

Abstract
Introduction
The purpose of this study was to determine if inhaled carbon monoxide (CO) can ameliorate skeletal muscle injury, modulate endogenous heme oxygenase-1 (HO) expression, improve indices of tissue integrity and inflammation following hind limb ischemia reperfusion(IR).
Methods
C57BL6 mice inhaling CO (250ppm) or room air were subjected to 1.5 hrs of ischemia followed by limb reperfusion for either 3 or 6 hours (total treatment time of 4.5 or 7.5 hrs). After the initial period of reperfusion, all mice breathed only room air until 24 hours after the onset of ischemia. Mice were sacrificed at either the end of CO treatment or at 24 hours reperfusion. Skeletal muscle was subjected to histologic and biochemical analysis.
Results
CO treatment for 7.5 hours protected skeletal muscle from histologic and structural evidence of skeletal muscle injury. Serum and tissue cytokines were significantly reduced (p<0.05) in mice treated with CO for 7.5 hours. Tubulin, Heme Oxygenase, and ATP levels were higher in CO treated mice.
Conclusions
Inhaled CO protected muscle from structural injury and energy depletion following IR.
Keywords: Carbon Monoxide, Reperfusion Injury, Heme Oxygenase, Skeletal Muscle
Introduction
Acute limb ischemia is a complication of advanced peripheral vascular disease. Therapeutic interventions are geared toward restoration of blood flow to the affected extremity and can result in the development of ischemia reperfusion injury (I/R). The local manifestations of extremity I/R injury often results in limb loss or dysfunction. Systemic manifestations of I/R injury include cardiopulmonary dysfunction and shock associated with release of metabolic byproducts and proinflammatory mediators from skeletal muscle. I/R injury is a complex physiologic scenario which is initially triggered by stasis, depletion of energy substrates and acidosis in skeletal muscle during ischemia. Upon reperfusion, there is a paradoxical increase in muscle injury which results in ongoing metabolic dysfunction, local thrombosis and a severe inflammatory response. Despite major improvements in the diagnosis of limb ischemia and interventions to successfully reperfuse ischemic extremities, the incidence of limb loss and mortality following acute limb ischemia have remained relatively unchanged over past decades. Pharmacologic interventions to ameliorate the local and systemic injury have been confined to thrombolytic, anticoagulant and anti-platelet agents.
Recent data suggest that non-toxic concentrations of inhaled carbon monoxide (CO) can reduce IR mediated injury in the heart (1), liver (2), and the kidney (3, 4). It has been proposed that CO activates cytoprotective pathways including expression of hemeoxygenase, which can decrease inflammation, thrombosis and atherosclerosis.
These experiments were designed to test the hypothesis that inhaled CO may reduce tissue injury in a murine model of hind limb ischemia reperfusion (8). Since there is substantial evidence to indicate that the basis for successful CO therapy for reperfusion injury is based on its pleiotropic effects, these experiments utilized biochemical analysis of inflammation, metabolism and morphologic evidence of muscle injury. Biochemical analysis of metabolism was confined to skeletal muscle ATP levels. To assess local skeletal muscle inflammation, skeletal muscle extracts were assayed for Keratinocyte chemoattractant protein (KC, a murine chemokine analogue of human IL-8)(9–11) and Interleukin-6 (IL-6) a cytokine known to mediate tissue injury in humans(12–14). Serum levels of KC and tail blood pressure monitoring were used to detect evidence of systemic stress and hypotension during skeletal muscle reperfusion. Tissue myeloperoxidase (MPO) levels were also assessed as an index of neutrophil infiltration and activation. Muscle levels of hemeoxygenase -1 protein were evaluated to determine whether exogenous administration of CO could increase expression of this cytoprotective protein during reperfusion. α-Tubulin expression in skeletal muscle was assessed to determine whether CO treatment preserved skeletal muscle levels of this important structural protein during reperfusion.
Methods
Hind Limb Ischemia Reperfusion
All experimental procedures were approved by the Massachusetts General Hospital’s Institutional Review Board and were in accordance with Principles of Laboratory Animal Care. Male C57BL6 mice (23–28 g) (Jackson, Bar Harbor, ME) were anesthetized via intraperitoneal administration of pentobarbital solution (60 mg/kg in 0.5 ml normal saline). Mice were warmed to 36±1 ºC on a warming blanket. An Orthodontic rubber band (ORB) was applied to the left hind limb to induce ischemia as previously described using a McGivney applicator (8). After induction of ischemia, the anesthetized mice were then kept in an airtight chamber breathing 20% oxygen and 80% nitrogen with or without 250 ppm CO. CO levels in the chamber were continuously monitored using a CO detector (T40 Rattler; Industrial Scientific Corporation, PA). Limbs were reperfused after 90 minutes by cutting the ORB. During reperfusion, mice recovered from anesthesia while they were kept in their respective chambers for either an additional 3 or 6 hours of either CO or room air therapy resulting in a cumulative CO treatment time of either 4.5 or 7.5 hours. After CO or room air therapy in the airtight chamber, one set of mice were sacrificed at 4.5 or 7.5 hours; another set of identically treated mice were then returned to their cages and allowed free access to water and food. The second set of mice were euthanized at 24 hours of reperfusion. Mice were euthanized with a lethal dose of pentobarbital. Serum was collected from whole blood for ELISA analysis. Skeletal muscle was harvested from the posterior calf compartment of injured and control hind limbs; the tissue was either fixed for histologic evaluation or snap frozen in liquid nitrogen, then stored at −80°C for biochemical analysis. Frozen muscle samples were homogenized in test tubes containing 1 mL Radioimmunoassay Precipitation Assay Buffer and 10-μL of protease inhibitor cocktail (Sigma, St. Louis, MO). Each homogenate was sonicated for 20 seconds and then centrifuged at 13,000 × g for 10 minutes. The supernatant (skeletal muscle protein extract) was aspirated, dispensed into test tubes and frozen at 80°C until ELISA analysis for cytokines and myeloperoxidase.
