

Abstract
Water-soluble octa-acid cavitands (OA) form dimeric capsules suitable for guest incorporation. Our studies reveal that the mechanism of pyrene (Py) binding follows the rapid (< 1 ms) formation of the Py•OA complex, followed by slower binding with the second OA. The dissociation of the capsular OA•Py•OA complex occurs with a lifetime of 2.7 s, which is five orders of magnitude slower than the microsecond opening-closing (`breathing') previously observed to provide access of small molecules to the encapsulated guest. These different dynamics of the capsules have a potential impact on how the chemistry of included guests could be altered.
Self-assembled container molecules provide ready access to a wide range of phenomena arising through controlled compartmentalization, including unusual chemical reactivity, separation technologies, and storage and/or transport. A growing focus of the field is the formation of containers in aqueous solution,1–4 and in this regard metal coordination5–11 and the hydrophobic effect12–16 have proven useful as methods to drive assembly. Regarding the latter, the Gibb group has developed deep-cavity cavitands that dimerize via the hydrophobic effect into capsules. These hosts, for example the so-called octa-acid (OA), possess water solubilizing outer coats, deep hydrophobic pockets, and a hydrophobic rim to the cavity that promotes self-assembly.
OA forms mono-dispersed and kinetically stable dimeric assemblies that – by dint of its water-free, low-polarity inner space17,18 – can encapsulate molecules and act as yocto-liter reaction vessels for photochemical transformations,17,19–22 bring about the separation of hydrocarbon gases,13 engender molecular protection for the kinetic resolution of structurally similar molecules,23 engender unusual self-sorting properties,24 and lead to the electrochemical modulation of encapsulated guests.25–27 Nevertheless weakening of the hydrophobic effect by the addition of co-solvents denatures these assemblies.28
In part, the properties of the supramolecular complexes formed by OA are dictated by the nature of the guest. An understanding of the complicated relationship between host and guest can be garnered using many different physicochemical techniques. For example, the study of the dynamics of encapsulated guest movement have been probed on both the millisecond (1H NMR) and the nanosecond timescale (EPR).29,30 Furthermore, a microsecond time-scale opening of the dimeric capsule sufficient to allow access of small molecules, without dissociation of one of the OAs or guest release, has been inferred from excited state quenching studies.20,31 Nevertheless, amiss from the current state-of-affairs are studies providing knowledge of the formation/dissociation dynamics of these encapsulation complexes. Indeed the formation/dissociation dynamics of host-guest complexes in general has been underexploited because of the lack of suitable methodology.32,33 Towards addressing this, here we measure in real time the kinetics for the formation of 1:1 and 2:1 host-guest complexes formed between OA and the guest pyrene (Py). Our results reveal the different rates of each of the processes shown in Scheme 1, and demonstrate for the first time that complex disassembly and concomitant guest release are kinetically very different from the `breathing' of the complex that allows small molecule entry and egression.
Scheme 1.
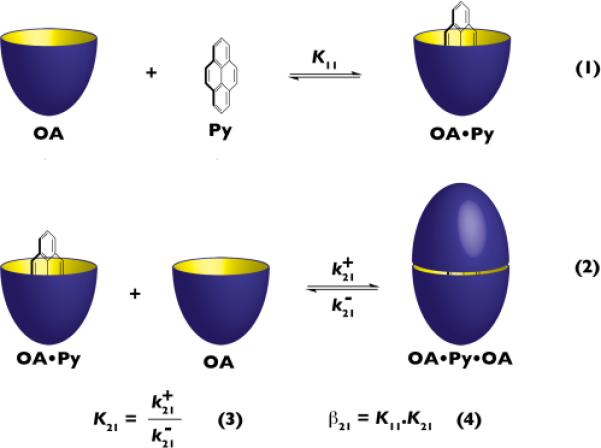
Cartoon representation for the formation of OA•Py (Equation 1) and OA•Py•OA (Equation 2), and definitions of K21 and β21 (Equations 3 and 4).
