

Synemin is an intermediate filament protein present in glioblastomas (GBMs) but not in normal brain. In GBM cells synemin interacts with and antagonizes PP2A, which is the phosphatase dephosphorylating Akt. This maintains the phosphorylation status of Akt sites that are substrates for PDPK1 and mTORc2, thereby fostering proliferation.
Abstract
The intermediate filament protein synemin is present in astrocyte progenitors and glioblastoma cells but not in mature astrocytes. Here we demonstrate a role for synemin in enhancing glioblastoma cell proliferation and clonogenic survival, as synemin RNA interference decreased both behaviors by inducing G1 arrest along with Rb hypophosphorylation and increased protein levels of the G1/S inhibitors p21Cip1 and p27Kip1. Akt involvement was demonstrated by decreased phosphorylation of its substrate, p21Cip1, and reduced Akt catalytic activity and phosphorylation at essential activation sites. Synemin silencing, however, did not affect the activities of PDPK1 and mTOR complex 2, which directly phosphorylate Akt activation sites, but instead enhanced the activity of the major regulator of Akt dephosphorylation, protein phosphatase type 2A (PP2A). This was accompanied by changes in PP2A subcellular distribution resulting in increased physical interactions between PP2A and Akt, as shown by proximity ligation assays (PLAs). PLAs and immunoprecipitation experiments further revealed that synemin and PP2A form a protein complex. In addition, treatment of synemin-silenced cells with the PP2A inhibitor cantharidic acid resulted in proliferation and pAkt and pRb levels similar to those of controls. Collectively these results indicate that synemin positively regulates glioblastoma cell proliferation by helping sequester PP2A away from Akt, thereby favoring Akt activation.
INTRODUCTION
Synemin is an intermediate filament (IF) protein four times larger than most IF proteins due to a large C-terminal domain that contains binding sites for actin-associated proteins (Bellin et al., 1999, 2001; Mizuno et al., 2001). Although synemin does not assemble into filaments by itself, it can integrate into vimentin networks (Bellin et al., 1999; Jing et al., 2007). Phylogenetic analysis suggests that synemin evolved from neurofilament protein genes and provides the basis for its classification as a type IV IF protein (Guérette et al., 2007).
Most IF proteins represent differentiation markers due to their tissue- or cell-type–specific expression. This is not the case, however, for synemin, as in the adult it is present in cell types belonging to distinct lineages, including myocytes, endothelial, lens, and hepatic stellate cells (Granger and Lazarides, 1984; Hirako et al., 2003; Tawk et al., 2003; Schmitt-Graeff et al., 2006). Furthermore, synemin is present in astrocyte progenitor cells but not in mature astrocytes (Sultana et al., 2000; Izmiryan et al., 2010). Pathological situations can induce synemin expression in diverse cell types, including reactive astrocytes, liver fibrotic cells (Schmitt-Graeff et al., 2006; Jing et al., 2007; Luna et al., 2010), and glioblastomas (Jing et al., 2005). Synemin expression is also altered in myoepithelial cells of breast carcinomas due to changes in the methylation of the synemin gene (Noetzel et al., 2010).
In myocytes, synemin connects the IF network with Z-lines and plasma membrane junctional complexes by virtue of its ability to integrate into desmin IFs and to interact with α-actinin and vinculin (Bellin et al., 2001; Mizuno et al., 2001). In glioblastoma cells and HeLa cells, synemin localizes in actin-rich regions such as lamellipodia and focal contacts (Jing et al., 2005; Sun et al., 2010). RNA interference (RNAi) studies established that synemin takes part in the motility of glioblastoma cells by influencing the amount of α-actinin associated with polymerized actin, as well as the amount of polymerized actin itself (Pan et al., 2008).
These RNAi studies also implicated synemin in the proliferation of glioblastoma cells (Pan et al., 2008). It can be hypothesized that this effect occurs through the interplay of synemin with signaling components, as other IF proteins influence diverse signaling pathways. Nestin, for instance, promotes the survival of neuronal progenitor cells by modulating the activity of p35, a regulator of cyclin-dependent kinase 5 (Sahlgren et al., 2006). The interaction of various keratins with proteins regulating apoptosis, such as TRADD, is well established (Kim and Coulombe, 2007; Omary et al., 2009). Furthermore, during epidermal wound healing, keratin 17 interacts with the adaptor protein 14-3-3σ to enhance protein synthesis through the mTOR pathway (Kim et al., 2006). Other examples include vimentin participation in integrin recycling mediated by protein kinase Cɛ in migratory fibroblasts (Ivaska et al., 2005) and in the translocation of activated MAP kinase to sites of neuronal injury (Perlson et al., 2005). Furthermore, vimentin is critical for the epithelial-to-mesenchymal transition that accompanies the progression of carcinoma cells (Mendez et al., 2010) and plays this role by directing Axl expression (Vuoriluoto et al., 2011).
Here we used RNAi to characterize the contribution of synemin to signaling pathways instrumental for the proliferation of glioblastoma cells. Immunoprecipitation and proximal ligation assays (PLAs) were also used to identify signaling proteins interacting with synemin. Taken together, the results of these studies outline a previously unrecognized cytoskeletal regulation of protein phosphatase type 2A (PP2A) that contributes to the proliferation of glioblastoma cells.
RESULTS
Synemin silencing alters the cell cycle distribution of A172 cells by influencing effectors of the G1/S transition
Synemin is expressed in several human glioblastoma cells, including A172 and U373 MG (Jing et al., 2005). To down-regulate synemin, cells were treated for 7 d with one of two different synemin short hairpin RNAs (shRNAs 1 and 2). Each of these shRNAs reduced by >90% the protein levels of the two synemin isoforms (α and β; Figure 1A). Synemin shRNAs 1 and 2 yielded similar results throughout our studies. Thus, for the sake of simplicity, the results shown will be those obtained with synemin shRNA 1.
FIGURE 1:

Effect of synemin down-regulation on the growth of A172 glioblastoma cell populations. (A) Western blots demonstrating the substantial reduction in synemin protein levels (top, α-synemin; bottom, β-synemin) in A172 cells treated for 8 d with control or two different synemin shRNAs (1 and 2). Blots were also incubated with anti-actin to verify equal loading. (B) Growth curves demonstrate that synemin shRNA sharply reduces the proliferation of synemin-expressing A172 and U373 glioblastoma cells but does not significantly influence the proliferation of PPC1, a prostate carcinoma cell line devoid of synemin. (C) Histogram of the number of colonies, expressed as percentage of control, obtained after clonogenic survival or soft-agar colony formation assays. (D) Histogram of the percentage of apoptotic A172 cells as determined by flow cytometry after annexin V and propidium iodide staining. The percentage of apoptotic cells was similar between A172 cells treated with control or synemin shRNAs, whether they were not subjected to additional treatment or were treated for 24 h with 10 mM campothecin or 50 mM H2O2. (E) Histogram of the percentage of control or synemin shRNA–treated A172 cells in the different phases of the cell cycle as determined by flow cytometry after propidium iodide staining. Note that synemin down-regulation increases the percentage of cells in G1 while decreasing the percentage of cells in S phase. (B, C) Bars represent means ± SEM of three to five independent experiments; asterisks indicate significance at p ≤ 0.001.
As previously demonstrated (Pan et al., 2008), synemin down-regulation reduced the number of A172 and U373 MG glioblastoma cells accumulating over 7 d by ∼80% (Figure 1B). PPC-1 prostate carcinoma cells do not express synemin (Pan et al., 2008), and here we demonstrate that synemin shRNA did not significantly alter the proliferation of these cells (Figure 1B). This indicates that the effect of synemin down-regulation on A172 and U373 MG cell accumulation is not likely to be caused by off-target effects.
Attachment-dependent and -independent clonogenic assays were performed to examine whether synemin silencing also hampered the ability of individual glioblastoma cells to proliferate indefinitely into colonies. For attachment-dependent clonogenic assays, A172 cells treated with control or synemin shRNAs were plated at low density. After 2 wk, the number of colonies of synemin-silenced cells was ∼20% that of cells treated with control shRNA (Figure 1C). Synemin down-regulation induced a similarly robust decrease in colony formation in A172 and U373 MG glioblastoma cells when anchorage-independent, soft agar growth assays were performed (Figure 1C; data not shown for U373 MG cells). As previously noted, these effects were independent of apoptosis, which was not increased by synemin down-regulation (Pan et al., 2008; Figure 1D). In addition, synemin down-regulation did not significantly increase the sensitivity of A172 cells to apoptosis inducing agents such as campothecin and H2O2 (Figure 1D).
