

Abstract
Motivation: Eukaryotic proteins are highly modular, containing multiple interaction interfaces that mediate binding to a network of regulators and effectors. Recent advances in high-throughput proteomics have rapidly expanded the number of known protein–protein interactions (PPIs); however, the molecular basis for the majority of these interactions remains to be elucidated. There has been a growing appreciation of the importance of a subset of these PPIs, namely those mediated by short linear motifs (SLiMs), particularly the canonical and ubiquitous SH2, SH3 and PDZ domain-binding motifs. However, these motif classes represent only a small fraction of known SLiMs and outside these examples little effort has been made, either bioinformatically or experimentally, to discover the full complement of motif instances.
Results: In this article, interaction data are analysed to identify and characterize an important subset of PPIs, those involving SLiMs binding to globular domains. To do this, we introduce iELM, a method to identify interactions mediated by SLiMs and add molecular details of the interaction interfaces to both interacting proteins. The method identifies SLiM-mediated interfaces from PPI data by searching for known SLiM–domain pairs. This approach was applied to the human interactome to identify a set of high-confidence putative SLiM-mediated PPIs.
Availability: iELM is freely available at http://elmint.embl.de
Contact: toby.gibson@embl.de
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Short linear motifs (SLiMs) are compact domain binding interfaces ubiquitous in eukaryotic proteomes. They mediate a range of important cellular processes including protein\vvp scaffolding [e.g. SOS1 SH3 motifs (Kaneko et al., 2008)], cell signalling [e.g. PDZ motifs (Lee and Zheng, 2010)], subcellular compartment targeting (e.g. nuclear localization signals (Fontes et al., 2003)], post-translational modification [e.g. sumoylation (Yang and Gregoire, 2006)] and cleavage [e.g. caspase 3 cleavage sites (Pop and Salvesen, 2009)]. SLiMs consist of ∼3–10 amino acids though usually only 2–4 residues are strictly required for binding. As a result of the limited number of residues contacting their binding partner, SLiMs bind with low affinity [usually between 1.0 and 150 micromolar (Diella et al., 2008)] distinguishing them from domain–domain interactions that often have an affinity in the nanomolar range (Neduva et al., 2005). This attribute of a weak-binding affinity renders SLiM-mediated interactions difficult to detect experimentally (Diella et al., 2008). A number of resource- and time-intensive experiments are therefore required to properly validate a SLiM, ranging from mutational analysis to structural studies (Davey et al., 2012). The use of bioinformatics is therefore an important technique to direct or augment the experimental elucidation of SLiMs.
A number of databases have been developed to facilitate our understanding of SLiMs. The Eukaryotic Linear Motif (ELM) resource (Dinkel et al., 2012) contains over 1600 experimentally validated SLiM instances while the Minimotif Miner (Mi et al., 2012) database has collected over 880 consensus sequences. These datasets generate insights into the attributes of SLiMs, such as their conservation among homologues and enrichment in disorder. This enables the development of prediction servers within both the ELM and Minimotif Miner resources to filter novel instances based on the attributes of the curated regular expressions. However, both servers have issues with over-prediction. The SLiMSearch resource (Davey et al., 2011) expands this methodology to whole proteome searches. This method scores a SLiM instance by assessing the sequence conservation of the motif in its orthologous proteins, however, disordered regions are often poorly aligned and this can lead to an artificially low score for some motifs (Perrodou et al., 2008). The Anchor (Meszaros et al., 2009) predictors rely on the propensity for SLiMs to undergo a disorder-to-order transition upon binding and α-MORF-Pred (Mohan et al., 2006) identifies patterns in a disorder prediction output. Other resources have focused on a subset of SLiMs (Hui and Bader, 2010; Li et al., 2008), for example, ScanSite (Obenauer et al., 2003) was established to identify short protein sequence motifs based on peptide library and phage display experiments.
The growth in the number of protein complexes with a determined 3D structure has facilitated the development of structural tools to predict SLiM specificities (Betel et al., 2007; Encinar et al., 2009; King and Bradley, 2010; Petsalaki et al., 2009; Stein and Aloy, 2010). The ADAN database (Encinar et al., 2009) utilizes the FoldX algorithm (Schymkowitz et al., 2005) to perform an assessment of the stability and affinity of peptide–domain complexes under in silico mutagenesis analysis. However, the requirement for extensive knowledge of these interfaces has generally curtailed this type of method to well-studied and ubiquitous domains, such as the SH3, SH2 and PDZ domains (Encinar et al., 2009; Stein and Aloy, 2010). The exception is PepSite (Petsalaki et al., 2009), which provides a generic method to predict peptide binding by using a position-specific scoring matrix to predict peptide binding though this all-encompassing approach lead to a decrease in accuracy when compared with domain-specific methods. SLiM prediction has also taken advantage of the recent advances in high-throughput proteomics (Beltrao and Serrano, 2005; Edwards et al., 2007; Linding et al., 2007; Neduva et al., 2005), for example, Dilimot (Neduva et al., 2005) and SLiMFinder (Edwards et al., 2007) identify novel SLiM classes by searching for enriched motifs within interaction data while NetworKIN (Linding et al., 2007) uses protein–protein interaction (PPI) data to elucidate the kinase associated with a particular phosphorylation site. However, the inherent noise within PPI networks hinders these methods. Despite these advances in the area of SLiM discovery tools, outside the intensively experimentally studied SH3, SH2 and PDZ domains, the expected deluge of new SLiM instances and classes has not occurred. Nevertheless, there is clearly signal in each of the methods described as demonstrated by the positive results produced in the analyses of Translin (Neduva et al., 2005), EH-1 (Copley, 2005) and KENBox (Michael et al., 2008) SLiM classes, as well as, the identification of kinases associated with particular phosphorylation sites by NetworKin (Linding et al., 2007).
