

Abstract
Transcriptional regulation in a tissue-specific and quantitative fashion is essential for developmental events, including those involved in cardiovascular morphogenesis. Tbx1 is a T-box containing transcription factor that is responsible for many of the defects observed in 22q11 deletion syndrome in humans. Tbx1 is expressed in the secondary heart field (SHF) and is essential for cardiac outflow tract (OFT) development. We previously reported that Tbx1 is regulated by sonic hedgehog via forkhead (Fox) transcription factors in the head mesenchyme and pharyngeal endoderm, but how it is regulated in the SHF is unknown. Here, we show that Tbx1 expression in the SHF is regulated by Fox proteins through a combination of two evolutionally conserved Fox binding sites in a dose-dependent manner. Cell fate analyses using the Tbx1 enhancer suggests that SHF-derived Tbx1-expressing cells contribute extensively to the right ventricular myocardium as well as the OFT during early development and ultimately give rise to the right ventricular infundibulum, pulmonary trunk and pulmonary valves. These results suggest that Fox proteins are involved in most, if not all, Tbx1 expression domains, and that Tbx1 marks a subset of SHF-derived cells, particularly those that uniquely contribute to the right-sided outflow tract and proximal pulmonary artery.
Keywords: Tbx1, secondary heart field, Fox proteins, cardiac outflow tract
Introduction
Development of the heart involves complex steps that are precisely regulated in a temporo-spatial manner. Several cell types including myocardial cells, endothelial cells and neural crest–derived cells participate in the development of the cardiac outflow tract. Regional differences in cell proliferation and programmed cell death result in elongation, rotation, and septation of the outflow tract, ultimately generating two separate vessels, the aorta and pulmonary trunk (Poelmann et al., 1998; Watanabe et al., 2001; reviewed in Srivastava and Olson, 2000). Abnormalities of this process result in outflow tract defects, which comprise approximately 30% of congenital cardiovascular malformations.
The primary heart field specified in the lateral plate mesoderm gives rise to the primitive linear heart tube. In recent years, there has been increasing evidence that a second cell lineage contributes to the cardiac outflow tract and right ventricular myocardium (Kelly et al., 2001; Mjaatvedt et al., 2001; Waldo et al., 2001). These cells are derived from a subset of precursor cells that arise from the pharyngeal mesoderm located antero-dorsal to the heart and have thus been termed the anterior heart field (AHF), or secondary heart field (SHF). The transcription factors essential for myocardial development, including Nkx2.5, Gata4, Mef2c and Hand are expressed in the SHF as well as in the primary heart field and are part of transcriptional complexes that cooperatively activate cardiac specific gene expression (Lee et al., 1998; Waldo et al., 2001; Garg et al., 2003). In addition, the transcription factors Isl1 (Cai et al., 2003; Dodou E et al., 2004) and Foxh1 (von Both et al., 2004) are specifically expressed in the SHF and activate Mef2c gene expression in the SHF (Verzi et al., in press). Null mutation of either Isl1 or Foxh1 in mice results in a lack of the outflow tract and right ventricular segments. Smarcd3, which encodes one of the subunits of BAF chromatin remodeling complexes, Baf60c, is also expressed in the developing cardiac outflow tract (Lickert et al., 2004). Smarcd3 knockdown embryos displayed hypoplasia of the outflow tract and right ventricle in a dose-dependent fashion, reflecting defective expansion of the SHF.
Tbx1, a member of the T-box family of transcription factors, is expressed in the SHF (Yamagishi et al., 2003) and is a major genetic determinant of 22q11.2 deletion syndrome (22q11DS) in humans (reviewed in Yamagishi and Srivastava, 2003). Outflow tract defects such as persistent truncus arteriosus and tetralogy of Fallot are characteristic cardiovascular features observed in patients with 22q11DS, in addition to craniofacial defects such as cleft palate (reviewed in Yamagishi, 2002). Tbx1-null mice phenocopy the 22q11DS phenotype (Jerome et al., 2001; Lindsay et al., 2001; Merscher et al., 2001) and Tbx1 hypomorphic mice display milder phenotype with cardiovascular defects but no cleft palate (Hu et al., 2004; Xu et al., 2004). Tissue-specific disruption of Tbx1 in the Nkx2.5 expression domain showed a single outflow tract with no evidence of aorto-pulmonary septum, suggesting that loss of Tbx1 function in the SHF or pharyngeal endoderm might result in outflow tract defects (Xu et al., 2004).
