

Abstract
The Escherichia coli protein DbpA is unique in its subclass of DEAD box RNA helicases, because it possesses ATPase-specific activity toward the peptidyl transferase center in 23S rRNA. Although its remarkable ATPase activity had been well defined toward various substrates, its RNA helicase activity remained to be characterized. Herein, we show by using biochemical assays and atomic force microscopy that DbpA exhibits ATP-stimulated unwinding activity of RNA duplex regardless of its primary sequence. This work presents an attempt to investigate the action of DEAD box proteins by a single-molecule visualization methodology. Our atomic force microscopy images enabled us to observe directly the unwinding reaction of a DEAD box helicase on long stretches of double-stranded RNA. Specifically, we could differentiate between the binding of DbpA to RNA in the absence of ATP and the formation of a Y-shaped intermediate after its progression through double-stranded RNA in the presence of ATP. Recent studies have questioned the designation of DbpA, in particular, and DEAD box proteins in general as RNA helicases. However, accumulated evidence and the results reported herein suggest that these proteins are indeed helicases that resemble in many aspects the DNA helicases.
Many putative RNA helicases are members of the DEAD box protein family, which catalyzes the hydrolysis of ATP in the presence of RNA presumably unwinding the duplex regions of RNA and RNA–DNA hybrids. They are characterized by the “DEAD” motif (Asp-Glu-Ala-Asp) as well as by seven other conserved amino acid motifs including two ATP binding domains A and B (1–3). These proteins are found in a wide range of organisms ranging from viruses to higher eukaryotes. Importantly, RNA helicases participate in many essential cellular processes such as transcription, translation, ribosome assembly, cell differentiation, cell development, RNA processing, and mRNA splicing (4–6). Furthermore, the unwinding of the RNA secondary structure is the rate-limiting step in obtaining the functional conformation of RNA, required in these biological processes (2). Therefore, helicases may play a key role in regulating these biological processes by controlling RNA structures. Although the unwinding activity has been demonstrated in vitro for a few RNA helicases (7–11), it has not been shown for many other members of the DEAD box family. It was suggested that, unlike DNA helicases (12), RNA helicases may not be required to unwind long stretches of double-stranded RNA (dsRNA; ref. 2). The mode of action of a DEAD box helicase was recently analyzed in detail in the vaccinia nucleoside-triphosphate phosphohydrolase-II (NPH-II) protein (11). NPH-II exhibited highly processive 3′ to 5′ helicase activity, in an ATP-dependent manner. These results raised the possibility that the mode of action of RNA and DNA helicases is similar and that the differences between them may be the result of their particular substrates and their interactions with other proteins (10).
DbpA was identified by its homology to the human RNA helicase p68 (13). Analysis of its RNA-dependent ATPase activity revealed an exceptional and high specificity toward bacterial 23S rRNA (14–17). Although most helicases do not display sequence-specific ATPase activity associated with their unwinding activity, the maximum activation of the ATP hydrolysis of DbpA can be triggered by a single helix region of 93 nucleotides located at the peptidyl transferase center of the 23S rRNA (15). Interestingly, kinetic analysis of DbpA in the presence of rRNA (16S + 23S) revealed a protein–cofactor rather than a protein–substrate relationship between DbpA and the tested rRNA (17). These results, as well as unsuccessful attempts to demonstrate unwinding activity by DbpA on artificial duplexes of RNA, suggested that DbpA is a unique RNA-dependent ATPase. Furthermore, because its pronounced ATPase activity was abolished in the presence of intact ribosomal particles, it was proposed that DbpA may be involved in ribosomal biogenesis (17). However, a direct coupling between its ATPase activity and its putative helicase activity remained to be shown.
Interestingly, in a recent report, it was shown that when DbpA is in a molar excess over RNA, it possesses helicase activity of duplex RNA that is highly dependent on a universally conserved hairpin (2,550 in 23S rRNA).‖ Herein we show that DbpA is an ATP-dependent helicase that exhibits unwinding activity on long linear dsRNA. In addition, we demonstrate its affinity and unwinding activity on artificial duplexes of RNA by using both biochemical and atomic force microscopy (AFM) methods. Our AFM results show that DbpA can bind and unwind long and nonspecific sequences of duplex RNA with 5′ overhang at a low protein:RNA ratio. Furthermore, its unwinding activity is stimulated by ATP.
AFM is becoming increasingly useful for biological research (18–22). This technique has the potential to assay protein–nucleic acid interactions at the molecular level for individual complexes. Examination of the helicase activity of DbpA by AFM provides means for directly visualizing its unwinding catalysis in a single-molecule resolution. Based on our biochemical and biophysical results, we propose tentative functional steps of the specific protein–RNA interactions that evolve during the unwinding of RNA by DbpA.
Materials and Methods
Reagents and Chemicals.
All reagents were purchased from Sigma. Enzymes were purchased from Roche Molecular Biochemicals. Milli-Q water was used in all AFM and RNA assays.
Protein Purification.
