

Abstract
Insulin-like growth factor binding protein-3 (IGFBP-3) is emerging as a critical regulator of cell survival. There has been no study which directly examined the potential role for this major growth factor in the programmed cell death (apoptosis) of insulin-secreting cells. To determine whether IGFBP-3 mediates apoptosis in insulin-secreting cells, we performed a rigorous series of experiments with the rat insulinoma (RIN) cell line m5F and the hamster insulin-secreting tumor (HIT) T-15.
Within 24 h exogenous IGFBP-3 induced significant DNA fragmentation in RIN and HIT cells, at doses ranging from 4.4 to 2000 ng/ml (P < 0.05) without a classic dose–response relationship (Fig. 3). DNA fragmentation induced by rhIGFBP-3 occurred in the presence of immunoglobulin to block the type 1 IGF receptor. As detected by flow cytometry for Annexin V exposure to the cell surface, rhIGFBP-3 treatment doubled the proportion of apoptotic HIT cells from 1.7 ± 0.4% (serum-free control) to 3.4 ± 0.2% (P < 0.02), an effect completely reversed by co-treatment with 1000 ng/ml rhIGF-I. Immunofluorescent microscopy disclosed that pro-inflammatory Th1 cytokines increased intranuclear aggregation of endogenous IGFBP-3. Cytokine-induced DNA fragmentation was completely blocked by relatively brief pre-treatment with antisense IGFBP-3 phosphorothioate oligodeoxynucleotides. In conclusion, we have presented the first evidence that IGFBP-3 contributes to cytokine-mediated apoptosis in insulin-secreting cells.
Keywords: IGF, IGFBP-3, Apoptosis, Diabetes mellitus, Cytokine
1. Introduction
Type 1 diabetes mellitus results from the autoimmune and non-immune destruction of the insulin-producing β-cells of the islets of Langerhans. This destruction results in part from apoptosis, a tightly controlled, multi-step process of cell death involving activation of specific intracellular pathways, including the cytosolic aspartic acid-specific proteases (caspases) [1]. Multiple lines of evidence suggest that cytokines are key mediators of β-cell growth and apoptosis, in particular, interleukin-1 α and β (IL-1 α and β) [1,2], interferon-γ (IFN-γ) [3], tumor necrosis factor-α (TNF-α) [4], and transforming growth factor-β1 (TGF-β1) [5]. The current concept of the impact of these cytokines in vivo posits an imbalance favoring the actions of proinflammatory Th1 cytokines over protective Th2 cytokines.
Two insulin-like growth factors (IGF-I and -II) and six closely related high affinity IGF binding proteins (IGFBP-1 through -6) comprise the insulin-like growth factor (IGF) superfamily [6]. IGFs have been shown to inhibit apoptosis in diverse cell types, mainly by counteracting the effects of agents which induce apoptosis [7]. IGF-I treatment has been reported to protect islets from cytokine-mediated apoptosis after isolation from pre-diabetic non-obese diabetic (NOD) mice [8] and to delay or prevent progression of insulitis in NOD in vivo [9].
The traditional model proposes that IGFBPs induce growth arrest by sequestration of IGFs, preventing IGF bioavailability to IGF receptors on the plasma membrane [10]. We and others have reported that IGFBP-3 induces apoptosis via an IGF receptor-independent mechanism in diverse cell types, including murine fibroblasts lacking type 1 IGF receptors [11,12], prostatic carcinoma [13] and breast carcinoma [14]. An IGFBP-3 fragment which lacks IGF binding affinity has been shown to inhibit IGF-stimulated and insulin-stimulated cell growth of chick embryo fibroblasts [15]. We have previously reported that the clonal insulin-secreting line HIT-T15 comprises an environment for the production and binding of IGFs and IGFBPs [16]. The present studies were designed to test our novel hypothesis that IGFBP-3 mediates cytokine-induced apoptosis in insulin-secreting cells.
2. Materials and methods
2.1. Cells and reagents
The American Type Tissue Collection (ATCC, Manassas, VA, USA) supplied RIN m5F cells and HIT T15 cells, derived, respectively, from β-cell tumors of Rattus norvegicus [17–19] and the Syrian golden hamster Mesocricetus auratus [20] transformed by the simian virus 40 large T antigen. RIN cells were grown in 90% RPMI 1640 media supplemented with 10% fetal bovine serum. HIT cells were grown in 87.5% Hamõs F12K media supplemented with 2.5% fetal bovine serum and 10% heat-inactivated horse serum. All media were supplemented with 1% penicillin and 1% streptomycin, and all cultures were maintained at 37 °C under 5% ambient CO2. Growth media was changed every third day.
Recombinant human (rh) IL-1β, IFN-γ, TNF-α, and TGF-β1 were purchased from Sigma (St. Louis, MO, USA). Genentech Inc. (South San Francisco, CA, USA) generously donated non-glycosylated rhIGFBP-3. Pharmacia Inc. (Peapack, NJ, USA) generously donated rhIGF-I. Anti-sense oligodeoxynucleotide designed to flank the initiation codon of murine IGFBP-3 [21] was 5′-GCGCGCGGGATGCATGGCGCCGGGTGGACG, with the corresponding sense oligo as 5′-CGTCCACCC GGCGCCATGCATCCCGCGCGC. Anti-sense oligo flanking the initiation codon of rat IGFBP-3 [22] was 5′-CGCGGGATGCATGGCG CTGG CGGAGGGCTC. Thioester bonds linked the first three and final three nucleotides of each oligo (Sigma-Genosys, Ltd., The Woodlands, TX, USA).
