

Abstract
Past autism research has often been dedicated to tracing the causes of the disorder to a localized neurological abnormality, a single functional network, or a single cognitive-behavioral domain. In this review, I argue that autism is a ‘distributed disorder’ on various levels of study (genetic, neuroanatomical, neurofunctional, behavioral). ‘Localizing’ models are therefore not promising. The large array of potential genetic risk factors suggests that multiple (or all) emerging functional brain networks are affected during early development. This is supported by widespread growth abnormalities throughout the brain. Interactions during development between affected functional networks and atypical experiential effects (associated with atypical behavior) in children with autism further complicate the neurological bases of the disorder, resulting in an ‘exponentially distributed’ profile. Promising approaches to a better characterization of neural endophenotypes in autism are provided by techniques investigating white matter and connectivity, such as MR spectroscopy, diffusion tensor imaging (DTI), and functional connectivity MRI. According to a recent hypothesis, the autistic brain is generally characterized by ‘underconnectivity’. However, not all findings are consistent with this view. The concepts and methodology of functional connectivity need to be refined and results need to be corroborated by anatomical studies (such as DTI tractography) before definitive conclusions can be drawn.
Keywords: Autism, brain imaging, connectivity, functional networks
INTRODUCTION
A conventional textbook approach to development is to first review mature brain organization in depth and then consider how the brain might have attained this mature state through developmental change. The approach is defensible on pragmatic grounds. Cognitive neuroscientists understand the adult brain much better than the developing brain and to venture out from safer ground to unsafe territory is good common sense. However, if the chronology of research is the inverse of actual biological development, our sense of causality may suffer. We may be tempted to believe that development works in certain ways because it is bound to reach the adult state. This implies that we could explain development by its endpoint. Such a teleological approach is, however, misleading. From a biological perspective, it becomes clear that development explains the mature state, not the inverse. For example, regional functional specialization in the brain emerges from the interaction of genetic and epigenetic factors, such as differential gene expression, activity of axonal afferents, and activity in response to environmental stimuli (O’Leary and Nakagawa 2002), i.e., processes that are present during development. It is this course of developmental events that will ultimately allow a brain region, such as inferior frontal cortex, to participate in a specific set of functions, such as language (for detailed discussion, see Müller, 2009).
Given this approach, developmental disorders cannot be understood using adult clinical models of lesion-symptom correspondences. The notion of ‘residual normality’, according to which a localized damage removes well-defined components from a functional system, leaving other components intact, has been applied to the mature nervous system, with some – probably debatable – success. Residual normality cannot, however, apply to the developing brain because interactive effects between brain regions and between functional systems are a known fact (see Thomas and Karmiloff-Smith 2002 for an extensive debunking). Therefore, it is not surprising that damage to left inferior frontal cortex (Broca’s area) or indeed the entire left hemisphere occurring early in life does not result in persistent language impairment, but may affect visuospatial functions (possibly through a developmental process called “crowding”), although these functions are not associated with this piece of cortex in the adult brain (Lidzba et al. 2006). A localized lesion in the developing brain will thus have a ‘non-local’ effect on the emerging cognitive system. This implies that developmental disorders, even those caused by relatively ‘simple’ localized damage, will be complex because of their impact on interactive processes across individual brain regions and functional systems. The complexity of the disorder to be discussed in this review is exponentially greater because it is not associated with localized damage, but primarily caused by the effects of multiple genetic risk factors on many or all functional brain systems. Autism, the disorder in question, can therefore be characterized as a ‘distributed’ disorder on all levels of study, from genes to anatomic brain development to functional brain organization and to behavioral sequelae. The purpose of this review is to consider the implications of this distributed nature for the study of autism, in particular the use of neuroimaging.
GENES
Contrary to earlier accounts of autism as caused by inappropriate parenting, it has been established for several decades that the disorder is neurological in nature and heavily based on genetic factors. Twin studies suggest a concordance for autism in monozygotic twins of about 70%, whereas concordance in dizygotic twins is below 10% (Folstein and Rosen-Sheidley 2001). Genome screening studies have identified a great number of loci that may be associated with risk for autism. A recent review by Vorstman and colleagues (2006) shows that linkage and association studies have identified loci on almost every single chromosome (with the exception of chromosomes 8, 9, 14, 18, and Y). The additional inclusion of cytogenetic evidence, which relates chromosomal aberrations to autistic symptomatology, implies that even genes on those latter chromosomes may be associated with some risk factors for autism.
Nonetheless, autistic disorder can be attributed to single defective genes in some cases. For example, fragile X syndrome is often associated with autistic disorder (Filipek 2005), possibly in over 50% of male fragile X patients (Clifford et al. 2007). Fragile X syndrome is caused by a mutation (excessive repeats) in the FMR1 gene on the long arm of the X chromosome (Terracciano et al. 2005). However, as argued by Belmonte & Bourgeron (2006), despite being accounted for by a single-gene mutation, fragile X syndrome nonetheless needs to be considered a network disorder on the neurological level. Furthermore, while FMR1 mutation may present a useful approach to studying one etiological pathway that frequently results in an autistic phenotype, it accounts for only a very small percentage of the general population with autism (Bailey et al. 1993).
The picture is further complicated by evidence for non-genetic factors that may additionally contribute to the risk. Among these may be viral infection (Tanoue et al. 1988) or neurotoxic exposure in utero (Edelson and Cantor 1998). It must also be noted that autism is often associated with other medical disorders (Ek et al. 1998), such as tuberous sclerosis (Gillberg et al. 1994). Using bioinformatics techniques, Herbert and colleagues (2006) recently showed an overlap between environmentally responsive genes and genes on linkage regions found in autism studies. This approach highlights the possibility that genetic and environmental risk factors may not be simply additive, but interactive; i.e., some genetic risk factors in ASD may make the fetus or child more susceptible to environmental toxins.
BRAIN ANATOMY
Given the historical bias towards an adult localizationist approach of lesion-symptom correspondences, mentioned earlier, it is not surprising that attempts to relate autism to damage or maldevelopment of a specific brain structure or a single functional network have been made. Some earlier reports based on structural MRI findings focused heavily on the cerebellum, specifically the posterior vermis (Courchesne et al. 1988). Since autism can be considered a specific impairment of socio-affective processing, it is further not surprising to see models such as the “amygdala theory of autism” (Baron-Cohen et al. 2000). This theory is based both on indirect evidence, such as the known involvement of the amygdala in social behaviors, as well as direct evidence for anatomical and functional compromise of the amygdala in autism (cf. Sweeten et al. 2002). Among the latter are findings of increased cell packing density in the medial temporal lobe (including the amygdala), which was observed in some postmortem studies (see reviews in Bauman and Kemper 2005; Palmen et al. 2004), but not in a recent study by Schumann and Amaral (2006). There is also evidence from some structural imaging studies of increased amygdala volume in autism (Schumann et al. 2004 and review in Brambilla et al. 2003), which again is not replicated in all studies (e.g., Palmen et al. 2006). Reduced amygdala activation has further been seen in a few functional imaging studies related to emotional processing (Baron-Cohen et al. 1999; Critchley et al. 2000), whereas some more recent fMRI studies of socio-emotional processing suggest normal levels of amygdalar activity (Pierce et al. 2004; Piggot et al. 2004). Intriguing correlations have also been found between amygdala volume and performance on socio-emotional tests (Dziobek et al. 2006).
The discussion of the amygdala theory of autism serves only as an example of a potential localizing model. As noted, evidence implicating the amygdala is surprisingly mixed. However, even if consistent evidence for some type of cellular or functional compromise of the amygdala could be identified, such evidence would not necessarily support a local model of autism. Indeed, considering the complex and intimate connectivity between amygdala and numerous subcortical, cerebral cortical, and limbic regions (LeDoux 1995), locally restricted compromise would be unexpected.
The view of distributed impairment in autism is supported by the structural imaging literature, which presents varied (and often not replicated) results, including abnormal hemispheric asymmetries in language-related regions (Herbert et al. 2005), and enlarged ventricles (Piven et al. 1995). Abnormalities have been reported for many regions, such as the frontal (Herbert et al. 2004) and parietal lobes (Courchesne et al. 1993), the cingulate region (Haznedar et al. 1997), the corpus callosum (Vidal et al. 2006), the hippocampus and amygdala (Otsuka et al. 1999), the thalamus (Tsatsanis et al. 2003), the basal ganglia (Hollander et al. 2005), and the brainstem (Rodier 2002).
Some inconsistencies in the structural MRI literature may be attributed to methodological differences. Besides these, the diversity of the findings could relate either to heterogeneity within the population meeting diagnostic criteria for autism or to the diffuse nature of brain involvement. Most likely it reflects both of these. The heterogeneity of autism is widely accepted by those who study the disorder. Today many researchers in the field therefore prefer to use the term ‘autism spectrum disorders’. Biological heterogeneity is not unexpected, given that the disorder is diagnosed in terms of a consensus-based set of behavioral symptoms. In clinical and diagnostic practice, autism is therefore a behavioral concept, while at the same time there is general consensus that it is a primarily gene-based neurobiological disorder. It is not likely that the behavioral concept of autism can be equated to a single pathological entity on the neurobiological level. As already established in the previous section, it appears that the genetic risk factors at work differ from individual to individual within the autistic population. Similar heterogeneity appears to be true with regard to brain anatomy. For example, some individuals with autism have very small cerebella (Courchesne et al. 1988), some have small parietal lobes (Courchesne et al. 1993), and some have cellular anomalies in frontal cortex (Bailey et al. 1998).