Recombinant DbpA was expressed in Escherichia coli BL21 (DE3) LysS. Pelleted cells were resuspended in 20 mM/Mops, pH 6.8/250 mM NaCl/1 mM dithioerythritol (DTE)/protease inhibitor mixture and sonicated on ice. Cells debris was removed by ultracentrifugation. The supernatant was loaded onto DEAE Sepharose (Amersham Pharmacia) equilibrated with 20 mM Mops pH 6.8/200 mM NaCl/1 mM DTE/1 mM PMSF/1 mM benzamidine, and the flow-through was collected. The flow-through was loaded onto a Hi-trap-SP column (Amersham Pharmacia) and eluted with a linear 0.2 M to 1 M NaCl gradient. The protein fractions were concentrated and further purified on HiLoad 16/26 Superdex 75 prep grade (Amersham Pharmacia), equilibrated with 50 mM Hepes, pH 7.5/200 mM NaCl/1 mM DTE. The protein concentration was determined by amino acid analysis and SDS/PAGE chromatography.
RNA for Gel-Based Studies.
RNA oligonucleotides were purchased from Dharmacom Research (Boulder, CO): top strand oligo, 5′-AAAAAAAAAAAAGGGGGAAUUAAUGAAU AAAGGGCUAGGAAUGG; bottom strand oligo, 5′-CCAUUCCUAGCCCUUUAUUCAAUUAAUUCCCCC. The bottom strand was 5′ end-labeled with γ-ATP (31) by using the standard procedure. The labeled RNA strand was hybridized to the bottom strand as described in ref. 11 and purified from gel. RNA concentrations were determined by quantification of the radiolabeled RNA with a β-counter (Bakman, Palo Alto, CA).
RNA for AFM studies.
A DNA fragment of 480 bp containing a sequence from Bacillus stearothermophilus 16S rRNA (925–1,391; accession no. K02663) was cloned into pBluescript (Stratagene) between the EcoRI and BamHI restriction sites. The RNA was prepared by in vitro transcription of BamHI- or EcoRI-digested pBluescript with T7 or T3 RNA polymerase, respectively, according to the standard protocol (Promega). The transcripts were treated with DNase I (Promega), phenol extracted, and passed through a G-50 minispin column (Roche Molecular Biochemicals). Both transcripts were coprecipitated in ethanol, resuspended in 20 mM Hepes, pH 7.5/150 mM KCl, then denatured at 85°C for 5 min, and slowly cooled in a thermal block to room temperature. RNA concentration was determined by optical spectroscopy at 260 nm.
Electrophoretic Mobility-Shift Assays.
The indicated concentrations of DbpA were incubated with 20 pmol of RNA duplexes in a final volume of 20 μl in 20 mM Tris⋅HCl, pH 7.5/40 mM KCl/2 mM Mg2Cl/1 mM DTE/5% (vol/vol) glycerol for 20 min on ice. The buffer composition of the electrophoretic mobility-shift assay was optimizing to obtain the maximum resolution in resolving RNA. The reaction products were separated by electrophoresis through native gels at 4°C containing 8% polyacrylamide (37.5:1 acrylamide:bisacrylamide), in 0.5× TBE buffer (45 mM Tris/45 mM boric acid/1.0 mM EDTA, pH 8.3). Autoradiographs of the dried gels were analyzed by densitometry with a Molecular Dynamics PhosphorImager. The Kd was calculated by quantifying the protein concentration at which half the RNA was shifted (23).
RNA Unwinding Gel-Based Assays.
For an RNA unwinding reaction, the indicated concentrations of DbpA were incubated with 20 pmol of RNA duplexes in a final volume of 20 μl in 20 mM Tris⋅HCl, pH 7.5/20 mM NH4Cl/2 mM Mg2Cl/1 mM DTE/2 mM ATP. The reaction was preincubated for 10 min on ice and then transferred to a 37°C bath for an additional 20 min. The reaction was quenched by 5 mM EDTA/0.4% SDS/5% (vol/vol) glycerol/100-fold of the labeled RNA oligo and loaded on native gels containing 10% polyacrylamide (29:1 acrylamide:bisacrylamide) in 0.5× TBE buffer. The percentage of unwinding (% UW) was calculated with the following formula
 |
1 |
where c denotes the densitometric value at the particular reaction at protein concentration [C]; ø is the densitometric value of the background unwinding without protein subtracted from the total unwinding (9); and ssRNA is single-stranded RNA.
AFM.