Blood gas measurement
To determine whether inhalation of 250 ppm CO resulted in increased blood carboxyhemoglobin levels, a group of non-ischemic animals was subjected to 20% oxygen, balance nitrogen with and without 250 ppm CO for a total of either 4.5 or 7.5 hours. These time intervals were chosen to reflect CO treatment during hind limb ischemia of 1.5 hours and either 3 or 6 hours CO treatment during reperfusion. Whole blood samples were collected and subjected to blood gas analysis using OSM3 blood gas analyzer (Radiometer; Copenhagen, Denmark).
Histology
Limbs from mice exposed to 90 minutes of ischemia reperfusion in the presence of air or CO ( a total of 4.5 or 7.5hrs treatment) were harvested at 24 hours of reperfusion, then fixed in 4% paraformaldehyde for at least 4 hours. The gastrocnemius muscle was dissected out, rinsed in Dulbecco’s phosphate buffered saline (PBS) for 1 hour and dehydrated. The samples were embedded in Jb-4 glycomethylmethacrylate, cut in cross-section at 2 μm thickness and stained with Masson’s trichrome stain. Slides were examined under microscopy at x200 magnification (NikonE600 upright microscope). Images were acquired from the entire muscle section and assigned a serial number using SPOT Insight microscope camera (Diagnostic Instruments, Sterling Heights, MI). A random numbers generator was used to obtain unique set of field numbers. Images were then processed with imaging software (Diagnostic Instruments, Sterling Heights, MI). A minimum of 1600 total fibers were counted per tissue section by an observer blinded to the treatment regimen. Muscle fibers were scored as uninjured or injured based on the morphology of the individual fibers and reported as percent injured muscle fibers(15).
ATP assay
200mg of frozen skeletal muscle samples were homogenized with a polytron homogenizer in 10% trichloroacetic acid. Samples were centrifuged for 10 minutes at 10,000 x g and supernatants were diluted in PBS. ATP levels were measured using ATPlite Luminescence Assay according to the manufacturer’s protocol (PerkinElmer Life, Boston, MA). Top counts were read using 1450 MicroBeta plate reader (PerkinElmer Life, Boston, MA). Concentrations of the unknowns were extrapolated off the standard curve and expressed as nmole per mg tissue weight. ATP levels of the ischemic limbs were then divided by ATP levels of the respective contralateral limbs and expressed as percent of contralateral limb ATP level.
IL-6, KC, Myeloperoxidase and Hemeoxygenase -1 Assays
KC, IL-6, Hemeoxygenase-1 (Quantikine mouse, R&D Systems, Minneapolis, MN), MPO (mouse MPO, Cell Sciences, Canton, MA) ELISA, employing quantitative sandwich enzyme immunoassay techniques were used to determine levels of KC, IL-6 and MPO levels. The ELISA plates were read with Spectromax-250 plate reader (Molecular Devices, Sunnyvale, CA). Values were extrapolated from the standard curve and normalized to the total protein concentration, which was determined with another assay using the BCA Protein Assay Reagent Kit (Pierce Biotechnology, Rockford, IL).
Total p38MAPK expression and activity (Thr180/Tyr 182 phosphorylation). Need methods, extraction buffer
Frozen skeletal muscle tissues were homogenized on ice with lysis buffer containing 20 nM Tris, pH 7.4, 100mM NaCl, 1mM EDTA, 1mM EGTA, 1mM NaF, 20mM sodium pyrophosphate, 2mM sodium orthovanadate, 1% Triton X-100, 10% glycerol, 0.1% SDS and 0.5% deoxycholate and protease inhibitor cocktail (Sigma-Aldrich, St Louise MO). tissue lysates were centrifuged at 10000xg at 4°C for 10 min. The total p38 MAPK and the phosphorylated 180 Threonine and 182 Tyrosine p38 MAPK residues were quantified using ELISA (Invitrogen Corporation, Camarillo, CA) data was normalized to total protein levels in each sample.
Western Blot for α-Tubulin
40 μg of total protein was solubilized with equal volume of Laemmli sample buffer, boiled for 5 min, loaded onto lanes in a 12% density Tris-HCl polyacrylamide/sodium dodecyl sulfate gel (BioRad, Hercules CA). Samples were subjected to electrophoresis followed by electro blotting transfer to a 0.45μm nitrocellulose membrane (BioRad, Hercules, CA). The blots were incubated with 1:1000 rabbit anti-mouse tubulin IgG peroxidase-conjugated IgG (Abcam, Cambridge, MA) in blocking buffer for one hour at room temperature. The membranes were then washed four times and incubated with peroxidase-conjugated goat anti-Rabbit IgG (Amersham) at 1:4000 dilutions in blocking for 1 hour at room temperature. Membranes were washed with PBS-T and developed with the advanced western blotting detection reagents enhanced chemiluminescence detection system (GE, Healthcare, Piscataway, NJ) at 1/20 dilution. Chemiluminescence light was visualized by exposing the blots into a BioMAX x-ray film. The generated specific proteins were quantified using and Integrated Data Viewer (IDV) gel-imaging system (Alpha Innotech Corporation, San Leandro, CA).