Previous 1H NMR experiments showed that OA forms a 2:1 host-guest complex with Py.17,18 Py encapsulation led to marked red shifts (5–6 nm) in its excitation (Figure 1) and absorption spectra (Figure S1; SI). Theoretically, such spectral changes can be due to the formation of Py dimers.34 However, there was no evidence of accompanying broadening of the absorption/excitation spectra, nor detection of any excimer emission (Figures S1 and S2; SI). Red shifts of 3–8 nm between the Py absorption in homogenous solution and when bound to hosts as a monomer were previously observed for Py binding to bile salt aggregates35 and a macrocycle.36 A consequence of the large absorption shift observed for OA-encapsulated Py is that depending on the excitation wavelength either a positive or negative change in the fluorescence is observed (inset Figure 1).
Figure 1.
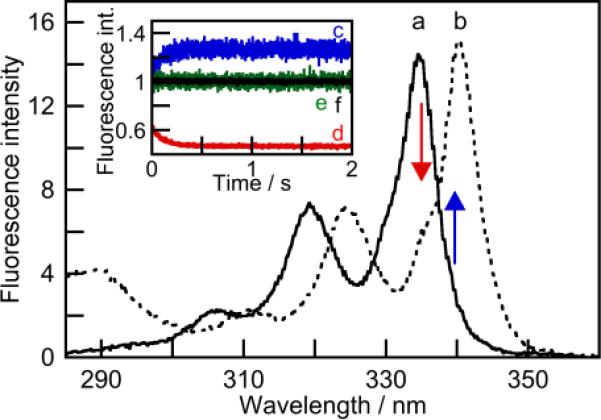
Fluorescence excitation spectra for Py (0.2 μM, λem = 390 nm) in the absence (a, solid line) and presence of OA (b, dashed line, 4 μM) at pH 8.9 (10 mM borate buffer). The arrows show the direction of the intensity change when Py is encapsulated into OA. The inset shows the kinetics for Py (0.2 μM) mixing with OA (3 μM) when excited at 340 nm (c, blue) or 335 nm (d, red). The intensities for the mixing of Py with buffer (e, green, λex = 340 nm; f, black, λex = 335 nm) were normalized to 1. The higher signal-to-noise ratio for excitation at 335 nm reflects the higher intensity of the Hg-Xe excitation lamp at this wavelength.
Time-resolved fluorescence measurements provide further evidence of monomeric Py complexation, its isolation from the aqueous phase in the dimeric OA capsule, and the absence of any significant amount at equilibrium of the 1:1 complex. The fluorescence lifetime of 361 ± 1 ns for Py (0.5 μM) in the presence of OA (10 μM) was much longer than in aqueous borate buffer (130 ± 1 ns), and is similar to that previously reported.18 Furthermore, the iodide anion (I−) quenching rate constant for singlet excited state Py in the capsule was measured to be (5.9 ± 0.7) × 105 M−1 s−1 (Figure S3; SI); a value much lower than in aqueous buffer ((1.0 ± 0.2) × 109 M−1 s−1). Both of these metrics confirm the efficient protection afforded by the capsule. Additionally, no other Py emission with an intermediate lifetime between free and encapsulated Py was observed, indicating that the 1:1 complex was not present in any appreciable amount.
The quenching rate constant for the singlet excited Py encapsulated in OA•Py•OA by iodide is similar to that previously determined for oxygen quenching of encapsulated triplet excited Py (5 × 105 M−1 s−1).31 This similarity supports the interpretation that the quenching rate constant for an excited guest in the capsule is determined by a partial opening (or `breathing') of the capsular complex that allows small guests to enter and egress the complex.31 Such a mechanism predicts similar rate constants for both different quenchers and different excited states of the same guest.
An understanding of the kinetics of assembly and disassembly of the host-guest complex requires knowledge of the types of complexes present at equilibrium, and their respective equilibrium constants. Fluorescence intensity change measurements as a function of OA concentration were carried out to determine the overall binding constant (Figure S4; SI). The resulting binding isotherm (Figure 2 and Figure S5; SI) fitted the expected 2:1 binding model and gave an overall β21 value of (3.19 ± 0.06) × 1012 M−2.
Figure 2.