Freshly cleaved mica (Electron Microscopy Sciences, Washington, PA) was soaked in 15 mM NiCl2/1 mM HCl before deposition of the samples. Either free RNA or protein–RNA solution (4 μl) was deposited on freshly treated mica for 1–5 min, rinsed with water, and dried with compressed Argon. Protein and RNA (1:1.5 pmol ratio) were incubated in a 20-μl reaction containing 20 mM Hepes (pH 7.5), 50 mM NH4Cl, 5 mM MgCl2, and 0.5 mM DTT. ATP (1 μl, 100 mM) was added to the mixture after a 0.5- to 1.5-min incubation on ice before deposition on the mica. The buffer composition for the unwinding reaction had to be optimized further for the AFM assay. Using Tris⋅HCl instead of Hepes or KCl instead of NH4Cl resulted in salt aggregates on the mica surface. Screening for suitable buffer and salt condition revealed the buffers described above. Nevertheless, the pH and the ionic strength conditions were maintained carefully for all of the assays. In addition, the unwinding reaction could be visualized by AFM by using the gel-based assay conditions. AFM imaging was performed in air with a Nanoscope III (Digital Instruments, Santa Barbara, CA) operating in tapping mode with silicon tapping mode tips (Olympus, Tokyo). Bungee cords were used for vibration isolation (24). Images were processed by flattening to remove the background slope with the Digital Instruments Nanoscope software package. End-to-end contour lengths, heights, and widths at half-heights of RNA and protein were measured with image sxm software (University of Liverpool, Liverpool, U.K.).
Results and Discussion
Most mechanistic studies performed with DEAD box proteins have been conducted on short RNA sequences. This experimental approach was developed, because (i) RNA helicases were believed to modulate only short duplex regions in RNA molecules (2), and (ii) this type of unwinding activity can be readily examined and resolved with gel-based assays (25). However, the precise unwinding mechanism is not fully understood, nor have the in vivo substrates of the DEAD box proteins been identified precisely. We developed the electrophoretic mobility-shift assay to characterize the formation of the complex between DbpA and nonspecific dsRNA. Furthermore, the helicase activity of DbpA was studied for a short and a long dsRNA by using gel-based assays and AFM techniques, respectively.
Affinity of DbpA Toward dsRNA.
Fig. 1b shows the results of a gel retardation assay that was developed to characterize the complex formation between DbpA and 33-bp dsRNA with a 12-nt overhang at its 5′ end (Fig. 1a). The DbpA–dsRNA complex began to form at a 0.2 μM protein concentration (Fig. 1b, lane 2). No complex was detected at a lower protein concentration. The minimum binding ratio between protein and RNA was 1:1.25. At a ratio of 1:5.2 RNA-to-protein, most of the RNA was bound to DbpA (1.3 μM DbpA; Fig. 1b, lane 9). Increasing the concentration of DbpA did not result in a significant change (data not shown). The apparent calculated Kd toward the dsRNA, used in our experiments, is 0.30 ± 0.02 μM. Comparison of Kd values, which are available for DbpA with several substrates, shows that the dissociation constants of the enzyme complexes dsRNAs belonging to the peptidyl transferase center of 23S rRNA are Kd = 0.15 μM (nucleotides 2,508–2,580; ref. 27), Kd = 0.34 nM (nucleotides 2,504–2,583), and Kd = 0.53 nM (nucleotides 2,454–2,606).** These values suggest that, although DbpA shows a great specificity toward the identified regions of rRNA, its binding efficiency to nonspecific RNA with sufficient single-strand overhang is comparable to the previously reported values. The discrepancy between the different Kds reported for the enzyme/peptidyl transferase center rRNA regions is explained by the use of different methods to obtain these values (ref. 26;**).
Figure 1.

Binding and unwinding activity of DbpA with short dsRNA. (a) Schematic of the short dsRNA substrate used for the following assays. (b) Gel retardation assay of DbpA and dsRNA substrate. Binding reactions were performed in a 20-μl reaction volume in the presence of 5 pmol of 32P-labeled dsRNA and increasing amounts of DbpA. The binding percentages are indicated. Free RNA, RNA-DbpA complexes, and the origins of the wells in the gel are indicated on the left. (c) Unwinding of dsRNA by DbpA. Lane 1 represents free dsRNA. Lane 2 shows the ssRNA that resulted after 3 min of incubation at 85°C. Lanes 3–10 show the unwinding reaction at a given protein concentration. The differences in the position of ssRNA on the gel between lane 2 and lanes 3–10 are caused by differences in ionic strength of the free dsRNA used for control and the product of the enzymatic reaction.
The unwinding activity of DbpA was examined with classical gel electrophoresis assays (25). The RNA used for these assays was dsRNA, studied by electrophoretic mobility-shift assay. Fig. 1c presents the unwinding activity of the same dsRNA by DbpA. In these assays, the shorter strand of the duplex RNA molecule was labeled with 32P to increase the electrophoretic separation gap between the displaced ssRNA and the unwound duplex molecule. Minimum unwinding activity of dsRNA was detected at 0.8 μM DbpA (Fig. 1c, lane 5). Increasing the protein concentration to 2.5 μM resulted in 37% unwinding compared with 8% ssRNA displacement without ATP. These results show that ATP stimulates unwinding activity by DbpA on this substrate. Displacement of ssRNA without ATP is fairly low but could still be detected for the highest protein:RNA ratio used in our gel-based assay (Fig. 1c, lane 10). The effect of strand displacement in the absence of ATP for lower protein:RNA ratios was less pronounced (data not shown). This result may be explained in terms of the nature of such assays in which a relatively high protein:RNA ratio is usually used to unwind short sequences of duplex nucleic acid substrates. Thus, partial strand displacement occurs because of melting as the consequence of the increase in protein concentration. Nevertheless, Fig. 1c demonstrates that DbpA exhibits a distinct unwinding activity in the presence of ATP.