2.2. Apoptosis assays
Prior to apoptosis assays, RIN cells were grown in 75 cm2 flasks to 80% confluence, then harvested with trypsin and plated at an approximate density of 100,000 cells/well (2 ml medium/well). Precisely 24 h later, growth media was removed and cells were washed twice with serum-free media. Media was replaced with equal volumes of serum-free media (SFM), with or without rhIGFBP-3 at a final concentration of 1000 ng/ml. Conditioned media were collected 24 h later as detailed elsewhere [23], and cell pellets were obtained by centrifugation. Floating and adherent cells were pooled for apoptosis analyses.
2.2.1. DNA fragmentation ELISA
Cells were plated at 70–90% confluence in 96-well plates and allowed to adhere overnight. Growth media were removed, and cells were washed twice with serum-free media. Each well contained a final volume of 200 μl for experimental conditions. To terminate each experiment, we centrifuged plates at 200g for 10 min at 25 °C to separate floating and attached cells from the conditioned media. Apoptosis in the floating and attached cells was quantified by photometric cell death ELISA for mono- and oligonucleosomes (Cell Death Detection Kit, Boehringer–Mannheim, Indianapolis, IN, USA), performed according to the manufacturerõs instructions. Mono- and oligonucleosomes represent histone-associated DNA fragments produced specifically during apoptosis.
For time course experiments with calphostin, cells were pre-treated for 90 min with 5 μM calphostin C (Sigma, St. Louis, MO, USA) before exposure to 1000 ng/ ml rhIGFBP-3 for 0.5, 2, or 16 h. To maintain the light-dependent activity of calphostin C, cells were incubated under 2.2-W incandescent flashlights (bulb PR2, Dura-cell, Chesapeake, VA, USA) positioned less than 5 cm above the surface of the media to assure equal exposure to all wells. Genistein was purchased from BIOMOL Research Laboratories (Plymouth Meeting, PA, USA).
2.2.2. Fluorescence activated cell sorting (FACS)
HIT cells were plated in 2 ml Vitacell F12K media (ATCC, Manassas, VA, USA), supplemented with 2.5% FBS and 10% dialyzed horse serum, at a density of 40,000 viable cells/ml, as determined by exclusion of tryphan blue. After adherence overnight cultures were washed twice with serum-free media, then exposed for 24 h to serum-free media containing 2000 ng/ml rhI-GFBP-3, with or without 1000 ng/ml rhIGF-1. Conditioned media were collected, and wells were washed with HEPES-buffered saline solution (HBSS, Clonetics, San Diego, CA, USA). This rinse was pooled with the conditioned media, then spun at 200g for 10 min at 25 °C to obtain floating cells. The adherent cells remaining in each well were detached by incubation with 1 ml of trypsin/EDTA solution at 25 °C for 5 min (Clonetics, San Diego, CA), then 2 ml of trypsin neutralizing solution (TNS, Clonetics, San Diego, CA) was added to each well for 1 min at 25 °C. The wells were then rinsed with HBSS, and this rinse was pooled with the TNS rinse prior to centrifugation at 200g for 10 min at 25 °C. These floating cells were washed in PBS, then spun at 200g for 10 min at 25 °C. Immediately prior to use, a reagent mixture was prepared containing 2 μl fluorescein-labeled Annexin V antibody, 2 μl (100 ng) propidium iodide, and 100 μl HEPES buffer using the Annexin-V-FLUOS kit according to the manufacturerõs instructions (Roche, Germany). Cells re-suspended in Annexin reagent were incubated under darkness for 15 min at 25 °C. Becton–Dickinson FACScan cytometer (1991 model) counted two hundred cells per second, for a total of at least 10,000 cells (i.e., gated events) per assay. Data were collected from forward and side scatter detectors, and autofluorescence was excluded by format on a four-quadrant, dual axis, logarithmic scale graph via dot-blot. Quadrant data were analyzed with Cell Quest software (version 3.2.1F1, ©1999, Becton–Dickinson, San Jose, CA).
2.3. Immunofluorescent microscopy
Cells plated at a density of 104/slide and cultured 24 h in complete media were rinsed twice with serum-free media, then exposed for 12 h to experimental serum-free conditions. After removal of conditioned media, cells were washed once with 1 ml phosphate-buffered saline (PBS), then fixed in −20 °C methanol, 0.5 ml/well, for 5 min. Air-dried cells were washed twice with PBS, then permeabilized with 0.5 ml of 0.2% Triton-X in PBS for 20 min on ice. After two washes with PBS, cells were incubated in PBS for 1 h at 25 °C with 5 ng/ll primary goat polyclonal anti-murine IGFBP-3 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Cells were then washed twice in PBS prior to incubation with 5 μg/μl of fluorescein-conjugated rabbit anti-goat IgG (Vector Laboratories, Inc., Burlingame, CA, USA) for 45 min at 25 °C in darkness. After two additional PBS washes, cells were incubated for 2 min at 25 °C with the nucleic acid stain 4′-6-diamidino-2-phenylindole (DAPI from Electron Microscopy Sciences, Ft. Washington, PA, USA). Cells were washed in ice-cold PBS twice for 10 min under darkness, then stored at 4 °C under darkness and FluoroGuard Antifade Reagent (Bio-Rad Laboratories Inc., Hercules, CA, USA) until performance of confocal microscopy. The 461 nm line from the Ar laser of our Leica TCS SP Inverted Confocal Microscope stimulated fluorescence, which was captured directly by Himamatsu digital camera and processed with Microscope Control Module Software (QED Imaging, Inc., Pittsburgh, PA, USA). Pixel analysis of confocal data utilized Photoshop® software (version 4.0 for Macintosh, Adobe Systems Inc., San Jose, CA, USA).