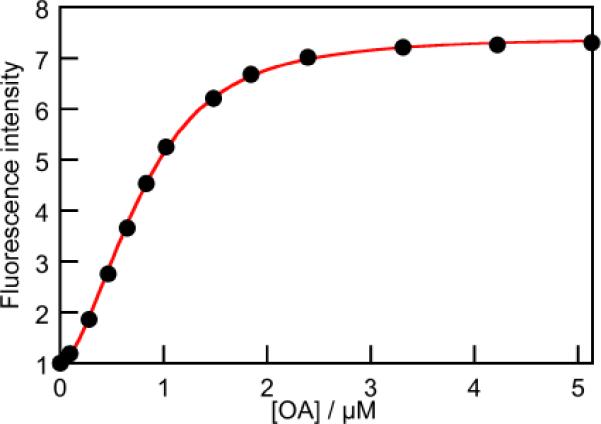
Binding isotherm for the complexation of Py (0.2 μM, λex = 340 nm) with OA obtained from steady-state fluorescence experiments. The data were fit to the sequential model shown in Scheme 1.
Because of the transient nature of the 1:1 complex, the equilibrium constant for this species (K11) can only be determined from kinetic studies. Stopped-flow experiments revealed that the kinetics for complex formation followed a two-phase behavior (Figure 3). First, an initial offset was observed between the intensity for Py in water (black line) and the intensities for Py in the presence of OA (colored lines). This result indicates that a reaction occurred within the 1 ms mixing time of the stopped-flow experiment. Furthermore, the magnitude of this offset intensity increased as the concentration of OA was raised. Consequently, a series of stopped-flow experiments in which the initially measured fluorescence intensity was plotted against [OA] revealed a 1:1 binding isotherm (Figure S6; SI). This fitting to a 1:1 model confirmed that the initial fast reaction was the binding of Py to OA, and revealed an equilibrium constant for this process of K11 = (4.5 ± 0.6) × 105 M−1. With K11 and β21 in hand a K21 value of 7 ± 1 × 106 M−1, was calculated using equation 4 (Scheme 1). The determination of both K11 and K21 for the guest Py represent the first glimpse of the effect of desolvating the rim defined by the `uppermost' aromatic rings around the portal of the host (see structure). In the formation of the 1:1 complex one hydrophobic pocket and half a guest are desolvated. In the capping of the 1:1 complex to form the capsular 2:1 complex there is this same desolvation, plus the additional desolvation of the rims as the hosts clamp together around the guest. This additional factor means that the assembly demonstrates positive cooperativity (K11 < K21), and explains why the 1:1 complex is not present at equilibrium in any appreciable amount.
Figure 3.
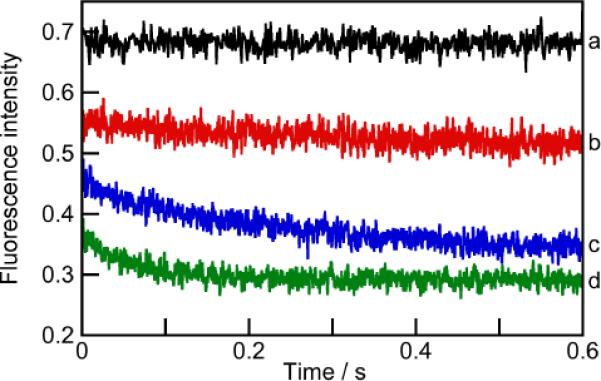
Kinetics for Py (0.2 μM, λex = 335 nm) mixing with buffer (a) or with OA (b: red, 1 μM; c: blue, 2 μM; d: green, 4 μM).
Returning to Figure 3, although these kinetic experiments revealed K11, these experiments cannot provide any kinetic information for the formation of OA•Py because the rate of the 1:1 complex formation is faster than the time-resolution of the stopped-flow experiments.