We have previously reported that Tbx1 is regulated by a signaling molecule, sonic hedgehog, via forkhead (Fox) transcription factors in the developing pharyngeal arch (Yamagishi et al., 2003). We also demonstrated that development of the cardiac outflow tract was more sensitive to Tbx1 dosage than craniofacial development (Hu et al., 2004). The increased dose-dependency in the outflow tract is likely due to amplification of Tbx1 via an autoregulatory loop involving Foxa2 and Tbx1 in the pharyngeal mesoderm resulting in upregulation of the downstream genes, Fgf8 and Fgf10 in the SHF. Here, we identified the regulatory elements necessary for Tbx1 expression in the developing cardiac outflow tract. Our data suggests that Tbx1 expression in the SHF is regulated through a combination of forkhead binding sites (Fox sites) upstream of Tbx1 and that the Tbx1-expressing cells in the SHF contribute largely to the right ventricular outflow tract and proximal pulmonary artery.
Materials and methods
Transgenic plasmid constructs
We previously identified an evolutionarily conserved Fox site approximately 12.8 kilo bases (kb) upstream of the Tbx1 translation start site (Yamagishi et al., 2003). We defined this site as Fox site #1. Another Fox site was identified approximately 6.6 kb upstream of Tbx1 translation start site in the present study, and defined as Fox site #2. A series of genomic fragments upstream of Tbx1 were used in lacZ reporter constructs as follows: 200 base pairs (bp) including Fox site #1 with 1.5 kb including Fox site #2, 200 bp including Fox site #1 with 200 bp including Fox site #2, 200 bp including mutated Fox site #1 with 200 bp including intact Fox site #2, and five tandem repeats of 68 bp containing Fox site #1 were cloned into the hsp68-lacZ constructs and designated as construct 1, 2, 3, and 4, respectively. In construct 3, Fox site #1 was mutated to a NotI restriction site as described previously (Yamagishi et al., 2003). F0 or F1 transgenic embryos were used for identifying the outflow tract enhancer element of Tbx1. A 1.1kb fragment including Fox site #1 for Tbx1 that was described elsewhere (Yamagishi et al., 2003) was subcloned upstream of a nuclear localizing signal (nls)-Cre expression plasmid without a basal promoter to generate transgenic mice (see below).
Generation of transgenic mice
All hsp68-lacZ reporter and nls-Cre transgenic constructs were linearized to remove the vector backbone and injected into fertilized oocytes as described previously (Yamagishi et al., 2003). In order to trace the fate of Tbx1-expressing cells, F0 nls-Cre transgenic mice were bred with ROSA26R (R26) mice (Soriano et al., 1999). Embryos were harvested based on the assumption that noon of the day of vaginal plugs in female mice was E0.5 to detect Cre-mediated recombination in those embryos using β-galactosidase staining. Genomic DNA was extracted from yolk sacs of embryos or tails of newborn pups. Transgene of lacZ or Cre was detected by PCR. Primers and reaction condition are available upon request.
β-galactosidase staining
Harvested embryos were fixed in 2% paraformaldehyde and 0.25% glutaraldehyde in phosphate buffered saline (PBS) at 4°C for 30 minutes to one hour and washed with PBS. Subsequently they were incubated in staining solution containing X-gal (1mg/ml), potassium ferricyanide (5mM), potassium ferrocyanide (5mM) and magnesium chloride (1mM) in PBS at room temperature overnight. After staining, they were post-fixed in 4% paraformaldehyde and 0.1% glutaraldehyde. For histology, stained and fixed embryos were embedded in paraffin, sectioned in 8µm thickness transversely and counterstained with nuclear Fast Red.
Luciferase reporter assay
COS-1 cells were cotransfected with 0.25µg to 0.75µg of Flag-tagged Foxa2 (a gift from K. Kaestner), Foxc1 or Foxc2 (gifts from T. Kume) in pcDNA3 (Invitrogen) in addition to 0.2µg of a promoterless luciferase reporter plasmid (pGL3-Basic Vector from Promega) fused to Tbx1 upstream regulatory fragment using FuGene6 (Roche) as follows: a 200 bp fragment including Fox site #1, a 200 bp fragment including Fox site #2, a 200 bp fragment including mutated Fox site #1, and a 200 bp fragment including mutated Fox site #2 were cloned into pGL3-Basic Vector and designated as Fox #1-luc, Fox #2-luc, Fox #1mt-luc, and Fox #2mt-luc, respectively. Fox #1 × 4 or Fox #1 × 8 was made of four or eight tandem repeats of a 68bp fragment containing Fox site #1. The total amount of DNA was adjusted with pcDNA plasmid in each sample. After 48 hours, cells were harvested and luciferase activity was assayed using Lucy2 luminometer (Rosys Anthos). 0.1µg of RSV-lacZ which expresses β-galactosidase was cotransfected in order to normalize luciferase activity by measuring β-galactosidase activity in each sample. Each experiment was duplicated and repeated at least three times. The results are shown as relative fold of luciferase activity to the reporter construct alone. Error bars represent two standard deviations.
