

Abstract
Structural biology places a high demand on proteins both in terms of quality and quantity. Although many protein expression and purification systems have been developed, an efficient and simple system which can be easily adapted is desirable. Here, we report a new system which combines improved expression, solubility screening and purification efficiency. The system is based on two newly constructed vectors, pEHISTEV and pEHISGFPTEV derived from a pET vector. Both vectors generate a construct with an amino-terminal hexahistidine tag (His-tag). In addition, pEHISGFPTEV expresses a protein with an N-terminal His-tagged green fluorescent protein (GFP) fusion to allow rapid quantitation of soluble protein. Both vectors have a tobacco etch virus (TEV) protease cleavage site that allows for production of protein with only two additional N-terminal residues and have the same multiple cloning site which enables parallel cloning. Protein purification is a simple two-stage nickel affinity chromatography based on the His tag removal. A total of seven genes were tested using this system. Expression was optimised using pEHISGFPTEV constructs by monitoring the GFP fluorescence and the soluble target proteins were quantified using spectrophotometric analysis. All the tested proteins were purified with sufficient quantity and quality to attempt structure determination. This system has been proven to be simple and effective for structural biology. The system is easily adapted to include other vectors, tags or fusions and therefore has the potential to be broadly applicable.
Keywords: Vector, pEHISGFPTEV, pEHISTEV, Protein expression, Purification protocol, TEV protease, AKTAxpress
Introduction
Techniques used for target selection, gene cloning, protein expression, purification and crystallization have developed rapidly [17, 34, 58]. As structural genomics projects have picked up in pace, the demand for rapid and simple production of pure proteins in milligram amounts has accelerated. In fact, the large-scale production of pure and active proteins is now regarded as the principle bottleneck in structural studies. As structure determination becomes a more routine technique, all laboratories will benefit from simple and effective systems for protein production.
Many different expression and purification systems have been developed to express a target protein in eukaryotic [15] or prokaryotic cells [11, 33, 43, 46, 48, 54]. The start of any project involves a bewildering set of choices which include expression vectors, purification schemes and fusion cleavage proteases. Among the factors affecting these choices are the level of the protein expression, solubility of the fusion protein, specificity of the protease, specificity and affinity of the purification resin, the associated time, material costs and the final yield. The E. coli expression systems are recombinant protein production workhorses with the various pET vector systems [54] being the most common. With pET vectors, the T7 promoter leads to high expression levels but optimisation of culture conditions is often required to balance soluble expression against overall expression. This is because proteins over-expressed in E. coli often misfold and aggregate [22, 31]. Despite extensive work trying to predict protein folding from sequences [41, 63], trial and error remains the principal method to obtain soluble proteins. A number of technologies for solubility screening have been reported [10, 20, 23, 34], however these methods increase the cost and are time consuming. The use of a green fluorescent protein (GFP)1 fusion [61] or, more recently, the translational incorporation of a fluorescent amino acid derivative fusion [8] have eliminated the need for any purification before quantitating soluble expression. These technologies have greatly simplified the evaluation of expression trials. However, the published vectors are often not compatible with the large scale expression and purification for crystallisation. As a result, re-cloning can be required and this introduces further steps with the potential for practical difficulty. The production of recombinant proteins as affinity-tagged fusions has become a routine technique. Available tags include His-tag fusions [26, 55], maltose-binding protein fusions [12, 30], glutathione S-transferase fusions [18, 19, 52] and chitin-binding domain fusions [16, 62]. In most cases N-terminal tags can be removed to yield recombinant proteins very similar to the native protein by using proteases. C-terminal tags often leave longer extensions if removed. Commercially available proteases include factor Xa [21, 32], enterokinase [26, 56], thrombin [35], Rhinovirus 3C [3, 24], Tobacco etch virus NIa proteases (TEV protease) [36] and more recently a self splicing element intein [6, 7]. Non-sequence specific cleavage [26, 56], materials cost, buffer sensitivity, contaminating E. coli proteins and removal of protease can present practical problems [1, 9] which require further intervention and / or optimisation. TEV protease recognises a specific heptapeptide sequence E-X-X-Y-X-Q↓S/G, is active under a wide range of conditions and in the presence of various protease inhibitors [13, 14, 36, 37, 42]. E. coli over-expression of a highly active form of the TEV protease has been reported [28, 59].
Here, we report a modified E. coli expression and purification system using two new pEHISGFPTEV and pEHISTEV vectors in which a gene can be cloned in parallel. Our approach combines the established advantages of the GFP-based solubility screening [8, 61], His-tag affinity isolation, TEV protease-mediated fusion partner removal with a powerful differentiated purification method. As a result, it provides a simple and efficient method for protein production that should be widely useful.
Materials and Methods
Plasmids, Bacterial Strains and Enzymes
Plasmid pET30a was obtained from Novagen. pEGFP-C1 vector was purchased from Clontech. Plasmid DNA containing Klebsiella pneumoniae UDP-galactopyranose mutase (Mutase-kp) [25], human nuclear pore complex protein RanBP2 (Nuporin358) [40], and tomato yellow leave curl virus (TYLCV) China isolate [60] were kindly provided by Dr. C. Whitfield (University of Guelph, Canada), Dr. M. S. Rodriguez (Laboratoire de Transport Nucléocytoplasmique, Institut Jacques Monod, CNRS UMR 7592, Tour 43, 2 Place Jussieu, 75251 Paris Cedex 05, France), and Dr. Yiguo Hong (Horticultural Research International, Wellesbourne, UK), respectively. Plasmids containing adenovirus precursor terminal protein (pTP), Nuclear factor III (NFIII) [4] and human nuclear body protein SP100 [47] and Sulfolobus solfataricus P2 (Sso) genomic DNA existed in house. E. coli strain DH5α and BL21 (DE3) were purchased from Stratagene. All the restriction endonucleases, T4 DNA ligase and Vent DNA polymerase were purchased from either Promega or New England BioLabs. His-tagged TEV protease was cloned as described [13, 14, 36, 37, 42] using a TEV cDNA clone pTEVGFP (SCRI, Dundee, UK), then expressed and purified [28, 59].