The fast initial intensity offset observed in the stopped-flow experiments was followed by a further slow decrease in the Py emission intensity (Figure 3). The kinetics leveled off within 10 s (only 0.6 s are shown in Figure 3, see Figure S7 in the SI for full data). The normalized intensity change measured at equilibrium in the kinetic experiment was the same as the intensity obtained from the steady-state experiments (Figure S8; SI), showing that the kinetics were completed within 10 s. This result eliminated the possibility of any change in the assembly occurring over longer periods of time. The kinetics of this slower process followed a mono-exponential decay (Figures S9 and S10; SI) that were assigned to the formation of OA•Py•OA from the bimolecular reaction between OA•Py and OA. Overall rate constants for relaxation processes always correspond to the sum of the rate constants for the forward and backward reactions, where an increase in the observed rate constant is expected when the concentrations of reagents involved in the bimolecular process are raised.32,33 The faster decay of the kinetics observed at higher OA concentrations (Figure 3) confirms that this process corresponds to a bimolecular reaction involving OA.
A global analysis method was employed in which kinetic traces at different OA concentrations were simultaneously fit to two models based on equations 1 and 2, where in both cases the value of K11 was fixed. In one model the dissociation rate constant was included, while in the second model this rate constant was considered infinitesimally small. Both models produced the same values for (Figure S11 and table S1; SI), supporting the conclusion that the contribution of the dissociation processes to the observed rate constant is negligible. The average value for determined from two independent experiments was (2.6 ± 0.2) × 106 M−1 s−1, whilst a value for of (0.37 ± 0.06) s−1 was calculated from equation 3. This latter rate constant corresponds to a lifetime for the dissociation of the OA•Py•OA capsule of 2.7 s.
As discussed above, the opening-closing of OA•Py•OA to provide access of Py to small guests, such as O2 or I−, but without release of Py was estimated to be in the microsecond timescale. Hence, even for ill-fitting Py37 the `breathing' dynamic is at least 100,000 times faster than the release of Py from the capsule. It is important to note that the value for OA•Py•OA does not correspond to the dissociation of the guest-free OA•OA dimer since there is no evidence to suggest that OA forms significant amounts of capsule in the absence of guest at such low concentrations of OA and Na+.14,29
The association of 1:1 host-guest complexes where the guest fits into the cavity of hosts is frequently fast and close to a diffusioncontrolled process. For example, the association rate constant for guests binding to cyclodextrins38 or the binding of a napththalene derivative to cucurbit[7]uril39 were determined to be 4 – 10 × 108 M−1 s−1. A similar rate constant for the Py•OA formation would explain the fast (< 1 ms) equilibration observed. The slower rate for the binding of the second OA is analogous to the slower rates observed for the formation of the 2:2 β-cyclodextrin-Py complex.40 Similarly, our results here demonstrate that the dynamics of the OA capsule system is defined by the dissociation rate constant of the higher order complex, i.e. OA•Py•OA. Where OA capsules differ from cyclodextrins is in the slow release of the guest from the OA capsule compared to the much faster `breathing' dynamics. These two kinds of opening processes suggest that it should be possible to selectively enhance `bimolecular' reactions involving the ingress of small molecules whilst maintaining the general structure of the complex. Previous results showed that capsule confinement affects the bimolecular reactivity of guests,41,42 and reactions with encapsulated guests can occur with small external molecules entering the capsule.20 Our results suggest that it may be possible to filter out competing reactions with larger reagent molecules attempting to enter the complex. The engendering of this separate dynamic `breathing' process resides in the fact that the OA capsule leads to true compartmentalization; a phenomenon that cannot occur with simple macrocyclic hosts. Consequently, the use of OA capsules as host systems offers more versatility to differentially influence competitive reaction pathways of guests. Future exploitation of the differences in the capsule dynamics will aid the rational design of functional OA supramolecular systems, and are currently underway.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the Natural Sciences and Engineering Council of Canada (NSERC) and by the National Institutes of Health (GM074031).