Tissue Edema
Immediately after harvest, posterior calf muscle samples were blotted, weighed and placed in a drying oven at 55°C until a constant weight was obtained. Muscle edema was determined by calculating the wet to dry weight ratio.
Analysis of Blood Pressure and Heart Rate
On three separate 20 minute intervals 48 hours prior to the IR experiments, mice were acclimatized to measurement of tail blood pressure without anesthesia by placing them inside the heated holder (37°C) of the Coda2 blood pressure system (Kent Scientific, Torrington, CN). Using this system, murine blood pressure was recorded non-invasively utilizing a tail blood pressure cuff at 6 and 24 hours reperfusion.
Statistical Analysis
All data was expressed as mean ± SEM and all analyses were performed with the Instat software (version 3, Graph Pad, San Diego, CA). Comparisons between groups were performed with parametric and non-parametric (Welch) unpaired, two-tailed Student’s t test. A value of p< 0.05 was considered significant. One way ANOVA with Tukey’s post test was used to analyze the carboxyhemoglobin levels.
Results
Blood Gas Analysis
Mice subjected to inhaled CO had significantly higher carboxyhemoglobin levels compared to mice breathing room air at 4.5 (4.5±0.7 vs 24.6±3.0 percent, p<0.001, n=4) and 7.5 (29.5 ± 1.8 percent, p<0.001, n=5) hours.
Skeletal Muscle Injury
Histological evaluation of skeletal muscle in mice subjected to hind limb ischemia reperfusion with inhalation of CO during 1.5 hours of ischemia and 3 hours reperfusion (total 4.5 hours of CO treatment) showed no significant difference in injured fibers by 24 hours reperfusion( 39±7 vs 45±9 % injured fibers, p=0.61, n=6). In contrast, skeletal muscle of mice subjected to CO treatment during 1.5 hours of ischemia and 6 hours of reperfusion (i.e. total inhaled CO treatment interval of 7.5 hours)showed a marked reduction of injured fibers at 24 hours reperfusion (16.8%±3 vs. 29.2%±2.7, p=0.01, n = 6). Based on these findings, all further biochemical analysis of skeletal muscle response to CO treatment were made in mice subjected to CO for 1.5 hours ischemia and 6 hours reperfusion.
Skeletal Muscle ATP Content
Skeletal muscle of mice subjected to CO treatment during 1.5 hours of ischemia and 6 hours of reperfusion showed a marked preservation of ATP at 24 hours reperfusion (21.8±2.67 vs. 8.5%±2.7 percent contralateral, p=0.002, n=10–13).
Serum and Skeletal Muscle Cytokine Levels
The levels of pro-inflammatory cytokine KC was markedly reduced in serum and skeletal muscle tissue from CO treated group when compared to Control: KC muscle at 24 hours reperfusion- 11.6±1.5 vs. 26.3±3.7 pg/mg total protein,, p=0.01, n=6 Fig.1A.; KC serum 125.2±19.8 vs. 382.5±70.9 pg/ml, p=0.02, n=6, Figure 1B); Skeletal muscle levels of IL-6 muscle at 24 hours reperfusion were markedly decreased in CO vs. room air treated mice: 32±4 vs. 90±11 pg/mg total protein, p=0.0032, n=6 , Fig. 2.
Figure 1. Muscle and Serum KC levels.
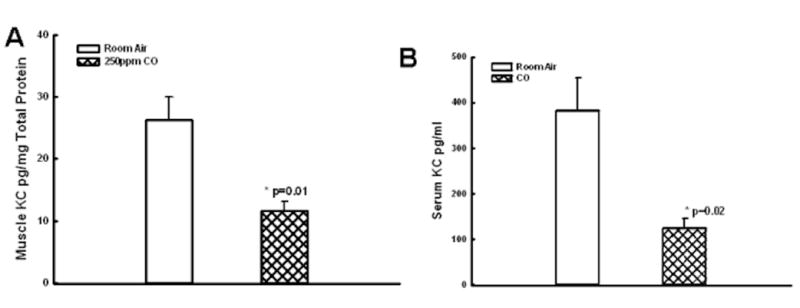
CO breathing markedly decreased KC levels in skeletal muscle (A,*p=0.01) and serum (B, **p=0.02) as compared to mice breathing room air alone.
Figure 2. Muscle IL-6 levels.
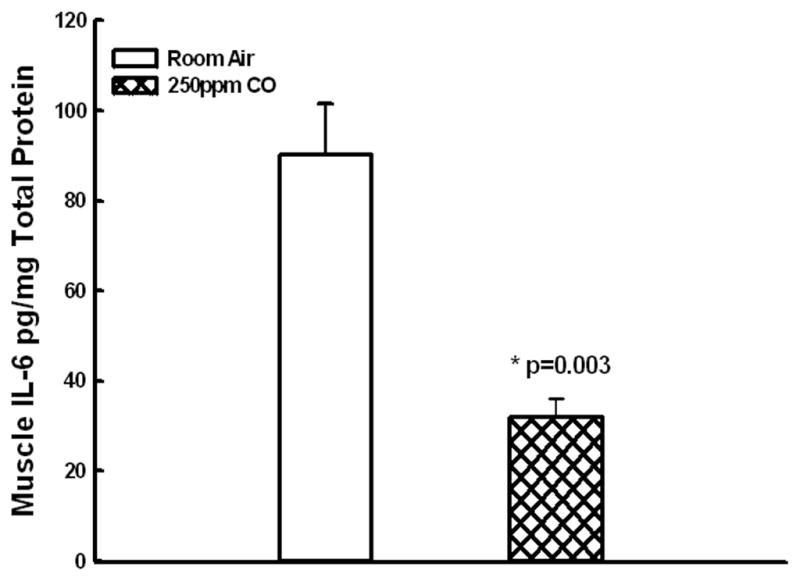
CO breathing markedly decreased IL-6 levels in skeletal muscle (A,*p=0.003) as compared to mice breathing room air alone.