Construction of pEHISTEV and pEHISGFPTEV vector
DNA manipulation was carried using established procedures [45]. To construct pEHISTEV vector, pET30a vector DNA was digested with NdeI/BamHI and the digested DNA was isolated using gel extraction kid (Qiagen). Oligonucletides (CODSEQ) encoding His-tag, TEV protease recognition site and the multiple cloning site (MCS) were synthesised. The double-stranded DNA was generated by PCR using these as the template with forward primer HITEVF and reverse primer HISTEVR (Table 1). The PCR-generated DNA fragment was digested by NdeI/BamHI and then ligated into the NdeI/BamHI–digested pET30a vector. To construct pEHISGFPTEV vector, the GFP encoding sequence was PCR amplified using a forward primer HISGFPF and a reverse primer HISGFPR. The amplified DNA fragments were digested by NdeI and NcoI and then ligated into a similarly digested pEHISTEV vector. The vectors were sequenced to confirm their integrity.
Table 1.
Primers used for gene cloning
| Primersa | Sequences | Res. enzymesb |
|---|---|---|
| CODSEQ | 5′CCGAACATATGTCGTACTACCATCACCATCACCATCA- CGATTACGACATCCCAACGACCGAAAACCTGTATTTTC- AGGGCGCCATGGCTGATATCGGATCCGGAA3′ |
NdeI/Ba mHI |
|
| ||
| HISTEV-F | 5′CCGAACATATGTCGTACTAC 3′ | NdeI |
| HISTEV-R | 5′TTCCGGATCCGATATCAGCC 3′ | BamHI |
| HISGFP-F | 5′GCGACATATGTCGTACTACCATCACCATCACCATCAC GATTACGATATGGTGAGCAAGGGCGAGG 3′ |
NdeI |
| HISGFP-R | 5′TCAGCCATGGCGCCCTGAAAATACAGGTTTTCGGTCG TTGGGATATCGTAATCCTTGTACAGCTCGTCC 3′ |
NcoI |
| NFIIIhd-F | 5′GGCGCCATGGAGGGCTTGAGCCGTAGGAGG3′ | NcoI |
| NFIIIhd-R | 5′AATTCGGATCCTCATGGTGGGTTGATTCTTTTTTC3′ | BamHI |
| SP100-F | 5′CTGGTGTCATGACTTTTCGAAGC3′ | BspHI |
| SP100-R | 5′TTATTTTCGAATTCTTATGGCTG3′ | EcoRI |
| RanBP2-F | 5′GTGAAACCATGGAACCATTTGC3′ | NcoI |
| RanBP2-R | 5′GTTATGGATCCTTACTGAGATTTTTG3′ | BamHI |
| Mutasekp-F | 5′ATACCCATGGAAAGTAAAAAAATATTG3′ | NcoI |
| Mutasekp-R | 5′CCAAGGATCCTCATCGTACAGAAAC3′ | BamHI |
| pTP-F | 5′CATGGTCATGAACCTTGAGCGTCAAC3′ | BspHI |
| pTP-R | 5′ATGCGAATTCCTAAAAGCGGTGACGC3′ | EcoRI |
| SSO2226-F | 5′GGCGCCATGGTAAAATATTCTATGCATTTAAAC3′ | NcoI |
| SSO2226-R | 5′AATTCGGATCCCTAAGGCCAACCTAAAATAG3′ | BamHI |
| TYLCVC2-F | 5′GGCGTCATGAGATCTTCGTCACCATCGACCAGCC3′ | BspHI |
| TYLCVC2-R | 5′AATTCGGATCCTTAAAGACTCTTAAAAAATGACC3′ | BamHI |
Forward primer (F) and reverse primer (R) used for PCR amplification.
Restriction enzymes used for cloning, the recognition sequence are underlined, BspHI-digested fragments were compatibly ligated into NcoI-digested vectors.
Construction of the pEHISTEV and pEHISGFPTEV carrying NFIIIhd, SP100181-480, RanBP22532-2767, Mutase-kp, pTP, Sso2226 and TYLCV C2 gene
The coding sequences of a functional domain of NFIII (NFIIIhd) which enhances adenovirus DNA replication, SP100 (SP100181-480) a human protein associated with nuclear bodies [53], RanBP2 (RanBP22532-2767) a protein modified by SUMO in human cells [29], UDP-galactropyranose mutase from Klebsiella pneumoniae (Mutase-kp), the precursor of adenovirus terminal protein (pTP), tomato yellow leave curl virus protein C2 (TYLCV C2) and protein 2226 from the archaon Sulfolobus sulfatariucus (Sso2226) were amplified by PCR using the primers specified in Table 1 with the appropriate templates described above. The PCR fragments were digested using the restriction enzymes (Table 1) and in parallel ligated into appropriately digested pEHISTEV and pEHISGFPTEV vectors. The insertions were sequenced to confirm their integrities.
Protein expressions and solubility screening
pEHISGFPTEV and pEHISTEV constructs containing the target protein coding sequences were transformed into E. coli strain BL21(DE3). The transformed E. coli cells were spread on L-agar plates containing 50μg/ml kanamycin and 0.4mM IPTG. After incubation overnight at 37°C, the plate was imaged under a UV light and the density of fluorescence was visually assessed. For solubility screening, an overnight culture was prepared by growing a single fluorescent colony of each transformed E. coli in 10 ml of Lysogeny broth (LB) medium [45] containing 50μg/ml of kanamycin overnight at 37°C. Small-scale (10 ml) expression evaluations were carried out in a 25 ml universal bottle. Cells were grown at 37 °C in an incubator with a shaking speed of 180 rpm and protein expression was induced with 0.4 mM IPTG at a cell density of 0.6 of OD600 for 8 hours at 37°C, or overnight at 20°C or 25°C. After induction, cells were collected by centrifugation, resuspended in 0.5 ml of Buffer A (Table 2) and sonicated for 3 × 10 seconds on ice with 1 minute interval to prevent overheat using a small probe at 15 amplitude microns (Soniprep 150, MSE). Lysozyme can be added but in our hands did not improve the yield. The soluble fraction was collected by centrifugation for 5 min at 14000 rpm in a microfuge. The expressed target protein was quantified by measuring the absorption of HisGFP-fusion at 488nm using a NanoDrop Spectrophotometer (ND-1000 UV-Vis, Labtech). The absorbance of HisGFP at 488 nm was measured using purified HisGFP protein and the extinction coefficient was calculated based on Beer’s Law. The expressed soluble protein was estimated as: Target protein (g/L−1) = 0.05 x OD488 /AbsHisGFP 0.1% (=1g/L−1) x MWtarget protein/MWHisGFP. The volume of scale-up culture used for production of 15 mg of target proteins was estimated based on the solubility screening results. This culture was inoculated with 1% of overnight-cultured inoculum. Expression was carried out under the conditions identified in the small expression trials. After induction, the cells expressing the recombinant proteins were harvested in 50 ml tubes by centrifugation and kept at -80°C.