Footnotes
Supporting Information. Experimental details, absorption and fluorescence spectra, quenching plots, model for binding isotherms, analysis of stopped-flow kinetics. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Pluth MD, Raymond KN. Chem. Soc. Rev. 2007;36:161–171. doi: 10.1039/b603168b. [DOI] [PubMed] [Google Scholar]
- (2).Biros SM, Rebek J., Jr. Chem. Soc. Rev. 2007;36:93–104. doi: 10.1039/b508530f. [DOI] [PubMed] [Google Scholar]
- (3).Yoshizawa M, Klosterman JK, Fujita M. Angew. Chem. Int. Ed. 2009;48:3418–3438. doi: 10.1002/anie.200805340. [DOI] [PubMed] [Google Scholar]
- (4).Laughrey Z, Gibb BC. Chem. Soc. Rev. 2011;40:363–386. doi: 10.1039/c0cs00030b. [DOI] [PubMed] [Google Scholar]
- (5).Takeda N, Umemoto K, Yamagushi K, Fujita M. Nature. 1999;398:794–796. [Google Scholar]
- (6).Pluth MD, Bergman RG, Raymond KN. Science. 2007;316:85–88. doi: 10.1126/science.1138748. [DOI] [PubMed] [Google Scholar]
- (7).Pluth MD, Bergman RG, Raymond KN. J. Org. Chem. 2009;74:58–63. doi: 10.1021/jo802131v. [DOI] [PubMed] [Google Scholar]
- (8).Suzuki K, Sato S, Fujita M. Nat. Chem. 2010;2:25–29. doi: 10.1038/nchem.446. [DOI] [PubMed] [Google Scholar]
- (9).Chakrabarty R, Mukherjee PS, Stang PJ. Chem. Rev. 2011;111:6810–6918. doi: 10.1021/cr200077m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Sun Q-F, Murase T, Sato S, Fujita M. Angew. Chem. Int. Ed. 2011;50:10318–10321. doi: 10.1002/anie.201104670. [DOI] [PubMed] [Google Scholar]
- (11).Wang ZJ, Brown CJ, Bergman RG, Raymond KN, Toste FD. J. Am. Chem. Soc. 2011;133:7358–7360. doi: 10.1021/ja202055v. [DOI] [PubMed] [Google Scholar]
- (12).Gibb CLD, Gibb BC. J. Am. Chem. Soc. 2004;126:11408–11409. doi: 10.1021/ja0475611. [DOI] [PubMed] [Google Scholar]
- (13).Gibb CLD, Gibb BC. J. Am. Chem. Soc. 2006;128:16498–16499. doi: 10.1021/ja0670916. [DOI] [PubMed] [Google Scholar]
- (14).Liu S, Gibb BC. Chem. Commun. 2008:3709–3716. doi: 10.1039/b805446k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hiraoka S, Nakamura T, Shiro M, Shiomoya M. J. Am. Chem. Soc. 2010;132:13223–13225. doi: 10.1021/ja1069135. [DOI] [PubMed] [Google Scholar]
- (16).Gan H, Benjamin CJ, Gibb BC. J. Am. Chem. Soc. 2011;133:4770–4773. doi: 10.1021/ja200633d. [DOI] [PubMed] [Google Scholar]
- (17).Kaanumalle LS, Gibb CLD, Gibb BC, Ramamurthy V. J. Am. Chem. Soc. 2004;126:14366–14367. doi: 10.1021/ja0450197. [DOI] [PubMed] [Google Scholar]
- (18).Porel M, Jayaraj N, Kaanumalle LS, Maddipatla MVSN, Parthasarathy A, Ramamurthy V. Langmuir. 2009;25:3473–3481. doi: 10.1021/la804194w. [DOI] [PubMed] [Google Scholar]
- (19).Kaanumalle L, Gibb CLD, Gibb BC, Ramamurthy V. Org. Biomol. Chem. 2007;5:236–238. doi: 10.1039/b617022f. [DOI] [PubMed] [Google Scholar]
- (20).Natarajan A, Kaanumalle LS, Jockusch S, Gibb CLD, Gibb BC, Turro NJ, Ramamurthy V. J. Am. Chem. Soc. 2007;129:4132–4133. doi: 10.1021/ja070086x. [DOI] [PubMed] [Google Scholar]