Expression of Cytoskeletal Muscle Proteins
Ischemia reperfusion significant altered α-Tubulin expression in skeletal muscle in the presence of CO (p=0.008 ANOVA) and inhaled room air (p=0.014 ANOVA). Under sham conditions, 7.5 hours of inhaled CO increased α-Tubulin expression (CO: 60,077±5156 vs Air: 42511±7469 IDV, *p=0.04) in skeletal muscle compared to mice exposed only to room air after 24 hours. However, at 7.5hrs IR (CO:55,262±5118 vs Air:36,331±7,065 IDV, **p=0.03, n=10, Figure 3) and 24hrs reperfusion (CO:32,741±6392 vs Air: 17,768±3.294 IDV, +p=0.04, n=10, Figure 3), α-Tubulin levels were relatively preserved in mice treated with CO inhalation compared to mice exposed to room air.
Figure 3. α-Tubulin in Skeletal Muscle.

CO breathing for 7.5 hours of sham ischemia reperfusion increased skeletal muscle α-tubulin level (*p=0.03). CO breathing preserved IR induced decreases in α-Tubulin at 7.5 hours ischemia reperfusion (**p=0.03) and 24 hours reperfusion (+p=0.04).
Expression of Hemeoxygenase-1 Protein
After 7.5 hours of IR in the presence of inhaled CO or room air followed by 24 hours reperfusion in room air, HO-1 protein expression was markedly increased by 24 hours reperfusion compared to sham (CO: 1.4±0.2 vs CO sham: 0.19±0.01 ng/mg, **p<0.001, n=10; Air: 0.83±0.13 vs air sham: 0.17±0.006 ng/mg protein, +p<0.001, n=10, Figure 4). At this 24 hour reperfusion interval, there was greater HO-1 protein expression in mice that inhaled CO during IR compared to mice that inhaled room air (CO: 1.4±0.2 vs. Air: 0.83±0.13, *p=0.03). Under 7.5 hour IR conditions, there was no difference in the level of HO-1 expression in mice treated with CO vs room air (CO: 0.33±0.02 vs. Air: 0.33±0.02 ng/mg protein, p=0.96, n=10). 7.5 hours of inhaled CO treatment under sham conditions did not alter levels of HO-1 expression when compared to levels measured in mice treated with room air alone at 24 hours (CO: 0.19±0.011 vs. Air: 0.17±0.006 ng/mg protein, p=0.23, n=10).
Figure 4. Heme Oxygenase Expression during IR.

IR stimulated HO expression in skeletal muscle by 24 hours reperfusion during IR (*,+p<0.01). CO breathing during IR resulted in significantly greater HO expression by 24 hours reperfusion as compared to mice who breathed room air during IR(*p=0.03).
Total p38MAPK expression and activity (Thr180/Tyr 182 phosphorylation)
There was no significant difference in total p38 MAPK expression in mice treated with CO or room air for 7.5 hours sham conditions or 7.5 hours I/R (Figure 5A). By 24 hours reperfusion, there was significant preservation of total p38MAPK expression in the CO vs room air treated mice (159.4 + 19.9 vs 51.6 + 8.8 pg/mg protein, p<0.05, n=7). Similarly, by 24 hours reperfusion, the pattern of Thr180/Tyr phosphorylation was significantly preserved in the CO vs room air treated mice (Figure 5B, 41.4 + 5.7 vs 21.4 + 2.7pg/mg protein, p<0.05 ,n=7).
Figure 5. Total p38MAPK expression and activity (Thr180/Tyr 182 phosphorylation).

CO treatment preserved total p38MAPK expression (5A, p<0.05) and phosphorylated p38MAPK (5B, p<0.05) by 24 hours reperfusion.
Skeletal Muscle Myeloperoxidase Levels
Myeloperoxidase levels were markedly increased in CO treated mice as compared to mice exposed to IR during ischemia reperfusion (48.8±6.3 vs 25.8±3.3 ng/mg total protein, p=0.007, n=10, Figure 6).
Figure 6. Skeletal Muscle Myeloperoxidase.
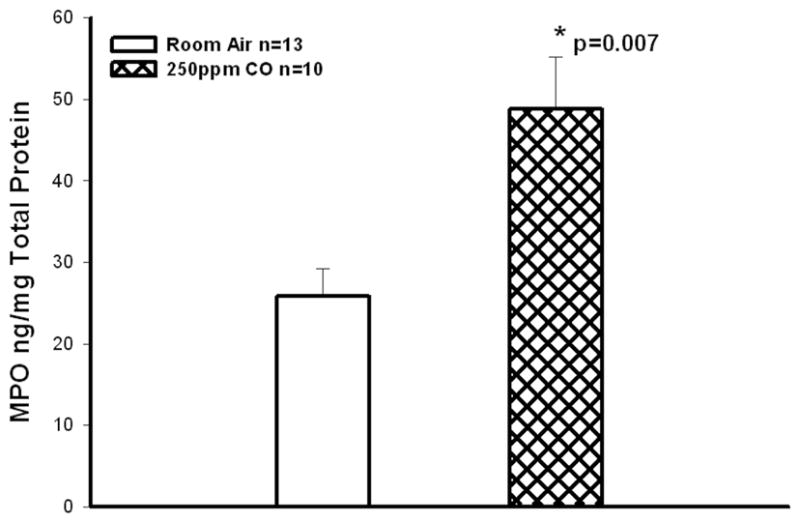
CO breathing increased levels of skeletal muscle MPO as compared to mice breathing room air alone(*p=0.007).