Therefore, to study the unwinding catalysis of DbpA in a lower protein:RNA ratio and to examine its activity on long stretches of dsRNA molecules, we performed single-molecule AFM imaging of DbpA and a ≈500-bp dsRNA.
AFM Imaging of DbpA.
AFM imaging is carried out without staining, shadowing, or labeling and, therefore, is particularly attractive for studying biological systems (18, 19, 22). A unique advantage of AFM is the ability to visualize single molecules directly. In contrast to AFM, electrophoretic methods provide information averaged over the distribution of conformations of molecular states in a given reaction condition. We used AFM to visualize directly unwinding catalysis of DbpA on long dsRNA molecules at single-molecule resolution. Furthermore, the overall structural organization of the protein–RNA interactions could be examined. A dsRNA was constructed to contain a double-stranded region of 480 bp and 5′ single-stranded overhangs of ≈50 nt at both ends (Fig. 2a). DbpA was incubated with its substrates on ice, rapidly deposited on mica, and imaged in air. This procedure allowed us to trap a random collection of complexes of enzyme–RNA that evolved during the unwinding catalysis.
Figure 2.

AFM images of free dsRNA. (a) Schematic of the RNA construct used for the AFM imaging containing two 5′ (≈50-nt) single-stranded overhangs at both ends of the molecule. The double-stranded central region (480 bp) was generated by hybridization of the complementary regions of both strands. (b) AFM images of long dsRNA in air on mica. (Bar = 100 nm.) The rod-shaped molecules possess a mean end-to-end contour length of 134.5 ± 4 nm, which is consistent with an A form dsRNA.
Fig. 2 shows the rod-shaped RNA substrate before binding to DbpA. Fig. 5a (see below) presents the contour length distribution of free duplex RNA molecules. The RNA rise per base pair, as determined from the ratio of the measured contour length of duplex RNA and the number of base pairs of the RNA construct (only dsRNA region), is 0.28 nm/bp. This value is smaller than the canonical value of 0.34 nm/bp that is assigned to B form DNA; however, it is in a good agreement with the values obtained for duplex RNA measured by AFM (27) and A form RNA (28, 29). A deviation in the measured contour length of a worm-like nucleic acid polymer from B form DNA by AFM measurement is often observed (30, 31). This observation can be explained by the limitation of AFM in resolving DNA bends in the range of ±4 nm and by the polynomial fitting used to measure the contour length, which induces a systematic error in rounding up sharp bends of the DNA trace. This effect seems to be enhanced in RNA because of the greater flexibility of the molecule (27, 31). Moreover, x-ray fiber data and crystallography of dsRNA indicate that dsRNA adopts the A conformation in solution (28, 29). The rise per base pair for A form conformation is 0.27 nm/bp, which is in good agreement with our results. Table 1 shows the mean end-to-end distribution of the measured contour length of free dsRNA (134.5 ± 4 nm) vs. the calculated value (130 nm). The calculated value contains the 480 bp of the dsRNA (Fig. 2a). It is expected for the 100 bases of ssRNA not to contribute to the contour length, because the persistence length of single-stranded nucleic acids is of the order of 5 nm. Therefore, they tend to fold around forming planar globular structures. These ssRNA segments can be seen in Fig. 2 as a broadening at the ends of most of the images. The discrepancy between the measured and calculated contour length values is within the error range of the measurement and can be explained by the induced systematic errors as described (30). It is therefore reasonable to assume that the measured end-to-end distance values obtained for the dsRNA molecules are not affected from sample preparation and drying.
Figure 5.

Statistical data analysis of AFM images. (a) Histogram of free dsRNA (Fig. 2a). The mean end-to-end length distribution is 134 ± 4 nm, which is consistent with A form dsRNA. (b) Same analysis of the prebound DbpA–RNA complex. The mean end-to-end contour length is similar to that shown in a. (c) Histogram of the Y-shaped contour length of the trapped DbpA–RNA–ATP complexes during unwinding catalysis. Comparison of the mean end-to-end length of the Y-shaped complexes to the free RNA and RNA + DbpA (a and b) reveals a significant shift to a longer value, which results from ssRNA formation.
Table 1.