In this study, we produce a high-confidence list of human SLiM-mediated interfaces by creating a method (iELM) that identifies SLiM–domain partners from interaction data. A dataset of SLiM-binding domains and SLiM-mediated interactions was manually curated from the literature. These annotated domains were used to train Hidden Markov Models (HMMs) to specifically recognize SLiM-binding domains associated with a particular ELM class. To identify true SLiM instances a combination of methods, relying on known SLiM attributes, were incorporated allowing the assessment of a binary interaction for a complimentary SLiM-domain partnership. This association is also assessed for structural feasibility by the structural bioinformatics tool, PepSite. The iELM method enables the analysis of the human interactome for SLiM-mediated interfaces and interactions. A list of high-confidence SLiM-mediated interfaces for the human interactome is produced and can be accessed at http://elmint.embl.de.
2 METHODS
iELM assesses a binary interaction for a SLiM–domain interface and, if present, outputs the SLiM sequence and the globular domain putatively responsible for binding.
2.1 Datasets
The SLiM functional classes used in iELM were extracted, in the form of a regular expression, from the ELM database (2011-03). The ELM resource annotation did not include information about the binding partners and binding domain for each ELM class. To identify this information, the 3DID resource (Stein et al., 2011) was parsed for the SLiM-binding domains in complex with a peptide from an ELM class; however, this search only identified 28% (44) of the binding domains for the ELM classes. To identify the remaining 72% (112) of SLiM-binding domains a literature search was undertaken. The annotation process recorded the UniProt ID, the binding domain and the domain's position within the sequence as well as, when possible, the affinity of the binding (see Supplementary Table S3).
2.1.1 Annotation of true positive SLiM-mediated interface dataset
The true positive dataset is the experimentally annotated dataset of SLiM–domain interaction interfaces (SLiMDoM dataset) based on the aforementioned literature survey and the crystal structures retrieved from the 3DID database. The SLiMDom test dataset consists of 1080 SLiM–domain-mediated interactions and the training set comprises of 434 SLiM–domain-mediated interactions. This dataset was divided for each ELM class in a 3:1 divide with respect to testing and training.
A second true positive dataset based on the annotation from the Domino (Ceol et al., 2007) resource (version 2009-10) was also assembled. The Domino database annotates the sequences of peptides experimentally shown to bind to a particular globular domain. With our a priori knowledge of the Pfam domain (Finn et al., 2010) that binds an ELM class, the appropriate ELM regular expression (Dinkel et al., 2012) was used to search within the binding peptides. The results were recorded and are referred to as the Domino dataset (Supplementary Table S4) consisting of 1684 interactions.
2.1.2 False positive or control SLiM-mediated interface datasets
Experimentally validated negative instances are too rare to be used as a control group. Instead a false positive dataset of SLiM-mediated interfaces unlikely to be true was constructed. The majority of these interfaces are likely to be true negatives, however, since our knowledge of SLiMs and PPIs is incomplete, this set will undoubtedly contain functional instances and true interactions.
Two false positive datasets (SLiMDoM- and Domino-False Positive Datasets) were created to be specific controls for each of the aforementioned true positive datasets and the same procedure was applied to each. First, all proteins in these datasets were collected along with their associated ELM class(es). These proteins were combined in all possible combinations such that in a dataset of 10 proteins, each protein would have nine interactions. This list was then filtered for proteins associated with the same ELM class as well as for known interactions [using STRING resource v9.0 (Szklarczyk et al., 2011)]. After these filtering steps, 211 600 protein pairs were present for the false positive SLiMDoM dataset and 111 156 pairs were present within the false positive Domino dataset. These datasets were pruned to produce two datasets each containing 30 000 interactions. The datasets used to train the support vector machine (SVM) algorithm are described in the Supplementary Material.
A final test dataset was constructed to assess the performance of the iELM method on ‘real-world’ PPI data from the BioGrid (Stark et al., 2011) database (version 3.1.70). This PPI network was randomized by node degree conservation using the Neat web server (Brohee et al., 2008) to ensure the underlying structure of the network remained intact.