Tissue Edema
CO treatment did not alter the amount of edema found in reperfused skeletal muscle 24 hours following limb ischemia (6.3±0.11 vs. 6.4 ± 0.3, p =0.75, n= 10).
Blood Pressure and Heart Rate
There was no difference in blood pressure or heart rate (Table One) in the CO vs. control treated mice at 6 and 24 hours reperfusion.
Table One
| Treatment | 6 Hours Reperfusion | 24 Hours Reperfusion | ||
|---|---|---|---|---|
| Heart Rate Beats/minute | Blood Pressure mm Hg | Heart Rate Beats/minute | Blood Pressure mm Hg | |
| Room Air | 775.2 + 53.1 | 112.3 + 4.7 | 813.0 + 44.9 | 131.9 + 8.6 |
| CO | 716.9 + 41.2 | 118.9 + 3.2 | 727.3 + 17.9 | 134.2 + 4.7 |
Discussion
The central hypothesis of this study was that CO inhalation of 4.5 and 7.5 hours can improve muscle damage caused by ischemia reperfusion in a hind limb model in the mouse. We report that CO inhalation decreased markers of tissue injury and components of the local and systemic inflammatory response. It is important to note that the CO protocol used for these experiments was substantially different from the organ transplant IR models. In the transplant models of liver and renal tissue IR, CO treatment has been usually initiated one hour prior to the onset of tissue ischemia and is continued for up to 24 hours reperfusion (16) . Such preemptive therapy is probably not relevant for patients at risk for acute hind limb ischemia reperfusion since these individuals cannot be identified prior to the onset of ischemia, whereas transplantation is an urgent life saving planned event. However, it seems clinically feasible to begin CO inhalation therapy after the diagnosis of limb ischemia is established, in a manner similar to beginning intravenous heparin therapy.
The level of carboxyhemoglobin in the blood of mice subjected to CO treatment in this experimental protocol was comparable to levels reported in studies of rodent liver and myocardial ischemia reperfusion where administration of CO proved to provide cytoprotection (17, 18). There is a time dependent relationship between CO treatment and protection against fiber injury since the 4.5 hour treatment protocol did not provide protection, whereas a 7.5 hour treatment protocol did provide protection against fiber injury. Since the 7.5 hour treatment protocol provided protection against I/R induced muscle fiber injury, all subsequent biochemical analysis was performed using this duration of CO treatment. Subsequent biochemical analysis in mice treated for 7.5 hours and confirmed that CO treatment preserved tissue levels of ATP essential for maintaining cellular membrane integrity and contractility. This finding may suggest that CO therapy provides some degree of metabolic rescue. ATP levels have been previously shown to be proportional to muscle viability(19, 20). Work studying myocardial IR reported a marked improvement in myocardial energetics associated with inhaled CO (1). Similarly, in a rodent model of hemorrhagic shock, ATP levels in liver were preserved by treatment with inhaled CO (21). Thus our findings showing preserved ATP levels in skeletal muscle following IR and CO treatment are consistent with previous reports from other tissues.
Additional analysis of local and systemic cytokine release, which has been associated with mortality and morbidity during vascular reconstruction in humans(22–24), revealed a marked decrease in serum and skeletal muscle KC at 24 hours reperfusion(Figure 1AB). There was also a significant decrease in muscle IL-6 (Figure 2). An important component of this observation is the fact that the levels of cytokines were reduced in both serum and skeletal muscle 18 hours after cessation of therapy, indicating the ability of CO to modulate inflammation in tissue after therapy is stopped. The finding of decreased local and systemic cytokines associated with inhaled CO treatment is also consistent with successful experimental therapeutic interventions from our and other laboratories (25, 26).
α-Tubulin levels were preserved in the skeletal muscle of mice that inhaled CO during ischemia reperfusion when compared to mice exposed to room air under the same conditions (Figure 3). α-Tubulin is a major component of microtubules which are present in all eukaryotic cells where they facilitate several biologic processes including mitosis, intracellular transport and mechanical response to the environment. Decreases in the cytoskeletal tubulin have been observed in atrophic skeletal muscle in rats (27). A specific highly conserved isoform of α-Tubulin has been cloned and sequenced from a human adult skeletal muscle cDNA library. Sequence comparison of these isoforms show that there is cross species preservation of these genes, consistent with the functional importance of this molecule (28). It has been proposed that Tubulin, a major component of muscle cytoskeleton contributes to maintenance of cell structure during degenerative processes by providing a cytostructural framework around which the regenerative processes may be initiated (29). Preservation of α-Tubulin in skeletal muscle of mice treated with CO during ischemia reperfusion suggests that the reparative characteristics and the functional capability of the muscle to respond to mechanical stimuli, such as contraction may also be preserved.