Table 2.
Buffers used for protein purification
| Buffers | Detailsa |
|---|---|
| Buffer A | 50mM NaH2PO4, 400mM NaCl, 1mM β-mercaptoethanol, 10mM imidazole, pH7.5 |
| Buffer B | 50mM NaH2PO4, 400mM NaCl, 30mM imidazol, 1mM PMSF, pH7.5 |
| Buffer C | 50mM NaH2PO4, 400mM NaCl, 300mM imidazol, 1mM PMSF, pH7.5 |
| Buffer D | 50mM NaH2PO4, 50mM Tris-HCl pH7.5, 400m M NaCl and 1mM DTT |
| Buffer E | 50mM NaH2PO4, 50mM Tris-HCl pH7.5, 400m M NaCl and 30mM imidazol |
| Low salt GF buffer | 10mM Tris pH7.5, 150mM NaCl. |
| Cellular extraction buffer (CEB) |
50mM NaH2PO4, 10mM Tris-HCl, 400mM NaCl, 1mM DTT, pH8.0 |
| Denaturing sample buffer (DSB) |
100mM NaH2PO4, 10mM Tris-HCl, 6M GuHCl pH8.0 |
| Denature washing buffer (DWB) |
100mM NaH2PO4, 10mM Tris-HCl, 6M GuHCl pH6.3 |
| Denature elution buffer (DEB) |
100mM NaH2PO4, 10mM Tris-HCl, 6M GuHCl pH4.5 |
| Refolding buffer I | 100mM NaH2PO4, 10mM Tris-HCl, 3M GuHCl pH8.0 |
| Refolding buffer II | 100mM NaH2PO4, 10mM Tris-HCl, 1.5M GuHCl pH8.0 |
PMSF: Phenylmethylsulfonyl Fluoride; DTT: dithiothreitol; GuHCl: guanidine hydrochloride.
Manual protein purification using nickel affinity column under native conditions
All the buffers used for protein purification are detailed in Table 2. To purify the expressed target protein under native conditions, cell pellet from one litre culture was thawed on ice in 10 ml of Buffer A plus protease inhibitors (Complete, Mini, EDTA-free, Roche) and optionally 200μg/ml of lysozyme and then sonicated three times, each for 30 seconds with 1 minute interval to prevent overheat using a large probe at 15 amplitude microns. The supernatant was collected by centrifugation at 18000 rpm for 30 min at 4°C. After filtering through a 0.2μm acrodisc filter, the supernatant was loaded onto a nickel affinity sepharose (NiAC) column pre-equilibrated with Buffer A with a flow rate of 1 ml/min. After washing the column with 10 bed volumes of Buffer B the target protein was eluted with Buffer C. The protein-containing fractions were pooled and dialysed against Buffer D. After dialysis, the purified protein was diluted to 1 mg/ml and mixed with His-tagged TEV protease (10μg of protease for 1mg of proteins). The digestion was carried out in the dialysis tubing in Buffer D for 8 hrs at 20°C or overnight at 4°C. After digestion, the protein sample was dialysed against the Buffer E for 1 hr. It was then loaded onto a NiAC column pre-equilibrated with Buffer E and followed by adding 2 bed volumes of Buffer B. The flow through fractions containing the expressed protein were collected, analysed by SDS-PAGE, and then applied to a gel filtration (GF, Superdex 75 or 200, Amersham Biosciences) column. The proteins were then concentrated using ultrafiltration and the concentration determined using ultraviolet-visible spectroscopy. The integrity and identity of all proteins were verified by mass spectrographic analysis.
Manual protein purification under denaturing conditions
The cell pellet was thawed in 10 ml of CEB buffer (Table 2) and then sonicated three times, each for 30 seconds with 1-minute interval to prevent overheat. The pellet was collected by centrifugation at 18000 rpm for 15 min and then resuspended in the 10ml of CEB buffer and sonicated again. The pellet was dissolved in DSB buffer (Table 2) and clarified by centrifugation at 18000 rpm for 30 min. After being filtered through a 0.2μm acrodisc filter, the sample was loaded onto a NiAC column pre-equilibrated with DSB buffer with a flow rate of 1 ml/min. The column was washed with 3 bed volumes of DWB buffer and eluted with DEB buffer (Table 2). The protein-containing fractions were pooled. To refold the protein, the pooled fractions were first dialysed against refolding buffer I, and then against refolding buffer II. Finally the protein sample was slowly diluted 10 times with CEB buffer containing 1% Triton X-100, 2mM DTT and 15% glycerol and stirred at 4°C overnight. Insoluble protein was removed by centrifugation and the soluble protein dialysed extensively against CEB buffer containing 0.5% Triton X-100 and 10% glycerol.
Automated multistep protein purification
An AKTAxpress together with the software UNICORN version 5.01 (GE Healthcare, Amersham Biosciences, Uppsala, Sweden) was used for automated multistep protein purifications at 20°C of Sso2226 and Mutase-kp expressed using pEHISTEV. The protocol uses a NiAC column (HisTrap HP, 5ml), a desalting (DS, Hiprep 26/10, Amersham Biosciences) column, a superloop, a second NiAC (HisTrap HP, 5ml) column and GF (Superdex 200, Amersham Biosciences) column in a linear series using the default settings (AC-DS-GF) preinstalled in the AKTAxpress. The proteins were detected by monitoring the absorbance at 280 nm and fractions above the designated threshold were collected into internal storage loops or directed onto the next column. His-tagged protein was eluted using 5 column volumes of Buffer C at 1 ml/min pooled then injected onto to the desalting column to remove imidazole. The protein was then injected into a superloop (preloaded with 1 ml of His-tagged TEV protease, 5 mg/ml) at 10ml/min. This higher flow rate ensured that the sample was mixed efficiently with the protease. After 8 hours the mixture in the superloop was applied onto the second 5-ml His-Trap FF (NiAC) column attached to the upper tubing of a GF column. The second NiAC column removed undigested protein, cleaved His-tag, His-tagged TEV protease and non-specific E. coli resin-binding proteins. The flow-through was applied directly onto the GF column. The process can be run without manual intervention, all fractions not collected as target protein were stored for off line analysis but no significant amount of target protein was detected.