2.2 HMM production
The HMMs were trained on a multiple sequence alignment consisting of the experimentally annotated SLiM-binding domain instance and its orthologous proteins. The underlying assumption of this being that the orthologous domains of the annotated domain would also bind the motif. The orthologous sequences of the annotated protein were identified using the Gopher programme (Davey et al., 2007) to search the UniProt database (UniProt release 2011-05) (UniProt Consortium, 2010) by BLAST reciprocal best hit for each species (Altschul et al., 1990). These orthologous proteins were aligned using the multiple sequence alignment programme Muscle (Edgar, 2004) and the position of the SLiM-binding domain identified within the alignment. To remove poorly sequenced and/or incorrectly identified orthologues, aligned domains with indels covering >10% of the reference domain sequence were removed. The sequences were then iteratively realigned and poorly aligned sequences removed until a set of orthologues were identified with <10% indel coverage compared with the curated reference SLiM-binding domain. The HMMs were trained on this alignment using the HMMer programme's (Eddy, 1998) HMMBuild. The HMMs produced by this process are the ‘domain identifier’ HMMs. For the benchmarking, only the 434 HMMs made from the SLiMDom training set were used.
2.3 Modelling domains for PepSite
PepSite requires a Protein Data Bank (PDB) structure in order to predict the binding position of a peptide. The sequences of all the 3D structures from the PDBe database (Velankar and Kleywegt, 2011) were blasted against the human UniProt (UniProt Consortium, 2010) sequences for matches with a sequence identity of >30%. For all the non-identical matches detected, structural models of the domain were produced using the MODELLER programme (Eswar et al., 2006) (see Supplementary Fig. S4 for receiver-operating characteristic curve (ROC) for PepSite benchmarking on models).
2.4 Training SVM kernel
The score for the iELM resource is calculated using a SVM learning algorithm (Joachims, 2002). The SVM algorithm was trained on the SVM true positive and SVM false positive datasets (see Supplementary Material). The iELM method was run with 75% of the data used as a training dataset and 25% as a test dataset and a SVM trained model produced.
2.5 Method outline
2.5.1 Domain identifier
The HMMer package's HMMSearch programme was used to search a sequence using the domain identifier HMMs. The domain identifier uses an E-value cut-off of 0.01 (Finn et al., 2010) and, in order to remove fragment hits, all hits with a length of <80% of the annotated SLiM-binding domain's length were also rejected; if a result is returned, the E-value score(s) is converted into a domain score. The domain score is a similarity score to the optimal score of an annotated SLiM-binding domain of similar length. This calculation was based on the equation of the regression line calculated from the optimal E-value hit for each domain against the length of the annotated HMM (Pearson's correlation value 0.96). The HMM_length is the length of the HMM used to make the prediction and the E-value is the estimated likelihood calculated by the HMMSearch programme:
2.5.2 iELM method
iELM predicts the SLiM-mediated interfaces of a single binary interaction by combining the domain identifier with the motif discovery programme SLiMSearch (Davey et al., 2011), the disorder predictor IUPred (Dosztanyi et al., 2005) and the structural analysis programme PepSite (Petsalaki et al., 2009) (see workflow in Fig. 1).
Fig. 1.
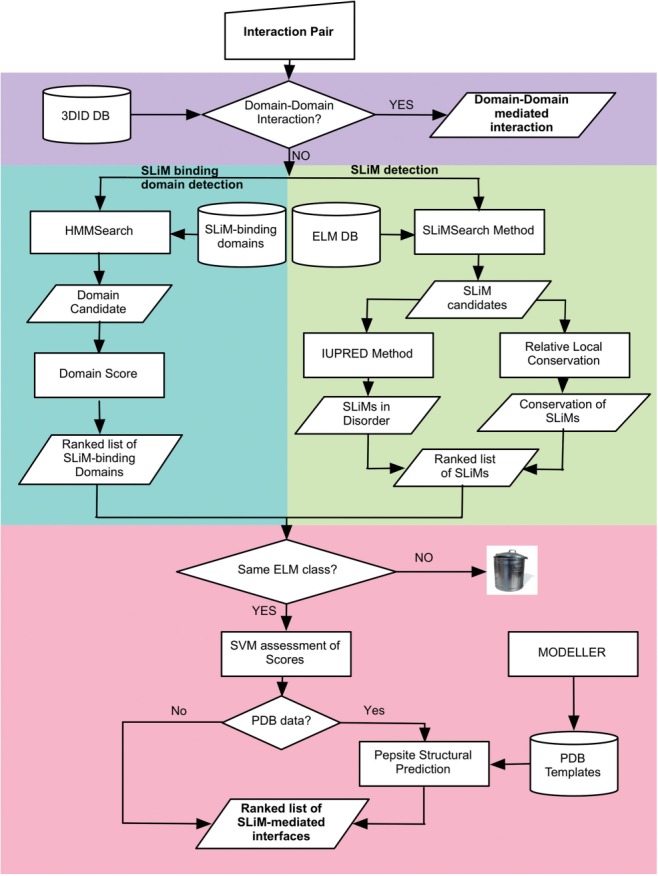
A workflow for the iELM method. The pipeline proceeds through four major stages utilizing the 3DID resource (purple), the SLiM-binding domain identification method (green), the SLiMSearch methods (yellow) and the PepSite structural bioinformatic methods (red). After this step, the bioinformatic pipeline ends and laboratory verification is required.