After 7.5 hours of IR, there was no difference in the level of heme oxygenase-1 expression in skeletal muscle of CO treated and untreated mice (Figure 4). By 24 hours reperfusion, heme oxygenase-1 protein expression was markedly stimulated in CO treated and untreated mice (Figure 4). In the CO treated mice, heme oxygenase-1 protein expression was significantly greater than levels detected in mice breathing room air during 7.5 hrs IR. This finding suggests that CO inhalation only during the 7.5 IR period stimulated ongoing expression of heme oxygenase-1 during the next 17.5 hours of reperfusion after CO therapy had ceased. Since CO is a product of HO-1 activity, it was possible that exogenous administration of CO might have decreased the expression of HO-1 protein through negative feed back. The fact that exogenous CO did not down regulate HO-1 protein expression is an important observation since HO is known to suppress microvascular thrombus formation (5). Thrombosis is a major component of reperfusion injury as it contributes to the no reflow period (30). Increased HO expression during hind limb ischemia reperfusion may enhance tissue healing since HO is also know to promote progenitor cell mobilization, neovascularization and functional recovery after critical hind limb ischemia in mice (31). In concert with increased expression of HO-1 at 24 hours reperfusion following inhaled CO therapy, there was preservation of total p38 MAPKinase and phosphorylated p38 MAPKinase (Figure 5A and 5B). These observations provide additional evidence that CO treatment influenced downstream signaling pathways know to be cytoprotective and modulated by heme Oxygenase (17).
Despite the decrease in muscle fiber injury, preserved cytoskeletal muscle protein, decreased skeletal muscle and serum cytokines in mice exposed to IR in the presence of CO, skeletal muscle MPO levels were actually higher in the CO treated mice than in the control mice (Figure 6). Inadvertent CO exposure has been associated with activation of neutrophils in lung tissue (16). Based on histologic assessment of skeletal muscle fiber injury, muscle ATP and cytokine analysis, we were not able to identify solid macroscopic or biochemical signs of skeletal muscle fiber cytotoxicity in the hind limbs of mice subjected to IR while breathing CO. It appears that the increase in muscle MPO did not reflect an increase in detectable tissue damage.
CO inhalation therapy in our model did not lower skeletal muscle edema. However, decreased skeletal muscle edema is not always associated with decreased skeletal muscle injury (25, 32). It is possible that the edema associated with CO treatment observed in these studies is related in part to the activation of neutrophils(33) as suggested by the paradoxical increase in MPO in CO treated mice (figure 6). However, the amount of edema detected in CO treated mice was no different than untreated mice, even though the amount of MPO in the muscle of reperfused mice treated with CO was twice that of untreated mice. This finding seems to indicate that as a single variable, edema does may not contribute to muscle fiber injury in the murine model.
Similar to the results of the analysis of skeletal muscle edema, there was no significant difference in blood pressure in the CO treated vs. control treated mice at 6 and 24 hours (Table One). It is possible that the blood pressure at earlier time points might have been different, however the intervals of 6 and 24 hours were selected to be sure the data was not complicated by the hemodynamic effects of anesthesia administered during ischemia and early reperfusion. While there is evidence that administration of CO can reverse the effects of shock and hemorrhage (21), the cytoprotective effects of CO on skeletal muscle ischemia reperfusion do not appear to be related to a long lasting effect on the blood pressure and heart rate associated with unilateral hind limb ischemia followed by reperfusion.
The precise mechanisms contributing to CO mediated cytoprotection of skeletal muscle exposed to IR have not been defined at this point. The breakdown of heme via hemeoxygenase naturally produces CO. CO has a variety of postulated effects including vasorelaxation, anti-apoptotic, anti-proliferative, and anti-inflammatory effects (34–36). The result of the experiments in this report indicates that exogenous CO preserved skeletal muscle ATP, and induced a predominantly anti-inflammatory/cytoprotective response following I/R injury. These findings parallel results reported in experimental transplantation and shock models (21).
In summary, we conclude that CO inhalation may have therapeutic potential to protect skeletal muscle from I/R injury in a model of acute hind limb ischemia reperfusion injury. However, examination into the precise mechanisms on how CO protects skeletal muscle from IR injury requires further investigation.
Acknowledgments