Taken together, these results suggest that synemin silencing reduces the accumulation of A172 cells by decreasing their proliferation. Flow cytometry analysis of A172 cells stained with propidium iodide was thus performed to examine whether synemin silencing affected the cell-cycle distribution of A172 cells. These experiments revealed that 57% of control cells were in the G1 phase and that synemin silencing increased that fraction to 81% (Figure 1E). Concomitantly, the fraction of cells in S phase decreased from 36% in controls to 12% in synemin-silenced cells (Figure 1E).
Next we examined by Western blotting whether the shift in cell-cycle distribution induced by synemin silencing also affected key biochemical events responsible for the G1/S transition. These include 1) Rb phosphorylation, which dissociates Rb from E2F transcription factors, thereby enabling the transcription of mRNAs encoding proteins necessary for the G1/S progression (Sun et al., 2007), and 2) the accumulation of the cyclin-dependent kinase inhibitors (CDIs) p21Cip1 and p27Kip1, as this inhibits cyclin-dependent kinases from phosphorylating Rb (Child and Mann, 2006). The results show that synemin silencing decreased Rb phosphorylation by ∼50%, whereas total Rb levels remain steady (Figure 2A). In addition, p21Cip1 and p27Kip1 protein levels were elevated by 1300 and 180%, respectively, in synemin-silenced A172 cells as compared with controls (Figure 2, B and C). Synemin shRNAs induced a similar pRb decrease and p21Cip1 and p27Kip1 accumulation in U373 MG cells but not in PPC1 prostate carcinoma cells (unpublished data).
FIGURE 2:

Western blots of total proteins from A172 cells treated with control or synemin shRNAs show the effect of synemin silencing on the levels of total Rb and pRb (A), p21 (B), p27 (C), pT145-p21 (D), and pS146-p21 (E). Blots were also incubated with anti-actin to verify equal loading. Histograms show the results of densitometric analysis of the blots; results were normalized to total Rb for pRb (A), to actin for p21 (B) and p27 (C), and to total p21 for pT145-p21 (D) and pS146-p21 (E). Bars represent means ± SEM of three to five independent experiments; asterisk indicates significance at p ≤ 0.001.
Synemin intervenes in the Akt pathway downstream of PI3K and mTOR complex 2
The foregoing results indicate that synemin regulates the G1/S transition of glioblastoma cells through a pathway involving p21Cip1 and p27Kip1. The potential involvement of Akt in eliciting p21Cip1 accumulation in synemin-silenced cells was investigated by determining the phosphorylation levels of p21Cip1 at residues T145 and S146. The rationale for these experiments is that the phosphorylation of these residues leads to p21Cip1 degradation and that Akt directly phosphorylates these sites (Child and Mann, 2006). Thus, if Akt is involved in p21Cip1 accumulation in synemin-silenced cells, one expects decreased pT145-p21Cip1 and pS146-p21Cip1. Western blot experiments with site-specific phosphoantibodies showed this to be the case, since when normalized to total p21Cip1 levels, pT145-p21Cip1 and pS146-p21Cip1 were decreased by 96 and 97%, respectively, upon synemin silencing (Figure 2, D and E).
Because these data suggested a decrease in Akt activity after synemin silencing, Akt was immunoprecipitated from A172 cells treated with either control or synemin shRNAs to examine its ability to incorporate 32P into one of its substrate, GSK-3. The results demonstrate that synemin silencing significantly lowered Akt activity by ∼50% relative to controls (Figure 3A). Akt activation is effected by phosphorylation at S473 by the Rictor-mTOR complex 2 (mTORc2) and at T308 by phosphatidylinositol (3,4,5)-triphosphate (PIP3)–dependent protein kinase 1 (PDPK1; previously referred to as PDK1; Alessi et al., 1996; Sarbassov et al., 2005; Bayascas, 2008). After synemin silencing in A172 cells, total Akt protein levels were steady, but pS473-Akt and pT308-Akt were lower by 46 and 72%, respectively (Figure 3B). Similar results were obtained in U373 MG glioblastoma cells, but synemin shRNAs did not affect Akt phosphorylation in synemin-free PPC1 prostate carcinoma cells (unpublished data).
FIGURE 3:

Akt kinase activity and phosphorylation levels in control or synemin shRNA–treated A172 cells. (A) Total Akt: Western blot showing the amount of Akt immunoprecipitated from A172 cells treated with control or synemin shRNAs. pGSK: autoradiograph showing the amount of 32P incorporated into GSK after in vitro phosphorylation with immunoprecipitated Akt in the presence of [γ-32P]ATP. (B) Western blots of total proteins from A172 cells treated with control or synemin shRNAs incubated with antibodies against Akt, pS473-Akt, and pT308-Akt. (A, B) Histograms show the results of densitometric analysis of autoradiographs (A) and blots (B) after normalization of pGSK to immunoprecipitated Akt (A) and of pS473-Akt and pT308-Akt to total Akt levels (B). Bars represent means ± SEM of three to five independent experiments; asterisks indicate significance at p ≤ 0.001.
Next we examined whether decreased Akt phosphorylation and activity in synemin-silenced cells paralleled reduced activation of Akt direct upstream activators mTORc2 and PDPK1. mTORc2 activity was similar in control and synemin-silenced cells, as shown by immunoprecipitating mTOR and measuring its capacity to phosphorylate Akt in vitro in the presence of [γ-32P]ATP (Figure 4A). Similarly, neither PDPK1 protein level nor pS241-PDPK1, which represents activated PDPK1 after PIP3-induced autophosphorylation (Bayascas, 2008), was significantly modified by synemin silencing. This was determined by densitometric analysis of Western blots of control and synemin-silenced A172 cells incubated with antibodies against total PDPK1 or pS241-PDPK1 (Figure 4B).
FIGURE 4:

Activity of Akt upstream activators in control or synemin shRNA–treated A172 cells. (A) Rictor: Western blot showing the amount of Rictor pulled down with mTOR antibodies. pS473-Akt: autoradiograph showing the amount of 32P incorporated into Akt after in vitro phosphorylation with immunoprecipitated mTORc2 in the presence of [γ-32P]ATP. (B) Western blots of total proteins from A172 cells incubated with antibodies against total PDPK1 and pS241-PDPK1. (A, B) Histograms show the results of densitometric analysis of autoradiographs (A) and blots (B) of pS473-Akt (A) and pS241-PDPK1 (B) after normalization to immunoprecipitated Rictor (A) and total PDPK1 (B). (C) Total PTEN: Western blot of total proteins from A172 cells incubated with PTEN antibodies. IP PTEN: Western blot showing the amount of PTEN immunoprecipitated with PTEN antibodies. Histogram shows the activity of IP PTEN as determined with malachite green to measure at OD 620 the amount of Pi released after incubating PIP3 with IP PTEN. Results were normalized to the amount of IP PTEN. (D) p85: Western blot showing the amount of p85/PI3K immunoprecipitated with p85 antibodies. 32P-PIP3: autoradiograph of TLC plate showing the amount of 32P-PIP3 produced after incubating PIP2 with immunoprecipitated p85/PI3K in the presence of [γ-32P]ATP. Histogram shows the results of densitometric analysis of autoradiographs after normalization to immunoprecipitated p85. Bars represent means ± SEM of three to five independent experiments. Statistical analysis of the data in A–D shows that synemin silencing does not significantly affect the levels and activities of Akt upstream activators.
PIP3 participates in Akt activation not only by activating PDPK1 but also by recruiting Akt from the cytosol to the plasma membrane (Manning and Cantley, 2007). PIP3 levels are determined primarily by phosphatidylinositol 3-kinase (PI3K), which converts PIP2 to PIP3, and by the phosphoinositol phosphatase PTEN, which reverses that reaction (Manning and Cantley, 2007). Synemin silencing did not significantly alter PTEN protein levels, as determined by Western blotting, nor did it affect PTEN phosphatase activity as determined with a colorimetric assay measuring the release of Pi from PIP3 in the presence of immunoprecipitated PTEN (Figure 4C). Immunoprecipitation experiments also demonstrated that the phosphotyrosine levels of PI3K (unpublished data) and the capacity of PI3K to catalyze the conversion of PIP2 into PIP3 in the presence of γ32P-ATP were similar in control and synemin shRNA-treated cells (Figure 4D). Similar results were obtained with cells maintained in serum-free medium overnight and stimulated with serum for 10 min (unpublished data).