2.5.3 Interface-pair identification
A binary interaction is first queried for interacting domains as annotated in the 3DID resource (Stein et al., 2011). The identification of a putative domain–domain interaction between the binary partners leads to the search being discontinued and the domain–domain interaction being returned. Otherwise, the two proteins in the binary interaction are searched using the following two procedures. The Domain identifier searches a sequence using the domain identifier HMMs in order to identify putative SLiM-binding domains. If a putative SLiM-binding domain is present, a search is undertaken for the corresponding SLiM of the same ELM class in the interacting protein. The SLiMSearch programme uses a regular expression, annotated within the ELM resource, to identify potential SLiMs and assigns a Relative Local Conservation (RLC) score of the residues based on a multiple alignment of the sequence and its orthologues [see Davey et al. (2011) for details]. The SLiM and its surrounding residues are then assessed for their propensity to be in a region of intrinsic disorder using IUPred. The SLiMSearch programme also outputs a score for the Conservation Score (Chica et al., 2008) and a RLC variance score indicating the differences in conservation between the individual amino acids of the SLiM instance. Contextual information such as overlapping Pfam Domains and PDB structures (Velankar and Kleywegt, 2011) is also included.
2.5.4 Interface-pair scoring
If a complimentary SLiM–domain association is found then the score from the domain identifier and the SLiM detection methods are assessed using a SVM trained model, otherwise the search discontinues. The following scores are considered using SVMlight classify programme (Joachims, 2002) for assessment: Domain score, RLC score, RLC variance, IUPred disorder score, the Conservation score and HMM length. Finally, the SLiM–domain interface is assessed using PepSite, to test whether or not the binding is biophysically feasible. This requires a PDB structure (or a model) of the putative SLiM-binding domain. If such a 3-dimensional structure is available, PepSite analyses the SLiM-binding domain for the likely binding position of the peptide, producing a putative binary complex and a score for the likelihood of the interaction. This score is not included in the iELM score calculated by the SVM, because identified SLiM-binding domains often do not have known 3D structures with >30% sequence identity and therefore cannot be assessed using PepSite.
2.6 Method assessment
2.6.1 Dataset assessment
The datasets were split into training and test datasets and assessed for sensitivity and specificity:
The true hits are considered correct if the annotated SLiM and SLiM-binding domain positions were predicted to bind with a score above the set threshold.
3 RESULTS
3.1 Features of SLiM–domain interfaces
The annotated SLiMDom dataset reveals that many globular domain classes bind to multiple ELM classes (156 ELM classes annotated to 85 globular domain functional classes). Those globular domain Pfam families that bind multiple ELM classes can be broadly divided into two categories. The first type, have an over-arching canonical SLiM with subgroups, in general, defined by slight differences in flanking residues of the motif. These classes partially overlap with changes in binding affinity distinguishing closely related subgroups [e.g. Huang et al. (2008); Kay et al. (2000)]. For example, the core constituent of the canonical SH3-binding SLiM is PxxP (x = any amino acid) with the specificity of the subgroups of this domain class arising from the flanking residues (e.g. YxxPxxP as compared to PxxPxR) (Li, 2005). The second category can also be divided into subgroups, however in contrast to the first type, no over-arching canonical SLiM can be defined, as the SLiMs associated with this type of domain family are too diverse. These subgroups often contain only paralogous proteins and have SLiM specificities that are very definitive and often exclusive to each subgroup. For example, the WD40 domains of beta-TrCP (uniprot: Q9Y297) bind to a phospho-dependent degron SLiM (LIG_SCF_TrCP_1 - DSGxxS) while the WD40 repeats of PEX7 (uniprot: O00628) binds to a seemingly unrelated SLiM (TRG_PTS2 – Rxxx[LIV]xx[HQ][LIF]) (Stirnimann et al., 2010).
A method for identifying SLiM-domains must be able to distinguish between the aforementioned subgroups. The use of HMMs to identify globular domains and transmembrane regions is well established (Eddy, 1998; Finn et al., 2010) and incorporated into resources such as Pfam. The HMMs trained by Pfam recognize functional domain groups and could therefore be used to identify SLiM-binding domains. However, these HMMs are not able to distinguish the aforementioned intra-domain binding specificities, since the training of Pfam HMMs does not take into account the subcategorization of a domain family by SLiM specificities. We therefore used the annotated and experimentally validated SLiM-binding domains (and their orthologues) to train HMMs. By incorporating known binding specificities, those HMMs trained to recognize SLiM-binding domain should distinguish the subgroups of those functional globular domains that bind multiple ELM classes (see Supplementary Material for details).
3.2 Benchmarking the domain identifier
Two types of HMMs were used: those extracted from Pfam (version 25.0) and those that we generated based on the experimentally validated SLiM-binding domains (domain identifier HMMs) (from the training set—see Section 2 for details). For each of the SLiM–domain interactions from the SLiMDoM dataset, the benchmarking assessed whether either the Pfam- or domain identifier HMMs identified the known binding domain. The domain identifier HMMs achieved a sensitivity of 84.0% (907/1080) and a specificity of 90.1% [false positive rate (FPR): 2696/30 000]. Pfam HMMs accomplished a sensitivity and specificity of 65.1% (703/1080) and 72.1% (FPR: 8370/30 000), respectively (see ROC curves in Fig. 2a) suggesting that the use of HMMs trained on SLiM-binding domains is a more effective way of identifying putative SLiM-binding domains. The domain identifier HMMs were also assessed for intra-domain specificities using the annotated SH2 and SH3 domains. The domain identifier HMMs achieved a specificity of 83.9% and a sensitivity of 80.3% (see Supplementary Fig. S2).