The authors acknowledge funding from the Pacific Vascular Research Foundation, The Geneen Fund of the Massachusetts General Hospital, the Division of Vascular and Endovascular Surgery, and the National Institutes of Health (1R01AR055843). Dr. Watkins is the Isenberg Fellow in Academic Surgery at the Massachusetts General Hospital. Faraz Hashmi received a student research award from the AVA Foundation. Wolfgang Steudel has been supported by the Department of Anesthesia and Critical Care at Massachusetts General Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lavitrano M, Smolenski RT, Musumeci A, Maccherini M, Slominska E, Di Florio E, Bracco A, Mancini A, Stassi G, Patti M, Giovannoni R, Froio A, Simeone F, Forni M, Bacci ML, D'Alise G, Cozzi E, Otterbein LE, Yacoub MH, Bach FH, Calise F. Carbon monoxide improves cardiac energetics and safeguards the heart during reperfusion after cardiopulmonary bypass in pigs. FASEB J. 2004;18 :1093–1095. doi: 10.1096/fj.03-0996fje. [DOI] [PubMed] [Google Scholar]
- 2.Ott MC, Scott JR, Bihari A, Badhwar A, Otterbein LE, Gray DK, Harris KA, Potter RF. Inhalation of carbon monoxide prevents liver injury and inflammation following hind limb ischemia/reperfusion. FASEB J. 2005;19 :106–108. doi: 10.1096/fj.04-2514fje. [DOI] [PubMed] [Google Scholar]
- 3.Martins PN, Reuzel-Selke A, Jurisch A, Atrott K, Pascher A, Pratschke J, Buelow R, Neuhaus P, Volk HD, Tullius SG. Induction of carbon monoxide in the donor reduces graft immunogenicity and chronic graft deterioration. Transplant Proc. 2005;37 :379–381. doi: 10.1016/j.transproceed.2004.11.079. [DOI] [PubMed] [Google Scholar]
- 4.Nakao A, Neto JS, Kanno S, Stolz DB, Kimizuka K, Liu F, Bach FH, Billiar TR, Choi AM, Otterbein LE, Murase N. Protection against ischemia/reperfusion injury in cardiac and renal transplantation with carbon monoxide, biliverdin and both. Am J Transplant. 2005;5 :282–291. doi: 10.1111/j.1600-6143.2004.00695.x. [DOI] [PubMed] [Google Scholar]
- 5.Lindenblatt N, Bordel R, Schareck W, Menger MD, Vollmar B. Vascular heme oxygenase-1 induction suppresses microvascular thrombus formation in vivo. Arterioscler Thromb Vasc Biol. 2004;24 :601–606. doi: 10.1161/01.ATV.0000118279.74056.8a. [DOI] [PubMed] [Google Scholar]
- 6.Orozco LD, Kapturczak MH, Barajas B, Wang X, Weinstein MM, Wong J, Deshane J, Bolisetty S, Shaposhnik Z, Shih DM, Agarwal A, Lusis AJ, Araujo JA. Heme oxygenase-1 expression in macrophages plays a beneficial role in atherosclerosis. Circ Res. 2007;100:1703–1711. doi: 10.1161/CIRCRESAHA.107.151720. [DOI] [PubMed] [Google Scholar]
- 7.True AL, Olive M, Boehm M, San H, Westrick RJ, Raghavachari N, Xu X, Lynn EG, Sack MN, Munson PJ, Gladwin MT, Nabel EG. Heme oxygenase-1 deficiency accelerates formation of arterial thrombosis through oxidative damage to the endothelium, which is rescued by inhaled carbon monoxide. Circ Res. 2007;101:893–901. doi: 10.1161/CIRCRESAHA.107.158998. [DOI] [PubMed] [Google Scholar]
- 8.Crawford RS, Hashmi FF, Jones JE, Albadawi H, McCormack M, Eberlin K, Entabi F, Atkins MD, Conrad MF, Austen WG, Jr, Watkins MT. A novel model of acute murine hindlimb ischemia. Am J Physiol Heart Circ Physiol. 2007;292 :H830–837. doi: 10.1152/ajpheart.00581.2006. [DOI] [PubMed] [Google Scholar]
- 9.Feng Y, Zhao H, Xu X, Buys ES, Raher MJ, Bopassa JC, Thibault H, Scherrer-Crosbie M, Schmidt U, Chao W. Innate immune adaptor MyD88 mediates neutrophil recruitment and myocardial injury after ischemia-reperfusion in mice. Am J Physiol Heart Circ Physiol. 2008;295 :H1311–H1318. doi: 10.1152/ajpheart.00119.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and Kupffer cells. Hepatology. 1998;27 :507–512. doi: 10.1002/hep.510270226. [DOI] [PubMed] [Google Scholar]
- 11.Liu M, Liang Y, Chigurupati S, Lathia JD, Pletnikov M, Sun Z, Crow M, Ross CA, Mattson MP, Rabb H. Acute kidney injury leads to inflammation and functional changes in the brain. J Am Soc Nephrol. 2008;19 :1360–1370. doi: 10.1681/ASN.2007080901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersen K, Pedersen BK. The role of inflammation in vascular insulin resistance with focus on IL-6. Horm Metab Res. 2008;40 :635–639. doi: 10.1055/s-0028-1083810. [DOI] [PubMed] [Google Scholar]
- 13.Szekanecz Z. Pro-inflammatory cytokines in atherosclerosis. Isr Med Assoc J. 2008;10 :529–530. [PubMed] [Google Scholar]
- 14.Franke A, Lante W, Kupser S, Becker HP, Weinhold C, Markewitz A. Procalcitonin levels after different types of conventional thoracic surgery. Thorac Cardiovasc Surg. 2008;56:46–50. doi: 10.1055/s-2007-989250. [DOI] [PubMed] [Google Scholar]
- 15.McCormack MC, Kwon E, Eberlin KR, Randolph M, Friend DS, Thomas AC, Watkins MT, Austen WG., Jr Development of reproducible histologic injury severity scores: skeletal muscle reperfusion injury. Surgery. 2008;143 :126–133. doi: 10.1016/j.surg.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 16.Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, Otterbein LE, Ning W, Oury TD, Lee PJ, McCurry KR, Choi AM. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol. 2003;163:231–242. doi: 10.1016/S0002-9440(10)63646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujimoto H, Ohno M, Ayabe S, Kobayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R. Carbon monoxide protects against cardiac ischemia--reperfusion injury in vivo via MAPK and Akt--eNOS pathways. Arterioscler Thromb Vasc Biol. 2004;24 :1848–1853. doi: 10.1161/01.ATV.0000142364.85911.0e. [DOI] [PubMed] [Google Scholar]
