to resuscitation; R30, 30 min post-ROSC; R60, 60 min post-ROSC.
Discussion
Our study reveals multiple specific defects in mitochondrial function following cardiac arrest as demonstrated by (a) reduced electron flow activity in specific complexes and loss of cytochrome c (b) increased ROS generation leading to H2O2 production from specific sites, and (c) oxidation of some mitochondrial proteins by tyrosine nitration. The earliest time point we examined during cardiac arrest was after 8 minutes and without reperfusion. Already at this very early time point we observed an increased generation of ROS from Complex I, a significant tyrosine nitration of mitochondrial proteins, and loss of mitochondrial cytochrome c. Within 30 minutes following reperfusion, maximal H2O2 generation from Complex I was detected, while impairments in electron flow at Complex IV became very pronounced after 60 minutes of reperfusion.
What intramitochondrial sites are most affected by ischemia and reperfusion?
Identifying the primary sites most affected by ischemia within the mitochondria remains a topic of some debate. While our in vivo model data demonstrates that Complexes I and IV appear more vulnerable to ischemia and reperfusion, many prior studies have presented similar and some dissimilar findings. The activity of Complex I, composed of 46 subunits, has been shown to be decreased by 35% after only 10 min of myocardial ischemia in a swine model (7, 9). Rapid loss of Complex I activity was also reported in the isolated heart subjected to ischemia and reperfusion (13). In addition to Complex I, it was noted that Complex IV, with 13 subunits and 3 redox centers, showed also significantly diminished activity at a later time point, between 30 and 60 minutes in this study and in work by others (7). Dysfunction of Complex I and IV can be related to loss and/or peroxidation of cardiolipin, the most abundant lipid in the inner mitochondrial membrane, which is required for the stabilization of both complexes (23). Relevant to our study, Paradies et al. have demonstrated that re-addition of cardiolipin-containing liposomes to heart mitochondria isolated after ischemia/reperfusion is able to restore the function of both Complex I and IV (13). Notwithstanding, the loss of cardiolipin is mainly attained during the ischemic phase rather than post-reperfusion, but previous studies using isolated heart employ significantly longer periods of ischemia (usually 30 min vs. 8 min). Thus, although fast cardiolipin loss during cardiac arrest could explain the early defects we found, we can not ascertain its causality in our experimental setting. More likely, the detection of increased tyrosine nitration of mitochondrial proteins during and early after resuscitation suggests that ONOO− formation might be responsible for Complex I reduced activity. ONOO−is a highly reactive oxidant formed primarily by the reaction of NO and O2−. During ischemia, if there is no total anoxia, and reperfusion, the increase in intracellular calcium leads to activation of constitutive nitric oxide synthases (NOS), principally nNOS, eNOS, and mtNOS in the cardiac muscle. The three isoforms are localized in the cytosol, plasma membrane, and mitochondria, respectively. NO generated by these enzymes blocks mitochondrial respiration transiently, but promotes the formation of O2− and consequently ONOO− (8). The last is an irreversible inhibitor of Complex I, relatively sparing Complex II and Complex III (22). It is therefore reasonable to believe that the conditions of ischemia and reperfusion provide these reactive species in significant concentrations, taking into account the small volume of a mitochondrion, which can be directly associated with the Complex I defect that we have found. We would like to emphasize that the mitochondrial changes we observed occur very fast during a short ischemic period (8 min), in contrast to studies with isolated organs or focal myocardial infarction in which ischemia is prolonged between 30 minutes to 2 hours (24). Our findings are then of high impact for human cardiac arrest and point to the necessity of a mitochondrial-targeted therapy administered if possible, prior to resuscitation.
Significant ROS leakage at Complex III, a surrogate for dysfunction, has also been extensively observed with progressive myocardial ischemia (24) and was prominent in our own prior cellular models of simulated ischemia/reperfusion (25). The fact that we were unable to demonstrate this in our present study suggests that in vivo Complex I may be affected prior to Complex III. Alternatively, it may be a methodological bias due to inherent differences in O2− production at each mitochondrial site. Complex I releases O2− vectorially into the mitochondrial matrix where active antioxidant defenses act to neutralize the ROS (26). Although the inner mitochondrial membrane is impermeable to O2−, its dismutation product H2O2 can readily cross lipid membranes and be released to the cytosol. By contrast, Complex III can deliver O2− both to the matrix and the intermembrane space, a small fraction of which moves through anionic channels and can be detected outside of the mitochondria. Thus, production of O2− by Complex III is echoed by increased O2− and H2O2 release to the rest of the cellular environment while production at Complex I leads only to H2O2 release. Indeed, O2− measurements in studies using intact cells were performed with a specific probe (DHE) that upon oxidation to ethidium binds to nuclear DNA to give a strong fluorescent signal. Since Complex I-generated O2− is not released from mitochondria, it might be barely detectable by DHE.