Fig. 2.

ROC curves and SVM kernel weights. Plots describing the properties of the domain identifier and iELM methods. (a), (b) and (c) are ROC curves. These curves are a graphical plot of sensitivity versus 1 - specificity as compared with random (the grey line). The ROC curves demonstrate that for detecting SLiM-binding domains and SLiM-mediated interfaces, respectively, the two methods are a considerable improvement over random. Furthermore, they illustrate the advantages of training HMMs on annotated SLiM-binding domains. (a) Benchmark dataset results for domain identifier method. (b) The iELM method as benchmarked against SLiMDom dataset. (c) The iELM method as benchmarked against Domino data. (d) A bar plot of the percentage of the total weight as assigned by the SVM kernel. (Domain = Domain Score, RLC_var = RLC variance, length = domain-length, IDR = intrinsic disordered regions, CS = conservation score). The ratio of weights was consistent during multiple testing with a standard deviation of 0.0087, 0.019, 0.012, 0.0068, 0.016 and 0.017 for the domain score, RLC, RLC_var, length, IDR and CS, respectively.
3.3 iELM benchmark
The iELM method was benchmarked using two separate datasets. The first consists of experimentally validated SLiM-mediated interaction data (SLiMDoM dataset) and the second is based on the Domino dataset, which is curated from the Domino database's experimentally annotated peptide–domain interactions (for full results see Supplementary Table S5). The performance of iELM on the SLiMDoM dataset using the domain identifier HMMs (cut-off = −1.0) was a sensitivity of 84.8% (916/1080) and a specificity of 86.5% (FPR: 4050/30 000) while using the Pfam HMMs decreased both the sensitivity and specificity scores to 76.1% (822/1080) and 80.4% (FPR: 5880/30 000), respectively (Fig. 2b). Using iELM (cut-off = −1.0) with the domain identifier HMMs on the Domino benchmark dataset achieved a sensitivity of 75.5% (1272/1684) and a specificity of 83.4% (FPR: 4980/30 000). In comparison, the use of Pfam HMMs managed a sensitivity and specificity of 60.9% (1025/1684) and 79.4% (FPR: 6180/30 000), respectively (Fig. 2c). The application of the SVM was contrasted to using a cut-off system, based on the recommendations in the respective papers. The cut-off version of iELM (IUPred: 0.4; Motif score: 0.5; Domain score: 0.4) on the SLiMDoM dataset achieved a slightly better specificity 89.3% (FPR: 3111/30 000) but a much lower sensitivity of 70.4% (760/1080) than the SVM-based method.
The iELM method was also benchmarked on ‘real world’ data whose interactions were collected independently of whether or not they were SLiM mediated. The BioGrid interaction dataset and a randomized version of this dataset (both containing 46 676 interactions) were assessed using iELM (cut-off = −1.0) with the domain identifier HMMs. Within the BioGrid interaction dataset, 11 153 SLiM-mediated interactions were identified compared to 1112 in the randomized network suggesting a FPR of 9.97%.
3.4 Human interactome analysis
The interfaces for the majority of PPIs are still unknown and it is therefore of interest to detect novel motif-mediated interfaces on a proteome-wide scale. A human PPI network comprising 306 211 interactions [extracted from STRING (Szklarczyk et al., 2011) v9.0; PPIs; cut-off = 0.6] was assessed using the iELM method (cut-off = −1.0). In total, 12 562 PPIs and 35 476 interfaces were predicted as SLiM-mediated by iELM, including 7251 predicted structures (PepSite score < 0.25) (Fig. 3b and Supplementary Table S2). A large number of these PPIs are mediated by multiple SLiM classes or SLiM instances, for example, in the interaction between GRB2 (uniprot:P62993) and SOS1 (uniprot:Q07889); SOS1 has seven putative SH3 motifs and GRB2 has two SH3 domains, potentially this can equate to 14 binding interfaces for a single PPI. The putative motif interface map of the human interactome, produced by the iELM method, identified a large number of potentially novel SLiM-mediated interfaces as well as demonstrating the ability of iELM to automatically annotate the edges of interactions within a PPI network. To explore the interactome produced by iELM, the putative SLiM-mediated-interaction interfaces associated with the cell division cycle protein 20 (CDC20; uniprot: Q12834) were studied. CDC20 is a regulatory subunit of the anaphase-promoting complex (APC/C) that targets proteins for ubiquitination and subsequent degradation by the 26S proteasome (Peters, 2006). In early mitosis, CDC20 joins the APC/C complex and targets substrates for ubiquitination containing either a destruction box SLiM (Glotzer et al., 1991) (D-box – RxxLxxφ − φ = hydrophobic amino acid) or a KEN-box (Pfleger and Kirschner, 2000) (xKENx). The iELM method identified 34 PPIs (from 246 binary interactions) with 41 putative SLiM-mediated interfaces that bind to CDC20 via a D-box motif. All the experimentally annotated (seven instances) ELM instances of D-box SLiMs (including human orthologues of non-human instances) were identified as well as five additional experimentally validated SLiMs (Peters, 2006). iELM identified a number of interesting candidate interfaces binding to CDC20 including the sperm-associated antigen 5 (SPAG5), a protein necessary for spindle formation during mitosis, a process whose completion synchronizes with the formation of the APC/C complex (Song and Rape, 2010) (see Supplementary Fig. S3).