Results
pEHISTEV and pEHISGFPTEV Vector
Vector pEHISTEV and pEHISGFPTEV were constructed identically except for the GFP coding sequence. This allows parallel cloning one gene into both vectors for solubility screening and large scale purification. pEHISTEV expresses N-terminal TEV protease cleavable His-tagged protein while pEHISGFPTEV expresses an N-terminal, TEV protease cleavable His-GFP fusion protein. The sequences of the MCS of pEHISTEV/pEHISGFPTEV are shown in Fig. 1. Both vectors use a T7 promoter and T7 terminator and are suitable for expressing a target protein in E. coli strains expressing T7 RNA polymerase such as BL21 (DE3) and its derivatives.
Fig. 1. Multiple cloning sites of pEHISTEV /pEHISGFPTEV.

Both vectors were constructed containing the f1 replication origin, kanamycin resistance gene, pB332 DNA replication origin, and lac repressor gene. The sequence details of the cloning/expression region show the T7 promoter, lac operator, 6xHis-tag (pEHISTEV) or 6xHis-tagged GFP (pEHISGFPTEV), TEV protease recognition site, restriction edonuclease sites for cloning, and the T7 terminator.
Expression and purification of the target proteins
Using our expression system, the fluorescence of single colonies showed the accumulation of the expressed target proteins (Fig. 2A) as reported previously [5, 61]. Spectrophotometric analysis of the purified HisGFP revealed that the extinction coefficient in units of M−1 cm−1 at 488 nm is 44934 and Abs 0.1%, 1gL−1 is 1.446 similar to that reported previously [38]. Based on the obtained extinction coefficient, the soluble target proteins in the cell are calculated and shown in Fig. 2B. In most cases, scaling up to two litres of cell culture was enough to produce 10 mg of pure protein.
Fig. 2. HisGFP mediated soluble expression screening.

A). Colonies of E. coli cells transformed with pEHISGFPTEV constructs (top) expressing GFP-NFIIIhd, RanBP22532-2767, Mutase-kp or Sso2226 and with their pEHISTEV constructs (bottom) on the L-agar plate containing 0.4 mM IPTG. We found that the His-tagged and His-GFP colonies could be combined on the same plate and selection was still straightforward. B) Quantitative analysis of soluble expression of the target proteins under the optimised conditions. Measurement of OD488 of purified HisGFP is shown on the bottom and the extinction coefficient was calculated based on Beer’s Law. The concentration of the expressed target proteins (after His-GFP removal) was calculated as described in the Materials and Methods section.
To test the effect of His-GFP fusion on the target protein expression levels, the target proteins were expressed and purified using both vector systems. The results showed that only NFIIIhd had significant difference in soluble expression, with the GFP fusion being more highly expressed in the soluble form than the simple His-tag (10mgL−1 vs 4mgL−1) indicating that the GFP improved the folding of this truncated NFIII functional domain. The His-GFP construct of NFIIIhd purified as easily as the smaller His-tagged construct (Fig. 3). In the other cases the yield of soluble protein obtained using pEHISTEV constructs (Fig. 4) was similar to that obtained using GFP-fusion (Fig. 5). However, in the case of pTP, more protein (10mgL−1) was obtained using pEHISTEV in comparison with that (7mgL−1) using pEHISGFPTEV. It is possible that the GFP fusion increased the molecular mass of pTP and reduced the expression efficiency. The proteins purified using both vectors showed a similar elution profiles in gel filtration indicating all the proteins expressed have proper conformation. The purified pTP showed activity in DNA initiation assay (data not shown) similar to that expressed using baculoviruses, confirming the protein is folded and functional. Expression of RanBP22532-2767 and SP100181 480 yielded 18 mgL−1 - and 8 mgL−1 respectively and the purified proteins were used for further biological analyses [57].
Fig. 3. Manual purification of NFIIIhd expressed using pEHISGFPTEV vector.

Green fluorescence of purified HisGFPNFIIIhd in elution fractions from first NiAC column (A); SDS-PAGE analysis of the accumulation of HisGFPNFIIIhd (hd) in comparison with the total cellular proteins (B); Elution fraction from the first NiAC column (C) and flow through fractions of second NiAC column after His-GFP removal (D). Numbers on the top of each image indicate the elution fractions. PM: protein molecular weight markers.
Fig. 4. SDS-PAGE analysis of expression and purification of pTP (A), RanBP22532-2767 (B) and SP100181-480 (C) using pEHISTEV vector.

pTP, RanBP22532-2767 and SP100181-480 were expressed using pEHISTEV vector in E. coli strain BL21(DE3) and purified as described in the Materials and Methods. Proteins were analysed using a 12% polyacrylamide gel and stained with 0.25% Coomassie blue. The positions of the proteins with His-tag, without His-tag and His-TEV protease are indicated. PM: protein markers, TCP-un, TCP-in: total cellular proteins from un-induced and induced cells, Ni1-pur: protein purified after the first NiAC column, Pro-C: protein after TEV protease cleavage and Ni2-pur: protein purified after the second NiAC.
Fig. 5. SDS-PAGE analysis of expression and purification of Mutase-kp (A), pTP (B) SP100181-480 (C) RanBP22532-2767 (D) and Sso2226 (E) using pEHISGFPTEV vector.
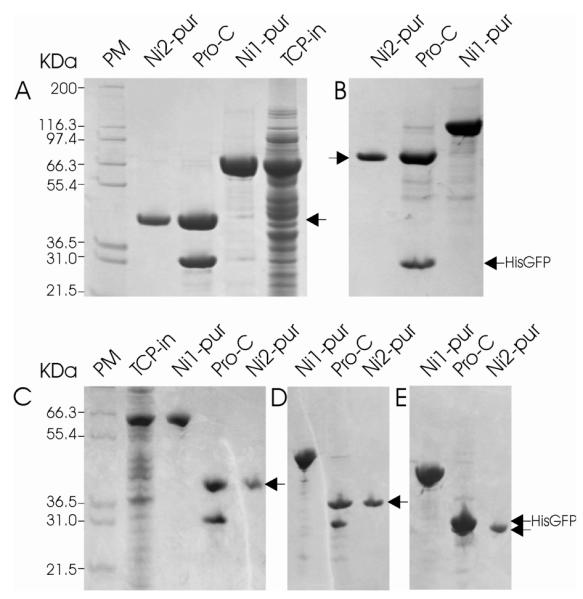
Mutase-kp, pTP, SP100181-480, Sso2226 and RanBP22532-2767 were expressed using pEHISGFPTEV vector in E. coli strain BL21(DE3) and purified as described in the Materials and Methods. Proteins were analysed using a 12% polyacrylamide gel and stained with 0.25% Coomassie blue. The positions of the purified proteins and His-GFP are marked by arrows. PM, TCP-un, TCP-in, Ni1-pur, Pro-C and Ni2-pur as for Fig. 4.