However, there is also evidence that brain anatomical involvement within autism spectrum disorders is not only varied, but also diffuse. This evidence comes from studies attempting to identify age-dependent changes in the autistic brain. (Note that very little truly longitudinal evidence on brain growth in ASD is currently available). Courchesne and colleagues (2001) studied 60 boys with autism aged 2–16 years, using structural MRI, and found abnormal brain growth patterns in comparison to matched typically developing children. Two to four year-old children with autism had significantly bigger brains than their controls, with regard to both gray and white matter. This was not true for children ages 10 years and older, who tended to have reduced gray and white matter volumes. These findings suggest early overgrowth – more recently corroborated by other groups (Hazlett et al. 2005; Sparks et al. 2002) – followed by reduced white matter growth and atypically early loss of gray matter volume in autism. Interestingly, evidence of these age-dependent effects has been found for all four forebrain lobes, with the possible exception of the occipital lobe (Carper et al. 2002). Diffuse brain growth abnormalities may be related to atypical profiles of brain growth factors found in neonates who were later diagnosed with autism (Nelson et al. 2006).
The evidence of diffuse or widespread growth anomalies is relevant, not only because it may explain some of the inconsistencies in the regional findings reviewed above, but more so because it indicates that autism is very likely to reflect impairment of many, maybe almost all, brain networks.
FUNCTIONAL BRAIN ORGANIZATION
In this section, I will consider only task-induced activation studies, such as oxygen-15 positron emission tomography (PET) and functional MRI (fMRI), not studies examining brain function in the absence of an experimental challenge (as for example, PET studies of regional glucose metabolism at rest). Many of these functional mapping studies of autism have focused on domains of known impairment, such as face perception and theory of mind.
It must be noted that functional neuroimaging evidence on autism pertains almost exclusively to individuals at the higher-functioning end of the spectrum. It is beyond the scope of this review to examine the potential impact of diagnostically heterogeneous samples, which may combine participants with autism, Asperger’s disorder, and even PDD-NOS (American Psychiatric Association 2000), on the results of each of the studies discussed here.
In functional mapping studies, many factors in addition to those discussed above in the context of anatomical studies (such as heterogeneity in patient samples) will affect the detected regional patterns of effects (activation, group difference). Among those additional factors are the specifics of the experimental task and the control condition, the timing and ordering of trials on each condition, the details of data processing (e.g., motion correction, spatial smoothing), and the statistical power (depending heavily on sample sizes, but also on the number of data points within each participant). Given all these methodological sources of variance, it is not surprising that non-replication in studies of brain function are even more common than in anatomical studies.
A field of intense functional imaging research concerns face perception and the fusiform gyrus. Reduced activity in the fusiform gyrus during face perception has been observed in several autism studies (Hubl et al. 2003; Pierce et al. 2001; Schultz et al. 2000), including a single case study (Grelotti et al. 2005), with additional reports of reduced fusiform activity related to the processing of emotional facial expressions (Critchley et al. 2000; Hall et al. 2002; Piggot et al. 2004). Interestingly however, two studies of face perception in autism (Hadjikhani et al. 2004; Pierce et al. 2004) found normal levels of activity in the fusiform gyrus. Differential fusiform activity in the cited studies could relate to gaze fixation in individuals with ASD. In a study by Dalton and colleagues (2005), activity in the right fusiform gyrus was correlated with the time of fixation on the eyes of presented faces. This effect was seen in individuals with ASD for strange and familiar faces, and with neutral or emotional expressions.
Dalton and colleagues (2005) suggest that gaze fixation may be a “proximal cause” of fusiform hypoactivation observed in many studies. However from a developmental perspective, gaze fixation may be considered more than a proximal cause. Avoidance of eye contact is one of the core diagnostic criteria for autism (American Psychiatric Association 2000; Baird et al. 2000). This implies that children with autism look at faces much less frequently than typically developing children, as supported by studies in adolescents and adults (Klin et al. 2002), which specifically indicate reduced fixation on the eyes (Dalton et al. 2005; Pelphrey et al. 2002). Electrophysiological findings from young children with autism suggest that such anomalies are present early in development (Grice et al. 2005; cf. also Senju et al. 2005). These results imply diminished domain-specific stimulation in autism.
Such evidence is important with regard to the known effects of experience (or absence thereof) in the fine-tuning of cerebral cortical functional organization during development (Kandel et al. 2000), as demonstrated in animal (O’Leary and Nakagawa 2002; Sur and Leamey 2001) and human studies (Elbert et al. 1995; Sadato et al. 2002). Atypical activity seen in adolescents or adults, who have lived with autism for many years, may not provide an explanation of behavioral impairments (face processing deficits), but rather reflect the brain’s normal plasticity in response to atypical perceptual input that is due to a reduced tendency in children with autism to look at faces (see Müller, 2007 for detailed discussion). Caution is therefore warranted when attributing causality to functional imaging findings. Indeed, similar reservations apply to anatomical findings, such as atypical volumetric asymmetries in perisylvian cortex potentially related to language impairment (Herbert et al. 2005).
Given the many potential sources of non-replication in functional mapping studies, it is remarkable that there may be relative consistency with regard to a few findings. For example, in the very first functional mapping study using oxygen-15 PET in ASD, Happé and colleagues (1996) found that activity in medial frontal cortex associated with the comprehension of stories requiring ‘theory of mind’ in typical adults was displaced inferiorly in five adults with Asperger’s disorder. While this specific displacement was not replicated, it is remarkable that at least one other study tapping into ‘theory of mind’ by means of a very different task paradigm also identified abnormality in the medial prefrontal cortex. In this study by Castelli and colleagues (2002) participants watched moving triangles on a screen in ways that either evoked attribution of mental states to the triangles (when they appeared to be kissing, for example) or did not (when they were simply bouncing around in straight trajectories). Attribution of mental states was associated with activation in medial frontal cortex in typical adults, which was significantly reduced in participants with ASD. Two other brain areas showed concordant effects: the superior temporal sulcus (STS) and a site close to the amygdala. These latter regions are further examples of partial consistency in the functional mapping of social cognition in autism (see review in Pelphrey et al. 2004).
The examples from studies of social cognition in ASD are encouraging because they suggest some moderate degree of consensus on brain regions that may be functioning abnormally in autism. However, a caveat regarding potential developmental and experience-driven effects of relatively normal plasticity, as brought up in the discussion on face processing, applies here as well. It cannot be ruled out that individuals with ASD activate the mentioned regions less because they process stimuli that elicit mentalizing in neurotypical individuals in different, non-mentalizing ways. The underlying causes of this may not at all be dysfunction of medial frontal cortex, STS, or the amygdala. For a hypothetical (but not entirely improbable) example, early-onset impairments in mentalizing may be related to sensorimotor integration and action understanding. It has been argued that the ‘mirror neuron system’ provides neural mechanisms crucial for imitation, but also for relating another person’s action to one’s own identical action. This implies that the roots for the ability to mentalize (assume that another person’s behavior or facial expression implies similar mental states as those felt during one’s own corresponding behavior or facial expression) may be found in the mirror neuron system (cf. Iacoboni et al. 2005). Early-onset dysfunction of this system in ASD, as proposed by Williams and coauthors (2001), may result in reduced mentalizing abilities, which will in turn affect the brain regions typically participating in mentalizing processes. In this model, medial prefrontal cortex, STS, and amygdala might only be secondarily affected because the autistic brain rarely engages in a type of activity they would typically be involved in.
The above scenario relates to the theme of this review. If the role of a brain region and the relevance of its atypical function in ASD can only be understood when all interactions with other functional systems during development are taken into account, it becomes obvious that neural compromise in ASD is distributed. Even if at some very early point of brain development the impairment was local, brain maturational and activity-related interaction between functional systems during development would result in a more distributed profile of regional effects. However as discussed above, the scattered picture of genetic risk factors makes a pathogenic starting point of local involvement rather unlikely. Instead, there is probably a distributed ontogenetic starting point affecting several emerging brain regions and functional systems (e.g., neurotrophic and transmitter systems, such as serotonin; Chugani 2004) and each of these will in turn affect additional regions and functional systems throughout development. A developmental disorder like autism may thus be ‘exponentially distributed’.