Fig. 3.

SLiM-mediated Human Interface Interactome. A summary of the iELM results for the human interactome. (a) A cytoscape image (Cline et al., 2007) of a subset of the interactions found to be motif-mediated within the human interactome. The heavily-shaded and highly connected nodes (in dark purple) are the SLiM-binding-domain-containing proteins (in a clockwise order from the top left are): NEDD4, TS101, GGA3 and CLH1. In a slightly lighter shading are highlighted those nodes, identified by iELM as, containing SLiMs binding to the aforementioned SLiM-binding domains. (b) Statistics for the number of interactions and interfaces for all the SLiM-mediated interactions and then divided by type using ELM resource distinctions (LIG = ligand, MOD = modification, TRG = targeting, CLV = cleavage). (c) A table derived from the interactome shown in (a) depicting those ELM classes found with the number of times they occur. (d) The modular interactions of HGS found from the previous network. Also mapped on are interactions found from the 3DID resource (in orange or lighter shading).
In addition, we investigated a subnetwork of the human SLIM-mediated PPI network associated with four SLiM binding proteins: Clathrin heavy chain 1 (CLH1) (uniprot: Q00610), ADP-ribosylation factor-binding protein GGA3 (GGA3) (uniprot: Q9NZ52), E3 ubiquitin-protein ligase NEDD4 (NEDD4) (uniprot: P46934) and tumour susceptibility gene 101 protein (TSG101) (uniprot: Q99816), and their interactions (Fig. 3a). This subnetwork contains 810 interactions, 173 of which are predicted by iELM as SLiM-mediated interactions. This number includes SLiM interfaces from three different categories of ELM (LIG or ligand, MOD or modification, and TRG or targeting) and 14 different classes (Fig. 3c). Of these 173 putative interactions, approximately half are predicted to bind to NEDD4 via a WW-binding motif associated with ubiquitinating substrates. The remainder of the putative protein interfaces function within endocytic-related pathways; for example, the Clathrin-Box motif-mediated interactions are associated with clathrin-mediated vesicular trafficking. The protein hepatocyte growth factor-regulated tyrosine kinase substrate (HGS) (uniprot: O14964) was extracted from this network and its module architecture investigated (Fig. 3d). This protein contains putative SLiMs for targeting HGS for ubiquitination (via NEDD4), clathrin-mediated endocytosis (via CLH1), signalling via Grb2 and P85A (uniprot: P27986) as well as an annotated PTAP SLiM, involved in the ESCRT signalling. Furthermore, 3DID data predict a domain–domain interaction with Tom1 (uniprot: O60784). This subnetwork highlights the information about functionality and directionality that can be garnered by mapping SLiM-predictions onto PPI networks.
4 DISCUSSION
SLiM-mediated binding interfaces are key components of the human proteome (Jorgensen and Linding, 2008) and are abundant within the signalling pathways of the cell (Pawson, 2007). In this article, we manually annotated domain-binding partners for 156 ELM classes and curated 1514 SLiM-mediated interfaces, thus generating a high-quality dataset for studying the interfaces between specific ELM classes and their interacting domains. This dataset enabled us to train HMMs for identifying SLiM-binding domains. These models were then incorporated into a novel method called iELM with the aim of detecting SLiM-mediated interfaces. iELM was able to distinguish specificities within SLiM-binding domains (see Supplementary Fig. S2), as well as identify SLiM-mediated interactions from a background of PPIs (Fig. 3). The iELM method uses an SVM algorithm in preference to a simple cut-off system due to our wish to develop a method with the best ratio between sensitivity and specificity. A comparison of these two techniques identifies the SVM model as having a higher sensitivity but a lower specificity, with the ratio weighted in favour of the SVM model. This suggests that using the SVM will identify a greater number of true positive interactions with only a slight increase in the FPR. iELM, so far, covers only linear motifs as they are annotated in the ELM resource, but is easily extendible to any SLiM, in the form of a regular expression, for which the interacting SLiM-binding domain is known.
The importance of a number of canonical and ubiquitous domains (e.g. SH2, SH3, PDZ and Pkinase) in signalling and regulatory networks has lead to a great deal of work focusing on their SLiM-binding properties (Beltrao and Serrano, 2005; Encinar et al., 2009; Gfeller et al., 2011; Huang et al., 2008; Hui and Bader, 2010; Li, 2005; Linding et al., 2007; Stein and Aloy, 2010). These domains are abundant in higher eukaryotes with small differences in amino acid composition leading to subtle shifts in specificities (Encinar et al., 2009; Gfeller et al., 2011; Huang et al., 2008). In the ELM resource, and by association in iELM, however, these subtle shifts in specificity are not necessarily fully explored. This is because for a particular SLiM functional class the ELM resource's annotation process aims to curate the full spectrum of variation within eukaryotes; potentially this can allow too broad a specificity for a SLiM and lead to false positive results. Despite these potential problems, the iELM method performed strongly on benchmarking datasets and was able to distinguish specificities for these ubiquitous domains. More importantly, iELM incorporates the less well-known SLiM classes (over two-thirds of those annotated in ELM) that do not have this overlapping intra-domain specificity enabling a more extensive array of SLiM-mediated interfaces to be predicted for the human interactome. This is illustrated by those interactions associated with targeting proteins for destruction, using D-box motifs, as well as by a subnetwork of interconnected SLiM-mediated interactions linked to endocytosis.