- 18.Kaizu T, Ikeda A, Nakao A, Tsung A, Toyokawa H, Ueki S, Geller DA, Murase N. Protection of transplant-induced hepatic ischemia/reperfusion injury with carbon monoxide via MEK/ERK1/2 pathway downregulation. Am J Physiol Gastrointest Liver Physiol. 2008;294:G236–244. doi: 10.1152/ajpgi.00144.2007. [DOI] [PubMed] [Google Scholar]
- 19.Lindgard A, Lundberg J, Rakotonirainy O, Elander A, Soussi B. Preservation of rat skeletal muscle energy metabolism by illumination. Life Sci. 2003;72:2649–2658. doi: 10.1016/s0024-3205(03)00176-0. [DOI] [PubMed] [Google Scholar]
- 20.Tsuchida T, Kato T, Yamaga M, Ikebe K, Oniki Y, Irie H, Takagi K. Effect of perfusion during ischemia on skeletal muscle. J Surg Res. 2001;101:238–241. doi: 10.1006/jsre.2001.6278. [DOI] [PubMed] [Google Scholar]
- 21.Zuckerbraun BS, McCloskey CA, Gallo D, Liu F, Ifedigbo E, Otterbein LE, Billiar TR. Carbon monoxide prevents multiple organ injury in a model of hemorrhagic shock and resuscitation. Shock. 2005;23 :527–532. [PubMed] [Google Scholar]
- 22.Dawson J, Cockerill G, Choke E, Loftus I, Thompson MM. Circulating cytokines in patients with abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:324–326. doi: 10.1196/annals.1383.010. [DOI] [PubMed] [Google Scholar]
- 23.Gol MK, Nisanoglu V, Iscan Z, Balci M, Kandemir O, Tasdemir O. Inhibition of systemic inflammatory response with sodium nitroprusside in open heart surgery. J Cardiovasc Surg (Torino) 2002;43 :803–809. [PubMed] [Google Scholar]
- 24.van Besouw NM, Baan CC, Holweg CT, de Groot-Kruseman HA, Peeters AM, Balk AH, Weimar W. Cytokine profiles as marker for graft vascular disease after clinical heart transplantation. Ann Transplant. 2000;5 :61–67. [PubMed] [Google Scholar]
- 25.Hua HT, Albadawi H, Entabi F, Conrad M, Stoner MC, Meriam BT, Sroufe R, Houser S, Lamuraglia GM, Watkins MT. Polyadenosine diphosphate-ribose polymerase inhibition modulates skeletal muscle injury following ischemia reperfusion. Arch Surg. 2005;140:344–351. doi: 10.1001/archsurg.140.4.344. discussion 351–342. [DOI] [PubMed] [Google Scholar]
- 26.Wakai M, Winter D, Street J, O'Sullivan R, Wang J, Redmond H. Inosine Attenuates Tourniquet Induced Skeletal Muscle Reperfusion Injury. J Surg Res. 2001;99:311–315. doi: 10.1006/jsre.2001.6192. [DOI] [PubMed] [Google Scholar]
- 27.Sakurai T, Fujita Y, Ohto E, Oguro A, Atomi Y. The decrease of the cytoskeleton tubulin follows the decrease of the associating molecular chaperone alphaB-crystallin in unloaded soleus muscle atrophy without stretch. FASEB J. 2005;19 :1199–1201. doi: 10.1096/fj.04-3060fje. [DOI] [PubMed] [Google Scholar]
- 28.Stanchi F, Corso V, Scannapieco P, Ievolella C, Negrisolo E, Tiso N, Lanfranchi G, Valle G. TUBA8: A new tissue-specific isoform of alpha-tubulin that is highly conserved in human and mouse. Biochem Biophys Res Commun. 2000;270 :1111–1118. doi: 10.1006/bbrc.2000.2571. [DOI] [PubMed] [Google Scholar]
- 29.Boudriau S, Cote CH, Vincent M, Houle P, Tremblay RR, Rogers PA. Remodeling of the cytoskeletal lattice in denervated skeletal muscle. Muscle Nerve. 1996;19 :1383–1390. doi: 10.1002/(SICI)1097-4598(199611)19:11<1383::AID-MUS2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 30.Conrad MF, Stone DH, Albadawi H, Hua HT, Entabi F, Stoner MC, Watkins MT. Local inflammatory and thrombotic responses differ in a murine model of partial and complete hindlimb ischemia/reperfusion. Surgery. 2005;138 :375–381. doi: 10.1016/j.surg.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 31.Tongers J, Knapp JM, Korf M, Kempf T, Limbourg A, Limbourg FP, Li Z, Fraccarollo D, Bauersachs J, Han X, Drexler H, Fiedler B, Wollert KC. Haeme oxygenase promotes progenitor cell mobilization, neovascularization, and functional recovery after critical hindlimb ischaemia in mice. Cardiovasc Res. 2008;78:294–300. doi: 10.1093/cvr/cvm107. [DOI] [PubMed] [Google Scholar]
- 32.Kauvar DS, Baer DG, Dubick MA, Walters TJ. Effect of fluid resuscitation on acute skeletal muscle ischemia-reperfusion injury after hemorrhagic shock in rats. J Am Coll Surg. 2006;202:888–896. doi: 10.1016/j.jamcollsurg.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 33.Crinnion JN, Homer-Vanniasinkam S, Parkin SM, Gough MJ. Role of neutrophil-endothelial adhesion in skeletal muscle reperfusion injury. Br J Surg. 1996;83 :251–254. [PubMed] [Google Scholar]
- 34.Lee S, Suk K. Heme oxygenase-1 mediates cytoprotective effects of immunostimulation in microglia. Biochem Pharmacol. 2007;74 :723–729. doi: 10.1016/j.bcp.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 35.Liu SH, Xu XR, Ma K, Xu B. Protection of carbon monoxide inhalation on lipopolysaccharide-induced multiple organ injury in rats. Chin Med Sci J. 2007;22:169–176. [PubMed] [Google Scholar]
- 36.Hasan RN, Schafer AI. Hemin upregulates Egr-1 expression in vascular smooth muscle cells via reactive oxygen species ERK-1/2-Elk-1 and NF-kappaB. Circ Res. 2008;102:42–50. doi: 10.1161/CIRCRESAHA.107.155143. [DOI] [PubMed] [Google Scholar]