It is clear that functional neuroimaging in older children or adults cannot directly elucidate the anticipated distributed nature of autism. Nonetheless, some puzzling results in fMRI studies of autism may appear in a different light from a perspective of ‘exponential distribution’. This applies in particular to studies that do not focus on a few regions of interest (as, for example, Castelli et al. 2002), but look for statistical effects throughout the brain. A study by Baron-Cohen and colleague (1999) may serve to illustrate the point. This group presented six adults with autism and 12 control participants with sections of faces showing eyes and eyebrows. In one condition, participants had to make mental state judgments by selecting one of two options underneath the stimulus (e.g., “unconcerned – concerned”). In a control condition, they had to choose a gender for the same type of stimuli. Comparing the two conditions, the investigators found widespread bilateral activation for the mental attribution task in frontal, parietal, and temporal lobes in the control group. Interestingly, although the autism group also showed activation on this comparison, there was only limited overlap between groups in left premotor cortex and Broca’s area. Most regions of activation for the control group showed no effects in the autism group and most activation clusters in the autism group showed no effects in the control group. These puzzling results could be attributed to problems with the task paradigm, such as a verbal confound (which may explain effects in left inferior frontal cortex). However, they may also reflect an unexpected, though truly interesting, finding. At least to some types of stimuli, atypical response of the autistic brain is not limited to a few local spots, but affects large portions of cerebral cortex, subcortex, and cerebellum.
Broadly atypical response can even occur when stimulus and task are exceedingly simple (not complex and socio-affectively charged as in the experiment by Baron-Cohen [1999]). In our study (Müller et al. 2001) of repetitive index finger button pressing in response to a simple visual stimulus (a dot appearing on the outline of a hand), extensive parts of the brain in all four cerebral lobes, basal ganglia, and cerebellum showed significant group differences. The aim of this study had been to confirm that the autistic brain functions normally on a very simple motor task. This expectation proved wrong: The brains of individuals with autism may be organized atypically even with regard to rather elementary processes.
It must be noted, however, that some studies, in particular those not examining autism fMRI findings on a single-subject level, have shown similar regional patterns of activation in ASD and in controls. For example, Just and colleagues (2004) found activation in left premotor, inferior frontal, superior temporal, and parieto-occipital cortex associated with sentence comprehension both in autism and control groups. However, they did identify reduced functional connectivity between various cerebral cortical regions of interest in ASD. The importance of connectivity studies will be discussed in detail in the final section.
BEHAVIOR
This review focuses on the neurobiology of autism and the discussion of behavioral symptomatology will thus be brief. Diagnostic criteria for autistic disorder in the DSM-IV (American Psychiatric Association 2000) relate primarily to social behavior and language. However, examples of criteria such as hand flapping are not obviously social in nature and instead appear to indicate problems in motor or proprioceptive systems. Furthermore, there are many other behavioral characteristics that are not included in the diagnostic criteria and may not be as consistently observed, but nonetheless underscore the impression of a distributed profile, i.e., one that affects numerous functional domains. Some of these characteristics pertain to additional impairments. For example, motor-related deficits have been identified in ASD, relating to a variety of aspects, such as dexterity (Ghaziuddin and Butler 1998), planning (Mari et al. 2003; Schmitz et al. 2003), posture (Minshew et al. 2004), gait (Hallett et al. 1993), and learning (Mostofsky et al. 2000). This catalogue implies that many parts of the motor networks may be affected, including premotor cortices, cerebellum, and basal ganglia.
Many studies have also shown that ASD is associated with attention deficits (for review, see Allen and Courchesne 2001). These go beyond joint attention (Bruinsma et al. 2004), which could be considered part of social cognition, and include slowed spatial orienting (Townsend et al. 1996) and impaired shifting of attention between auditory and visual modalities (Courchesne et al. 1994). The latter finding may relate to another set of results suggesting executive impairments (as reviewed in Hill 2004).
While social, linguistic, motor, attention, and executive domains are typically impaired in ASD, there are a few types of task at which individuals with autism tend to excel. One example is visual search. O’Riordan and colleagues (2004; 2001) showed that children and adults with autism perform faster and with greater accuracy – compared to matched controls – on simple visual search tasks, especially single-feature searches with moderate numbers of distracters. Our own studies (Brenner et al. 2005) further suggest that enhanced performance in ASD is not the result of greater effort; on the contrary, reduced response times in participants with ASD were associated with reduced pupil dilation, which is a physiological measure of cognitive activity and effort (Marshall in press). Individuals with ASD have also been found superior on a rather complex visual search task, the Embedded Figures Test (Jolliffe and Baron-Cohen 1997), and on the visuoconstructive Block Design (Caron et al. 2006). These findings are intriguing, given evidence for atypical spatial attention and oculomotor functions in ASD (as reviewed in Brenner et al., 2007).
For the purposes of the present review, two general conclusions can be drawn. First, on the cognitive-behavioral level, impairment and atypical function can be observed in many different functional systems in the social, linguistic, motor, attentional, executive, and visuospatial domains, making ASD a behaviorally distributed disorder. Second, in some domains atypical function is expressed partly in deficient, partly in superior performance compared to typically developing control participants. Besides visuospatial processing discussed above, partially superior performance may be found for auditory discrimination (O’Riordan and Passetti 2006).
INVESTIGATING AUTISM AS A DISTRIBUTED DISORDER
The basic conclusion from the above review is very simple: Autism cannot be described and much less explained as a localized defect, but needs to be modeled as a network disorder. This has several implications: (1) Any local findings, such as atypical cellular organization, reduced volume, or lack of expected activation, needs to be viewed in the context of the interaction of the region in question with other regions during development, as discussed above. (2) In addition to local findings, research on autism needs to examine the integrity of interregional connectivity. This can be done in a variety of ways, including the study of (a) white matter volume and integrity, (b) the study of anatomical fiber tracts, and (c) the study of functional cooperation between brain regions (functional connectivity).
The single most replicated finding of white matter volume decrease in ASD concerns the corpus callosum. While reduced callosal size has been observed repeatedly, regional patterns within the callosum are less established. A recent study by Vidal and colleagues (2006) suggests that in children with autism, reductions of callosal thickness are most pronounced in three of its sections, an anterior section in the genu, which connects orbitofrontal cortices of the two hemispheres, a midsection in the body, which connects perirolandic somatosensory and motor regions, and a posterior region in the splenium, which connects parahippocampal and extrastriatal visual cortices. Some of the region-specific findings are intriguing – as, for example, the anterior finding with respect to the known role of orbitofrontal cortex in socio-affective functions (Adolphs 2003). More generally, callosal reduction suggests impaired interhemispheric connectivity. As mentioned before, one should be cautious not to view callosal findings in older children and adults as evidence explaining atypical interhemispheric processing. To be sure, the findings might reflect early-onset abnormalities as part of gene-driven growth defects. Yet, the predominant effects of these growth defects have been described as early overgrowth (Courchesne et al. 2001), but not volume reduction.
In an alternative and more likely scenario, therefore, callosal reduction may be primarily driven by inefficient intra-hemispheric organization of emerging functional networks. In other words, gray matter growth abnormalities mentioned above are expected to affect the layered architecture of cortex locally and to reduce its processing efficiency. Early white matter overgrowth may provide the developing brain with abundant substrates for intrahemispheric signal transfer. However, such overgrowth probably reflects non-selective and insufficiently organized connectivity, which would negatively affect the fine-tuning of functional networks. Local (gray matter) organization and network connectivity will further be affected by the mismatched timing of maturational events and experiential effects. In the typical brain, initial growth and subsequent regression (neuronal loss, synaptic pruning) are timed in ways that allow activity and experience to support the organization of functional networks (Kandel et al. 2000). The growth profiles in autism do not seem to support such synergy between maturation and experience. Callosal volume reduction may be largely due to the repercussions of such a mismatch between brain growth and experience.
Volumetric MRI studies are limited to determining white matter (ab)normality based solely on size. Other MR modalities, which have been applied to ASD in a few studies, can assess white matter integrity regardless of size. One study by Hendry and colleagues (2006) found increased transverse (T2) relaxation times of white matter in parieto-occipital and some prefrontal regions, suggesting abnormally high water content. A number of studies have used MR spectroscopy (MRS), which detects various brain metabolites based on chemical resonance shift. Most of these studies examined gray matter or measured from large voxels that included both gray and white matter. Friedman and coworkers (2006) found that N-acetylaspartate (NAA) and Myo-inositol – considered markers of neuronal and membrane integrity, respectively – were reduced in the white matter of children with autism around age 4 years. This difference was, however, only found in comparison to typically developing children, not to those with developmental delay, suggesting that the finding is not specific to autism. Diagnostically more specific findings, such as reduced NAA, were limited to gray matter. Another MRS study of white matter in slightly older children with autism failed to detect reduced NAA in comparison to typical controls (Fayed and Modrego 2005). There is thus currently no clear evidence of dramatic chemical white matter compromise in ASD.
As mentioned, MRS detects only a limited set of metabolites and the above null findings are far from conclusive. Another approach to the study of white matter is provided by diffusion imaging, an MRI modality that takes advantage of the effects of the movement of water molecules on magnetic resonance. Since axonal membranes in white matter prevent water molecules from diffusing freely in all directions (isotropically), fractional anisotropy, which can be measured in diffusion-tensor imaging (DTI), reflects the integrity and organization of axons. In one study applying this technique in male adolescents with autism, Barnea-Goraly and colleagues (2004) found widespread reductions in fractional anisotropy in a comparison with matched control participants. Some of the effects found in all four lobes of the cerebrum were consistent with previous findings from other techniques, such as reductions in anterior portions of the corpus callosum and in ventromedial prefrontal cortex, whereas others in occipital and pericentral regions were less expected. DTI can also map out long-range fiber tracts between brain regions through computation of the predominant direction of diffusion in each voxel (Ramnani et al. 2004). While this technique offers great promise for the study of developmental disorders, little evidence relevant to ASD is currently available.