2.4. Western ligand blot
Equal volumes of conditioned media were lyophilized and subjected to 12.5% PAGE–SDS overnight. The next morning, each gel was electroblotted onto nitrocellulose, then sequentially washed with 3% NP40/Tris–HCl at 4 °C for 30 min, 1% bovine serum albumin/Tris–HCl at 4 °C for 2 hours, and 0.1% Tween/Tris–HCl at 4 °C for 10 min. Blots were incubated with 1.5 million c.p.m. 125I-IGF-I and 125I-IGF-II in 1% bovine serum albumin/ 0.1% Tween/Tris–HCl at 4 °C for 12 h. Membranes were sequentially washed three times with 0.1% Tween in PBS at 4 °C for 15 min and three times with PBS at 4 °C for 15 min, followed by air drying and exposure to film at −70 °C. Densitometry of autoradiographs was performed using ImageQuaNT software (Storm 840, Molecular Dynamics, Sunnyvale, CA, USA).
2.5. Statistical analysis
Making no assumption about the scatter of data in experiments which compared multiple interventions, we analyzed data by ANOVA and the Kruskal–Wallis test for ordinary, unmatched measures (InStat version 2.00; GraphPad Software Inc., San Diego, CA, USA) on a Macintosh PowerBook (Apple Computer, Cupertino, CA, USA). Graphs depict mean data ± SE except as noted. Probability values (P) below 5% were considered significant.
3. Results
3.1. Pro-apoptotic cytokines induce IGFBP-3 release
Compared to serum-free conditions, treatment of HIT cells with Th1 pro-inflammatory cytokines (48 h with IL-1β 100 ng/ml, TNF-α 100 ng/ml, IFN-γ 33 ng/ ml, or TGF-β 1 1 ng/ml) induced at least a 56% increase in DNA fragmentation (Fig. 1; *P < 0.01; n = 3). Western ligand blots revealed the predominant IGFBPs secreted from RIN cells under serum-free conditions were IGFBP-2 and IGFBP-4 (Fig. 2), consistent with our previous observations in HIT cells [16]. Th1 cytokine treatment which has been shown to induce apoptosis [1] (100 ng/ml IL-1β for 72 h) appeared to reduce slightly the quantity of IGFBP-2 released from RIN cells. Despite minimal amounts of IGFBP-3 detected in conditioned media from RIN cells under serum-free conditions, the quantity of IGFBP-3 released (in equal volumes of conditioned media) modestly increased after the treatment with IL-1β (Fig. 2). Release of IGFBP-3 from HIT cells to the extracellular media in response to Th1 cytokines was not observed.
Fig. 1.
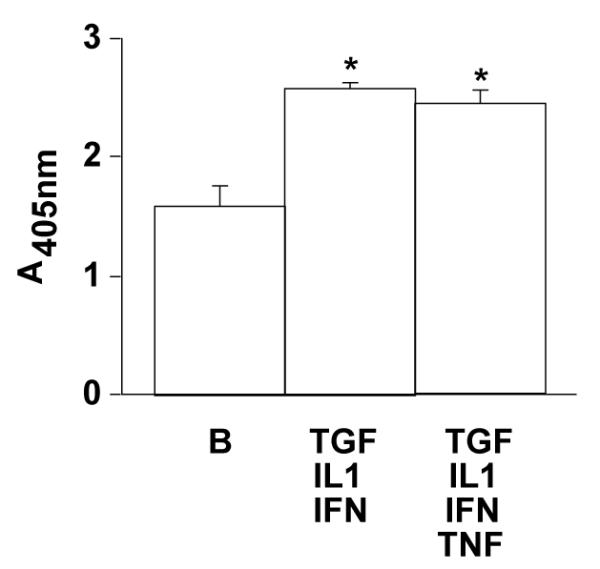
DNA fragmentation ELISA of HIT cells exposed to serum-free basal (B) conditions or cytokines.
Fig. 2.
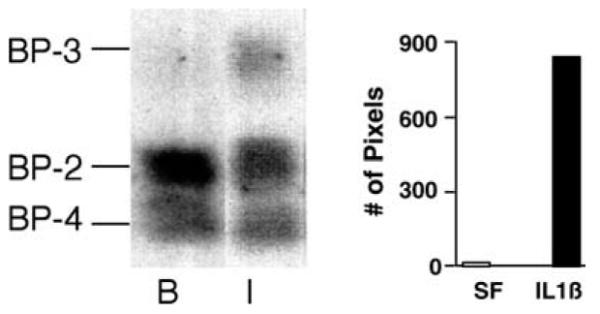
Western ligand blots for IGFBPs in conditioned media of RIN cultures after 72 h with serum-free baseline (B) conditions or IL-1β (I). Cytokine treatment induced release of IGFBP-3 into conditioned media. Densitometry was performed by quantification of all pixels in the IGFBP-3 bands using the black channel, and all values below a level of 190 were summated. Vertical bars represent the total black pixel count in the IGFBP-3 band for each experiment.