Synemin associates with PP2A and regulates its subcellular distribution and interaction with Akt
The finding that synemin silencing did not affect the two kinases directly phosphorylating Akt at residues S473 and T308 while greatly reducing the phosphorylation of these two residues suggested that synemin could influence PP2A because it is the major phosphatase controlling phosphate turnover at these residues (Arroyo and Hahn, 2005). PP2A holoenzyme comprises a structural A subunit, a regulatory B subunit, and a catalytic C subunit (Shi, 2009; Virshup and Shenolikar, 2009). Although many different types of B subunit exist, B55 determines PP2A specificity for Akt (Kuo et al., 2008). Synemin silencing did not affect PP2A A and B55 subunit total protein levels (Figure 5, A and B). Antibodies against PP2A A or B55 subunits were used to immunoprecipitate the holoenzyme from cells treated with control or synemin shRNAs, and the phosphatase activity of immunoprecipitated PP2A was determined by measuring the release of Pi from a phosphopeptide substrate. This assay revealed that the phosphatase activity of PP2A immunoprecipitated with antibodies against either the A or B55 subunits was 180 and 140% higher, respectively, in synemin-silenced cells when compared with controls (Figure 5, C and D).
FIGURE 5:

PP2A levels (A, B) and activity (C, D) in control or synemin shRNA–treated A172 cells. (A, B) Western blots of total cellular proteins incubated with antibodies against: PP2A A subunit (A) or PP2A B55 subunit (B). Blots were also incubated with anti-actin to verify equal loading. (C, D) Western blots showing the amount of PP2A immunoprecipitated with antibodies against PP2A A (C) or B55 subunit (D). Histograms show the amount of Pi released after incubation of a phosphopeptide substrate with immunoprecipitated PP2A. Phosphate release was measured at OD 620 with a malachite green assay and was normalized to the amount of immunoprecipitated PP2A. Histograms show results of densitometric analysis of blots (A, B) and of colorimetric malachite green assays (C, D). Statistical analysis of the data reveals that synemin silencing did not alter the cellular levels of PP2A A (A) and B55 (B) subunits but that it increased the phosphatase activity of PP2A (C, D). Bars represent means ± SEM of three to five independent experiments; asterisks indicate significance at p ≤ 0.001.
The change in PP2A activity is the most upstream event that could be identified with respect to the effect of synemin down-regulation on glioblastoma cell proliferation. This raises the possibility that synemin regulation of PP2A involves an interaction between the two proteins. This possibility was tested by a series of immunoprecipitation experiments in which naive A172 cells were lysed in a buffer containing 1% NP-40. Lysates were spun to separate insoluble structures from soluble protein complexes. Under these conditions, a pool of synemin was recovered in the supernatants. As expected, immunoprecipitation of these supernatants with synemin antibodies pulled down both α- and β-synemin as revealed by Western blotting (Figure 6). In addition, incubation of Western blots of the immunoprecipitation product with anti–PP2A A subunit revealed a band (Figure 6B) that comigrated with the band for that subunit on Western blots of A172 cell total proteins (Figure 6A). Similar results were obtained with antibodies against the 55-kDa PP2A B55 subunit (Figure 6B). Neither PP2A A nor PP2A B55 subunits could be detected on blots when the immunoprecipitation was carried out with control immunoglobulin G (IgG; unpublished data).
FIGURE 6:

Immunoprecipitation experiments with synemin and PP2A antibodies using A172 cell cytosolic proteins. (A) Western blots of A172 cells total protein extracts show the specificities of the antibodies used for immunoprecipitation; note that the synemin antibody recognizes two bands corresponding to α- and β-synemin. (B) Immunoprecipitation with synemin antibodies demonstrates that the A subunit and B55 subunit of PP2A are pulled down together with synemin. (C) Measurements of the phosphatase activity present in the proteins immunoprecipitated with control or synemin IgGs. In some experiments, 500 nM CA was added to the immunoprecipitated proteins. Immunoprecipitated proteins were incubated with a phosphopeptide substrate, and the Pi released was measured at OD 620 with a malachite green assay. Compared to control IgGs, synemin antibodies immunoprecipitated a phosphatase activity that can be inhibited with CA. Bars represent means ± SEM of three independent experiments; asterisks indicate significance at p ≤ 0.001. (D, E) Immunoprecipitation with antibodies against the A (D) and B55 (E) subunits of PP2A demonstrate that synemin pulls down together with these subunits.
To determine whether synemin antibodies immunoprecipitated PP2A A or B55 subunits individually or as part of the PP2A holoenzyme, we examined the immunoprecipitate obtained with synemin antibodies for phosphatase activity. For these experiments, the proteins obtained after immunoprecipitation with synemin antibodies or control IgGs were incubated with a phosphopeptide and the release of Pi was determined with a colorimetric assay. This demonstrated that the proteins immunoprecipitated with synemin antibodies contained a phosphatase activity that was substantially higher than when immunoprecipitating with control IgGs (Figure 6C). Incubation of the proteins immunoprecipitated with synemin antibodies with 500 nM of the specific PP2A inhibitor cantharidic acid (CA; Li et al., 1993; Swingle et al., 2007) reduced the PP2A activity in the synemin immunoprecipitate to the baseline levels obtained with control IgGs (Figure 6C). Further evidence that synemin and PP2A form protein complexes in A172 cells was obtained by using antibodies against PP2A A or B55 subunit in the immunoprecipitation step. Under these conditions, Western blots demonstrated that synemin associated with both the immunoprecipitated A and B55 subunits of PP2A (Figure 6, D and E).
Synemin and PP2A interactions were also evidenced in situ. First, immunofluorescence staining showed that, as reported earlier (Jing et al., 2005; Pan et al., 2008), synemin localizes at the periphery as well as in the nuclear area of control cells (Figure 7B) and that synemin down-regulation alters cell shape from polygonal (Figure 7A) to elongated with cellular processes (Figure 7B).
FIGURE 7:

Fluorescence staining of A172 cells treated with control (A–D) or synemin (E–H) shRNAs. Staining was performed with four fluorescent reagents: phalloidin Alexa 633 (to stain actin; purple) and DAPI (to stain DNA; blue; A, E), Alexa 488 synemin antibodies (green; B, F), and Alexa 568 PP2A antibodies (red; C, G). (A, E) Actin staining shows that control cells are polygonal in shape with prominent peripheral actin (A, arrowheads), whereas synemin-silenced cells have an elongated cell body with cytoplasmic processes (E). (B, F) Staining with synemin antibodies shows that in control cells synemin is localized at the cellular periphery (B, arrowheads), as well as in the nuclear area (B, arrows); synemin staining is decreased after synemin silencing (F). (C, G) Staining with PP2A antibodies shows that PP2A is present in the nuclear area of control cells (C, arrows); after synemin silencing, PP2A antibodies stain the cytoplasm as well (G, V-shaped arrowheads). (D, H) Overlay of synemin and PP2A staining appears yellow and reveals that the two proteins overlap in the nuclear area of control cells (D, arrows); little overlap is seen in synemin-silenced cells (H). Bars, 10 μm.
In controls, PP2A antibodies stained the nuclear area (Figure 7C), and in that area the staining of PP2A and synemin overlapped (Figure 7D). PLAs with synemin and PP2A antibodies indicated that the nuclear area was indeed the major site of synemin and PP2A interactions because most PLA reaction products concentrated in that region (Figure 8A). When compared with controls, there were few synemin and PP2A PLA reaction products in synemin-silenced cells (Figure 8B).
FIGURE 8:

Proximal ligation assays performed with antibodies against PP2A and synemin (A, B) or with antibodies against PP2A and Akt (C, D) in A172 cells treated with control (A, C) or synemin (B, D) shRNAs. PLA reaction products appear as red dots in cells where boundaries and nuclei were stained with phalloidin–Alexa 488 (green) and DAPI (blue), respectively. (A, B) PLA reaction products for PP2A and synemin localize primarily in the nuclear area of control cells (A), whereas fewer reaction products appear in the nuclear area of synemin-silenced cells (B). (C, D) PLA reaction products for PP2A and Akt are scarce in control cells (C) but numerous in synemin silenced cells (D), where they localize in the cytoplasm (D, arrowheads). Bars, 10 μm.