Contour length analysis of free dsRNA, DbpA–RNA, and DbpA–RNA–ATP complexes
| Nucleic acid | Mean of contour length, nm | Calculated mean of counter length, nm | Rise per base pair, nm/bp |
|---|---|---|---|
| B DNA | 0.34 | ||
| A RNA | 0.27 | ||
| dsRNA | 134 ± 4 | 130 | 0.28 |
| dsRNA–DbpA | 135 ± 4 | 130 | |
| dsRNA–DbpA–ATP | |||
| Y-shaped | 167 ± 4 | 230 (Lss = 0.6) | |
| Y-shaped | 143 (Lss = 0.3) |
The measured mean end-to-end length distribution of the various complexes is compared to a calculated value. The discrepancy in measured vs. calculated values is discussed in the text. Lss is the rise per base per nm/bp taken into calculation on the basis of previous reported values.
As was widely discussed by Rivetti et al. (30) and others (31, 32), nucleic acid molecules are able to equilibrate in two dimensions before their final attachment to the mica surface. However, protein–nucleic acid complexes may not have this ability, because the nature of protein–mica interactions may be affected by the surface potential of the protein and by the buffer conditions of the surface deposition. Nevertheless, a stable complex between DbpA and RNA could be detected in various reaction conditions.
Fig. 3 shows that in the absence of ATP, a stable protein–RNA complex forms at the single-stranded region of the RNA substrate. Protein signals are assigned to round globular structures observed at the end of the dsRNA molecule that possesses a mean height of 0.22 ± 0.3 nm. In higher protein:RNA ratios, two protein signals from both ends of the RNA molecule could be detected (data not shown). This result is caused by the nature of the RNA construct, which was designed to have 5′ polarity and ≈50 nt at its ssRNA region from both ends. We used this protein titration experiment as a control experiment for the characterization of the duplex RNA molecules and the assignment of protein signals. Fig. 5 (see below) presents the mean end-to-end length distribution of DbpA–RNA complexes. The mean of the length distribution is 135 ± 4 nm, which is very close to the free RNA value (Table 1). The stable protein–RNA complex obtained in various protein concentrations represents the initial DbpA–RNA interaction before binding to ATP and unwinding catalysis. The percentage of the unwounded RNA molecules that could be detected in these samples was less than 1%.
Figure 3.
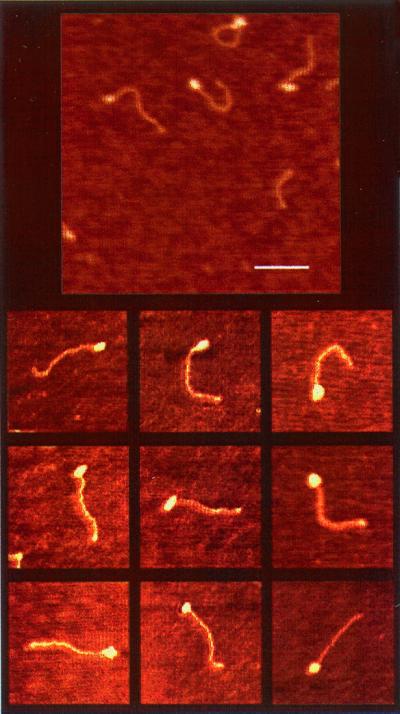
AFM images of DbpA–RNA complexes. (Bar = 100 nm.) Globular structures at the end of the RNA molecule represent protein signals. The mean end-to-end contour length is 135 ± 4 nm, which is consistent with the free dsRNA. Visualization of protein signals at the end of the RNA molecule indicates that DbpA requires an ssRNA or moderate fork junction for binding before performing the unwinding activity.
Visualization of Unwinding Activity of dsRNA by DbpA.
Fig. 4 shows a collection of DbpA–RNA complexes after the addition of ATP. In these assays, the protein concentration was maintained equal or slightly below the Kd. The ATP hydrolysis of DbpA in the presence of this dsRNA substrate was confirmed by measuring the amount of 32P release after incubation at 37°C by TLC (data not shown). Fig. 4 shows that after addition of ATP, the protein translocates along the substrate, and a Y-shaped structure, representing strand-separation, is observed. Unwinding activity was not detected when the nonhydrolyzable ATP analog 5′-adenylylimidophosphate (AMP-pNp) was used instead of ATP (data not shown). These results suggest that translocation of DbpA through duplex RNA regions is a direct consequence of the energy released by ATP hydrolysis. Protein signals are assigned to globular structures observed at the fork junction of the Y and often at the top of the Y arms (Fig. 4). The height of these protein signals is 2.1 nm ± 0.3. Regardless of how far the fork junction has progressed, the protein signals at the ends of the arms remain stationary. In contrast, DbpA bound at the fork junction does translocate. It seems likely that protein–ssRNA complexes left behind on the Y-shaped arms form because of the availability of additional ssRNA after unwinding catalysis and because of free protein in solution. However, we cannot rule out the possibility that the formation of these complexes keeps the RNA from reannealing after strand separation.
Figure 4.