Interregional connectivity in autism has instead been inspected using an off-shoot of functional MRI, commonly called functional connectivity MRI (fcMRI). Like conventional fMRI, fcMRI is based on the blood-oxygenation level dependent (BOLD) signal, which indirectly relates to local neuronal activity (Logothetis and Pfeuffer 2004). Whereas fMRI looks at the variance in BOLD time series that can be explained by task designs (“activation”, defined as greater signal during an experimental compared to a control task), fcMRI is based on interregional cross-correlations of the BOLD signal in the low-frequency domain, typically below 0.1 Hz (Cordes et al. 2001). These correlations may reflect low-frequency fluctuations in local field potentials (Leopold et al. 2003). Applications of fcMRI have quite impressively shown this technique’s potential to map out complex distributed functional networks, such as the motor network (Biswal et al. 1995) or perisylvian language areas (Hampson et al. 2002). Interestingly, interhemispheric BOLD correlations, typically seen between homotopic areas (e.g., left and right superior temporal cortex), were disrupted in patients with callosal agenesis (Quigley et al. 2003), further suggesting that these correlations are indirectly linked to anatomical connectivity.
The number of fcMRI studies of autism currently remains small. A frequently cited model of neurofunctional organization in autism derived from fcMRI studies is the ‘underconnectivity theory’, as put forth by Just and colleagues (2004) in the context of a study on sentence comprehension. In this study, BOLD signal cross-correlations associated with task performance (determining the agent or recipient in a sentence by pressing a button) was found to be slightly reduced (though generally present) between a number of cortical regions of interest (Fig. 1). Largely consistent findings have been reported in additional studies by Just and colleagues on verbal working memory (Koshino et al. 2005), semantic judgments of sentences (Kana et al. 2006), and executive processing on the Tower of London task (Just et al. 2006). Evidence from PET studies appears consistent with the underconnnectivity theory. In an early PET study, Horwitz and colleagues (1988) found that positive correlations in glucose metabolic rates between frontal and parietal regions, as seen in a group of typical adults, were reduced in men with autism. Castelli et al. (2002), in their theory-of-mind PET activation study described earlier, found regional blood flow correlations between extrastriate cortex and superior temporal sulcus reduced in adults with autism.
Fig. 1.
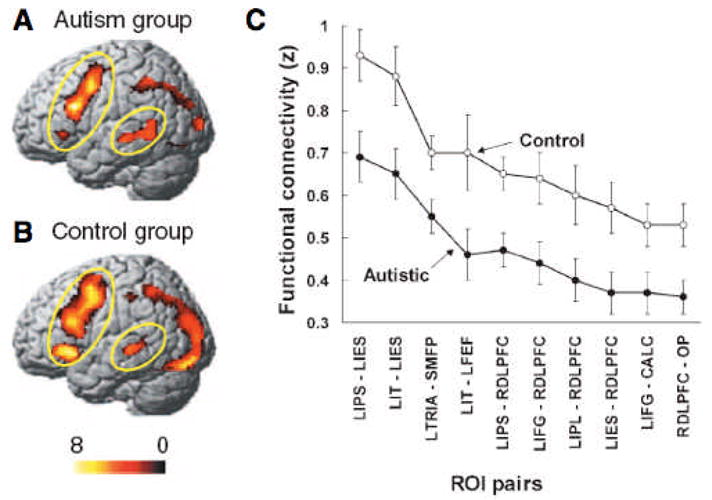
Activation effects for sentence comprehension in autism group (A) and control group (B), showing overall reduced effects in the former. (C) Correlation of mean time series between diverse cortical regions of interest (ROIs) shows generally reduced functional connectivity in the autism group. Abbreviations: L = left; R = right; CALC = calcarine fissure; DLPFC = dorsolateral prefrontal cortex; FEF = frontal eye field; IES = inferior extrastriate cortex; IFG = inferior frontal gyrus; IPL = inferior parietal lobe; IPS = intraparietal sulcus; IT = inferior temporal gyrus; TRIA = pars triangularis; OP = occipital pole; SMFP = superior medial frontal paracingulate gyrus. From Just et al. (2004).
In the above mentioned study by Just and colleagues (2006) on executive function, fcMRI findings were strengthened by correlations with structural and diagnostic measures. Thus frontoparietal connectivity was correlated positively with the size of the anterior portion (genu) of the callosum and negatively with the total score on the Autism Diagnostic Observation Schedule (Lord et al. 2001). The callosal finding is consistent with results from Vidal et al. (2006) discussed earlier. Taken together, these results may indicate that inefficient functional connectivity between cortical regions is linked to reduced callosal volume as well as greater symptom load.
It should be noted, however, that the concept of functional connectivity is not well defined – or “elusive” in the words of Horwitz (2003) – and that methodological decisions heavily impact results. Functional connectivity has been defined as “observed temporal correlations between spatially remote neurophysiological events” (Friston et al. 1993; see also Rippon et al. 2006). While the conventional dichotomy with effective connectivity, defined as “the influence one neural system exerts over another” (Friston et al. 1993), is conceptually clear, the lines between the two may be blurred in actual applications and due to a large number of differences in methodological approaches (Horwitz 2003).
One conceivable confound in autism fMRI studies is head motion. In activation studies, a result according to which activation effects are simply weaker or absent – with little inverse effects (activation being stronger than normal) in other parts of the brain – needs to be treated with great caution, as such differences may simply be the effect of head motion, which will result in noisier BOLD time series. An analogous argument can be made for fcMRI studies.i If time series in one region of interest are slightly noisier in an autism group than in a control group, correlations of time series across regions can also be expected to be lower. It is therefore important to consider data on detected motion.
Even if there are no significant differences in head motion, alternative explanations that do not relate to connectivity may be possible. Many early fcMRI studies (e.g., Biswal et al. 1995) were acquired in the resting state. Even in this state, uncontrolled cognitive processing might drive interregional correlations. This possibility is much greater, however, when data are acquired during task performance. If interregional correlations are reduced, for example, during a sentence comprehension task, the argument could be made that such finding is still related to activation, rather than functional connectivity. The reasons for this are related to our incomplete knowledge of functional differentiation in autistic cerebral cortex. Very few studies to date have examined autism fMRI data on a single-subject level. In one study briefly mentioned before (Müller et al. 2001), we found that what appeared to be slightly reduced activation in perirolandic regions during simple finger movement on a groupwise analysis could be explained in qualitatively different ways after inspecting results from single participants. These suggested that the reasons for the group finding were twofold. First, activation maps tended to be more distributed and often scattered in autistic cortex (Fig. 2). For fcMRI analyses, this implies that activity in a seed volume (or region of interest) will be less coherent across different voxels (or volume elements) within the seed. Incoherence of time series within a region will likely reduce correlations of time series between regions. Secondly, there was greater variability of activation maps across individuals with autism (compared to control individuals). To date, any study that examined single-subject data has supported this latter finding of atypical individual variability, which is also reasonable on theoretical grounds given the genetic and neuroanatomical findings discussed in earlier sections.
Fig. 2.
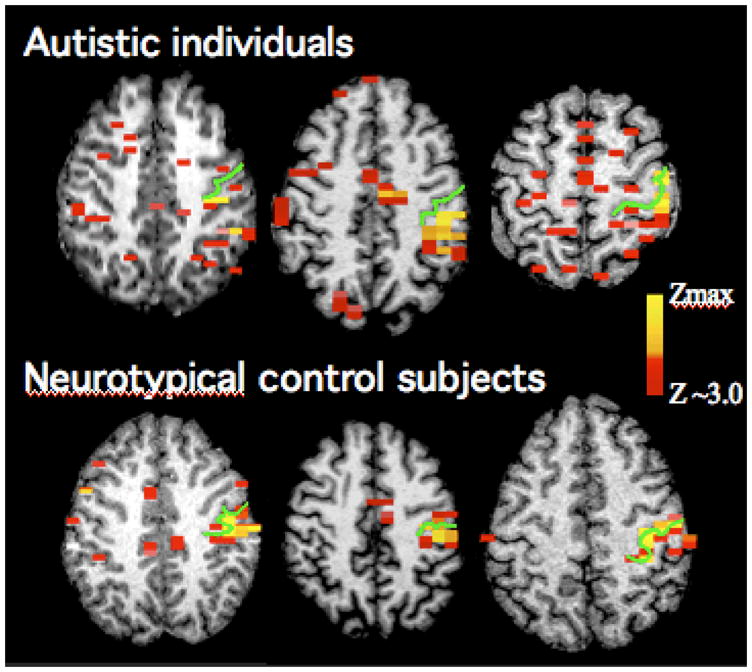
Activation patterns for repetitive index finger movement in three men with autism and three gender and age-matched control participants. All participants show activation clusters in the vicinity of the central sulcus contralateral to the side of movement (indicated by the green line). However, in individuals with autism activity tends to be widely scattered across fronto-parietal regions, whereas in control participants additional activity is largely limited to supplementary motor cortex in the medial frontal lobe and a few sites in ipsilateral motor and premotor cortex. Adapted from Müller et al. (2001).