3.2. RhIGFBP-3 induces apoptosis of RIN and HIT cells
Treatment of RIN cells for 24 h with 1000 ng/ml rhIGFBP-3 induced a 30% increase of caspase 3 activity (P < 0.02 vs. SFM; data not shown). Treatment of HIT cells for 24 h with 2000 ng/ml rhIGFBP-3 induced a 42% increase in DNA fragmentation (Fig. 3(A), **P < 0.0001 vs. serum-free; n = 8); whereas, complete serum (S) suppressed apoptosis (*P < 0.006 vs. serum-free). Exposure of HIT or RIN cells to rhIGFBP-3 at a concentration as low as 4.4 ng/ml (10−10 M) for 24 h significantly induced DNA fragmentation above basal serum-free conditions (n = 8; P < 0.03 vs. SF for HIT; P < 0.007 vs. SF for RIN; data not shown). Immunoglobulin to the alpha subunit of the type I IGF receptor (aIR) at a concentration of 1000 ng/ml has been previously shown to block the growth stimulatory effects of exogenous insulin or IGF [24]. However, the presence of aIR antibody at a concentration of 1000 ng/ml alone exerted no significant effect on DNA fragmentation in HIT cultures under basal serum-free conditions (Fig. 3(A); P > 0.05 vs. SF; n = 8). RhIGFBP-3-induced DNA fragmentation was enhanced further above baseline from 42% to 78% in the presence of this neutralizing antibody (Fig. 3(A); ***P < 0.007 vs. Ab alone; ***P < 0.0001 vs. serum-free or vs. rhIGFBP3 alone). RhIGFBP-3 induced at least 63% increase in DNA fragmentation within RIN cells in a dose-sensitive manner (Fig. 3(B); **P < 0.005 vs. serum-free; n = 8). Nucleosome production induced in RIN cells by 1000 ng/ml rhIGFBP-3 treatment (Fig. 3(C); *P < 0.02 vs. serum-free) was suppressed by co-treatment with 1000 ng/ml rhIGF-I (**P < 0.0004 vs. rhIGFBP-3 alone; **P < 0.03 vs. serum-free; n = 6).
Fig. 3.

DNA fragmentation ELISA of HIT and RIN cultures in the presence of rhIGFBP-3 (BP3), rhIGF-I (IGF), neutralizing antibody to the type 1 IGF receptor (aIR), or genistein (G).
The phytoestrogen genistein has been demonstrated to inhibit tyrosine kinase activity specifically in multiple cell types. The pro-apoptotic action of rhIGFBP-3 at a relatively high dose of 5000 ng/ml was equivalent to the effect of 5 μg/ml genistein at 24 h (Fig. 3(D); *P < 0.05 vs. serum-free). Exogenous IGFBP-3 induced DNA fragmentation despite inactivation of the IGF receptor tyrosine kinase activity by pre-treatment with genistein for 90 min (Fig. 3(D); *P < 0.05 vs. serum-free). RhI-GFBP-3 induced apoptosis in RIN cells as rapidly as 30 min after exposure via a mechanism independent of protein kinase C activation (Fig. 4; *P < 0.05 vs. calphostin alone and vs. serum-free; †P < 0.05 vs. SF; n = 4).
Fig. 4.

DNA fragmentation ELISA of RIN cells exposed to rhIGFBP-3 in the presence of calphostin (CAL).
To quantify the percentage of cells undergoing apoptosis, we analyzed HIT cultures by FACS after exposure for 24 h to 2000 ng/ml rhIGFBP-3 (Fig. 5). The proportion of HIT cells staining positive for Annexin V, but not propidium iodide, doubled from 1.73 ± 0.36% to 3.36 ± 0.16% upon exposure to IGFBP-3 for 24 h (Fig. 5(D); *P < 0.02 vs. serum-free; triplicate experiments, each measuring 10,000 gated events). This apoptotic effect of exogenous IGFBP-3 was completed blocked by simultaneous addition of rhIGF-I, which decreased the proportion of Annexin V-positive cells to 1.83 ± 0.43%, similar to results under the basal serum-free conditions (Fig. 5(D); †P < 0.02 vs. rhIGFBP-3 alone; triplicate experiments, each measuring 10,000 gated events).
Fig. 5.

Representative FACS for HIT cultures. Histogram compiles mean (±SE) data of Annexin V-positive cell populations (lower right panels) from triplicate FACS experiments, each counting 10,000 gated events per condition.
3.3. Cytokines induce nuclear localization of endogenous IGFBP-3
Endogenous IGFBP-3 was not detected in the nuclei of HIT cells under serum-free conditions (Fig. 6(A–C)). Treatment with TNF-α 100 ng/ml for 24 h induced dramatic nuclear localization of endogenous IGFBP-3 (Fig. 6(H); **P < 0.0001; n = 9 cells). Under serum-free conditions, endogenous IGFBP-3 in RIN cells localized to the cytosol as well as the nucleus (data not shown) and did not appear to associate significantly with the nuclear heterochromatin, as indicated by DAPI stain. Treatment of RIN cells for 24 h with 10 ng/ml TNF-α was associated with notable aggregation of endogenous IGFBP-3 on the nuclear heterochromatin.
Fig. 6.

Immunofluorescent microscopy of HIT cell cultures under serum-free conditions (A–C) and after treatment with TNF-α (D–F). Green stain depicts endogenous murine IGFBP-3, and DAPI stains heterochromatin blue. Histograms (G–H) depict means ± SD.
3.4. Anti-sense IGFBP-3 oligos inhibit cytokine-induced apoptosis
As expected, treatment with TNF-α 20 ng/ml for 24 h induced DNA fragmentation in RIN cells (Fig. 7(A); *P < 0.003). Pre-treatment with sense oligo for murine IGFBP-3 had no apparent effect on TNF-α-induced apoptosis; whereas, pre-treatment for 30 min with anti-sense IGFBP-3 oligo completely blocked the DNA fragmentation induced by exposure to TNF-α (Fig. 7(A); **P < 0.002 vs. TNF-α alone; **P < 0.03 vs. serum-free; †P < 0.008 vs. serum-free; n = 6). Pre-treatment for 90 min with anti-sense oligo to rat IGFBP-3 inhibited DNA fragmentation induced in HIT cultures by either IL-1β 20 ng/ml (Fig. 7(B); *P < 0.008 vs. serum-free; **P < 0.008 vs. IL-1β alone; n = 8) or serum withdrawal for 24 h (†P < 0.0001 vs. serum-free; n = 8).
Fig. 7.

DNA fragmentation ELISA of RIN (A) and HIT (B) cultures.