AFM images of DbpA–RNA complexes in the presence of ATP (DbpA–RNA–ATP). The Y structure, observed in all images, represents the most dominant intermediate trapped during unwinding catalysis. It comprises two single ssRNA regions at the arms and dsRNA at the trunk of the Y. Protein signals were observed as globular structures at the fork junction (a--d and f) and on the tips of the Y-shaped arms (a, b, d, and f). Because of the structure of the RNA construct, two proteins can sometimes be seen entering the dsRNA region from opposite sides, as shown in d. On the other hand, e shows a Y-shaped molecule without a protein signal at the fork junction. The formation of this complex may result from substrate reannealing, or alternatively, the central protein may be washed away during sample preparations. The measured end-to-end contour length of the Y-shaped molecules shows a significant elongation (see Fig. 5c) because of the ssRNA formation during unwinding catalysis. In addition, the variation in the amount of unwinding among the Y-shaped complexes are snapshots representing the distribution of structural conformations during catalysis.
Although the unwinding activity of DbpA can be observed directly by direct visualization (Fig. 4), this activity still should be subjected to statistical data analysis. Therefore, the following strategy of data analysis was developed. Each sample was examined for the amount of Y-shaped structures that could be detected after the addition of ATP to the prebound DbpA–RNA complex. For fresh protein batches, the amount of Y-shaped complexes was between 40% and 50%. Y-shaped structures could not be detected when inactive protein was used.
End-to-End Distance Distribution of Y-Shaped Functional DbpA–RNA Molecules.
To exclude the possibility that the Y-shaped protein–RNA structures are the product of random associations or nonspecific interactions, we developed a specific scoring method. In this procedure, we scored only those molecules that contained double- and single-stranded regions and a protein signal at their fork junction or at the tips of the Y-shaped arms. Double- and single-stranded regions were identified by their height and contour lengths. Analysis of height distribution of dsRNA and ssRNA was consistent with 0.9 nm ± 0.1 and 0.6 nm ± 0.1 (n = 200), respectively. Although the height of the molecule is influenced directly by the surface treatment of the mica (33), these values are in good agreement with the reported height of dsRNA and ssRNA measured by AFM (31). After scoring all of the Y-shaped DbpA–RNA complexes, we measured the total contour length of the molecules and compared it to the expected calculated mean contour length value that was obtained from Eq. 2.
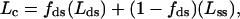 |
2 |
where Lc is the expected contour length of the molecule that contains a fraction of the dsRNA region and a residual fraction of a fully extended ssRNA region; Lds is the measured end-to-end mean contour length of the free RNA molecule; fds is the measured fraction of dsRNA; (1 − fds) is the fraction of the residual ssRNA fraction at the Y-shaped arms; and Lss is the optimum contour length of an ssRNA. Lss was calculated by using two different rises per base values (0.3 and 0.6 nm/base) obtained from crystallography (29) and AFM (31).
The mean contour length distributions of the measured and calculated values are 164 ± 3 nm, 143 ± 4 nm (for 530 nt × 0.3 nm/base), and 230 ± 4 nm (for 530 nt × 0.6 nm/base), respectively (Fig. 5c and Table 1). A rather longer rise per base value was obtained by AFM measurements (0.2–0.6 nm/base) for different sizes of ssRNA molecules deposited on various types of treated mica (31). It can be explained by the interaction of the flexible ssRNA molecule with the mica. Calculations of the rise per base of the ssRNA regions in the Y-shaped arms are consistent with a value of 0.45 nm/base, which is in a good agreement with AFM measurements of ssRNA of 400–600 bases long (31). Comparisons of the mean end-to-end distance distribution of free RNA, prebound DbpA–RNA, and the DbpA–RNA–ATP complex show a pronounced elongation of the Y-shaped complexes. This elongation is expected if unwinding is taking place, because the ssRNA molecule deviates from an A conformation and retains a somewhat more flexible nature. Therefore, each Y-shaped molecule maintains its integrity and represents a functional DbpA–RNA–ATP complex.
The imaging of ssDNA/RNA is problematic, because the molecule tends to fold on itself (31). Such folded structures were often observed as background or when the reaction went to completion. The single-stranded regions within the Y-shaped molecule could be detected, because they were stabilized by the dsRNA region and proteins that “pinned” the end of the Y-shaped molecule to the surface and prevented the folding of ssRNA. However, the allocation of single complexes on the surface of the mica was difficult, because the single stranded regions of the Y-shaped molecule tend to form head-to-head or side-to-side interactions with other molecules. However, sufficient separation of Y-shaped complexes could be achieved after diluting and vortexing the samples before depositing them on the mica. However, these results provided an additional indication that unwinding catalysis is taking place on the addition of ATP, because this effect was not observed for free RNA and the prebound DbpA–RNA complex.
Despite the importance of RNA helicases in cellular processes, little is known about their molecular mechanism of translocation and unwinding (2, 10). Recent work by Jankowsky et al. (11) demonstrates that the DEAD box NPH-II is a highly processive 3′ → 5′ helicase. NPH-II was compared with the DNA helicase UvrD (34), because both enzymes showed similar modes of processivity. Therefore, it is suggested that the basic determinants of the motor activity and the unwinding mechanism of the DNA and RNA helicases may share common themes. Differences between RNA and DNA helicases may be caused by their particular biological function, which may be controlled by interactions of other proteins or specific substrates (10).