In control cells PP2A antibodies stained only the nuclear area (Figure 7C), whereas in synemin-silenced cells they stained the cytoplasm as well (Figure 7G). Of interest, this appeared to modify PP2A and Akt interactions as determined by PLA. In control cells, PLA reaction products obtained with PP2A and Akt antibodies were scarce and localized primarily in the nuclear area (Figure 8C). In contrast, in synemin-silenced cells, PP2A and Akt antibodies and PLA reaction products abounded and were distributed throughout the cytoplasm (Figure 8D). Control experiments were carried out by omitting one of the two primary antibodies from the PLA protocol; under these conditions, PLA fluorescence amplification products were absent (unpublished data).
PP2A inhibition with CA restores the proliferation of synemin-silenced cells
Finally, the involvement of PP2A in the phenotype of synemin-silenced cells was further examined by treating these cells with 500 nM CA. This treatment increased the number of synemin-silenced cells by 200% relative to synemin-silenced cells treated with vehicle over 72 h (Figure 9A). This increase in cell number was comparable to that obtained with A172 cells plated at a similar cell density and treated for the same time duration with control shRNA and vehicle (Figure 9A).
FIGURE 9:

Effect of treatment with 500 nM CA on the proliferation (A) and levels of pT308-Akt (B) and pRb (C) in synemin-silenced A172 cells. (A) Equal numbers of control and synemin shRNA–treated A172 cells were plated at day 0. CA was added to some dishes of synemin shRNA–treated cells 18 h after plating. Cells counts demonstrate that after 3 d in culture, the number of synemin-silenced cells is lower than in controls and that the number of synemin-silenced cells treated with CA is similar to that of controls. (B, C) Western blots of total proteins from A172 cells treated with control shRNA, synemin shRNA, or synemin shRNA and CA were incubated with antibodies against (B) pT308-Akt, Akt, and actin and (C) pRb and actin. (B, C) Histograms show the results of densitometric analysis of blots after normalization to total actin levels. After treatment of synemin-silenced cells with CA, pT308-Akt (B) and pRb (C) levels were significantly increased compared with synemin-silenced cells treated with vehicle and were similar to those of control cells. Bars represent means ± SEM of three independent experiments; asterisk indicates significance at p ≤ 0.001.
Western blots also demonstrated that treatment of synemin-silenced cells with 500 nM CA for 24 h increased the phosphorylation levels of Rb (Figure 9B) and Akt (Figure 9C) by 150 and 200%, respectively, thereby bringing them to the levels present in cells treated with control shRNAs and vehicle (Figure 9, B and C).
DISCUSSION
A role for the IF protein synemin in the malignant phenotype of glioblastoma cells is suggested by its abundance in glioblastoma tumors relative to normal brain (Jing et al., 2005) and by RNAi experiments revealing that synemin positively influences glioblastoma cell motility and proliferation (Pan et al., 2008). Synemin contribution to the malignant phenotype of glioblastoma cells is further substantiated by the present findings that its silencing vigorously reduces attachment-dependent and -independent clonogenic survival. Our results also demonstrate that synemin facilitates the G1/S transition, as its silencing shifts cell cycle distribution toward the G1 phase at the expense of the S phase. This is accompanied by telltale modifications in biochemical markers specific for the G1/S transition, including decreased Rb phosphorylation and the accumulation of two upstream regulators of Rb phosphorylation, the cyclin-dependent kinase inhibitors p21Cip1 and p27Kip1. Synemin facilitation of the G1/S transition appears to be the principal mechanism by which synemin supports the expansion of glioblastoma cell populations, since synemin-silencing did not influence glioblastoma cell apoptosis.
A major activator of the G1/S transition is Akt, which plays that role by antagonizing the accumulation of two G1/S transition inhibitors, p21Cip1 and p27Kip1 (Vervoorts and Lüscher, 2008; Lee and Kim 2009). This occurs through transcriptional and posttranslational controls, including p21Cip1 phosphorylation and ensuing degradation. p21Cip1 can be phosphorylated either directly by Akt or by kinases activated by Akt (Child and Mann, 2006). Thus our findings that Akt activity and Akt-dependent phosphorylation of p21Cip1 are significantly lower in synemin-silenced cells show that Akt mediates the effects of synemin on the cell cycle distribution and proliferation of glioblastoma cells.
Decreased Akt activity after synemin silencing was paralleled by reduced phosphorylation of two sites essential for Akt activity. Phosphorylation of these two sites is determined by the balance between phosphate addition by PDPK1 and mTORc2 (Alessi et al., 1996; Sarbassov et al., 2005), on one hand, and phosphate removal by PP2A, on the other (Resjö et al., 2002; Andrabi et al., 2007; Kuo et al., 2008). In addition, PI3K and PTEN are major upstream activators of Akt, as they determine the production of PIP3, which is responsible for PDPK1 and Akt activation through conformational changes and/or translocation to the plasma membrane (Manning and Cantley, 2007; Bayascas, 2008). Our results demonstrate that synemin silencing did not significantly affect the activities of PDPK1, mTORc2, PI3K, and PTEN. In contrast, synemin silencing significantly increased PP2A phosphatase activity. Treatment of synemin-silenced cells with CA restored proliferation along with Akt and Rb phosphorylation. Taken together, these results clearly identify increased PP2A activity as the major, if not sole, determinant of decreased Akt phosphorylation and of the resultant reduced proliferation of synemin-silenced cells. Thus, in glioblastoma cells, synemin-dependent negative regulation of PP2A is instrumental for maintaining Akt phosphorylation and cell proliferation (Figure 10).
FIGURE 10:
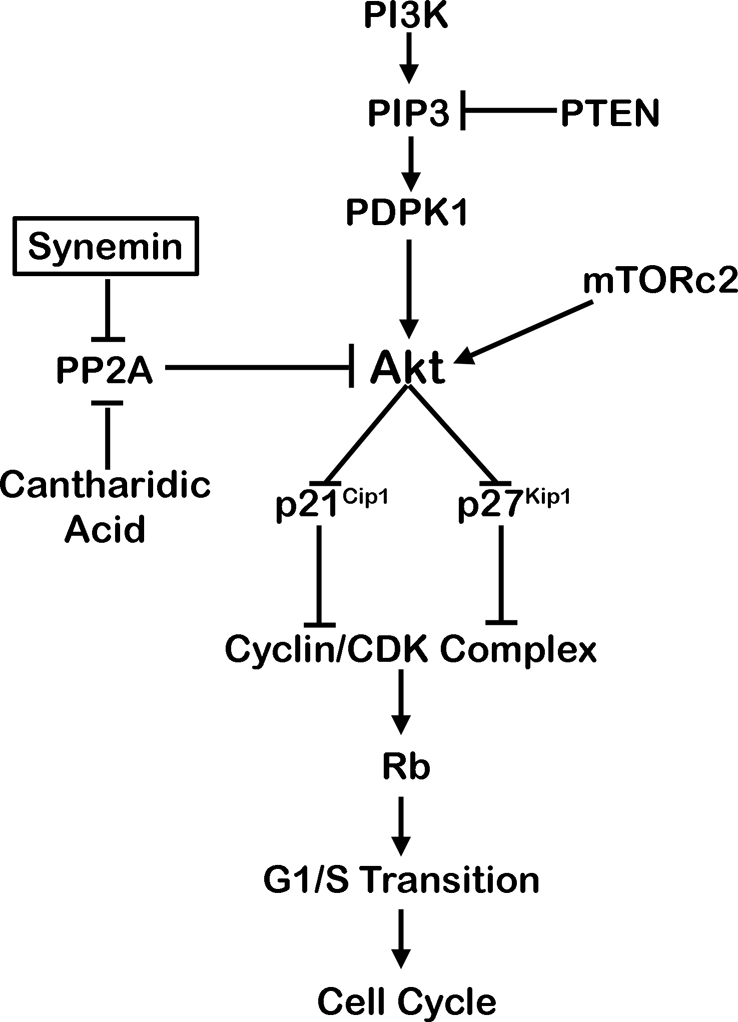
Proposed mechanism by which synemin affects the proliferation of glioblastoma cells. Synemin shelters PP2A away from Akt, which helps to maintain the phosphorylation status of Akt activation sites phosphorylated by PDPK1 and mTORc2. In turn, this promotes the G1/S transition and proliferation.