4. Discussion
Multiple lines of evidence support the concept that IGFBP-3 modulates apoptosis via both IGF receptor-dependent and IGF receptor-independent mechanisms [11–13,25]. Although the mechanisms relating IGFBPs and their binding partners (or putative receptors) remain unclear, two compatible models for IGFBP action have been proposed. The most widely accepted mechanism is based on the receptor-dependent model which describes sequestration of IGFs from binding to IGF receptors. Support for this model arose from reports that addition of IGFBP-3 or IGFBP-2 to diverse cell cultures resulted in dose-dependent inhibition of IGF-induced mitogenesis [26,27]. In support of the alternate but compatible IGF receptor-independent model, IGFBP-3 has been shown in human breast cancer to induce apoptosis independently of its cell surface binding or its nuclear actions [28]. We now present the first evidence that IGFBP-3 mediates cytokine-induced apoptosis in insulin-secreting cells.
Both exogenous IGFBP-3 (Figs. 3 and 4) and endogenous IGFBP-3 (Fig. 7) mediated apoptosis in the insulin-secreting cell lines studied. IGFBP-3 secretion from RIN and HIT cells appeared to be minimal upon cytokine stimulation compared to serum-free conditions. However, IGFBP-3 aggregated in the nuclei of both RIN and HIT nuclei upon exposure to Th1 cytokines (Fig. 6), supporting a predominantly intracellular action, consistent with data from other systems [29,30]. Observations that anti-sense IGFBP-3 oligos inhibit Th1 cytokine-induced apoptosis, coupled with confocal data demonstrating cytokine-induced nuclear aggregation, are also consistent with an intracellular (paracrine or autocrine) mechanism for IGFBP-3-induced apoptosis in these insulin-secreting cells. A recent report that IGFBP-3 activates phosphotyrosine phosphatase activity [31] provides one specific and novel intracellular mechanism by which IGFBP-3 modulates the tyrosine kinase activity of the IGF receptor, although the mechanism in insulin-secreting cells may differ. It is logical to deduce that rapid, non-genomic actions of IGFBP-3 represent modulation of phosphorelays, whereas genomic actions occur over longer time courses.
HIT cells possess atypical IGF receptors which bind IGFs with very low affinity [16]. Passive blockade of IGF ligand binding by the aIR antibody was insufficient by itself to induce apoptosis in HIT cells (Fig. 3(A)). We observed enhanced IGFBP-3-mediated apoptosis of insulin-secreting cells despite passive blockade of anti-apoptotic signaling via the IGF receptor with aIR antibody (Fig. 3(A)), confirming that, at least in part, IGFBP-3-induced apoptosis in these cells involves the IGF receptor. Our data demonstrate that high extracellular concentrations of IGF block IGFBP-3-induced apoptosis (Figs. 3(D) and 5(D)), presumably by extracellular sequestration of IGFBP-3 and by ligand activation of the IGF receptor. If IGF receptor activation is absolutely required for IGFBP-3 to induce apoptosis (in other words, if IGFBP-3 can induce apoptosis only via the modulation of the IGF receptorsõ tyrosine kinase activity), then apoptosis resulting from inactivation of the tyrosine kinase would resemble that induced by IGF sequestration with IGFBP-3. We observed such similarity (Fig. 3(D)). We also observed rapid induction of apoptosis within 30 min of exposure to IGFBP-3 (Fig. 4). Thus, our data are most consistent with the model that IGFBP-3 induces apoptosis in these insulin-secreting cells primarily via an IGF receptor-dependent mechanism, although IGFBP-3 can induce apoptosis independently of IGF sequestration. Our data are compatible with the emerging concept that IGFBP-3 modulates key phosphorelays critical to cell survival via intracellular (paracrine or autocrine) mechanisms [31]. In separate experiments to test the hypothesis that intracellular IGFBP-3 directly mediates apoptosis, we did not detect intrinsic caspase-like activity for rhIGFBP-3 using in vitro assays for caspases 1 through 9.
Radulescu first reported that IGFBP-3 possesses a nuclear localization sequence [32], and subsequent investigations demonstrated IGFBP-3 localization to the nucleus of the dividing cell [33,34]. We observed that cytokine-induced apoptosis is associated with translocation of IGFBP-3 to the nuclei of insulin-secreting cells, an effect which persists as long as 24 h after exposure (Fig. 6). These data support the emerging concept that endogenous IGFBP-3 can exert direct nuclear actions to promote apoptosis. We have recently reported that IGFBP-3 specifically binds nuclear retinoic X receptor-a (RXR-a) in prostatic and embryonal carcinomas to regulate RXR-specific response elements which play a critical role in cell cycle control [35]. Chertow and colleagues [36] demonstrated that all-trans retinoic acid – an agonist for both nuclear retinoic X receptor-a and retinoic acid receptors – induces apoptosis in RIN cells, observations which concur with our novel hypothesis that IGFBP-3 can regulate apoptosis in insulin-secreting cells via nuclear receptors. Recent evidence obtained in COS cells demonstrates that IGFBP-3 import into the nucleus is a tightly controlled process, which displays saturation kinetics, depends on ATP, and involves nuclear β-importins [37]. Putative nuclear IGFBP-3 receptors were not saturated in the insulin-secreting cell lines we studied using concentrations similar to the physiologic concentrations of total IGFBP-3 observed in human serum.
Although insulin at high concentrations can activate the IGF receptor, IGFBP-3 does not bind insulin, so the effects we have reported are insulin receptor-independent. Moreover, the pro-inflammatory cytokines we studied, particularly in combination, have repeatedly been shown to decrease insulin synthesis or release from RIN m5F cells, without increasing expression of insulin receptors or type 1 IGF receptors [38–41]. Although IL-6 has been shown to increase insulin secretion in HIT cells [42], no evidence exists to our knowledge that IL-1β, IFN-γ, TNF-α, or TGF-β1 increase IGF synthesis by or release from RIN and HIT cells. Activation of protein kinase C has been implicated as a major survival pathway in RIN cells [43]; however, we have presented the first evidence that IGFBP-3 can exert its pro-apoptotic action independently of protein kinase C activity (Fig. 4).