A highly specific substrate of the E. coli DEAD box RNA helicase DbpA was identified. DbpA can stimulate nearly to its maximum ATPase activity in the presence of a segment within the peptidyl transferase center in 23S rRNA (15). In addition, recent biochemical results suggest that DbpA may possess a specific helicase activity that depends on a conserved hairpin sequence.‖ Herein, we show that DbpA can unwind nonspecific sequences of long RNA duplexes. Our results suggest that despite its specificity to rRNA, DbpA can act as a nonspecific RNA helicase, providing the availability of a sufficient number of bases in the 5′ single-stranded overhang and ATP. However, we cannot rule out that substrate possessing 3′ overhang could be unwound by DbpA. The main Y-shaped protein–RNA–ATP complex, which evolves during translocation of the enzyme on RNA, suggests that the unwinding of dsRNA by DbpA is consistent with the “inch-worm”-type helicase progression during unwinding catalysis (12, 35). Although not conclusive, this proposal was made based on our AFM images. In all of the functional complexes that were visualized, we could not detect a specific RNA bending or looping by DbpA, as suggested by the model of the “rolling” mechanism. Furthermore, we suggest that the presence of the conserved motifs (36) in DbpA and other helicases provides the structural basis for these motor enzymes to be capable of translocating and unwinding long stretches of duplex nucleic acid by using ATP.
DbpA is believed to have a role in rRNA biogenesis based on its specificity to the peptidyl transferase center region (15). In addition, this enzyme can act as an ATPase and use rRNA as a cofactor (17); it can unwind specific rRNA sequences‖ and, as we show here, long RNA stretches. On the basis of this accumulated knowledge, we suggest that DbpA may use its general and specific activities for two separate functions during rRNA biogenesis. Therefore, we propose that the unique interaction of DbpA with the peptidyl transferase center depends on the accessibility of this conserved RNA region with a specific RNA-binding domain within DbpA. The specific ATPase and helicase activity of DbpA could control the correct RNA structure, as well as the binding, and release of other cellular factors involved in the assembly of the peptidyl transferase center. On the other hand, the nonpeptidyl transferase center-related helicase activity of DbpA could serve to modulate different rRNA structures during ribosomal biogenesis.
Acknowledgments
We thank Dr. Ernesto Joselevich for critical reading of this manuscript and Profs. Ada Yonath and Arthur Grollman for useful discussions. We also thank Dr. Sophie Matlis from the AFM service unit at the Weizmann Institute of Science for providing excellent technical assistance. This research was supported by the Minerva Foundation.
Abbreviations
- dsRNA
double-stranded RNA
- ssRNA
single-stranded RNA
- AFM
atomic force microscopy
- DTE
dithioerythritol
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Diges, C. M. & Uhlenbech, O. C., Fifth Annual Meeting of the RNA Society, May 30–June 4, 2000, Madison, WI, p. 306 (abstr.).
Polach, K. J. & Uhlenbech, O. C. Fifth Annual Meeting of the RNA Society May 30–June 4, 2000, Madison, WI, p. 565 (abstr.).
References
- 1.Linder P, Lasko P F, Ashburner M, Leroy P, Nielsen P J, Nishi K, Schnier J, Slonimski P P. Nature (London) 1989;337:121–122. doi: 10.1038/337121a0. [DOI] [PubMed] [Google Scholar]
- 2.de la Cruz J, Kressler D, Linder P. Trends Biochem Sci. 1999;24:192–198. doi: 10.1016/s0968-0004(99)01376-6. [DOI] [PubMed] [Google Scholar]
- 3.Gorbalenya A E, Koonin E V, Donchenko A P, Blinov V M. Nature (London) 1988;333:22. doi: 10.1038/333022a0. (lett.). [DOI] [PubMed] [Google Scholar]
- 4.Egelman E H. J Struct Biol. 1998;124:123–128. doi: 10.1006/jsbi.1998.4050. [DOI] [PubMed] [Google Scholar]
- 5.Luking A, Stahl U, Schmidt U. Crit Rev Biochem Mol Biol. 1998;33:259–296. doi: 10.1080/10409239891204233. [DOI] [PubMed] [Google Scholar]
- 6.Schmid S R, Linder P. Mol Microbiol. 1992;6:283–291. doi: 10.1111/j.1365-2958.1992.tb01470.x. [DOI] [PubMed] [Google Scholar]