PP2A is a trimer comprising a structural A subunit, a regulatory B subunit, and a catalytic C subunit (Shi, 2009; Virshup and Shenolikar, 2009). B subunits belong to a large, highly diverse protein family arising from different genes and alternative splicing (Kuo et al., 2008). B subunits primarily dictate PP2A specificity toward distinct substrates, and, in particular, B55 determines PP2A specificity toward Akt (Kuo et al., 2008). Synemin antibodies immunoprecipitated the A as well as the B55 subunit, whereas antibodies against either PP2A A or B55 subunit immunoprecipitated synemin. In addition, PP2A phosphatase activity was discernible in the product of synemin immunoprecipitation. These results indicate that synemin associates with the PP2A holoenzyme containing the B subunit type responsible for targeting PP2A toward Akt.
PLA investigations validate immunoprecipitation evidence of synemin and PP2A interactions and demonstrate furthermore that they take place in the nuclear area. This result is consistent with immunofluorescence showing that in control cells PP2A and synemin colocalize in the nuclear area. We cannot exclude at this point the presence of a pool of synemin in the nucleus, and therefore more work is needed to determine whether synemin and PP2A interact in the nucleus and/or in the perinuclear area. Immunofluorescence also showed that synemin silencing altered PP2A localization, as a pool of PP2A assumed a cytoplasmic distribution. The fact that some of the PP2A staining remained in the nuclear area after synemin silencing suggests that synemin may interact with a subset of PP2A, perhaps one with a particular isoform composition. In any case, PLA demonstrated that synemin silencing increased PP2A and Akt cytoplasmic interactions. Obviously, this increase accounts for the decreased Akt phosphorylation following synemin down-regulation. Taken together, these results indicate that synemin regulates PP2A activity by helping specify its subcellular distribution in a manner that shelters it away from Akt. This is in keeping with recent reports showing that changes in PP2A subcellular distribution are instrumental in regulating its activity during mitotic progression (Lee et al., 2010; Rossio and Yoshida, 2011).
PP2A can also be regulated by the isoform composition of the holoenzyme, posttranslational modifications, and various protein interactions (Shi, 2009). Several viral proteins, for instance, antagonize PP2A phosphatase activity by interacting with the catalytic subunit and/or displacing the B subunit (Pallas et al., 1990; Yang et al., 1991). Of interest, similar to synemin, this negative PP2A regulation promotes proliferation (Arroyo and Hahn, 2005). Also of interest, cytoskeletal microtubules interact also with PP2A to reduce its phosphatase activity (Sontag et al., 1995). This regulates microtubule stability by maintaining the phosphorylation of tau, which is a microtubule-stabilizing protein and PP2A substrate (Sontag et al., 1995, 1999; Gong et al., 2000).
In addition to synemin, the IF proteins vimentin (Turowski et al., 1999), NF-L (Saito et al., 1995), and keratins 8 and 18 (Tao et al., 2006) also associate with PP2A. These interactions, however, functionally differ from those between synemin and PP2A because they do not regulate PP2A activity. Instead, they target PP2A toward these IF proteins to control their phosphate turnover, a phenomenon central to the dynamics of IF networks (Eriksson et al., 1992, 2004). Synemin is also a phosphoprotein (Sandoval et al. 1983) and, therefore, in addition to modulating Akt signaling, synemin/PP2A interactions may participate in synemin phosphate turnover.
Various keratins have been implicated in proliferation, but through mechanisms differing from those outlined here for synemin. Keratin 10 (K10) inhibits epithelial cell proliferation through the ability of its end domain to sequester and antagonize Akt and PKCζ (Paramio et al., 2001). In addition, K10 influences the proliferation of basal epithelial cells through c-Myc and 14-3-3 proteins (Reichelt and Magin, 2002). The latter proteins also interact with K8 and K18 to affect hepatocyte proliferation (Toivola et al., 2001; Ku et al., 2002). Of interest, keratins influence Akt signaling to regulate protein synthesis and cellular growth during epithelial wound healing (Kim et al., 2006) and liver and pancreas injury (Ku et al., 2010).
In conclusion, high Akt activity is frequent in glioblastomas, where it results often from PI3K overexpression and/or PTEN inactivation, leading to the accumulation of Akt major activator, PIP3 (Parsons et al., 2008). Our study delineates an alternate and potentially synergistic mechanism by which Akt is maintained active by antagonizing its inhibitor, PP2A, through PP2A association with synemin. This mechanism of PP2A regulation may be particular to glioblastomas, since synemin expression is widespread in these tumors (Jing et al., 2005) but uncommon in other tumor types (Schmitt-Graff et al., 2006; Noetzel et al., 2010).
MATERIAL AND METHODS
Cell culture
Human A172 and U373 MG glioblastoma cells and PPC1 prostate carcinoma cells (American Type Culture Collection, Rockville, MD) were grown as described (Pan et al., 2008). In some experiments, cells were serum deprived for 18 h to reduce basal PI3K levels and stimulated for 10 min with serum. In other experiments, 500 nM of the PP2A inhibitor CA (EMD Biosciences, San Diego, CA) was added to the medium 18 h after plating. For the duration of the experiment, the medium was replaced daily with medium containing fresh CA.
RNAi
RNAi of synemin was performed by lentiviral delivery of shRNAs cloned into pLKO.1-puro as described (Pan et al. 2008). The shRNAs targeted the human synemin sequences CGCTTACAGTACCATTTCATT (synemin shRNA 1) and GCCGTCAGAATTCAGAAACAA (synemin shRNA 2). Control shRNA was represented by the sequence CAACAAGATGAAGAGCACCAA, which is not present in the human genome. Puromycin selection (1 μg/ml for A172 and PPC1 cells and 2 μg/ml for U373 MG cells) was applied for 8 d to select for stable incorporation events. At that time, the cells were used for the assays described.
Proliferation, clonogenic, and soft agar survival assays
For proliferation assays, cells were plated into six-well plates (105 cells/well). Cells were trypsinized 2, 4, and 6 d after plating, resuspended in complete medium, and counted with a hemacytometer or with the Vi-CELL XR Cell Viability Analyzer (Beckman Coulter, Brea, CA).
For clonogenic survival assays, cells were trypsinized and plated at low density (50 cells/cm2). After 2 wk, cells were fixed with methanol and stained with 1% crystal violet for 10 min. For soft agar assays, equal cell numbers were plated in 0.35% agarose prepared in Iscove's modified Dulbecco's medium (IMDM; Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum, 50 IU/ml penicillin, and 50 μg/ml streptomycin over a bottom cushion of 0.7% agarose made in IMDM with the same supplementation. After 2 wk of incubation, colony counts were performed with an inverted Olympus CK2 microscope (Olympus, Center Valley, PA) equipped with phase contrast optics.
Apoptosis
Control or synemin-silenced A172 cells were serum starved for 24 h and treated with 10 μM camptothecin or 50 μM H2O2 for another 24 h. Both adherent and floating cells were collected by trypsinization and/or centrifugation at 1000 rpm for 5 min. After phosphate-buffered saline (PBS) washes, cells were stained with annexin V–fluorescein isothiocyanate and propidium iodide following the manufacturer's instructions (BD Biosciences PharMingen, San Diego, CA). Apoptotic cell counts were performed with a FACSCalibur flow cytometer (BD Biosciences, San Diego, CA).
Cell cycle analysis
Cells (2 × 106) were trypsinized and centrifuged at 1000 rpm for 5 min, and cell pellets were resuspended in PBS (0.5 ml). Seventy percent ethanol (5 ml) was added dropwise to the cell suspension while vortexing. After 2 h of fixation at 4°C, cells were washed twice with PBS and incubated 30 min at 20°C in PBS containing 5 U of RNase (Sigma-Aldrich) and 50 μg/ml propidium iodide (Sigma-Aldrich). Cell cycle analysis was performed with a FACSCalibur Flow Cytometer, using ModFit software (Verity Software House, Topsham, ME).