If task-driven activity in a region of interest is slightly reduced in an autism group for the two above reasons, cross-correlations across regions are likely to be similarly reduced. It is unclear, however, whether this may truly inform us about underconnectivity. Fortunately, there are some methodological decisions that can address the issues raised above, at least in part. Scanning in the resting state may appear a solution, but it should be noted that the mind is not usually ‘blank’ during rest and participants tend to engage in cognitive activities (Raichle et al. 2001). Since this activity is uncontrolled experimentally and may be systematically different in ASD (Kennedy et al. 2006), the resting state appears undesirable. An alternative is the statistical removal of effects driven primarily by a specific task. Are functional connectivity results based on interregional BOLD cross-correlation affected by this procedure?
One study by Villalobos and colleagues (2005) examined functional connectivity between primary visual cortex and other parts of the brain during two visuomotor conditions, one of which was extremely easy (pressing a button with an index finger each time a blue dot appeared on a hand outline on a screen) whereas the second one was slightly harder (pressing fingers in 6-digit sequences prompted by the dot appearing on corresponding fingers on the screen). An adapted boxcar model for this design was used as an orthogonal regressor, thus removing effects in BOLD time series that were primarily driven by activation (i.e., the alternation of blocks in the harder and easier conditions). In this study, time series of detected head motion (for each axis and rotation) were also used as orthogonal regressors to minimize effects of head motion. Finally, BOLD time series were low-pass filtered at 0.1 Hz given that functional connectivity effects of interest are known to occur below this frequency (Cordes et al. 2001). A main finding from the study by Villalobos et al. (2005) showed reduced functional connectivity between primary visual cortex and inferior frontal cortex (Fig. 3A,B). Since inferior frontal cortex is the presumed site of mirror neurons (Rizzolatti and Craighero 2004), the finding is of interest in the context of recent proposals of impairment of the mirror neuron system in autism (Williams et al. 2001). Although the autism group in the study by Villalobos et al. showed many regions of significant fcMRI effects, especially in parietal and subcortical regions, direct group comparisons yielded only effects of greater interregional correlations in the control group, but no inverse effects. These results would thus appear consistent with the underconnectivity theory.
Fig. 3.
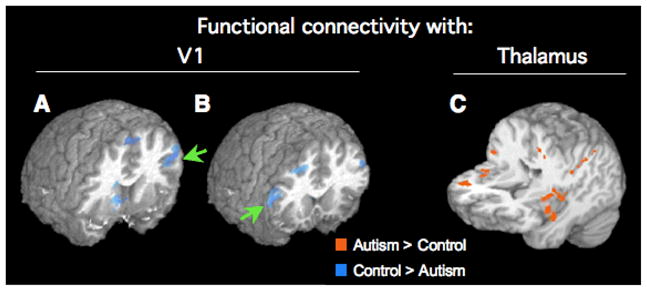
Effects of functional connectivity (correlation of BOLD time series) with primary visual cortex (A, B) and thalamus (C). Overlays are color-coded showing results of direct statistical group comparisons. Connectivity with V1 is reduced for a group of 8 men with autism (compared to age-matched neurotypical men) in bilateral inferior frontal area 44 (arrows). In the same sets of participants, functional connectivity between thalamus and several cerebral cortical regions in frontal and parietal lobes is, however, increased, in particular in insular and perirolandic regions. Adapted from Villalobos et al., 2005 (A, B) and Mizuno et al., 2006 (C).
Not all of the few autism fcMRI studies have been able to replicate generalized underconnectivity in terms of reduced interregional BOLD correlations, however. Welchew and colleagues (2005) examined Pearson correlation matrices for 90 cortical and subcortical regions of interest in 13 adults ASD participants. Although they did find regionally specific disconnectivity for the amygdala and parahippocampal gyrus (in their comparison with matched controls), no evidence of generalized underconnectivity was seen. It should be noted that participants were scanned during viewing of faces at different levels of fearful expressions. The findings in the medial temporal lobe may have therefore been driven by group differences in activation effects, rather than by functional connectivity effects in the strict sense (as explained above). The overall picture of group comparisons for all 90 regions was a mixture of increased and reduced correlations, which is inconsistent with general underconnectivity.
Two studies from our group have recently explored subcortico-cortical functional connectivity in autism. Participants performed the simple visuomotor tasks described previously. One study examined functional connectivity of the thalamus (Mizuno et al. 2006). Our expectation of reduced thalamocortical fcMRI effects – based on some previous findings implicating the thalamus (Tsatsanis et al. 2003) and thalamo-cortical connections (Chugani et al. 1997) – was not confirmed. On the contrary, functional connectivity between thalamus and several fronto-parietal regions (including perirolandic and insular cortices) was greater than in a matched control group (Fig. 3C). A second study investigating functional connectivity between caudate nuclei and cerebral cortex (Turner et al. 2006) was also inconsistent with general underconnectivity, yielding instead a picture of widespread and distributed clusters of greater caudato-cortical connectivity in frontal and parietal lobes of participants with autism.
One tempting interpretation of the current findings would be generally reduced cortical connectivity in autism, both within and between hemispheres, but partly increased functional connectivity between subcortical structures, such as basal ganglia and thalamus, and cerebral cortex. However, as much as there are methodological caveats related to the cortical underconnectivity findings, such caveats also apply to the subcortical fcMRI data, which were acquired in a small sample of eight high-functioning male participants with autism, for a single visuomotor task paradigm, and using a specific set of data processing techniques. For example, it cannot be ruled out that task performance was associated with generally greater arousal in participants with autism, which could result in overall greater interregional correlations of the BOLD signal.
CONCLUSIONS
Functional connectivity studies in autism have to be viewed as work in progress. Nonetheless, a very general and surely oversimplified scenario can be sketched from the evidence reviewed above. In early postnatal development the autistic brain is characterized by widespread white matter overgrowth. It is unclear at this point whether increased volume of white matter reflects a greater number of axons or increased myelination. Even if one were to assume that this white matter overgrowth reflects ‘early overconnectivity’ of the autistic brain, it is doubtful that such abundant connectivity could be fully functional, since the establishment of efficient functional networks requires domain-specific experience and organized activity. The presumed phase of early overconnectivity would thus be ill-timed. Secondary effects of this mismatch between white matter growth of experientially-based differentiation may be reflected in the flatter than normal growth curves in older children with autism. A most parsimonious account would relate to normal effects of synaptic pruning on afferent axons, which will strongly affect inefficiently organized circuits. These secondary events result in mostly reduced functional connectivity between cortical regions, as seen in some studies of adolescents and adults with autism.
As mentioned, this scenario is consistent with the bulk of the current evidence. Even if it is corroborated, it is highly likely that additional developmental events will contribute to atypical connectivity. For example, cellular abnormalities in the medial temporal lobe or the cerebellum may specifically affect efferent and afferent connectivity of these regions; behavioral characteristics, such as repetitive motor acts or avoidance of eye contact, may strengthen or weaken some motor and visual circuits in very specific ways. Although long-range connectivity in autism may thus not be captured by general accounts of underconnectivity, the study of autism as a distributed impairment of multiple networks nonetheless appears more promising than the search for localized defects.
Acknowledgments
This review was supported by the National Institutes of Health, grants R01-NS43999 and R01-DC6155.
Footnotes
Head motion may also be associated with correlated noise, resulting in false positive fcMRI effects especially on the edges of cortex, ventricles, etc.