- 7.Hirling H, Scheffner M, Restle T, Stahl H. Nature (London) 1989;339:562–564. doi: 10.1038/339562a0. [DOI] [PubMed] [Google Scholar]
- 8.Liang L, Diehl-Jones W, Lasko P. Development (Cambridge, UK) 1994;120:1201–1211. doi: 10.1242/dev.120.5.1201. [DOI] [PubMed] [Google Scholar]
- 9.Iost I, Dreyfus M, Linder P. J Biol Chem. 1999;274:17677–17683. doi: 10.1074/jbc.274.25.17677. [DOI] [PubMed] [Google Scholar]
- 10.Linder P, Daugeron M C. Nat Struct Biol. 2000;7:97–99. doi: 10.1038/72464. [DOI] [PubMed] [Google Scholar]
- 11.Jankowsky E, Gross C H, Shuman S, Pyle A M. Nature (London) 2000;403:447–451. doi: 10.1038/35000239. [DOI] [PubMed] [Google Scholar]
- 12.Lohman T M, Bjornson K P. Annu Rev Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 13.Iggo R, Picksley S, Southgate J, McPheat J, Lane D P. Nucleic Acids Res. 1990;18:5413–5417. doi: 10.1093/nar/18.18.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuller-Pace F V, Nicol S M, Reid A D, Lane D P. EMBO J. 1993;12:3619–3626. doi: 10.1002/j.1460-2075.1993.tb06035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicol S M, Fuller-Pace F V. Proc Natl Acad Sci USA. 1995;92:11681–11685. doi: 10.1073/pnas.92.25.11681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boddeker N, Stade K, Franceschi F. Nucleic Acids Res. 1997;25:537–545. doi: 10.1093/nar/25.3.537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsu C A, Uhlenbeck O C. Biochemistry. 1998;37:16989–16996. doi: 10.1021/bi981837y. [DOI] [PubMed] [Google Scholar]
- 18.Lal R, John S A. Am J Physiol. 1994;266:C1–C21. doi: 10.1152/ajpcell.1994.266.1.C1. [DOI] [PubMed] [Google Scholar]
- 19.Yang J, Shao Z. Micron. 1995;26:35–49. doi: 10.1016/0968-4328(94)00041-n. [DOI] [PubMed] [Google Scholar]
- 20.Lyubchenko Y L, Jacobs B L, Lindsay S M, Stasiak A. Scanning Microsc. 1995;9:705–724. [PubMed] [Google Scholar]
- 21.Bustamante C, Rivetti C. Annu Rev Biophys Biomol Struct. 1996;25:395–429. doi: 10.1146/annurev.bb.25.060196.002143. [DOI] [PubMed] [Google Scholar]
- 22.Hansma H G, Kim K J, Laney D E, Garcia R A, Argaman M, Allen M J, Parsons S M. J Struct Biol. 1997;119:99–108. doi: 10.1006/jsbi.1997.3855. [DOI] [PubMed] [Google Scholar]
- 23.Carey J. Methods Enzymol. 1991;208:103–117. doi: 10.1016/0076-6879(91)08010-f. [DOI] [PubMed] [Google Scholar]
- 24.Hansma H G, Laney D E, Bezanilla M, Sinsheimer R L, Hansma P K. Biophys J. 1995;68:1672–1677. doi: 10.1016/S0006-3495(95)80343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matson S W, Bean D W. Methods Enzymol. 1995;262:389–405. doi: 10.1016/0076-6879(95)62031-1. [DOI] [PubMed] [Google Scholar]
- 26.Pugh G E, Nicol S M, Fuller-Pace F V. J Mol Biol. 1999;292:771–778. doi: 10.1006/jmbi.1999.3112. [DOI] [PubMed] [Google Scholar]
- 27.Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, Nellen W. RNA. 2000;6:563–570. doi: 10.1017/s1355838200992318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arnott S, Hukins D W, Dover S D, Fuller W, Hodgson A R. J Mol Biol. 1973;81:107–122. doi: 10.1016/0022-2836(73)90183-6. [DOI] [PubMed] [Google Scholar]
- 29.Saenger W. In: Principles of Nucleic Acid Structures. Cantor C R, editor. New York: Springer; 1984. pp. 298–330. [Google Scholar]
- 30.Rivetti C, Guthold M, Bustamante C. J Mol Biol. 1996;264:919–932. doi: 10.1006/jmbi.1996.0687. [DOI] [PubMed] [Google Scholar]
- 31.Hansma H G, Revenko I, Kim K, Laney D E. Nucleic Acids Res. 1996;24:713–720. doi: 10.1093/nar/24.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansma H G. Biophys J. 1995;68:3–4. doi: 10.1016/S0006-3495(95)80155-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansma H G, Laney D E. Biophys J. 1996;70:1933–1939. doi: 10.1016/S0006-3495(96)79757-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ali J A, Lohman T M. Science. 1997;275:377–380. doi: 10.1126/science.275.5298.377. [DOI] [PubMed] [Google Scholar]
- 35.Soultanas P, Wigley D B. Curr Opin Struct Biol. 2000;10:124–128. doi: 10.1016/s0959-440x(99)00059-7. [DOI] [PubMed] [Google Scholar]
- 36.Bird L E, Subramanya H S, Wigley D B. Curr Opin Struct Biol. 1998;8:14–18. doi: 10.1016/s0959-440x(98)80004-3. [DOI] [PubMed] [Google Scholar]