Western blotting
Comparison of protein or site-specific phosphorylation levels was performed on Western blots of polyacrylamide gels loaded with equal amounts of proteins as determined with the bicinchoninic acid assay (Pan et al., 2008). The primary antibodies used and their dilution were as follows: mouse anti-p21 (1:100; Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-T145p21 (1:100; Abcam, Cambridge, MA), rabbit anti-S146p21 (1:100; Abcam), mouse anti-p27 (1:10; Santa Cruz Biotechnology), rabbit anti-phosphoRb (1:500; Cell Signaling, Beverley, MA), rabbit anti-Rb (1:500; Santa Cruz Biotechnology), rabbit anti-Akt (1:500; Cell Signaling), rabbit anti-pS473Akt (1:200; Cell Signaling), rabbit anti-pT308Akt (1:100; Cell Signaling), rabbit anti-PDPK1 (Sigma-Aldrich), rabbit anti–pS241PDPK1 (Cell Signaling), rabbit anti-PI3k/p85 (1:100; Millipore, Billerica, MA), rabbit anti-PTEN (1:500; Cell Signaling), mouse anti-phosphotyrosine (1:50; BD Transduction Laboratories, Lexington, KY), rabbit anti–PP2A A subunit (1:100; Cell Signaling), rabbit anti–PP2A B55 subunit (1:100; Cell Signaling), rabbit anti-actin (1:1000; Sigma-Aldrich), and affinity-purified rabbit anti-synemin (1:500; Jing et al., 2005). Peroxidase-conjugated protein G (1:500; Pierce, Thermo Fisher Scientific, Rockford, IL) was used as secondary. Peroxidase activity was detected with ECL (Pierce) or tetramethylbenzidine (Kirkegaard and Perry Laboratories, Gaithersburg, MD).
Quantitation of protein ratios between control and experimental samples was performed by densitometry of Western blots, using a ratio standard curve as described (Pitre et al., 2007). Statistical analysis (t test or analysis of variance) was performed with InStat software (GraphPad Software, La Jolla, CA).
Akt activity assay
Cells were lysed in 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM ethylene glycol tetraacetic acid (EGTA), 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml leupeptin. Lysates were spun at 14,000 × g for 15 min, and the supernatants were incubated at 4°C for 18 h with Akt antibodies immobilized on agarose beads (Cell Signaling). Beads were washed twice with lysis buffer and twice with reaction buffer (25 mM Tris pH 7.5, 5 mM β-glycerophosphate, 2 mM dithiothreitol [DTT], 0.1 mM Na3VO4, 10 mM MgCl2). Next the beads were resuspended in 50 μl of reaction buffer containing 1 μg of GSK-3 fusion protein (Cell Signaling), 0.2 mM ATP, and 3 μCi of [γ-32P]ATP (4500 Ci/mmol; MP Biomedicals, Solon, OH). The substrate, GSK-3, was omitted in control reactions. Reactions proceeded for 30 min at 30°C and were terminated by adding an equal volume of SDS–PAGE sample buffer. GSK-3 phosphorylation levels were determined by autoradiography after SDS–PAGE. Immunoprecipitated Akt levels were measured by densitometry of Western blots and used for normalization of GSK-3 phosphorylation levels.
PI3K activity assay
PI3K activity was determined as described (Whitman et al., 1985; Orr et al., 2003). Briefly, cells were lysed in 20 mM Tris/HCl (pH 7.4), 1% NP-40, 140 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, 0.1 mM sodium orthovanadate, and protease inhibitors (Sigma-Aldrich). Lysates were spun at 14,000 × g for 10 min, and the supernatants were incubated with anti–p85/PI3K antibodies covalently conjugated to agarose beads (Millipore). Next the beads were washed twice with lysis buffer, twice with wash buffer (0.1 M Tris-HCl, pH 7.4, 5 mM LiCl, 0.1 mM sodium orthovanadate) and twice with TNE (10 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 0.1 mM sodium orthovanadate). Finally, the beads were suspended in 50 μl of TNE containing 1 mM MgCl2, 10 μg of phosphatidyl serine, 10 μg of PIP2, 10 μM ATP, and 30 μCi of [γ-32P]ATP (4500 Ci/mmol; MP Biomedicals). PIP2 phosphorylation proceeded for 10 min at 25°C. The kinase was omitted in control reactions.
The beads were then collected by centrifugation, and the bound kinase was eluted with SDS–PAGE sample buffer and analyzed by Western blot. PIP3 was extracted from the supernatants as described (Whitman et al., 1985; Orr et al. 2003) and analyzed by TLC on silica gel plates impregnated with 1% oxalate (Analtech, Newark NJ) and using chloroform/acetone/methanol/acetic acid/water (40/15/13/12/7, vol/vol). Autoradiography of the plates was performed and the densitometric values of the PIP3 spots were normalized to the amount of p85 recovered from the beads as determined by densitometry of Western blots.
mTORc2 activity assay
mTORc2 activity was determined as described (Beevers et al., 2009). Cells were lysed in ice-cold 40 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; pH 7.4), 120 mM NaCl, 1 mM EDTA, 1% Triton X-100, 10 mM pyrophosphate, 10 mM glycerophosphate, 50 mM NaF, 1.5 mM Na3VO4, and protease inhibitors (Sigma-Aldrich). Cell lysates were spun for 10 min at 14,000 × g. An aliquot of the supernatant was saved for total protein measurements for the purpose of normalization. Supernatants were incubated for 18 h at 4°C with anti-mTOR (FRAP N-19 antibody; Santa Cruz Biotechnology) and protein G–agarose beads. Next the beads were washed twice with lysis buffer and twice with reaction buffer (25 mM HEPES/NaOH, pH 7.4, 50 mM NaCl, 10% glycerol, 10 mM MnCl2, 1 mM DTT). For the phosphorylation reaction, the beads were resuspended in 30 μl of reaction buffer containing 1 μg of inactive Akt (Millipore), 5 μM ATP and 3 μCi of [γ-32P]ATP (4500 Ci/mmol; MP Biomedicals). Incubation was for 15 min at 25°C. Akt was omitted from control reactions. Reactions were terminated with SDS–PAGE sample buffer, and Akt phosphorylation levels were analyzed by autoradiography of SDS–PAGE gels.
PTEN and PP2A phosphatase assay
Cells were lysed in 10 mM imidazole-HCl (pH 7.0), 2 mM EDTA, 2 mM EGTA, 0.2% Triton X-100, and protease inhibitors (Sigma-Aldrich). Lysates were centrifuged at 2000 × g for 5 min. Supernatants were incubated for 3 h at 4°C with antibodies against either PTEN or PP2A (Cell Signaling) and then with protein G conjugated to agarose beads (Santa Cruz Biotechnology). Beads were washed three times with TBS and twice with reaction buffer (100 mM Tris, pH 8.0, for PTEN and buffer obtained from Millipore for PP2A). PIP3 (Echelon Bioscience, Salt Lake City, UT) or phosphopeptide KRpTIRR (Millipore) were added to beads bearing immunoprecipitated PTEN or PP2A, respectively. Incubation was for 10 min at 30°C. Control reactions did not include the substrate or the phosphatase. Inorganic phosphate released from the substrate was quantified with malachite green (Baykov et al., 1988) using an assay kit (Cayman Chemical Company, Ann Arbor, MI) and by measuring the optical density of the reaction product at 620 nm. Inorganic phosphate was used for the standard curve.
Immunoprecipitation
Cells were lysed in 20 mM Tris-HCl, pH 7.4, 1% NP-40, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2 mM NaF, 10 mM β-glycerophosphate, 2 mM Na4P2O4, and protease inhibitors (Sigma-Aldrich). Lysates were centrifuged at 5000 × g for 5 min, and supernatants were cleared with protein A/G–agarose beads (Pierce) for 30 min at 25°C. Supernatants were then mixed for 3 h at 25°C with the appropriate antibodies covalently linked to protein A/G–agarose beads (Pierce) following the manufacturer's instructions. Beads were washed five times with lysis buffer. Immunoprecipitated proteins were eluted from the beads with SDS–PAGE sample buffer and analyzed by Western blotting.