Complex II dysfunction has also been cited in some papers on post-ischemia (10). A 30 min coronary artery ligation followed by 24 hours of reperfusion was noted to decrease electron flow by 42% in a rat model of focal ischemia (10). Future studies will be required to advance our understanding in these normal and pathological states of dysfunction if we hope to create optimal therapies that prevent the consequences of reperfusion in our patients.
Limitations of current methods for determination of mitochondrial dysfunction
We believe that is fair to note that the lack of a straightforward, reliable mitochondrial measurement method for in vivo studies still confounds our progress in this field. Our current methods for the measurements of mitochondrial dysfunction and mitochondrial isolation methods remain somewhat primitive and unsatisfactory. To advance our understanding and to develop new therapies, better methods of measuring mitochondria in vivo should be prioritized and developed. These may include the use of intra-vital microscopy, in vivo optical sensors, specific mitochondrial probes, or transgenic animals that allow better measurements of electron flow and ROS generation in vivo. Attempts to improve methods for mitochondrial studies are also important given recent data to support the notion that mitochondria within a single heart cell have differing subpopulations with distinct subsarcolemmal and interfibrillatory mitochondria where the subsarcolemmal mitochondria are preferentially affected at complex IV during ischemia (11). Our current methods may be quite biased or even obscure proper identification of the in vivo physiology.
Roles of cytochrome c and tyrosine nitration
Our data further support the notion that cytochrome c release and tyrosine nitration occur very rapidly and can further progress during reperfusion. While the decrease of cytochrome c was only in the range of 10–20% this may have a significant effect on initiation of caspase activation and apoptosis within cardiomyocytes. Our data is some of the first data derived from a cardiac arrest model to demonstrate this consistent loss of cytochrome c so early in ischemia and reperfusion. The physiology that is emerging suggests that mitochondria can initiate cascades that amplify injury in reperfusion. However, there may be pro-survival signals that promote healing and repair as a response to injury coincident with the injury amplification pathways. It is likely that a delicate balance between those responses determines whether the cells, and ultimately the organism are going to survive and repair or die. An understanding of these signaling pathways may likewise provide opportunities for new therapeutic agents to attenuate reperfusion mediated injury.
Conclusions
Taken collectively our results along with the prior literature reveal several dynamic concepts. While there is not 100% agreement, a surprisingly “orderly” process of progressive mitochondrial dysfunction occurs during ischemia and reperfusion. Significant dysfunction in electron flow and ROS generation can be observed during ischemia and this dysfunction may be further amplified or progress during reperfusion. Specific therapies including hypothermia, exogenous administration of cardiolipin or antioxidants, specific mitochondrial inhibitors, and mitochondrial uncoupling agents have all been demonstrated to improve mitochondrial dysfunction. Because specific sites are likely to be responsible for this dysfunction, research in the coming years should help further elucidate the specific sites and mechanisms behind mitochondrial dysfunction following ischemia – as they present potentially important clinical therapies for human cardiac arrest victims. The literature collectively supports the notion that an effective mitochondrial targeted post ischemia therapy is possible; with continued research it may be helpful in the near future for treatment of human disease.
Acknowledgments
The work detailed in this manuscript was supported by a grant from the National Institutes of Health (R01 HL 71734-01).
We thank Dr. Alvaro Estevez, Weil Cornell University, for a generous gift of monoclonal anti-nitrotyrosine antibody.
References
- 1.Becker LB. New concepts in reactive oxygen species and cardiovascular reperfusion physiology. Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- 2.Mayevsky A, Chance B. Oxidation-reduction states of NADH in vivo: from animals to clinical use. Mitochondrion. 2007;7:330–339. doi: 10.1016/j.mito.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Borutaite V, Mildaziene V, Brown GC, et al. Control and kinetic analysis of ischemia-damaged heart mitochondria: which parts of the oxidative phosphorylation system are affected by ischemia? Biochim Biophys Acta. 1995;1272:154–158. doi: 10.1016/0925-4439(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 4.Vanden Hoek TL, Li C, Shao Z, et al. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. J Mol Cell Cardiol. 1997;29:2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 5.Qin Y, Vanden Hoek TL, Wojcik K, et al. Caspase-dependent cytochrome c release and cell death in chick cardiomyocytes after simulated ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2004;286:H2280–2286. doi: 10.1152/ajpheart.01063.2003. [DOI] [PubMed] [Google Scholar]