The apoptosis induced with exogenous IGFBP-3 appears modest in these transformed cell lines: 3.4% vs. 1.7% under serum-free conditions (Fig. 5). Our exclusive use of transformed cell lines limits the conclusions one can draw about the effects of IGFBP-3 on β-cells in vivo. Transformed insulin-secreting cell lines lack the normal pattern of glucose-stimulated insulin secretion and are often resistant to the induction of apoptosis by cytokines. It is known that these transformed cells lines can secrete significant quantities of IGF-I or -II, which could bind extracellular IGFBP-3, preventing its uptake [16,30]. Nevertheless, multiple experiments presented here support the concept that exposure of insulin-secreting cells to high, paracrine concentrations of IGFBP-3 can result in their apoptotic destruction.
In conclusion, we have presented the first evidence that IGFBP-3 contributes to cytokine-mediated apoptosis of insulin-secreting cells. Further in vivo investigations of this novel pathway for β-cell destruction are warranted to determine the feasibility of IGFBP-3 antagonists, such as IGF-I or specific IGFBP-3 proteases, to preserve residual β-cell mass in the face of autoimmune destruction.
Acknowledgements
The authors wish to thank Ms. Hsiao-Wang Chen, Kurtis J. Haas, Ms. Mish Mizrahi, Ms. Heather L. Tienson, and Daniel Afzali as well as Stephen Carbonniere of the Cytometry Core Facility for their technical assistance. The views expressed herein are solely those of the authors and do not reflect an official position of the Texas State Army Guard.
Footnotes
This work was supported in part by grants from the NIH (Grants T32 HD07512 to M.S., R01 DK47591 and R01 AI40203 to P.C., the Clinical Associate Physician Award and University of Pennsylvania Diabetes Endocrinology Research Center Grant P30 DK19525 to L.K., and K08 DK02876 to R.F.), Glaser Foundation (to M.S.), Lawson Wilkins Pediatric Endocrine Society (Genentech Clinical Scholars Award to R.F.), UCLA Child Health Research Center (to R.F. from NIH Grant P30 HD34610), Howard Hughes Medical Institute (Grant 53000245 to R.F.), Juvenile Diabetes Research Foundation (to P.C.), and a summer research fellowship from The Endocrine Society (to W.D.). Portions of this work were presented in abstract form at The 82nd and 83rd Annual Meetings of The Endocrine Society held June 2000 (Toronto, Canada) and June 2001 (Denver, Colorado).
References
- [1].Vassiliadis S, Dragiotis V, Protopapadakis E, Athanassakis I, Mitlianga P, Konidaris K, et al. The destructive action of IL-1alpha and IL-1beta in IDDM is a multistage process: evidence and confirmation by apoptotic studies, induction of intermediates and electron microscopy. Mediat. Inflamm. 1999;8:85–91. doi: 10.1080/09629359990577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dunger A, Augstein P, Schmidt S, Fischer U. Identification of interleukin-1 induced apoptosis in rat islets using in situ specific labeling of fragmented DNA. J. Autoimmun. 1996;3:309–313. doi: 10.1006/jaut.1996.0042. [DOI] [PubMed] [Google Scholar]
- [3].Wang B, Andre I, Gonzalez A, Katz JD, Aguet M, Benoist C, et al. Interferon-gamma impacts at multiple points during the progression of autoimmune diabetes. Proc. Natl. Acad. Sci. USA. 1997;94:13844–13849. doi: 10.1073/pnas.94.25.13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Delaney CA, Pavlovic D, Hoorens A, Pipeleers DG, Eizirik DL. Cytokines induce deoxyribonucleic strand breaks and apoptosis in human pancreatic islet cells. Endocrinology. 1997;138:2610–2614. doi: 10.1210/endo.138.6.5204. [DOI] [PubMed] [Google Scholar]
- [5].Sanvito F, Nichols A, Herrera PL, Huarte J, Wohlwend A, Vassalli JD, et al. TGF-beta 1 overexpression in murine pancreas induces chronic pancreatitis and, together with TNF-alpha, triggers insulin-dependent diabetes. Biochem. Biophys. Res. Commun. 1995;217:1279–1286. doi: 10.1006/bbrc.1995.2906. [DOI] [PubMed] [Google Scholar]
- [6].Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 1999;20:761–787. doi: 10.1210/edrv.20.6.0382. [DOI] [PubMed] [Google Scholar]
- [7].Blakesley VA, Butler AA, Koval AP, Okubo Y, Le Roith D. IGF-I receptor function: transducing the IGF-I signal into intraacellular events. In: Rosenfeld RG, Roberts CT Jr., editors. Contemporary Endocrinology: The IGF System. Humana Press; Totowa, NJ: 1999. pp. 143–163. [Google Scholar]
- [8].Hill DJ, Petrik J, Arany E, McDonald TJ, Delovitch TL. Insulin-like growth factors prevent cytokine-mediated cell death in isolated islets of Langerhans from pre-diabetic non-obese diabetic mice. J. Endocrinol. 1999;161:153–165. doi: 10.1677/joe.0.1610153. 1999. [DOI] [PubMed] [Google Scholar]