References
- Adolphs R. Cognitive neuroscience of human social behaviour. Nat Rev Neurosci. 2003;4(3):165–178. doi: 10.1038/nrn1056. [DOI] [PubMed] [Google Scholar]
- Allen G, Courchesne E. Attention function and dysfunction in autism. Front Biosci. 2001;6:D105–119. doi: 10.2741/allen. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders - IV -TR. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Bailey A, Bolton P, Butler L, et al. Prevalence of the fragile X anomaly amongst autistic twins and singletons. J Child Psychol Psychiatry. 1993;34(5):673–688. doi: 10.1111/j.1469-7610.1993.tb01064.x. [DOI] [PubMed] [Google Scholar]
- Bailey A, Luthert P, Dean A, et al. A clinicopathological study of autism. Brain. 1998;121:889–905. doi: 10.1093/brain/121.5.889. [DOI] [PubMed] [Google Scholar]
- Baird G, Charman T, Baron-Cohen S, et al. A screening instrument for autism at 18 months of age: a 6-year follow-up study. J Am Acad Child Adolesc Psychiatry. 2000;39(6):694–702. doi: 10.1097/00004583-200006000-00007. [DOI] [PubMed] [Google Scholar]
- Barnea-Goraly N, Kwon H, Menon V, et al. White matter structure in autism: preliminary evidence from diffusion tensor imaging. Biol Psychiatry. 2004;55(3):323–326. doi: 10.1016/j.biopsych.2003.10.022. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Ring HA, Bullmore ET, et al. The amygdala theory of autism. Neurosci Biobehav Rev. 2000;24(3):355–364. doi: 10.1016/s0149-7634(00)00011-7. [DOI] [PubMed] [Google Scholar]
- Baron-Cohen S, Ring HA, Wheelwright S, et al. Social intelligence in the normal and autistic brain: an fMRI study. European Journal of Neuroscience. 1999;11(6):1891–1898. doi: 10.1046/j.1460-9568.1999.00621.x. [DOI] [PubMed] [Google Scholar]
- Bauman ML, Kemper TL. Neuroanatomic observations of the brain in autism: a review and future directions. Int J Dev Neurosci. 2005;23(2–3):183–187. doi: 10.1016/j.ijdevneu.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9(10):1221–1225. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- Biswal B, Yetkin FZ, Haughton VM, et al. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn Reson Med. 1995;34(4):537–541. doi: 10.1002/mrm.1910340409. [DOI] [PubMed] [Google Scholar]
- Brambilla P, Hardan A, di Nemi SU, et al. Brain anatomy and development in autism: review of structural MRI studies. Brain Res Bull. 2003;61(6):557–569. doi: 10.1016/j.brainresbull.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Brenner LA, Ramos AI, Turner KC, et al. Autism and visual search: cognitive effort is related to response time. Society for Neuroscience Abstracts. 2005:564, 515. [Google Scholar]
- Brenner LA, Turner KC, Müller R-A. Eye movement and visual search: Are there elementary abnormalities in autism? Journal of Autism and Developmental Disorders. 2007;37:1289–1309. doi: 10.1007/s10803-006-0277-9. [DOI] [PubMed] [Google Scholar]
- Bruinsma Y, Koegel RL, Koegel LK. Joint attention and children with autism: A review of the literature. Ment Retard Dev Disabil Res Rev. 2004;10(3):169–175. doi: 10.1002/mrdd.20036. [DOI] [PubMed] [Google Scholar]
- Caron MJ, Mottron L, Berthiaume C, et al. Cognitive mechanisms, specificity and neural underpinnings of visuospatial peaks in autism. Brain. 2006;129(Pt 7):1789–1802. doi: 10.1093/brain/awl072. [DOI] [PubMed] [Google Scholar]
- Carper RA, Moses P, Tigue ZD, et al. Cerebral lobes in autism: early hyperplasia and abnormal age effects. Neuroimage. 2002;16(4):1038–1051. doi: 10.1006/nimg.2002.1099. [DOI] [PubMed] [Google Scholar]
- Castelli F, Frith C, Happe F, et al. Autism, Asperger syndrome and brain mechanisms for the attribution of mental states to animated shapes. Brain. 2002;125(Pt 8):1839–1849. doi: 10.1093/brain/awf189. [DOI] [PubMed] [Google Scholar]
- Chugani DC. Serotonin in autism and pediatric epilepsies. Ment Retard Dev Disabil Res Rev. 2004;10(2):112–116. doi: 10.1002/mrdd.20021. [DOI] [PubMed] [Google Scholar]
- Chugani DC, Muzik O, Rothermel RD, et al. Altered serotonin synthesis in the dentato-thalamocortical pathway in autistic boys. Annals of Neurology. 1997;14:666–669. doi: 10.1002/ana.410420420. [DOI] [PubMed] [Google Scholar]
- Clifford S, Dissanayake C, Bui QM, et al. Autism Spectrum Phenotype in Males and Females with Fragile X Full Mutation and Premutation. J Autism Dev Disord. 2007;37:738–747. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- Cordes D, Haughton VM, Arfanakis K, et al. Frequencies contributing to functional connectivity in the cerebral cortex in “resting-state” data. AJNR Am J Neuroradiol. 2001;22(7):1326–1333. [PMC free article] [PubMed] [Google Scholar]
- Courchesne E, Karns CM, Davis HR, et al. Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology. 2001;57(2):245–254. doi: 10.1212/wnl.57.2.245. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Press GA, Yeung-Courchesne R. Parietal lobe abnormalities detected with MR in patients with infantile autism. American Journal of Roentgenology. 1993;160(2):387–393. doi: 10.2214/ajr.160.2.8424359. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Townsend J, Akshoomoff NA, et al. Impairment in shifting attention in autistic and cerebellar patients. Behavioral Neuroscience. 1994;108(5):848–865. doi: 10.1037//0735-7044.108.5.848. [DOI] [PubMed] [Google Scholar]
- Courchesne E, Yeung-Courchesne R, Press GA, et al. Hypoplasia of cerebellar vermal lobules VI and VII in autism. New England Journal of Medicine. 1988;318(21):1349–1354. doi: 10.1056/NEJM198805263182102. [DOI] [PubMed] [Google Scholar]
- Critchley HD, Daly EM, Bullmore ET, et al. The functional neuroanatomy of social behavior. Changes in cerebral blood flow when people with autistic disorder process facial expressions. Brain. 2000;123:2203–2212. doi: 10.1093/brain/123.11.2203. [DOI] [PubMed] [Google Scholar]
- Dalton KM, Nacewicz BM, Johnstone T, et al. Gaze fixation and the neural circuitry of face processing in autism. Nat Neurosci. 2005;8(4):519–526. doi: 10.1038/nn1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziobek I, Fleck S, Rogers K, et al. The ‘amygdala theory of autism’ revisited: linking structure to behavior. Neuropsychologia. 2006;44(10):1891–1899. doi: 10.1016/j.neuropsychologia.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Edelson SB, Cantor DS. Autism: xenobiotic influences. Toxicology and Industrial Health. 1998;14(4):553–563. doi: 10.1177/074823379801400406. [DOI] [PubMed] [Google Scholar]
- Ek U, Fernell E, Jacobson L, et al. Relation between blindness due to retinopathy of prematurity and autistic spectrum disorders: a population-based study. Developmental Medicine and Child Neurology. 1998;40(5):297–301. [PubMed] [Google Scholar]
- Elbert T, Pantev C, Wienbruch C, et al. Increased cortical representation of the fingers of the left hand in string players. Science. 1995;270:305–307. doi: 10.1126/science.270.5234.305. [DOI] [PubMed] [Google Scholar]
- Fayed N, Modrego PJ. Comparative study of cerebral white matter in autism and attention-deficit/hyperactivity disorder by means of magnetic resonance spectroscopy. Acad Radiol. 2005;12(5):566–569. doi: 10.1016/j.acra.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Filipek PA. Medical aspects of autism. In: Volkmar FR, Paul R, Klin A, et al., editors. Handbook of Autism and Pervasive Developmental Disorders. 3. New York: Wiley; 2005. pp. 534–578. [Google Scholar]
- Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2(12):943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- Friedman SD, Shaw DW, Artru AA, et al. Gray and white matter brain chemistry in young children with autism. Arch Gen Psychiatry. 2006;63(7):786–794. doi: 10.1001/archpsyc.63.7.786. [DOI] [PubMed] [Google Scholar]
- Friston KJ, Frith CD, Frackowiak RSJ. Time-dependent changes in effective connectivity measured with PET. Human Brain Mapping. 1993;1:69–79. [Google Scholar]
- Ghaziuddin M, Butler E. Clumsiness in autism and Asperger syndrome: a further report. Journal of Intellectual Disability Research. 1998;42(Pt 1):43–48. doi: 10.1046/j.1365-2788.1998.00065.x. [DOI] [PubMed] [Google Scholar]
- Gillberg IC, Gillberg C, Ahlsen G. Autistic behaviour and attention deficits in tuberous sclerosis: a population-based study. Developmental Medicine & Child Neurology. 1994;36(1):50–56. doi: 10.1111/j.1469-8749.1994.tb11765.x. [DOI] [PubMed] [Google Scholar]
- Grelotti DJ, Klin AJ, Gauthier I, et al. fMRI activation of the fusiform gyrus and amygdala to cartoon characters but not to faces in a boy with autism. Neuropsychologia. 2005;43(3):373–385. doi: 10.1016/j.neuropsychologia.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Grice SJ, Halit H, Farroni T, et al. Neural correlates of eye-gaze detection in young children with autism. Cortex. 2005;41(3):342–353. doi: 10.1016/s0010-9452(08)70271-5. [DOI] [PubMed] [Google Scholar]