Essentially, all TYLCV C2 [60] accumulated in inclusion bodies irrespective of vector although a trace of soluble protein was detected when expressed as a GFP fusion (Fig. 2). Purification of this protein was carried out under denaturing conditions and the results are shown in Fig. 5. The purified His-tagged C2 protein was then refolded and the His-tag was cleaved by TEV protease after a 4 hour incubation at 37°C. The His-tag and TEV protease were removed by a second NiAC column step with a final protein yield of 5 mgL−1.
Automated purification of the proteins expressed using our new vector systems proved successful. Chromatograms of fully automated purification of Sso2226 and Mutase-kp are shown in Fig. 6. The final purification yields were 13 mgL−1 of Sso2226 protein and 25 mgL−1 of Mutase-kp with a recovery rate of 78% for Sso2226 and 64.2% for Mutase-kp.
Fig. 6. SDS-PAGE analysis of the expression and purification of TYLCV C2 under denaturing conditions.
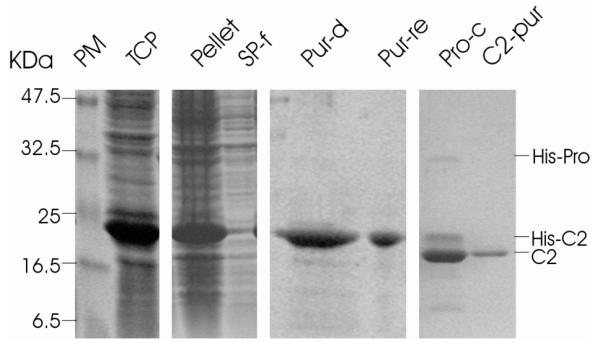
TYLCV C2 gene was expressed using pEHISTEV vector in E. coli strain BL21(DE3). Proteins were analysed using a 15% polyacrylamide gel and stained with 0.25% Coomassie blue. The purified C2 proteins, TEV protease and the cleaved His-tag are indicated. PM: protein markers, TCP: total cellular proteins, SP-f: soluble proteins, Pur-D: purified denatured C2, Pur-re: refolded C2, Pro-C: C2 after TEV protease cleavage and C2-pur: C2 after second NiAC column.
Discussion
The use of GFP fusions to monitor the solubility [5, 8, 39, 44, 61] and His tags to aid the purification of recombinant proteins have been widely used. The difficulties in recloning often faced by smaller laboratories may have restricted the use of some of these technologies. We now report a system which allows for parallel cloning into both His-tagged and His-GFP fusion vectors by engineering a common multiple cloning site. Irrespective of the choice of vector, the downstream expression and purification steps are identical. The system builds in a very powerful differentiated purification method that gives pure protein in a highly reproducible, relatively cheap and standardised protocol. We have shown that, in favourable cases, this procedure can be carried out entirely automatically. While fully automated procedures may not be cost effective for uncharacterised or poorly expressing proteins, they are potentially very effective for multiple repeat experiments for well characterised proteins. The final protein product has only two additional amino acids when compared to native (after cleavage by TEV protease which is itself removed as part of the protocol). Using the proteins produced in this study, only the structure of Mutase-kp has been determined so far [2]. Several structures have been determined using this system but their expression not studied in depth, these included structure of human quinolinate phosphoribosyltransferase [27], structure of NEDP1 in isolation and in a transition state complex with NEDD8 [51], structure of SENP1-SUMO-2 complex [50] and structure of VC1805 [49].
The ability to do small-scale experiments using GFP to monitor expression has been shown to be extremely useful in identifying either optimal constructs or conditions for expression [5, 39, 44]. Our vectors are based on very common pET vectors and present no major change in expression or sequencing protocol. Our results showed that the fluorescence of single colonies indicated the accumulation of the expressed soluble target proteins (Fig. 2A). The GFP constructs even when used on a small scale are very robust indicators of expression of the expression level of the simpler His-tagged construct. It should be noted that the yield of HisGFP is expressed as molar concentration based on fluorescence density. The yields of the expressed target proteins are described as mg ml−1 (following the current convention). Thus a high yield (expressed as mg ml−1) of a large target protein can be obtained even when the fluorescent density of its HisGFP-fusion (Fig 2B) is lower than another smaller (in terms of molecular weight) fusion protein. In addition, a large target protein attached to the GFP fusion can quench the fluorescence of HisGFP and result in an underestimation of the concentration. C-terminal tagged GFP fusion has been reported as a “folding reporter” for solubility screening [61], our N-terminal His-GFP fusion expression system produces a final protein product with only two additional amino acids after tag removal. In most cases, we did not detect any difference between the His tag at the N-terminus and the larger HisGFP at the N-terminus. NFIIIhd, the N-terminal HisGFP fusion showed enhanced expression with HisGFP (compared to His alone) whereas pTP showed lower soluble expression. We therefore have insufficient evidence to offer a definitive finding on what vector should be used and suspect that it may be protein dependent. For proteins smaller than around 50kDa, we did not observe any loss of yield or problem in purifying the His-tagged GFP fusion. For larger target proteins, the additional mass of the GFP fusion could conceivably reduce the expression level. Since our system has two vectors which can be processed in parallel, the scale up experiment can be performed with the simpler His-tag system where desirable. It is worth noting that for TLCV C2 protein, although trace amount of soluble expression was obtained using pEHISGFPTEV vector (Fig 2B), none of these two vectors produced sufficient soluble protein for purification. In these cases which will always occur, other vectors or refolding procedures may be more suitable.
Our new system brings together established protocols and technologies in a single simple format. The system is suitable for both high throughput laboratories but more importantly can be readily used by smaller laboratories without recourse to cloning. Based on our approach, new vectors for yeast and baculovirus expression with a variety of tags can be constructed using the same multiple cloning sequence and TEV cleavage site. This would preserve the robust purification procedure and should mean that our system is broadly useful.
Fig. 7. Automated purification of Sso2226 and Mutase-kp using default setting of protocol AC-DS-GF.