Electrophoretic mobility shift assay (EMSA)
Double strand oligonucleotides containing Fox site #1 and Fox site #2 were used as 32P radioactive probes or non-radioactive competitors. The nucleotide sequences of Fox site #2 were as follows; 5’-GGGCCATTTGTTTGTTTTTGAGAAATTCCAAG-3’, 3’-GGTAAACAAACAAAAACTCTTTAAGGTTCGGG-5’. Mutated Fox site #2 oligonucleotides contained a NotI restriction site (5’-GCGGCCGC-3’) instead of underlined sequence. Flag-tagged Foxa2, Foxc1, or Foxc2-pcDNA was translated to its corresponding protein using TNT Coupled Reticulocyte Lysate Systems (Promega). EMSA was performed as described previously (Scott et al., 1994).
Results
Expression of Tbx1 in the cardiac outflow tract is regulated through multiple consensus Fox binding sites
Using VISTA software to compare genomic sequence upstream of Tbx1 we have identified regions of high homology between human and mouse that contain conserved cis-regulatory elements (Hu et al., 2004). A combination of a 200 bp region (−12.8 kb to −12.6 kb from Tbx1 translation start site) with a 1.5 kb region (−8.1 kb to −6.6 kb) could direct all domains of Tbx1 expression, namely head mesenchyme, pharyngeal endoderm and mesoderm, and the cardiac outflow tract (Fig. 1, construct 1, A–C) (Hu et al., 2004). We have previously shown that a Fox site in the 200 bp region is essential for Tbx1 expression in the head mesenchyme and pharyngeal endoderm (Yamagishi et al., 2003), and define this site as Fox site #1 in the present study (Fig. 1 upper column). Expression directed by the combined enhancers was dependent on the 200bp region containing Fox site #1 since the 1.5kb region alone did not direct lacZ expression in any tissue as shown previously (Hu et al., 2004).
Fig. 1. Fox cis-elements are essential for Tbx1 expression in the cardiac outflow tract.
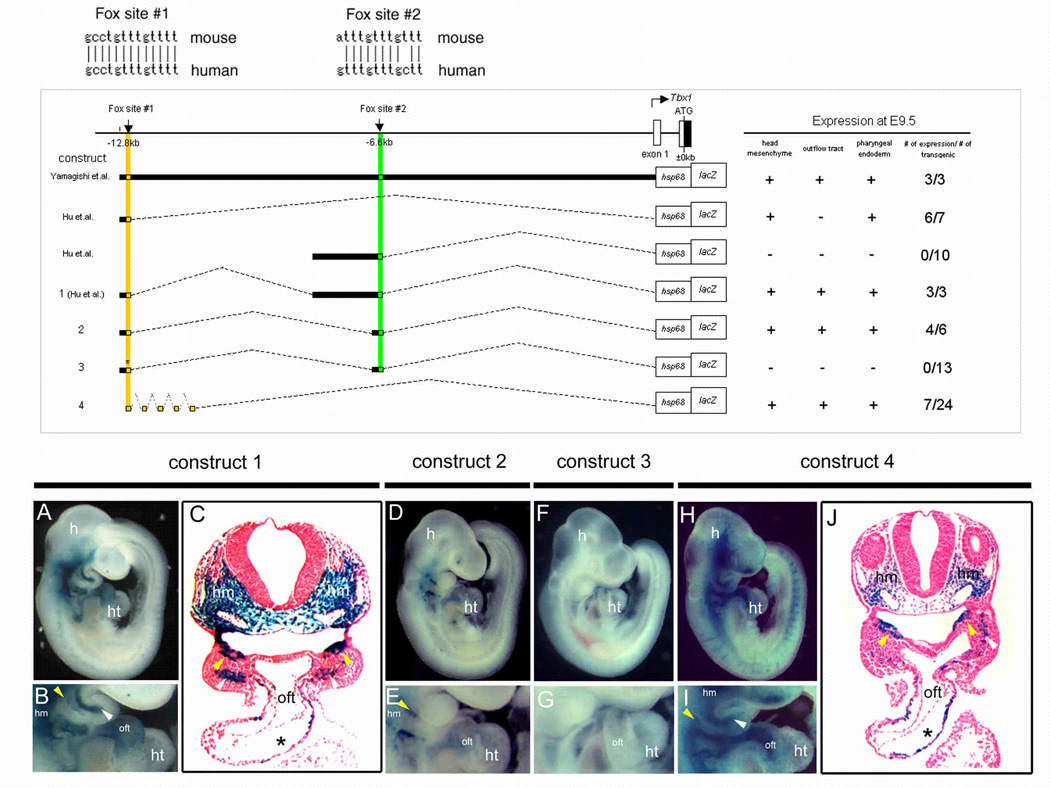
(Upper panel) The sequence comparison of Fox site #1 and #2 between mouse and human showing 100% and 83% conservation across species, respectively. (Middle panel) Schematic diagram of Tbx1-lacZ constructs 5’ of the mouse Tbx1 locus. Numbers below the horizontal line represent kilobase pairs (kb) from the translation site. The location of Fox site #1 or Fox site #2 is shown as a small yellow or green square, respectively. The first four constructs and their lacZ expression patterns were previously described (Yamagishi et al., 2003; Hu et al., 2004). Construct number is indicated on the left and the representative lacZ expression pattern is summarized on the right. The star indicates mutation. Construct 4 has five tandem repeats of 68 base pairs (bp) fragment of Fox site #1. (Lower panel) Right lateral view of whole mount lacZ staining of E9.5 transgenic embryos (A, D, F, H) and magnified view of the heart (B, E, G, I). Construct number is indicated above the corresponding images. Transverse sections of A and H are shown in C and J, respectively. All transgenic constructs except construct 3 directed lacZ in the head mesenchyme (hm) and pharyngeal endoderm (yellow arrowhead). Construct 1, 2, and 4 demonstrate lacZ expression in the cardiac outflow tract (oft). Construct 1 and 4 show lacZ expression in pharyngeal mesodermal core (white arrowhead). Stars in C and J indicate the endocardial cushion. h, head; ht, heart.