- 6.Vanden Hoek TL, Qin Y, Wojcik K, et al. Reperfusion, not simulated ischemia, initiates intrinsic apoptosis injury in chick cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;284:H141–150. doi: 10.1152/ajpheart.00132.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rouslin W. Mitochondrial complexes I, II, III, IV, and V in myocardial ischemia and autolysis. Am J Physiol Heart Circ Physiol. 1983;244:H743–748. doi: 10.1152/ajpheart.1983.244.6.H743. [DOI] [PubMed] [Google Scholar]
- 8.Poderoso JJ, Lisdero C, Schöpfer F, et al. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem. 1999;274:37709–37716. doi: 10.1074/jbc.274.53.37709. [DOI] [PubMed] [Google Scholar]
- 9.Rouslin W, Millard RW. Mitochondrial inner membrane enzyme defects in porcine myocardial ischemia. Am J Physiol Heart Circ Physiol. 1981;240:H308–313. doi: 10.1152/ajpheart.1981.240.2.H308. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Chen C, Pfeiffer DR, et al. Mitochondrial complex II in the post-ischemic heart: oxidative injury and the role of protein S-glutathionylation. J Biol Chem. 2007;282:32640–32654. doi: 10.1074/jbc.M702294200. [DOI] [PubMed] [Google Scholar]
- 11.Lesnefsky EJ, Tandler B, Ye J, et al. Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria. Am J Physiol. 1997;273:H1544–1554. doi: 10.1152/ajpheart.1997.273.3.H1544. [DOI] [PubMed] [Google Scholar]
- 12.Lavani R, Chang W, Anderson T, et al. Altering CO2 during reperfusion of ischemic cardiomyocytes modifies mitochondrial oxidant injury. Crit Care Med. 2007;35:1709–1716. doi: 10.1097/01.CCM.0000269209.53450.EC. [DOI] [PubMed] [Google Scholar]
- 13.Paradies G, Petrosillo G, Pistolese M, et al. Decrease in Mitochondrial Complex I Activity in Ischemic/Reperfused Rat Heart: Involvement of Reactive Oxygen Species and Cardiolipin. Circ Res. 2004;94:53–59. doi: 10.1161/01.RES.0000109416.56608.64. [DOI] [PubMed] [Google Scholar]
- 14.Kern KB, Berg RA, Hilwig RW, et al. Myocardial cytokine IL-8 and nitric oxide synthase activity during and after resuscitation: Preliminary observations in regards to post-resuscitation myocardial dysfunction. Resuscitation. 2008;77:401–409. doi: 10.1016/j.resuscitation.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeh C, Chen T, Wu Y, et al. Inhibition of NFkappaB activation with curcumin attenuates plasma inflammatory cytokines surge and cardiomyocytic apoptosis following cardiac ischemia/reperfusion. J Surg Res. 2005;125:109–116. doi: 10.1016/j.jss.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Radhakrishnan J, Wang S, Ayoub IM, et al. Circulating levels of cytochrome c after resuscitation from cardiac arrest: a marker of mitochondrial injury and predictor of survival. Am J Physiol Heart Circ Physiol. 2007;292:H767–775. doi: 10.1152/ajpheart.00468.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gomez L, Thibault H, Gharib A, et al. Inhibition of mitochondrial permeability transition improves functional recovery and reduces mortality following acute myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2007;293:H1654–1661. doi: 10.1152/ajpheart.01378.2006. [DOI] [PubMed] [Google Scholar]
- 18.Poderoso JJ, Carreras MC, Lisdero C, et al. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys. 1996;328:85–92. doi: 10.1006/abbi.1996.0146. [DOI] [PubMed] [Google Scholar]
- 19.Starkov AA, Fiskum G. Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem. 2003;86:1101–1107. doi: 10.1046/j.1471-4159.2003.01908.x. [DOI] [PubMed] [Google Scholar]
- 20.Schuh RA, Kristián T, Gupta RK, et al. Methoxychlor inhibits brain mitochondrial respiration and increases hydrogen peroxide production and CREB phosphorylation. Toxicol Sci. 2005;88:495–504. doi: 10.1093/toxsci/kfi334. [DOI] [PubMed] [Google Scholar]
- 21.Clementi E, Brown GC, Feelisch M, et al. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riobó NA, Clementi E, Melani M, et al. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem J. 2001;359:139–145. doi: 10.1042/0264-6021:3590139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lesnefsky EJ, Slabe TJ, Stoll MSK, et al. Myocardial ischemia selectively depletes cardiolipin in rabbit heart subsarcolemmal mitochondria. Am J Physiol Heart Circ Physiol. 2001;280:H2770–2778. doi: 10.1152/ajpheart.2001.280.6.H2770. [DOI] [PubMed] [Google Scholar]
- 24.Lesnefsky EJ, Gudz TI, Migita CT, et al. Ischemic injury to mitochondrial electron transport in the aging heart: damage to the iron-sulfur protein subunit of electron transport complex III. Arch Biochem Biophys. 2001;385:117–128. doi: 10.1006/abbi.2000.2066. [DOI] [PubMed] [Google Scholar]
- 25.Vanden Hoek TL, Shao Z, Li C, et al. Mitochondrial electron transport can become a significant source of oxidative injury in cardiomyocytes. J Mol Cell Cardiol. 1997;29:2441–2450. doi: 10.1006/jmcc.1997.0481. [DOI] [PubMed] [Google Scholar]
- 26.Chen Q, Vazquez EJ, Moghaddas S, et al. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]