- Hadjikhani N, Joseph RM, Snyder J, et al. Activation of the fusiform gyrus when individuals with autism spectrum disorder view faces. Neuroimage. 2004;22(3):1141–1150. doi: 10.1016/j.neuroimage.2004.03.025. [DOI] [PubMed] [Google Scholar]
- Hall DA, Johnsrude IS, Haggard MP, et al. Spectral and temporal processing in human auditory cortex. Cereb Cortex. 2002;12(2):140–149. doi: 10.1093/cercor/12.2.140. [DOI] [PubMed] [Google Scholar]
- Hallett M, Lebiedowska MK, Thomas SL, et al. Locomotion of autistic adults. Archives of Neurology. 1993;50(12):1304–1308. doi: 10.1001/archneur.1993.00540120019007. [DOI] [PubMed] [Google Scholar]
- Hampson M, Peterson BS, Skudlarski P, et al. Detection of functional connectivity using temporal correlations in MR images. Hum Brain Mapp. 2002;15(4):247–262. doi: 10.1002/hbm.10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happé F, Ehlers S, Fletcher PC, et al. ‘Theory of mind’ in the brain. Evidence from a PET scan study of Asperger syndrome. Neuroreport. 1996;8:197–201. doi: 10.1097/00001756-199612200-00040. [DOI] [PubMed] [Google Scholar]
- Hazlett HC, Poe M, Gerig G, et al. Magnetic resonance imaging and head circumference study of brain size in autism: birth through age 2 years. Arch Gen Psychiatry. 2005;62(12):1366–1376. doi: 10.1001/archpsyc.62.12.1366. [DOI] [PubMed] [Google Scholar]
- Haznedar MM, Buchsbaum MS, Metzger M, et al. Anterior cingulate gyrus volume and glucose metabolism in autistic disorder. American Journal of Psychiatry. 1997;154:1047–1050. doi: 10.1176/ajp.154.8.1047. [DOI] [PubMed] [Google Scholar]
- Hendry J, DeVito T, Gelman N, et al. White matter abnormalities in autism detected through transverse relaxation time imaging. Neuroimage. 2006;29(4):1049–1057. doi: 10.1016/j.neuroimage.2005.08.039. [DOI] [PubMed] [Google Scholar]
- Herbert MR, Russo JP, Yang S, et al. Autism and environmental genomics. Neurotoxicology. 2006 doi: 10.1016/j.neuro.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Herbert MR, Ziegler DA, Deutsch CK, et al. Brain asymmetries in autism and developmental language disorder: a nested whole-brain analysis. Brain. 2005;128(Pt 1):213–226. doi: 10.1093/brain/awh330. [DOI] [PubMed] [Google Scholar]
- Herbert MR, Ziegler DA, Makris N, et al. Localization of white matter volume increase in autism and developmental language disorder. Ann Neurol. 2004;55(4):530–540. doi: 10.1002/ana.20032. [DOI] [PubMed] [Google Scholar]
- Hill EL. Executive dysfunction in autism. Trends Cogn Sci. 2004;8(1):26–32. doi: 10.1016/j.tics.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Hollander E, Anagnostou E, Chaplin W, et al. Striatal Volume on Magnetic Resonance Imaging and Repetitive Behaviors in Autism. Biol Psychiatry. 2005 doi: 10.1016/j.biopsych.2005.03.040. [DOI] [PubMed] [Google Scholar]
- Horwitz B. The elusive concept of brain connectivity. Neuroimage. 2003;19(2 Pt 1):466–470. doi: 10.1016/s1053-8119(03)00112-5. [DOI] [PubMed] [Google Scholar]
- Horwitz B, Rumsey JM, Grady CL, et al. The cerebral metabolic landscape in autism: intercorrelations of regional glucose utilization. Archives of Neurology. 1988;45:749–755. doi: 10.1001/archneur.1988.00520310055018. [DOI] [PubMed] [Google Scholar]
- Hubl D, Bolte S, Feineis-Matthews S, et al. Functional imbalance of visual pathways indicates alternative face processing strategies in autism. Neurology. 2003;61(9):1232–1237. doi: 10.1212/01.wnl.0000091862.22033.1a. [DOI] [PubMed] [Google Scholar]
- Iacoboni M, Molnar-Szakacs I, Gallese V, et al. Grasping the intentions of others with one’s own mirror neuron system. PLoS Biol. 2005;3(3):e79. doi: 10.1371/journal.pbio.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe T, Baron-Cohen S. Are people with autism and Asperger syndrome faster than normal on the Embedded Figures Test? J Child Psychol Psychiatry. 1997;38(5):527–534. doi: 10.1111/j.1469-7610.1997.tb01539.x. [DOI] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, et al. Functional and Anatomical Cortical Underconnectivity in Autism: Evidence from an fMRI Study of an Executive Function Task and Corpus Callosum Morphometry. Cereb Cortex. 2006 doi: 10.1093/cercor/bhl006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just MA, Cherkassky VL, Keller TA, et al. Cortical activation and synchronization during sentence comprehension in high-functioning autism: evidence of underconnectivity. Brain. 2004;127(Pt 8):1811–1821. doi: 10.1093/brain/awh199. [DOI] [PubMed] [Google Scholar]
- Kana RK, Keller TA, Cherkassky VL, et al. Sentence comprehension in autism: thinking in pictures with decreased functional connectivity. Brain. 2006 doi: 10.1093/brain/awl164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER, Jessell TM, Sanes JR. Sensory experience and the fine tuning of synaptic connections. In: Kandel ER, Schwartz JH, Jessell TM, editors. Principles of Neural Science. 4. New York: Elsevier; 2000. pp. 1115–1130. [Google Scholar]
- Kennedy DP, Redcay E, Courchesne E. Failing to deactivate: Resting functional abnormalities in autism. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0600674103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klin A, Jones W, Schultz R, et al. Visual fixation patterns during viewing of naturalistic social situations as predictors of social competence in individuals with autism. Arch Gen Psychiatry. 2002;59(9):809–816. doi: 10.1001/archpsyc.59.9.809. [DOI] [PubMed] [Google Scholar]
- Koshino H, Carpenter PA, Minshew NJ, et al. Functional connectivity in an fMRI working memory task in high-functioning autism. Neuroimage. 2005;24(3):810–821. doi: 10.1016/j.neuroimage.2004.09.028. [DOI] [PubMed] [Google Scholar]
- LeDoux J. Emotion: Clues from the brain. Annu Rev Psychol. 1995;46:209–235. doi: 10.1146/annurev.ps.46.020195.001233. [DOI] [PubMed] [Google Scholar]
- Leopold DA, Murayama Y, Logothetis NK. Very slow activity fluctuations in monkey visual cortex: implications for functional brain imaging. Cereb Cortex. 2003;13(4):422–433. doi: 10.1093/cercor/13.4.422. [DOI] [PubMed] [Google Scholar]
- Lidzba K, Staudt M, Wilke M, et al. Visuospatial deficits in patients with early left-hemispheric lesions and functional reorganization of language: Consequence of lesion or reorganization? Neuropsychologia. 2006;44(7):1088–1094. doi: 10.1016/j.neuropsychologia.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Logothetis NK, Pfeuffer J. On the nature of the BOLD fMRI contrast mechanism. Magn Reson Imaging. 2004;22(10):1517–1531. doi: 10.1016/j.mri.2004.10.018. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, DiLavore P, et al. Autism Diagnostic Observation Schedule. Los Angeles: Western Psychological Services; 2001. [Google Scholar]
- Mari M, Castiello U, Marks D, et al. The reach-to-grasp movement in children with autism spectrum disorder. Philos Trans R Soc Lond B Biol Sci. 2003;358(1430):393–403. doi: 10.1098/rstb.2002.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall SP. Measures of Attention and Cognitive Effort in Tactical Decision Making. In: Noyes J, editor. Human Factors of Decision Making in Complex Systems. Aldershot (Hampshire, UK): Ashgate Publishing; in press. [Google Scholar]
- Minshew NJ, Sung K, Jones BL, et al. Underdevelopment of the postural control system in autism. Neurology. 2004;63(11):2056–2061. doi: 10.1212/01.wnl.0000145771.98657.62. [DOI] [PubMed] [Google Scholar]
- Mizuno A, Villalobos ME, Davies MM, et al. Partially enhanced thalamo-cortical functional connectivity in autism. Brain Research. 2006;1104(1):160–174. doi: 10.1016/j.brainres.2006.05.064. [DOI] [PubMed] [Google Scholar]
- Mostofsky SH, Goldberg MC, Landa RJ, et al. Evidence for a deficit in procedural learning in children and adolescents with autism: implications for cerebellar contribution. Journal of the International Neuropsychological Society. 2000;6(7):752–759. doi: 10.1017/s1355617700677020. [DOI] [PubMed] [Google Scholar]
- Müller R-A. Functional neuroimaging of developmental disorders: Lessons from autism research. In: Hillary FD, DeLuca J, editors. Functional Neuroimaging in Clinical Populations. Guilford: 2007. pp. 145–184. [Google Scholar]
- Müller R-A. Language universals in the brain: How linguistic are they? In: Christiansen MH, Collins C, Edelman S, editors. Language Universals. Oxford University Press; 2009. pp. 224–252. [Google Scholar]
- Müller R-A, Pierce K, Ambrose JB, et al. Atypical patterns of cerebral motor activation in autism: a functional magnetic resonance study. Biological Psychiatry. 2001;49:665–676. doi: 10.1016/s0006-3223(00)01004-0. [DOI] [PubMed] [Google Scholar]