Inspection of the 1.5 kb region for binding sites of transcription factors using the MatInspector V.2.2 based on TRANSFAC4.0 (Quandt et al., 1995) identified another consensus Fox binding site approximately 6.6 kb upstream of the Tbx1 translation start site which was 83% conserved between mouse and human (Fig. 1 upper column). This site is defined as Fox site #2 in the present study.
A combination of the 200 bp region containing Fox site #1 and a 200 bp region containing Fox site #2 directed lacZ expression in the outflow tract along with the head mesenchyme and pharyngeal endoderm when cloned upstream of a lacZ reporter gene with an hsp68 heterologous basal promoter (Fig.1 construct 2, D, E). A point mutation of Fox site #1 in the context of its own 200 bp enhancer along with a 200 bp fragment including Fox site #2 abolished lacZ expression in all domains (Fig. 1, construct 3, F, G). These results, together with our previous results, indicate that Fox site #1 is necessary for outflow tract expression and can regulate this expression in combination with Fox site #2 although it is not sufficient for outflow tract expression at a level detectable by the reporter.
Requirement of Fox site #1 for Tbx1 expression in the outflow tract prompted us to examine whether it has weak outflow tract enhancer activity but simply needs additional Fox sites to elevate the level of activity. By generating five tandem repeats of a 68 bp region surrounding Fox site #1, we tested whether Fox site #1 could activate Tbx1 expression in the outflow tract without Fox site #2 if its function was amplified. Although this transgene is artificial, seven of twenty-four F0 transgenic embryos demonstrated lacZ expression in the outflow tract in addition to head mesenchyme and pharyngeal endoderm and mesoderm (Fig. 1, construct 4, H–J). Transverse sections of these embryos showed the same distribution of lacZ expressing cells as those under control of the 200 bp plus 1.5 kb Tbx1 enhancers (compare Fig. 1C and J). These results suggest that Fox site #1 might act as a subtle enhancer for outflow tract expression, and may require the additive enhancer activity of Fox site #2 in the endogenous situation.
Fox proteins activate the Tbx1 enhancer in a dose-dependent manner
Our previous data indicated that Foxa2, Foxc1 and Foxc2 could bind and activate transcription through Fox site #1 of Tbx1 in vitro (Yamagishi et al. 2003). Since Fox site #1 directed Tbx1 expression in the cardiac outflow tract in vivo in combination with Fox site #2 (Fig. 1, construct 2, D, E), we tested if the common Fox proteins could bind to not only Fox site #1, but also Fox site #2 using electrophoretic mobility shift assays (EMSA) with 32P-labeled oligonucleotides. Radioactive oligonucleotide probes including Fox site #2 were able to bind Foxa2, Foxc1 and Foxc2 (Fig. 2). These interactions were competed by non-radioactive oligonucleotide containing Fox site #2 in a dose-dependent manner, suggesting binding specificity of these Fox proteins to Fox site #2.