Immunofluorescence and proximal ligation assays
Control and synemin-silenced A172 cells were plated on glass coverslips at similar cell densities. For both immunofluorescence and PLA, cells were fixed in 4% paraformaldehyde and permeabilized in 0.1% NP-40 as detailed by Jing et al. (2005). Immunofluorescence with synemin and PP2A antibodies was performed following standard protocol (Jing et al., 2005). In situ PLA (Fredriksson et al., 2002) was carried out with the Duolink Detection Kit (Olink Bioscience, Uppsala, Sweden) according to the manufacturer's instructions. Briefly, fixed and permeabilized cells were incubated for 30 min at 37°C in blocking solution. Next they were incubated with the appropriate combination of affinity-purified goat anti-synemin (1:10), affinity-purified rabbit anti-synemin (1:10; Jing et al., 2005), mouse anti-Akt (1:10) (Cell Signaling), and rabbit anti–PP2A A subunit (1:10; Cell Signaling) for 60 min at 25°C. After washes, cells were incubated for 60 min at 37°C with appropriate PLA probes, consisting of secondary antibodies (anti-mouse, anti-rabbit, and anti-goat) conjugated to oligonucleotides (Fredriksson et al., 2002). Following washes, circularization and ligation of appropriate oligonucleotides were performed in ligase-containing solution for 30 min at 37°C. Cells were then rinsed briefly and incubated for 90 min at 37°C with the amplification solution prior to hybridizing the amplified product with complementary probe labeled with Alexa 568.
For immunofluorescence and PLA, cells were counterstained with phalloidin–Alexa 488 or 633 (1:500; Invitrogen, Carlsbad, CA). After PBS washes, coverslips were mounted with Prolong Antifade containing 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes, Invitrogen).
Observations were carried out with a Nikon A1 laser-scanning confocal microscope (Nikon, Melville, NY). Representative results are shown from experiments repeated at least three times.
Acknowledgments
This work was supported by a grant-in-aid to O.S. and a fellowship to A.P. from the Feist-Weiller Cancer Center at Louisiana State University Health Sciences Center in Shreveport and a graduate student assistantship from the University of Memphis to M.P.
Abbreviations used:
- CA
cantharidic acid
- IF
intermediate filament
- PLA
proximal ligation assay
- PP2A
protein phosphatase type 2A
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-08-0685) on February 15, 2012.
REFERENCES
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B. Protein phosphatase 2A regulates life and death decisions via Akt in a context-dependent manner. Proc Natl Acad Sci USA. 2007;104:19011–19016. doi: 10.1073/pnas.0706696104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005;24:7746–7755. doi: 10.1038/sj.onc.1209038. [DOI] [PubMed] [Google Scholar]
- Bayascas JR. Dissecting the role of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) signalling pathways. Cell Cycle. 2008;7:2978–2982. doi: 10.4161/cc.7.19.6810. [DOI] [PubMed] [Google Scholar]
- Baykov AA, Evtushenko OA, Avaeva SM. A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal Biochem. 1988;171:266–270. doi: 10.1016/0003-2697(88)90484-8. [DOI] [PubMed] [Google Scholar]
- Beevers CS, Chen L, Liu L, Luo Y, Webster NJ, Huang S. Curcumin disrupts the mammalian target of rapamycin-raptor complex. Cancer Res. 2009;69:1000–1008. doi: 10.1158/0008-5472.CAN-08-2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellin RM, Huiatt TW, Critchley DR, Robson RM. Synemin may function to directly link muscle cell intermediate filaments to both myofibrillar Z-lines and costameres. J Biol Chem. 2001;276:32330–3233. doi: 10.1074/jbc.M104005200. [DOI] [PubMed] [Google Scholar]
- Bellin RM, Sernett SW, Becker B, Ip W, Huiatt TW, Robson RM. Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J Biol Chem. 1999;274:29493–29499. doi: 10.1074/jbc.274.41.29493. [DOI] [PubMed] [Google Scholar]
- Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313–1319. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- Eriksson JE, Brautigan DL, Vallee R, Olmsted J, Fujiki H, Goldman RD. Cytoskeletal integrity in interphase cells requires protein phosphatase activity. Proc Natl Acad Sci USA. 1992;89:11093–11097. doi: 10.1073/pnas.89.22.11093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson JE, He T, Trejo-Skalli AV, Härmälä-Braskén AS, Hellman J, Chou YH, Goldman RD. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J Cell Sci. 2004;117:919–32. doi: 10.1242/jcs.00906. [DOI] [PubMed] [Google Scholar]
- Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Östman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. J Biol Chem. 2000;275:5535–5544. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- Granger BL, Lazarides E. Expression of the intermediate-filament-associated protein synemin in chicken lens cells. Mol Cell Biol. 1984;4:1943–1950. doi: 10.1128/mcb.4.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guérette D, Khan PA, Savard PE, Vincent M. Molecular evolution of type VI intermediate filament proteins. BMC Evol Biol. 2007;7:164. doi: 10.1186/1471-2148-7-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirako Y, Yamakawa H, Tsujimura Y, Nishizawa Y, Okumura M, Usukura J, Matsumoto H, Jackson KW, Owaribe K, Ohara O. Characterization of mammalian synemin, an intermediate filament protein present in all four classes of muscle cells and some neuroglial cells: co-localization and interaction with type III intermediate filament proteins and keratins. Cell Tissue Res. 2003;313:195–207. doi: 10.1007/s00441-003-0732-2. [DOI] [PubMed] [Google Scholar]
- Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 2005;24:3834–3845. doi: 10.1038/sj.emboj.7600847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izmiryan A, Peltekian E, Paulin D, Li ZL, Xue ZG. Synemin isoforms in astroglial and neuronal cells from human central nervous system. Neurochem Res. 2010;35:881–887. doi: 10.1007/s11064-009-0111-9. [DOI] [PubMed] [Google Scholar]
- Jing R, Pizzolato G, Robson RM, Gabbiani G, Skalli O. Intermediate filament protein synemin is present in human reactive and malignant astrocytes and associates with ruffled membranes in astrocytoma cells. Glia. 2005;50:107–120. doi: 10.1002/glia.20158. [DOI] [PubMed] [Google Scholar]
- Jing R, Wilhelmsson U, Goodwill W, Li L, Pan Y, Pekny M, Skalli O. Synemin is expressed in reactive astrocytes in neurotrauma and interacts differentially with vimentin and GFAP intermediate filament networks. J Cell Sci. 2007;120:1267–1277. doi: 10.1242/jcs.03423. [DOI] [PubMed] [Google Scholar]
- Kim S, Coulombe PA. Intermediate filament scaffolds fulfill mechanical, organizational, and signaling functions in the cytoplasm. Genes Dev. 2007;21:1581–1597. doi: 10.1101/gad.1552107. [DOI] [PubMed] [Google Scholar]
- Kim S, Wong P, Coulombe PA. A keratin cytoskeletal protein regulates protein synthesis and epithelial cell growth. Nature. 2006;441:362–365. doi: 10.1038/nature04659. [DOI] [PubMed] [Google Scholar]
- Ku NO, Michie S, Resurreccion EZ, Broome RL, Omary MB. Keratin binding to 14-3-3 proteins modulates keratin filaments and hepatocyte mitotic progression. Proc Natl Acad Sci USA. 2002;99:4373–4378. doi: 10.1073/pnas.072624299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku NO, Toivola DM, Strnad P, Omary MB. Cytoskeletal keratin glycosylation protects epithelial tissue from injury. Nat Cell Biol. 2010;12:876–885. doi: 10.1038/ncb2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. 2008;283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- Lee J, Kim SS. The function of p27 KIP1 during tumor development. Exp Mol Med. 2009;41:765–771. doi: 10.3858/emm.2009.41.11.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TY, Lai TY, Lin SC, Wu CW, Ni IF, Yang YS, Hung LY, Law BK, Wu CC. The B56γ3 regulatory subunit of protein phosphatase 2A (PP2A) regulates S phase-specific nuclear accumulation of PP2A and the G1 to S transition. J Biol Chem. 2010;285:21567–21580. doi: 10.1074/jbc.M109.094953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YM, Mackintosh C, Casida JE. Protein phosphatase 2A and its [3H]cantharidin/[3H]endothall thioanhydride binding site. Inhibitor specificity of cantharidin and ATP analogues. Biochem Pharmacol. 1993;46:1435–1443. doi: 10.1016/0006-2952(93)90109-a. [DOI] [PubMed] [Google Scholar]