- [9].Bergerot I, Fabien N, Maguer V, Thivolet C. Insulin-like growth factor-I (IGF-I) protects NOD mice from insulitis and diabetes. Clin. Exp. Immunol. 1995;102:335–340. doi: 10.1111/j.1365-2249.1995.tb03786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baserga R, Hongo A, Rubini M, Prisco M, Valentinis B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim. Biophys. Acta. 1997;1332:F105–F126. doi: 10.1016/s0304-419x(97)00007-3. [DOI] [PubMed] [Google Scholar]
- [11].Cohen P, Lamson G, Okajima T, Rosenfeld RG. Transfection of the human insulin-like growth factor binding protein-3 gene into Balb/c fibroblasts inhibits cellular growth. Mol. Endocrinol. 1993;7:380–386. doi: 10.1210/mend.7.3.7683373. [DOI] [PubMed] [Google Scholar]
- [12].Zadeh SM, Binoux M. The 16-kDa proteolytic fragment of insulin-like growth factor (IGF) binding protein-3 inhibits the mitogenic action of fibroblast growth factor on mouse fibroblasts with a targeted disruption of the type 1 IGF receptor gene. Endocrinology. 1997;138:3069–3072. doi: 10.1210/endo.138.7.5380. [DOI] [PubMed] [Google Scholar]
- [13].Rajah R, Valentinis B, Cohen P. Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J. Biol. Chem. 1997;272:12181–12188. doi: 10.1074/jbc.272.18.12181. [DOI] [PubMed] [Google Scholar]
- [14].Gill ZP, Perks CM, Newcomb PV, Holly JM. Insulin-like growth factor-binding protein (IGFBP-3) predisposes breast cancer cells to programmed cell death in a non-IGF-dependent manner. J. Biol. Chem. 1997;272:25602–25607. doi: 10.1074/jbc.272.41.25602. [DOI] [PubMed] [Google Scholar]
- [15].Lalou C, Lassarre C, Binoux M. A proteolytic fragment of insulin-like growth factor (IGF) binding protein-3 that fails to bind IGFs inhibits the mitogenic effects of IGF-I and insulin. Endocrinology. 1996;137:3206–3212. doi: 10.1210/endo.137.8.8754741. [DOI] [PubMed] [Google Scholar]
- [16].Katz LE, Bhala A, Camron E, Nunn SE, Hintz RL, Cohen P. IGF-II, IGF-binding proteins and IGF receptors in pancreatic beta-cell lines. J. Endocrinol. 1997;152:455–464. doi: 10.1677/joe.0.1520455. [DOI] [PubMed] [Google Scholar]
- [17].Chick WL, Warren S, Chute RN, Like AA, Lauris V, Kitchen KC. A transplantable insulinoma in the rat. Proc. Natl. Acad. Sci. USA. 1977;74:628–632. doi: 10.1073/pnas.74.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gazdar AF, Chick WL, Oie HK, Sims HL, King DL, Weir GC, et al. Continuous, clonal, insulin- and somatostatin-secreting cell lines established from a transplantable rat islet cell tumor. Proc. Natl. Acad. Sci. USA. 1980;77:3519–3523. doi: 10.1073/pnas.77.6.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Oie HK, Gazdar AF, Minna JD, Weir GC, Baylin SB. Clonal analysis of insulin and somatostatin secretion and L-dopa decarboxylase expression by a rat islet cell tumor. Endocrinology. 1983;112:1070–1075. doi: 10.1210/endo-112-3-1070. [DOI] [PubMed] [Google Scholar]
- [20].Santerre RF, Cook RA, Crisel RM, Sharp JD, Schmidt RJ, Williams DC, et al. Insulin synthesis in a clonal cell line of simian virus 40-transformed hamster pancreatic beta cells. Proc. Natl. Acad. Sci. USA. 1981;78:4339–4343. doi: 10.1073/pnas.78.7.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Schuller AG, Groffen C, van Neck JW, Zwarthoff EC, Drop SL. cDNA cloning and mRNA expression of the six mouse insulin-like growth factor binding proteins. Mol. Cell. Endocrinol. 1994;104:57–66. doi: 10.1016/0303-7207(94)90051-5. [DOI] [PubMed] [Google Scholar]
- [22].Albiston AL, Herington AC. Cloning and characterization of the growth hormone-dependent insulin-like growth factor binding protein (IGFBP-3) in the rat. Biochem. Biophys. Res. Commun. 1990;166:892–897. doi: 10.1016/0006-291x(90)90894-s. [DOI] [PubMed] [Google Scholar]
- [23].Powell DR, Rosenfeld RG, Baker BK, Liu F, Hintz RL. Serum somatomedin levels in adults with chronic renal failure: the importance of measuring insulin-like growth factor-I (IGF-I) and IGF-II in acid chromatographed uremic serum. J. Clin. Endocrinol. Metab. 1986;63:1186–1192. doi: 10.1210/jcem-63-5-1186. [DOI] [PubMed] [Google Scholar]
- [24].Van Wyk JJ, Graves DC, Casella SJ, Jacobs S. Evidence from monoclonal antibody studies that insulin stimulates deoxyribonucleic acid synthesis through the type I somatomedin receptor. J. Clin. Endocrinol. Metab. 1985;61:639–643. doi: 10.1210/jcem-61-4-639. [DOI] [PubMed] [Google Scholar]
- [25].Perks CM, McCaig C, Clarke JB, Clemmons DR, Holly JM. A non-IGF binding mutant of IGFBP-3 modulates cell function in breast epithelial cells. Biochem. Biophys. Res. Commun. 2002;294:988–994. doi: 10.1016/S0006-291X(02)00569-7. [DOI] [PubMed] [Google Scholar]
- [26].Le Roith D, Werner H, Burguera B, Roberts CT, Jr, Mulroney S, Haramati A. The insulin-like growth factor family of peptides, binding proteins and receptors: their potential role in tissue regeneration. Adv. Exp. Med. Biol. 1992;321:21–28. doi: 10.1007/978-1-4615-3448-8_3. [DOI] [PubMed] [Google Scholar]