A) The full chromatogram for Sso2226 with detection peaks of NiAC1 column (1), DS (2) and GF (3). B) The chromatogram obtained for the final gel filtration step of Sso2226 purification. C) SDS-PAGE analysis of purified Sso2226. Lanes 1-2: samples eluted from NiAC1, lanes 3-5: samples from the superloop and lanes 6-16: samples from FG fractions. D) The chromatogram obtained for the final GF step of Mutase-kp purification. E) SDS-PAGE analysis of purified Mutase-kp. Lanes1-9: samples from GF fractions, lane 10: sample from the superloop and lane 11: total cellular extract. PM: protein markers. F) The chromatogram obtained for the calibration of GF column with protein standards of thyroglobulin (670 kDa), gamma globulin (158 kDa), ovalbumin (44 kDa) and myoglobin (17 kDa).
Acknowledgements
We thank Dr. C. Whitfield, Dr. M. S. Rodriguez, and Dr. Yiguo Hong for providing the gene constructs, Dr. Catherine Botting for mass spectrographic analysis of the expressed proteins. The work was funded by the BBSRC and SFC.
Footnotes
Abbreviations used: GFP, green fluorescent protein; TEV protease, tobacco etch virus NIa protease; Mutase-kp, Klebsiella pneumoniae UDP-galactopyranose mutase; TYLCV, tomato yellow leave curl virus; pTP, precursor terminal protein; NFIII, nuclear factor III; Sso, Sulfolobus solfataricus; MCS, multiple cloning site; NiAC, nickel affinity sepharose column; DS, desalting; GF, gel filtration.
References
- [1].Baum EZ, Bebernitz GA, Palant O, Mueller T, Plotch SJ. Purification, properties, and mutagenesis of poliovirus 3C protease. Virology. 1991;185:140–150. doi: 10.1016/0042-6822(91)90762-z. [DOI] [PubMed] [Google Scholar]
- [2].Beis K, Srikannathasan V, Liu H, Fullerton SW, Bamford VA, Sanders DA, Whitfield C, Mcneil MR, Naismith JH. Crystal structures of Mycobacteria tuberculosis and Klebsiella pneumoniae UDP-galactopyranose mutase in the oxidised state and Klebsiella pneumoniae UDP-galactopyranose mutase in the (active) reduced state. J. Mol. Biol. 2005;348:971–982. doi: 10.1016/j.jmb.2005.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Birch GM, Black T, Malcolm SK, Lai MT, Zimmerman RE, Jaskunas SR. Purification of recombinant human rhinovirus 14 3C protease expressed in Escherichia coli. Protein Expr. Purif. 1995;6:609–618. doi: 10.1006/prep.1995.1080. [DOI] [PubMed] [Google Scholar]
- [4].Botting CH, Hay RT. Characterisation of the adenovirus preterminal protein and its interaction with the POU homeodomain of NFIII (Oct-1) Nucleic Acids Res. 1999;27:2799–2805. doi: 10.1093/nar/27.13.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cabantous S, Pedelacq JD, Mark BL, Naranjo C, Terwilliger TC, Waldo GS. Recent advances in GFP folding reporter and split-GFP solubility reporter technologies. Application to improving the folding and solubility of recalcitrant proteins from Mycobacterium tuberculosis. J. Struct. Funct. Genomics. 2005;6:113–119. doi: 10.1007/s10969-005-5247-5. [DOI] [PubMed] [Google Scholar]
- [6].Chong S, Mersha FB, Comb DG, Scott ME, Landry D, Vence LM, Perler FB, Benner J, Kucera RB, Hirvonen CA, Pelletier JJ, Paulus H, Xu MQ. Single-column purification of free recombinant proteins using a self-cleavable affinity tag derived from a protein splicing element. Gene. 1997;192:271–281. doi: 10.1016/s0378-1119(97)00105-4. [DOI] [PubMed] [Google Scholar]
- [7].Chong S, Xu MQ. Protein splicing of the Saccharomyces cerevisiae VMA intein without the endonuclease motifs. J. Biol. Chem. 1997;272:15587–15590. doi: 10.1074/jbc.272.25.15587. [DOI] [PubMed] [Google Scholar]
- [8].Coleman MA, Lao VH, Segelke BW, Beernink PT. High-throughput, fluorescence-based screening for soluble protein expression. J. Proteome Res. 2004;3:1024–1032. doi: 10.1021/pr049912g. [DOI] [PubMed] [Google Scholar]
- [9].Cordingley MG, Register RB, Callahan PL, Garsky VM, Colonno RJ. Cleavage of small peptides in vitro by human rhinovirus 14 3C protease expressed in Escherichia coli. J. Virol. 1989;63:5037–5045. doi: 10.1128/jvi.63.12.5037-5045.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cornvik T, Dahlroth SL, Magnusdottir A, Herman MD, Knaust R, Ekberg M, Nordlund P. Colony filtration blot: a new screening method for soluble protein expression in Escherichia coli. Nat. Methods. 2005;2:507–509. doi: 10.1038/nmeth767. [DOI] [PubMed] [Google Scholar]
- [11].Crowl R, Seamans C, Lomedico P, Mcandrew S. Versatile expression vectors for high-level synthesis of cloned gene products in Escherichia coli. Gene. 1985;38:31–38. doi: 10.1016/0378-1119(85)90200-8. [DOI] [PubMed] [Google Scholar]
- [12].Di Guan C, Li P, Riggs PD, Inouye H. Vectors that facilitate the expression and purification of foreign peptides in Escherichia coli by fusion to maltose-binding protein. Gene. 1988;67:21–30. doi: 10.1016/0378-1119(88)90004-2. [DOI] [PubMed] [Google Scholar]
- [13].Dougherty WG, Carrington JC, Cary SM, Parks TD. Biochemical and mutational analysis of a plant virus polyprotein cleavage site. Embo. J. 1988;7:1281–1287. doi: 10.1002/j.1460-2075.1988.tb02942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dougherty WG, Cary SM, Parks TD. Molecular genetic analysis of a plant virus polyprotein cleavage site: a model. Virology. 1989;171:356–364. doi: 10.1016/0042-6822(89)90603-x. [DOI] [PubMed] [Google Scholar]
- [15].Emery VC, Bishop DH. The development of multiple expression vectors for high level synthesis of eukaryotic proteins: expression of LCMV-N and AcNPV polyhedrin protein by a recombinant baculovirus. Protein Eng. 1987;1:359–366. doi: 10.1093/protein/1.4.359. [DOI] [PubMed] [Google Scholar]