- Luna G, Lewis GP, Banna CD, Skalli O, Fisher SK. Expression profiles of nestin and synemin in reactive astrocytes and Müller cells following retinal injury: a comparison with glial fibrillar acidic protein and vimentin. Mol Vis. 2010;16:2511–2523. [PMC free article] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez MG, Kojima S, Goldman RD. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010;24:1838–1851. doi: 10.1096/fj.09-151639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Thompson TG, Guyon JR, Lidov HGW, Brosius M, Imamura M, Ozawa E, Watkins SC, Kunkel LM. Desmuslin, an intermediate filament protein that interacts with α-dystrobrevin and desmin. Proc Natl Acad Sci USA. 2001;98:6156–6161. doi: 10.1073/pnas.111153298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel E, Rose M, Sevinc E, Hilgers RD, Hartmann A, Naami A, Knüchel R, Dahl E. Intermediate filament dynamics and breast cancer: aberrant promoter methylation of the synemin gene is associated with early tumor relapse. Oncogene. 2010;29:4814–4825. doi: 10.1038/onc.2010.229. [DOI] [PubMed] [Google Scholar]
- Omary MB, Ku NO, Strnad P, Hanada S. Toward unraveling the complexity of simple epithelial keratins in human disease. J Clin Invest. 2009;119:1794–1805. doi: 10.1172/JCI37762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr AW, Pedraza CE, Pallero MA, Elzie CA, Goicoechea S, Strickland DK, Murphy-Ullrich JE. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J Cell Biol. 2003;161:1179–1189. doi: 10.1083/jcb.200302069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallas DC, Shahrik LK, Martin BL, Jaspers S, Miller TB, Brautigan DL, Roberts TM. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60:167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- Pan Y, Jing R, Pitre A, Williams BJ, Skalli O. Intermediate filament protein synemin contributes to the migratory properties of astrocytoma cells by influencing the dynamics of the actin cytoskeleton. FASEB J. 2008;22:3196–3206. doi: 10.1096/fj.08-106187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paramio JM, Segrelles C, Ruiz S, Jorcano JL. Inhibition of protein kinase B (PKB) and PKCzeta mediates keratin K10-induced cell cycle arrest. Mol Cell Biol. 2001;21:7449–7459. doi: 10.1128/MCB.21.21.7449-7459.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Hanz S, Ben-Yaakov K, Segal-Ruder Y, Seger R, Fainzilber M. Vimentin-dependent spatial translocation of an activated MAP kinase in injured nerve. Neuron. 2005;45:715–726. doi: 10.1016/j.neuron.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Pitre A, Pan Y, Pruett S, Skalli O. On the use of ratio standard curves to accurately quantitate relative changes in protein levels by Western blot. Anal Biochem. 2007;361:305–307. doi: 10.1016/j.ab.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichelt J, Magin TM. Hyperproliferation, induction of c-Myc and 14-3-3sigma, but no cell fragility in keratin-10-null mice. J Cell Sci. 2002;115:2639–2650. doi: 10.1242/jcs.115.13.2639. [DOI] [PubMed] [Google Scholar]
- Resjö S, Göransson O, Härndahl L, Zolnierowicz S, Manganiello V, Degerman E. Protein phosphatase 2A is the main phosphatase involved in the regulation of protein kinase B in rat adipocytes. Cell Signal. 2002;14:231–238. doi: 10.1016/s0898-6568(01)00238-8. [DOI] [PubMed] [Google Scholar]
- Rossio V, Yoshida S. Spatial regulation of Cdc55-PP2A by Zds1/Zds2 controls mitotic entry and mitotic exit in budding yeast. J. Cell Biol. 2011;193:445–454. doi: 10.1083/jcb.201101134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlgren CM, Pallari HM, He T, Chou YH, Goldman RD, Eriksson JE. A nestin scaffold links Cdk5/p35 signaling to oxidant-induced cell death. EMBO J. 2006;25:4808–4819. doi: 10.1038/sj.emboj.7601366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Shima H, Osawa Y, Nagao M, Hemmings BA, Kishimoto T, Hisanaga S. Neurofilament-associated protein phosphatase 2A: its possible role in preserving neurofilaments in filamentous states. Biochemistry. 1995;34:7376–7384. doi: 10.1021/bi00022a010. [DOI] [PubMed] [Google Scholar]
- Sandoval IV, Colaco CA, Lazarides E. Purification of the intermediate filament-associated protein, synemin, from chicken smooth muscle. Studies on its physicochemical properties, interaction with desmin, and phosphorylation. J Biol Chem. 1983;258:2568–2576. [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the Rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Schmitt-Graeff A, Jing R, Nitschke R, Desmoulière A, Skalli O. Synemin expression is widespread in liver fibrosis and is induced in proliferating and malignant biliary epithelial cells. Hum Pathol. 2006;37:1200–1210. doi: 10.1016/j.humpath.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Bloom GS, Mumby MC. A novel pool of protein phosphatase 2A is associated with microtubules and is regulated during the cell cycle. J Cell Biol. 1995;128:1131–1144. doi: 10.1083/jcb.128.6.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag E, Nunbhakdi-Craig V, Lee G, Brandt R, Kamibayashi C, Kuret J, White CL, 3rd, Mumby MC, Bloom GS. Molecular interactions among protein phosphatase 2A, tau, and microtubules. Implications for the regulation of tau phosphorylation and the development of tauopathies. J Biol Chem. 1999;274:25490–25498. doi: 10.1074/jbc.274.36.25490. [DOI] [PubMed] [Google Scholar]
- Sultana S, Sernett SW, Bellin RM, Robson RM, Skalli O. Intermediate filament protein synemin is transiently expressed in a subset of astrocytes during development. Glia. 2000;30:143–153. doi: 10.1002/(sici)1098-1136(200004)30:2<143::aid-glia4>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Sun A, Bagella L, Tutton S, Romano G, Giordano A. From G0 to S phase: a view of the roles played by the retinoblastoma (Rb) family members in the Rb-E2F pathway. J Cell Biochem. 2007;102:1400–1404. doi: 10.1002/jcb.21609. [DOI] [PubMed] [Google Scholar]
- Sun N, Huiatt TW, Paulin D, Li Z, Robson RM. Synemin interacts with the LIM domain protein zyxin and is essential for cell adhesion and migration. Exp Cell Res. 2010;316:491–505. doi: 10.1016/j.yexcr.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao GZ, Toivola DM, Zhou Q, Strnad P, Xu B, Michie SA, Omary MB. Protein phosphatase-2A associates with and dephosphorylates keratin 8 after hyposmotic stress in a site- and cell-specific manner. J Cell Sci. 2006;19:1425–1432. doi: 10.1242/jcs.02861. [DOI] [PubMed] [Google Scholar]
- Tawk M, Titeux M, Fallet C, Li Z, Daumas-Duport C, Cavalcante LA, Paulin D, Moura-Neto V. Synemin expression in developing normal and pathological human retina and lens. Exp Neurol. 2003;183:499–507. doi: 10.1016/s0014-4886(03)00240-1. [DOI] [PubMed] [Google Scholar]
- Toivola DM, Nieminen MI, Hesse M, He T, Baribault H, Magin TM, Omary MB, Eriksson JE. Disturbances in hepatic cell-cycle regulation in mice with assembly-deficient keratins 8/18. Hepatology. 2001;34:1174–1183. doi: 10.1053/jhep.2001.29374. [DOI] [PubMed] [Google Scholar]
- Turowski P, Myles T, Hemmings BA, Fernandez A, Lamb NJ. Vimentin dephosphorylation by protein phosphatase 2A is modulated by the targeting subunit B55. Mol Biol Cell. 1999;10:1997–2015. doi: 10.1091/mbc.10.6.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Lüscher B. Post-translational regulation of the tumor suppressor p27(KIP1) Cell Mol Life Sci. 2008;65:3255–3264. doi: 10.1007/s00018-008-8296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virshup DM, Shenolikar S. From promiscuity to precision: protein phosphatases get a makeover. Mol Cell. 2009;33:537–545. doi: 10.1016/j.molcel.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Vuoriluoto K, Haugen H, Kiviluoto S, Mpindi JP, Nevo J, Gjerdrum C, Tiron C, Lorens JB, Ivaska J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene. 2011;30:1436–1448. doi: 10.1038/onc.2010.509. [DOI] [PubMed] [Google Scholar]
- Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315:239–242. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- Yang SI, Lickteig RL, Estes R, Rundell K, Walter G, Mumby MC. Control of protein phosphatase 2A by simian virus 20 small-T antigen. Mol Cell Biol. 1991;11:1988–1995. doi: 10.1128/mcb.11.4.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]