- [27].Katz J, Nasatzky E, Werner H, Le Roith D, Shemer J. Tumor necrosis factor alpha and interferon gamma-induced cell growth arrest is mediated via insulin-like growth factor binding protein-3. Growth Horm. IGF Res. 1999;9:174–178. doi: 10.1054/ghir.1999.0101. [DOI] [PubMed] [Google Scholar]
- [28].Butt AJ, Fraley KA, Firth SM, Baxter RC. IGF-binding protein-3-induced growth inhibition and apoptosis do not require cell surface binding and nuclear translocation in human breast cancer cells. Endocrinology. 2002;143:2693–2699. doi: 10.1210/endo.143.7.8876. [DOI] [PubMed] [Google Scholar]
- [29].Franklin SF, Jr., Ferry RJ, Cohen P. Rapid IGF-independent effects of IGFBP-3 on endothelial cell survival. J. Clin. Endocrinol. Metab. 2003;88:900–907. doi: 10.1210/jc.2002-020472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee KW, Liu B, Ma L, Li H, Bang P, Koeffler HP, Cohen P. Cellular internalization of insulin-like growth factor binding protein-3 (IGFBP-3): distinct endocytic pathways facilitate reuptake and nuclear localization. J. Biol. Chem. 2004;279:469–476. doi: 10.1074/jbc.M307316200. [DOI] [PubMed] [Google Scholar]
- [31].Ricort JM, Binoux M. Insulin-like growth factor-binding protein-3 activates a phosphotyrosine phosphatase. Effects on the insulin-like growth factor signaling pathway. J. Biol. Chem. 2002;277:19448–19454. doi: 10.1074/jbc.M200439200. [DOI] [PubMed] [Google Scholar]
- [32].Radulescu RT. Nuclear localization signal in insulin-like growth factor-binding protein type 3. Trends Biochem. Sci. 1994;19:278. doi: 10.1016/0968-0004(94)90004-3. [DOI] [PubMed] [Google Scholar]
- [33].Jaques G, Noll K, Wegmann B, Witten S, Kogan E, Radulescu RT, et al. Nuclear localization of insulin-like growth factor binding protein 3 in a lung cancer cell line. Endocrinology. 1997;138:1767–1770. doi: 10.1210/endo.138.4.5177. [DOI] [PubMed] [Google Scholar]
- [34].Wraight CJ, Liepe IJ, White PJ, Hibbs AR, Werther GA. Intranuclear localization of insulin-like growth factor binding protein-3 (IGFBP-3) during cell division in human keratinocytes. J. Invest. Dermatol. 1998;111:239–242. doi: 10.1046/j.1523-1747.1998.00258.x. [DOI] [PubMed] [Google Scholar]
- [35].Liu B, Lee HY, Weinzimer SA, Powell DR, Clifford JL, Kurie JM, et al. Direct functional interactions between IGFBP-3 and RXR-alpha regulate transcriptional signalling and apoptosis. J. Biol. Chem. 2000;275:33607–33613. doi: 10.1074/jbc.M002547200. [DOI] [PubMed] [Google Scholar]
- [36].Chertow BS, Goking NQ, Driscoll HK, Primerano DA, Matthews KA. Effects of all-trans-retinoic acid (ATRA) and retinoic acid receptor (RAR) expression on secretion, growth, and apoptosis of insulin-secreting RINm5F cells. Pancreas. 1997;15:122–131. doi: 10.1097/00006676-199708000-00003. [DOI] [PubMed] [Google Scholar]
- [37].Schedlich LJ, Le Page SL, Firth SM, Briggs LJ, Jans DA, Baxter RC. Nuclear import of insulin-like growth factor-binding protein-3 and -5 is mediated by the importin beta subunit. J. Biol. Chem. 2000;275:23462–23470. doi: 10.1074/jbc.M002208200. [DOI] [PubMed] [Google Scholar]
- [38].Campbell IL, Harrison LC, Colman PG, Papaioannou J, Ashcroft RG. Expression of class I MHC proteins on RIN-m5F cells is increased by interferon-gamma and lymphokine-conditioned medium. Diabetes. 1986;35:1225–1228. doi: 10.2337/diab.35.11.1225. [DOI] [PubMed] [Google Scholar]
- [39].Campbell IL, Oxbrow L, Harrison LC. Interferon-gamma: pleiotropic effects on a rat pancreatic beta cell line. Mol. Cell. Endocrinol. 1987;52:161–167. doi: 10.1016/0303-7207(87)90109-2. [DOI] [PubMed] [Google Scholar]
- [40].de-Mello MA, Flodstrom M, Eizirik DL. Ebselen and cytokine-induced nitric oxide synthase expression in insulin-producing cells. Biochem. Pharmacol. 1996;52:1703–1709. doi: 10.1016/s0006-2952(96)00520-5. [DOI] [PubMed] [Google Scholar]
- [41].Hong TP, Andersen NA, Nielsen K, Karlsen AE, Fantuzzi G, Eizirik DL, et al. Interleukin-18 mRNA, but not interleukin-18 receptor mRNA, is constitutively expressed in islet beta-cells and up-regulated by interferon-gamma. Eur. Cytokine Network. 2000;11:193–205. [PubMed] [Google Scholar]
- [42].Shimizu H, Ohtani K, Kato Y, Mori M. Interleukin-6 increases insulin secretion and preproinsulin mRNA expression via Ca2+-dependent mechanism. J. Endocrinol. 2000;166:121–126. doi: 10.1677/joe.0.1660121. [DOI] [PubMed] [Google Scholar]
- [43].Sanchez-Margalet V, Lucas M, Solano F, Goberna R. Sensitivity of insulin-secreting RIN m5F cells to undergoing apoptosis by the protein kinase C inhibitor staurosporine. Exp. Cell Res. 1993;209:160–163. doi: 10.1006/excr.1993.1297. 1993. [DOI] [PubMed] [Google Scholar]