- (21).Sundaresan AK, Ramamurthy V. Org. Lett. 2007;9:3575–3578. doi: 10.1021/ol0713990. [DOI] [PubMed] [Google Scholar]
- (22).Gibb CLD, Sundaresan AK, Ramamurthy V, Gibb BC. J. Am. Chem. Soc. 2008;130:4069–4080. doi: 10.1021/ja7107917. [DOI] [PubMed] [Google Scholar]
- (23).Liu S, Gan H, Hermann AT, Rick SW, Gibb BC. Nat. Chem. 2010;2:847–852. doi: 10.1038/nchem.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gan H, Gibb BC. Chem. Commun. 2012;48:1656–1658. doi: 10.1039/c2cc16603h. [DOI] [PubMed] [Google Scholar]
- (25).Podkoscielny D, Philip I, Gibb CLD, Gibb BC, Kaifer AE. Chem. Eur. J. 2008;14:4704–4710. doi: 10.1002/chem.200701961. [DOI] [PubMed] [Google Scholar]
- (26).Podkoscielny D, Gadde S, Kaifer AE. J. Am. Chem. Soc. 2009;131:12876–12877. doi: 10.1021/ja9045108. [DOI] [PubMed] [Google Scholar]
- (27).Qiu Y, Yi S, Kaifer AE. Org. Lett. 2011;13:1770–1773. doi: 10.1021/ol200258g. [DOI] [PubMed] [Google Scholar]
- (28).Liu S, Gibb BC. Chem. Commun. 2011;47:3574–3576. doi: 10.1039/c1cc10122f. [DOI] [PubMed] [Google Scholar]
- (29).Jayaraj N, Zhao Y, Parthasarathy A, Porel M, Liu RSH, Ramamurthy V. Langmuir. 2009;25:10575–10586. doi: 10.1021/la901367k. [DOI] [PubMed] [Google Scholar]
- (30).Kulasekharan R, Jayaraj N, Porel M, Choudhury R, Sundaresan AK, Parthasarathy A, Ottaviani MF, Jokusch S, Turro NJ, Ramamurthy V. Langmuir. 2010;26:6943–6953. doi: 10.1021/la904196g. [DOI] [PubMed] [Google Scholar]
- (31).Jayaraj N, Jokusch S, Kaanumalle L, Turro NJ, Ramamurthy V. Can. J. Chem. 2011;89:203–213. [Google Scholar]
- (32).Pace TCS, Bohne C. Adv. Phys. Org. Chem. 2008;42:167–223. [Google Scholar]
- (33).Bohne C. In: Supramolecular Photochemistry: Controlling Photochemical Processes. Ramamurthy V, Inoue Y, editors. John Wiley & Sons; Singapore: 2011. pp. 1–51. [Google Scholar]
- (34).Winnik FM. Chem. Rev. 1993;93:587–614. [Google Scholar]
- (35).Ju C, Bohne C. Photochem. Photobiol. 1996;63:60–67. [Google Scholar]
- (36).Diederich F, Dick K, Griebel D. J. Am. Chem. Soc. 1986;108:2273–2286. doi: 10.1021/ja00269a025. [DOI] [PubMed] [Google Scholar]
- (37).Models demonstrate that the host must distort from its C4v geometry to a C2v point group possessing a pseudo-elliptical rather than pseudo-circular binding site.
- (38).Bohne C. Langmuir. 2006;22:9100–9111. doi: 10.1021/la061227w. [DOI] [PubMed] [Google Scholar]
- (39).Tang H, Fuentealba D, Ko YH, Selvapalam N, Kim K, Bohne C. J. Am. Chem. Soc. 2011;133:20623–20633. doi: 10.1021/ja209266x. [DOI] [PubMed] [Google Scholar]
- (40).Dyck ASM, Kisiel U, Bohne C. J. Phys. Chem. B. 2003;107:11652–11659. [Google Scholar]
- (41).Kaanumalle LS, Ramamurthy V. Chem. Commun. 2007:1062–1064. doi: 10.1039/b615937k. [DOI] [PubMed] [Google Scholar]
- (42).Parthasarathy A, Kaanumalle LS, Ramamurthy V. Org. Lett. 2007;9:5059–5062. doi: 10.1021/ol702322u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.