- Nelson PG, Kuddo T, Song EY, et al. Selected neurotrophins, neuropeptides, and cytokines: developmental trajectory and concentrations in neonatal blood of children with autism or Down syndrome. Int J Dev Neurosci. 2006;24(1):73–80. doi: 10.1016/j.ijdevneu.2005.10.003. [DOI] [PubMed] [Google Scholar]
- O’Leary DD, Nakagawa Y. Patterning centers, regulatory genes and extrinsic mechanisms controlling arealization of the neocortex. Curr Opin Neurobiol. 2002;12(1):14–25. doi: 10.1016/s0959-4388(02)00285-4. [DOI] [PubMed] [Google Scholar]
- O’Riordan M, Passetti F. Discrimination in Autism Within Different Sensory Modalities. J Autism Dev Disord. 2006 doi: 10.1007/s10803-006-0106-1. [DOI] [PubMed] [Google Scholar]
- O’Riordan MA. Superior visual search in adults with autism. Autism. 2004;8(3):229–248. doi: 10.1177/1362361304045219. [DOI] [PubMed] [Google Scholar]
- O’Riordan MA, Plaisted KC, Driver J, et al. Superior visual search in autism. J Exp Psychol Hum Percept Perform. 2001;27(3):719–730. doi: 10.1037//0096-1523.27.3.719. [DOI] [PubMed] [Google Scholar]
- Otsuka H, Harada M, Mori K, et al. Brain metabolites in the hippocampus-amygdala region and cerebellum in autism: an 1H-MR spectroscopy study. Neuroradiology. 1999;41(7):517–519. doi: 10.1007/s002340050795. [DOI] [PubMed] [Google Scholar]
- Palmen SJ, Durston S, Nederveen H, et al. No evidence for preferential involvement of medial temporal lobe structures in high-functioning autism. Psychol Med. 2006;36(6):827–834. doi: 10.1017/S0033291706007215. [DOI] [PubMed] [Google Scholar]
- Palmen SJ, van Engeland H, Hof PR, et al. Neuropathological findings in autism. Brain. 2004;127(Pt 12):2572–2583. doi: 10.1093/brain/awh287. [DOI] [PubMed] [Google Scholar]
- Pelphrey K, Adolphs R, Morris JP. Neuroanatomical substrates of social cognition dysfunction in autism. Ment Retard Dev Disabil Res Rev. 2004;10(4):259–271. doi: 10.1002/mrdd.20040. [DOI] [PubMed] [Google Scholar]
- Pelphrey KA, Sasson NJ, Reznick JS, et al. Visual scanning of faces in autism. J Autism Dev Disord. 2002;32(4):249–261. doi: 10.1023/a:1016374617369. [DOI] [PubMed] [Google Scholar]
- Pierce K, Haist F, Sedaghat F, et al. The brain response to personally familiar faces in autism: findings of fusiform activity and beyond. Brain. 2004;127(Pt 12):2703–2716. doi: 10.1093/brain/awh289. [DOI] [PubMed] [Google Scholar]
- Pierce K, Müller R-A, Ambrose JB, et al. Face processing occurs outside the ‘fusiform face area’ in autism: evidence from functional MRI. Brain. 2001;124:2059–2073. doi: 10.1093/brain/124.10.2059. [DOI] [PubMed] [Google Scholar]
- Piggot J, Kwon H, Mobbs D, et al. Emotional attribution in high-functioning individuals with autistic spectrum disorder: a functional imaging study. J Am Acad Child Adolesc Psychiatry. 2004;43(4):473–480. doi: 10.1097/00004583-200404000-00014. [DOI] [PubMed] [Google Scholar]
- Piven J, Arndt S, Bailey J, et al. An MRI study of brain size in autism. American Journal of Psychiatry. 1995;152(8):1145–1149. doi: 10.1176/ajp.152.8.1145. [DOI] [PubMed] [Google Scholar]
- Quigley M, Cordes D, Turski P, et al. Role of the corpus callosum in functional connectivity. AJNR Am J Neuroradiol. 2003;24(2):208–212. [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, et al. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98(2):676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramnani N, Behrens TE, Penny W, et al. New approaches for exploring anatomical and functional connectivity in the human brain. Biol Psychiatry. 2004;56(9):613–619. doi: 10.1016/j.biopsych.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Rippon G, Brock J, Brown C, et al. Disordered connectivity in the autistic brain: Challenges for the ‘new psychophysiology’. Int J Psychophysiol. 2006 doi: 10.1016/j.ijpsycho.2006.03.012. [DOI] [PubMed] [Google Scholar]
- Rizzolatti G, Craighero L. The mirror-neuron system. Annu Rev Neurosci. 2004;27:169–192. doi: 10.1146/annurev.neuro.27.070203.144230. [DOI] [PubMed] [Google Scholar]
- Rodier PM. Converging evidence for brain stem injury in autism. Dev Psychopathol. 2002;14(3):537–557. doi: 10.1017/s0954579402003085. [DOI] [PubMed] [Google Scholar]
- Sadato N, Okada T, Honda M, et al. Critical period for cross-modal plasticity in blind humans: a functional MRI study. Neuroimage. 2002;16(2):389–400. doi: 10.1006/nimg.2002.1111. [DOI] [PubMed] [Google Scholar]
- Schmitz C, Martineau J, Barthelemy C, et al. Motor control and children with autism: deficit of anticipatory function? Neurosci Lett. 2003;348(1):17–20. doi: 10.1016/s0304-3940(03)00644-x. [DOI] [PubMed] [Google Scholar]
- Schultz RT, Gauthier I, Klin A, et al. Abnormal ventral temporal cortical activity during face discrimination among individuals with autism and Asperger syndrome [see comments] Archives of General Psychiatry. 2000;57(4):331–340. doi: 10.1001/archpsyc.57.4.331. [DOI] [PubMed] [Google Scholar]
- Schumann CM, Amaral DG. Stereological analysis of amygdala neuron number in autism. J Neurosci. 2006;26(29):7674–7679. doi: 10.1523/JNEUROSCI.1285-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann CM, Hamstra J, Goodlin-Jones BL, et al. The amygdala is enlarged in children but not adolescents with autism; the hippocampus is enlarged at all ages. J Neurosci. 2004;24(28):6392–6401. doi: 10.1523/JNEUROSCI.1297-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju A, Tojo Y, Yaguchi K, et al. Deviant gaze processing in children with autism: an ERP study. Neuropsychologia. 2005;43(9):1297–1306. doi: 10.1016/j.neuropsychologia.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Sparks BF, Friedman SD, Shaw DW, et al. Brain structural abnormalities in young children with autism spectrum disorder. Neurology. 2002;59(2):184–192. doi: 10.1212/wnl.59.2.184. [DOI] [PubMed] [Google Scholar]
- Sur M, Leamey CA. Development and plasticity of cortical areas and networks. Nat Rev Neurosci. 2001;2(4):251–262. doi: 10.1038/35067562. [DOI] [PubMed] [Google Scholar]
- Sweeten TL, Posey DJ, Shekhar A, et al. The amygdala and related structures in the pathophysiology of autism. Pharmacol Biochem Behav. 2002;71(3):449–455. doi: 10.1016/s0091-3057(01)00697-9. [DOI] [PubMed] [Google Scholar]
- Tanoue Y, Oda S, Asano F, et al. Epidemiology of infantile autism in southern Ibaraki, Japan: differences in prevalence in birth cohorts. Journal of Autism and Developmental Disorders. 1988;18(2):155–166. doi: 10.1007/BF02211943. [DOI] [PubMed] [Google Scholar]
- Terracciano A, Chiurazzi P, Neri G. Fragile X syndrome. Am J Med Genet C Semin Med Genet. 2005;137(1):32–37. doi: 10.1002/ajmg.c.30062. [DOI] [PubMed] [Google Scholar]
- Thomas M, Karmiloff-Smith A. Are developmental disorders like cases of adult brain damage? Implications from connectionist modeling. Behavioral and Brain Sciences. 2002;25(6):727–750. doi: 10.1017/s0140525x02000134. [DOI] [PubMed] [Google Scholar]
- Townsend J, Harris NH, Courchesne E. Visual attention abnormalities in autism: delayed orienting to location. Journal of the International Neuropsychological Society. 1996;2:541–550. doi: 10.1017/s1355617700001715. [DOI] [PubMed] [Google Scholar]
- Tsatsanis KD, Rourke BP, Klin A, et al. Reduced thalamic volume in high-functioning individuals with autism. Biol Psychiatry. 2003;53(2):121–129. doi: 10.1016/s0006-3223(02)01530-5. [DOI] [PubMed] [Google Scholar]
- Turner KC, Frost L, Linsenbardt D, et al. Atypically diffuse functional connectivity between caudate nuclei and cerebral cortex in autism. Behavioral and Brain Functions. 2006;2:34–45. doi: 10.1186/1744-9081-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal CN, Nicolson R, Devito TJ, et al. Mapping Corpus Callosum Deficits in Autism: An Index of Aberrant Cortical Connectivity. Biol Psychiatry. 2006 doi: 10.1016/j.biopsych.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Villalobos ME, Mizuno A, Dahl BC, et al. Reduced functional connectivity between V1 and inferior frontal cortex associated with visuomotor performance in autism. Neuroimage. 2005;25:916–925. doi: 10.1016/j.neuroimage.2004.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Staal WG, van Daalen E, et al. Identification of novel autism candidate regions through analysis of reported cytogenetic abnormalities associated with autism. Mol Psychiatry. 2006;11(1):1, 18–28. doi: 10.1038/sj.mp.4001781. [DOI] [PubMed] [Google Scholar]
- Welchew DE, Ashwin C, Berkouk K, et al. Functional disconnectivity of the medial temporal lobe in Asperger’s syndrome. Biol Psychiatry. 2005;57(9):991–998. doi: 10.1016/j.biopsych.2005.01.028. [DOI] [PubMed] [Google Scholar]
- Williams JH, Whiten A, Suddendorf T, et al. Imitation, mirror neurons and autism. Neurosci Biobehav Rev. 2001;25(4):287–295. doi: 10.1016/s0149-7634(01)00014-8. [DOI] [PubMed] [Google Scholar]