- [16].Evans TC, Jr., Benner J, Xu MQ. The in vitro ligation of bacterially expressed proteins using an intein from Methanobacterium thermoautotrophicum. J. Biol. Chem. 1999;274:3923–3926. doi: 10.1074/jbc.274.7.3923. [DOI] [PubMed] [Google Scholar]
- [17].Goh SL, Murthy N, Xu M, Frechet JM. Cross-linked microparticles as carriers for the delivery of plasmid DNA for vaccine development. Bioconjug. Chem. 2004;15:467–474. doi: 10.1021/bc034159n. [DOI] [PubMed] [Google Scholar]
- [18].Guan KL, Dixon JE. Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase. Anal. Biochem. 1991;192:262–267. doi: 10.1016/0003-2697(91)90534-z. [DOI] [PubMed] [Google Scholar]
- [19].Hakes DJ, Dixon JE. New vectors for high level expression of recombinant proteins in bacteria. Anal. Biochem. 1992;202:293–298. doi: 10.1016/0003-2697(92)90108-j. [DOI] [PubMed] [Google Scholar]
- [20].Hammarstrom M, Hellgren N, Van Den Berg S, Berglund H, Hard T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Heaphy S, Singh M, Gait MJ. Cloning and expression in E. coli of a synthetic gene for the bacteriocidal protein caltrin/seminalplasmin. Protein Eng. 1987;1:425–431. doi: 10.1093/protein/1.5.425. [DOI] [PubMed] [Google Scholar]
- [22].King J, Haase-Pettingell C, Robinson AS, Speed M, Mitraki A. Thermolabile folding intermediates: inclusion body precursors and chaperonin substrates. Faseb. J. 1996;10:57–66. doi: 10.1096/fasebj.10.1.8566549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Knaust RK, Nordlund P. Screening for soluble expression of recombinant proteins in a 96-well format. Anal. Biochem. 2001;297:79–85. doi: 10.1006/abio.2001.5331. [DOI] [PubMed] [Google Scholar]
- [24].Knott JA, Orr DC, Montgomery DS, Sullivan CA, Weston A. The expression and purification of human rhinovirus protease 3C. Eur. J. Biochem. 1989;182:547–555. doi: 10.1111/j.1432-1033.1989.tb14862.x. [DOI] [PubMed] [Google Scholar]
- [25].Koplin R, Brisson JR, Whitfield C. UDP-galactofuranose precursor required for formation of the lipopolysaccharide O antigen of Klebsiella pneumoniae serotype O1 is synthesized by the product of the rfbDKPO1 gene. J. Biol. Chem. 1997;272:4121–4128. doi: 10.1074/jbc.272.7.4121. [DOI] [PubMed] [Google Scholar]
- [26].Kroll DJ, Abdel-Malek Abdel-Hafiz H, Marcell T, Simpson S, Chen CY, Gutierrez-Hartmann A, Lustbader JW, Hoeffler JP. A multifunctional prokaryotic protein expression system: overproduction, affinity purification, and selective detection. DNA Cell Biol. 1993;12:441–453. doi: 10.1089/dna.1993.12.441. [DOI] [PubMed] [Google Scholar]
- [27].Liu H, Woznica K, Catton G, Crawford A, Botting N, Naismith JH. Structural and kinetic characterization of quinolinate phosphoribosyltransferase (hQPRTase) from Homo sapiens. J. Mol. Biol. 2007;373:755–763. doi: 10.1016/j.jmb.2007.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lucast LJ, Batey RT, Doudna JA. Large-scale purification of a stable form of recombinant tobacco etch virus protease. Biotechniques. 2001;30:544–546. 548, 550 passim. doi: 10.2144/01303st06. [DOI] [PubMed] [Google Scholar]
- [29].Mahajan R, Delphin C, Guan T, Gerace L, Melchior F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell. 1997;88:97–107. doi: 10.1016/s0092-8674(00)81862-0. [DOI] [PubMed] [Google Scholar]
- [30].Maina CV, Riggs PD, Grandea AG, Slatko BE, Moran LS, Tagliamonte JA, Mcreynolds LA, Guan CD. An Escherichia coli vector to express and purify foreign proteins by fusion to and separation from maltose-binding protein. Gene. 1988;74:365–373. doi: 10.1016/0378-1119(88)90170-9. [DOI] [PubMed] [Google Scholar]
- [31].Makrides SC. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol. Rev. 1996;60:512–538. doi: 10.1128/mr.60.3.512-538.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Markmeyer P, Ruhlmann A, Englisch U, Cramer F. The pAX plasmids: new gene-fusion vectors for sequencing, mutagenesis and expression of proteins in Escherichia coli. Gene. 1990;93:129–134. doi: 10.1016/0378-1119(90)90146-i. [DOI] [PubMed] [Google Scholar]
- [33].Melcher K. A modular set of prokaryotic and eukaryotic expression vectors. Anal. Biochem. 2000;277:109–120. doi: 10.1006/abio.1999.4383. [DOI] [PubMed] [Google Scholar]
- [34].Moy S, Dieckman L, Schiffer M, Maltsev N, Yu GX, Collart FR. Genome-scale expression of proteins from Bacillus subtilis. J. Struct. Funct. Genomics. 2004;5:103–109. doi: 10.1023/B:JSFG.0000029203.42187.20. [DOI] [PubMed] [Google Scholar]
- [35].Nishikawa S, Yanase K, Tokunaga-Doi T, Kodama K, Gomi H, Uesugi S, Ohtsuka E, Kato Y, Suzuki F, Ikehara M. Efficient cleavage by alpha-thrombin of a recombinant fused protein which contains insulin-like growth factor I. Protein Eng. 1987;1:487–492. doi: 10.1093/protein/1.6.487. [DOI] [PubMed] [Google Scholar]
- [36].Parks TD, Leuther KK, Howard ED, Johnston SA, Dougherty WG. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 1994;216:413–417. doi: 10.1006/abio.1994.1060. [DOI] [PubMed] [Google Scholar]
- [37].Parks TD, Smith HA, Dougherty WG. Cleavage profiles of tobacco etch virus (TEV)-derived substrates mediated by precursor and processed forms of the TEV NIa proteinase. J. Gen. Virol. 1992;73:149–155. doi: 10.1099/0022-1317-73-1-149. [DOI] [PubMed] [Google Scholar]
- [38].Patterson GH, Knobel SM, Sharif WD, Kain SR, Piston DW. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 1997;73:2782–2790. doi: 10.1016/S0006-3495(97)78307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pedelacq JD, Piltch E, Liong EC, Berendzen J, Kim CY, Rho BS, Park MS, Terwilliger TC, Waldo GS. Engineering soluble proteins for structural genomics. Nat. Biotechnol. 2002;20:927–932. doi: 10.1038/nbt732. [DOI] [PubMed] [Google Scholar]