The automatic annotation of the molecular detail of a protein–protein interface is an important step in understanding the function of many of the interactions identified by proteomic experiments. In this study, we developed a novel method enabling for the first time, to our knowledge, the fast and automatic annotation of SLiM-mediated interactions on large-scale datasets. The development of iELM permitted us to produce an edge-based interactome of 12 562 interactions with 35 476 interfaces representing ∼4% of the known human interactome. This number is likely to represent only a small fraction of the SLiM-mediated interactions within the interactome, as it is only based on 156 ELM classes and SLiM-mediated interactions are known to be under-represented in mass spectrometry-derived proteomic data (Gavin et al., 2006). The final percentage is difficult to estimate as the total number of SLiM classes is unknown but taking into consideration that there are over 13 000 globular domain classes annotated in Pfam, the potential influence of SLiM-mediated interactions is prodigious.
The annotation of the edges of PPI networks allows a more biologically realistic edge-based analysis of PPI networks to be implemented. This is important, as proteins are modular entities whose function can vary depending on their interaction partners. Furthermore, as proteins have a finite number of binding sites, an appreciation of the location of their interaction surface will facilitate models to consider mutually exclusive binding. The use of a node-based view generalizes these properties and therefore loses the subtleties of a protein's behaviour, while an edge-based view would distinguish this difference enabling a more accurate portrayal of cellular networks.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thanks members of the Gibson Team for their support and advice; in particular, Kim van Roey for his critical reading of the manuscript and Holger Dinkel for assistance with the website.
Funding: R.J.W. was supported by the EMBL international PhD programme. K.L. was supported by the Région Alsace and Collége Doctoral Européen. N.E.D. was supported by an EMBL Interdisciplinary Postdoctoral (EIPOD) fellowship.
Conflict of Interest: none declared.
REFERENCES
- Altschul S.F., et al. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Beltrao P., Serrano L. Comparative genomics and disorder prediction identify biologically relevant SH3 protein interactions. PLoS Comput. Biol. 2005;1:e26. doi: 10.1371/journal.pcbi.0010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betel D., et al. Structure-templated predictions of novel protein interactions from sequence information. PLoS Comput. Biol. 2007;3:1783–1789. doi: 10.1371/journal.pcbi.0030182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohee S., et al. NeAT: a toolbox for the analysis of biological networks, clusters, classes and pathways. Nucleic Acids Res. 2008;36:W444–W451. doi: 10.1093/nar/gkn336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol A., et al. DOMINO: a database of domain-peptide interactions. Nucleic Acids Res. 2007;35:D557–D560. doi: 10.1093/nar/gkl961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chica C., et al. A tree-based conservation scoring method for short linear motifs in multiple alignments of protein sequences. BMC Bioinformatics. 2008;9:229. doi: 10.1186/1471-2105-9-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline M.S., et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copley R.R. The EH1 motif in metazoan transcription factors. BMC Genomics. 2005;6:169. doi: 10.1186/1471-2164-6-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N.E., et al. The SLiMDisc server: short, linear motif discovery in proteins. Nucleic Acids Res. 2007;35:W455–W459. doi: 10.1093/nar/gkm400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N.E., et al. SLiMSearch 2.0: biological context for short linear motifs in proteins. Nucleic Acids Res. 2011;39(Suppl. 2):W56–W60. doi: 10.1093/nar/gkr402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey N.E., et al. Attributes of short linear motifs. Mol. Biosyst. 2012;8:268–281. doi: 10.1039/c1mb05231d. [DOI] [PubMed] [Google Scholar]
- Diella F., et al. Understanding eukaryotic linear motifs and their role in cell signaling and regulation. Front Biosci. 2008;13:6580–6603. doi: 10.2741/3175. [DOI] [PubMed] [Google Scholar]
- Dinkel H., et al. ELM–the database of eukaryotic linear motifs. Nucleic Acids Res. 2012;40:D242–D251. doi: 10.1093/nar/gkr1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z., et al. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics. 2005;21:3433–3434. doi: 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Eddy S.R. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R.J., et al. SLiMFinder: a probabilistic method for identifying over-represented, convergently evolved, short linear motifs in proteins. PLoS One. 2007;2:e967. doi: 10.1371/journal.pone.0000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinar J.A., et al. ADAN: a database for prediction of protein-protein interaction of modular domains mediated by linear motifs. Bioinformatics. 2009;25:2418–2424. doi: 10.1093/bioinformatics/btp424. [DOI] [PubMed] [Google Scholar]
- Eswar N., et al. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006 doi: 10.1002/0471250953.bi0506s15. Chapter 5Unit 5.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R.D., et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontes M.R., et al. Structural basis for the specificity of bipartite nuclear localization sequence binding by importin-alpha. J. Biol. Chem. 2003;278:27981–27987. doi: 10.1074/jbc.M303275200. [DOI] [PubMed] [Google Scholar]