- [40].Pichler A, Gast A, Seeler JS, Dejean A, Melchior F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell. 2002;108:109–120. doi: 10.1016/s0092-8674(01)00633-x. [DOI] [PubMed] [Google Scholar]
- [41].Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- [42].Polayes DA, Goldstein A, Ward G, Hughes AJ. TEV protease, recombinant: A site-specific protease for efficient cleavage of affinity tags from expressed proteins. Focus. 1994;16:2–5. [Google Scholar]
- [43].Ramos CR, Abreu PA, Nascimento AL, Ho PL. A high-copy T7 Escherichia coli expression vector for the production of recombinant proteins with a minimal N-terminal His-tagged fusion peptide. Braz. J. Med. Biol. Res. 2004;37:1103–1109. doi: 10.1590/s0100-879x2004000800001. [DOI] [PubMed] [Google Scholar]
- [44].Rucker E, Schneider G, Steinhauser K, Lower R, Hauber J, Stauber RH. Rapid evaluation and optimization of recombinant protein production using GFP tagging. Protein Expr. Purif. 2001;21:220–223. doi: 10.1006/prep.2000.1373. [DOI] [PubMed] [Google Scholar]
- [45].Sambrook J, Fristsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd ed CSH laboratry Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- [46].Schoepfer R. The pRSET family of T7 promoter expression vectors for Escherichia coli. Gene. 1993;124:83–85. doi: 10.1016/0378-1119(93)90764-t. [DOI] [PubMed] [Google Scholar]
- [47].Seeler JS, Marchio A, Losson R, Desterro JM, Hay RT, Chambon P, Dejean A. Common properties of nuclear body protein SP100 and TIF1alpha chromatin factor: role of SUMO modification. Mol. Cell Biol. 2001;21:3314–3324. doi: 10.1128/MCB.21.10.3314-3324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Sharrocks AD. A T7 expression vector for producing N- and C-terminal fusion proteins with glutathione S-transferase. Gene. 1994;138:105–108. doi: 10.1016/0378-1119(94)90789-7. [DOI] [PubMed] [Google Scholar]
- [49].Sheikh MA, Potter JA, Johnson KA, Sim RB, Boyd EF, Taylor GL. Crystal structure of VC1805, a conserved hypothetical protein from a Vibrio cholerae pathogenicity island, reveals homology to human p32. Proteins. 2008;71:1563–1571. doi: 10.1002/prot.21993. [DOI] [PubMed] [Google Scholar]
- [50].Shen LN, Dong C, Liu H, Naismith JH, Hay RT. The structure of SENP1-SUMO-2 complex suggests a structural basis for discrimination between SUMO paralogues during processing. Biochem. J. 2006;397:279–288. doi: 10.1042/BJ20052030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Shen LN, Liu H, Dong C, Xirodimas D, Naismith JH, Hay RT. Structural basis of NEDD8 ubiquitin discrimination by the deNEDDylating enzyme NEDP1. Embo. J. 2005;24:1341–1351. doi: 10.1038/sj.emboj.7600628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Smith DB, Johnson KS. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- [53].Sternsdorf T, Jensen K, Reich B, Will H. The nuclear dot protein sp100, characterization of domains necessary for dimerization, subcellular localization, and modification by small ubiquitin-like modifiers. J. Biol. Chem. 1999;274:12555–12566. doi: 10.1074/jbc.274.18.12555. [DOI] [PubMed] [Google Scholar]
- [54].Studier FW. Use of bacteriophage T7 lysozyme to improve an inducible T7 expression system. J. Mol. Biol. 1991;219:37–44. doi: 10.1016/0022-2836(91)90855-z. [DOI] [PubMed] [Google Scholar]
- [55].Studier FW, Moffatt BA. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986;189:113–130. doi: 10.1016/0022-2836(86)90385-2. [DOI] [PubMed] [Google Scholar]
- [56].Svetina M, Krasevec N, Gaberc-Porekar V, Komel R. Expression of catalytic subunit of bovine enterokinase in the filamentous fungus Aspergillus niger. J. Biotechnol. 2000;76:245–251. doi: 10.1016/s0168-1656(99)00191-1. [DOI] [PubMed] [Google Scholar]
- [57].Tatham MH, Kim S, Jaffray E, Song J, Chen Y, Hay RT. Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nat. Struct. Mol. Biol. 2005;12:67–74. doi: 10.1038/nsmb878. [DOI] [PubMed] [Google Scholar]
- [58].Tyler RC, Aceti DJ, Bingman CA, Cornilescu CC, Fox BG, Frederick RO, Jeon WB, Lee MS, Newman CS, Peterson FC, Phillips GN, Shahan MN, Singh S, Song J, Sreenath HK, Tyler EM, Ulrich EL, Vinarov DA, Vojtik FC, Volkman BF, Wrobel RL, Zhao Q, Markley JL. Comparison of cell-based and cell-free protocols for producing target proteins from the Arabidopsis thaliana genome for structural studies. Proteins. 2005;59:633–643. doi: 10.1002/prot.20436. [DOI] [PubMed] [Google Scholar]
- [59].Van Den Berg S, Lofdahl PA, Hard T, Berglund H. Improved solubility of TEV protease by directed evolution. J. Biotechnol. 2006;121:291–298. doi: 10.1016/j.jbiotec.2005.08.006. [DOI] [PubMed] [Google Scholar]
- [60].Van Wezel R, Liu H, Tien P, Stanley J, Hong Y. Gene C2 of the monopartite geminivirus tomato yellow leaf curl virus-China encodes a pathogenicity determinant that is localized in the nucleus. Mol. Plant Microbe Interact. 2001;14:1125–1128. doi: 10.1094/MPMI.2001.14.9.1125. [DOI] [PubMed] [Google Scholar]
- [61].Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat. Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- [62].Watanabe T, Ito Y, Yamada T, Hashimoto M, Sekine S, Tanaka H. The roles of the C-terminal domain and type III domains of chitinase A1 from Bacillus circulans WL-12 in chitin degradation. J. Bacteriol. 1994;176:4465–4472. doi: 10.1128/jb.176.15.4465-4472.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Wilkinson DL, Harrison RG. Predicting the solubility of recombinant proteins in Escherichia coli. Biotechnology (N Y) 1991;9:443–448. doi: 10.1038/nbt0591-443. [DOI] [PubMed] [Google Scholar]