- Gavin A.C., et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- Gfeller D., et al. The multiple-specificity landscape of modular peptide recognition domains. Mol. Syst. Biol. 2011;7:484. doi: 10.1038/msb.2011.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M., et al. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Huang H., et al. Defining the specificity space of the human SRC homology 2 domain. Mol. Cell. Proteomics. 2008;7:768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- Hui S., Bader G.D. Proteome scanning to predict PDZ domain interactions using support vector machines. BMC Bioinformatics. 2010;11:507. doi: 10.1186/1471-2105-11-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachims T. Learning to Classify Text Using Support Vector Machines: Methods, Theory and Algorithms. Germany: Springer; 2002. [Google Scholar]
- Jorgensen C., Linding R. Directional and quantitative phosphorylation networks. Brief. Funct. Genomic Proteomic. 2008;7:17–26. doi: 10.1093/bfgp/eln001. [DOI] [PubMed] [Google Scholar]
- Kaneko T., et al. The SH3 domain–a family of versatile peptide- and protein-recognition module. Front Biosci. 2008;13:4938–4952. doi: 10.2741/3053. [DOI] [PubMed] [Google Scholar]
- Kay B.K., et al. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000;14:231–241. [PubMed] [Google Scholar]
- King C.A., Bradley P. Structure-based prediction of protein-peptide specificity in Rosetta. Proteins. 2010;78:3437–3449. doi: 10.1002/prot.22851. [DOI] [PubMed] [Google Scholar]
- Lee H.J., Zheng J.J. PDZ domains and their binding partners: structure, specificity, and modification. Cell Commun. Signal. 2010;8:8. doi: 10.1186/1478-811X-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., et al. Prediction of phosphotyrosine signaling networks using a scoring matrix-assisted ligand identification approach. Nucleic Acids Res. 2008;36:3263–3273. doi: 10.1093/nar/gkn161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.S. Specificity and versatility of SH3 and other proline-recognition domains: structural basis and implications for cellular signal transduction. Biochem. J. 2005;390:641–653. doi: 10.1042/BJ20050411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linding R., et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B., et al. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009;5:e1000376. doi: 10.1371/journal.pcbi.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi T., et al. Minimotif Miner 3.0: database expansion and significantly improved reduction of false-positive predictions from consensus sequences. Nucleic Acids Res. 2012;40:D252–D260. doi: 10.1093/nar/gkr1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael S., et al. Discovery of candidate KEN-box motifs using cell cycle keyword enrichment combined with native disorder prediction and motif conservation. Bioinformatics. 2008;24:453–457. doi: 10.1093/bioinformatics/btm624. [DOI] [PubMed] [Google Scholar]
- Mohan A., et al. Analysis of molecular recognition features (MoRFs) J. Mol. Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- Neduva V., et al. Systematic discovery of new recognition peptides mediating protein interaction networks. PLoS Biol. 2005;3:e405. doi: 10.1371/journal.pbio.0030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer J.C., et al. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson T. Dynamic control of signaling by modular adaptor proteins. Curr. Opin. Cell. Biol. 2007;19:112–116. doi: 10.1016/j.ceb.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Perrodou E., et al. A new protein linear motif benchmark for multiple sequence alignment software. BMC Bioinformatics. 2008;9:213. doi: 10.1186/1471-2105-9-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J.M. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 2006;7:644–656. doi: 10.1038/nrm1988. [DOI] [PubMed] [Google Scholar]
- Petsalaki E., et al. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput. Biol. 2009;5:e1000335. doi: 10.1371/journal.pcbi.1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfleger C.M., Kirschner M.W. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 2000;14:655–665. [PMC free article] [PubMed] [Google Scholar]
- Pop C., Salvesen G.S. Human caspases: activation, specificity, and regulation. J. Biol. Chem. 2009;284:21777–21781. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J., et al. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388. doi: 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L., Rape M. Regulated degradation of spindle assembly factors by the anaphase-promoting complex. Mol. Cell. 2010;38:369–382. doi: 10.1016/j.molcel.2010.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C., et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A., Aloy P. Novel peptide-mediated interactions derived from high-resolution 3-dimensional structures. PLoS Comput. Biol. 2010;6:e1000789. doi: 10.1371/journal.pcbi.1000789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A., et al. 3DID: identification and classification of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2011;39:D718–D723. doi: 10.1093/nar/gkq962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnimann C.U., et al. WD40 proteins propel cellular networks. Trends Biochem. Sci. 2010;35:565–574. doi: 10.1016/j.tibs.2010.04.003. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D., et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010;38:D142–D148. doi: 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S., Kleywegt G.J. The Protein Data Bank in Europe (PDBe): bringing structure to biology. Acta Crystallogr. D Biol. Crystallogr. 2011;67:324–330. doi: 10.1107/S090744491004117X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.J., Gregoire S. A recurrent phospho-sumoyl switch in transcriptional repression and beyond. Mol. Cell. 2006;23:779–786. doi: 10.1016/j.molcel.2006.08.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.