Fig. 2. Foxa2, Foxc1 and Foxc2 specifically bind to Fox sites in the Tbx1 regulatory region.

Electrophoretic mobility shift assays demonstrated Foxa2, Foxc1 and Foxc2 protein specifically bound to radioactively labeled Fox site #2. Shifted bands were efficiently competed with excess non-radioactive competitors.
To test whether these Fox proteins could not only bind, but also activate transcription through these sites, we transiently transfected COS-1 cells with a luciferase reporter cloned downstream of the 200 bp fragment including Fox site#1 or Fox site #2 of the Tbx1 enhancer. Co-transfection with expression vectors of Foxa2, Foxc1, or Foxc2 activated the reporter with Fox site #1 and Fox site #2 to varying degrees (Fig. 3A). In contrast, mutations in Fox site #1 and #2 ablated activation by any of Foxa2, Foxc1 and Foxc2 (Fig. 3A). Furthermore, our in vitro assay revealed that the transactivation level through the Fox binding sites is dependent on the dose of Foxa2, Foxc1 or Foxc2 proteins (Fig. 3B).
Fig. 3. Forkhead proteins transactivate Tbx1 in a dose dependent manner.

Foxa2, Foxc1 or Foxc2 and Tbx1-luciferase reporter were cotransfected into COS-1 cells. The horizontal axis of bar graphs shows the relative fold of measured luciferase activity. (A) Fox proteins transactivate Tbx1 reporter with 200 bp fragment involving Fox site #1 (Fox #1-luc) as well as Tbx1 reporter with 200 bp involving Fox site #2 (Fox #2-luc). Both Fox site #1 with Fox mutation (Fox #1 mt-luc) and Fox site #2 with Fox mutation (Fox #2 mt-luc) abolish the activation. (B) 250ng to 750ng of expression plasmids of Fox proteins were cotransfected with Tbx1 reporter containing Fox site #2 (Fox #2-luc) and demonstrated transactivation in a dose dependent manner. (C) Increasing copies of 68 bp fragment of Fox site #1 using four (Fox #1 × 4-luc) or eight (Fox #1 × 8-luc) tandem repeats of Fox site #1 resulted in enhanced transactivation by Foxa2, Foxc1 and Foxc2 as the number of Fox site #1 increased.
To examine whether activation of the Tbx1 enhancer containing Fox site #1 is dependent on the number of cis-elements, we created four and eight tandem repeats of Fox site #1 cloned upstream of the luciferase reporter. Co-transfection with Foxa2, Foxc1 or Foxc2 resulted in linear increase in the luciferase activity corresponding to the number of Fox binding sites (Fig.3C). Together with the results of the in vivo assays, these data suggest that Fox proteins may regulate the specific domains of Tbx1 expression in a dose-dependent manner, through the combination of Fox site #1 and Fox site #2.
Tbx1-expressing cells contribute to the development of the cardiac outflow tract and right ventricle
We subcloned the previously described 1.1 kb region including Fox site #1 upstream of the Cre recombinase gene with a nuclear localization signal and generated Tbx1-Cre transgenic mice. We interbred these transgenic mice with ROSA26 reporter (R26R) mice, in which lacZ is transcribed in the presence of Cre recombinase activity, which allowed us to detect activity of the 1.1kb Tbx1 enhancer with greater sensitivity using β-galactosidase staining (Soriano, 1999). Examination of multiple F1 embryos at E9.5 derived from two independent lines of F0 transgenic mice demonstrated that the embryonic expression pattern of lacZ in the 1.1kb Tbx1 enhancer-Cre/R26R transgenic mice was similar to endogenous Tbx1 expression and that of the 12.8 kb Tbx1 upstream sequence fused to hsp68-lacZ reporter (Fig. 4A, Yamagishi et al. 2003). Transverse section of these embryos showed lacZ expression in the SHF in the pharyngeal mesoderm and in the outflow tract (Fig. 4I), indicating that at least a subset of Tbx1-expressing cells derived from the SHF can be marked with this Cre-mediated system.
Fig. 4. The fate of Tbx1-expressing cells during early cardiac development.
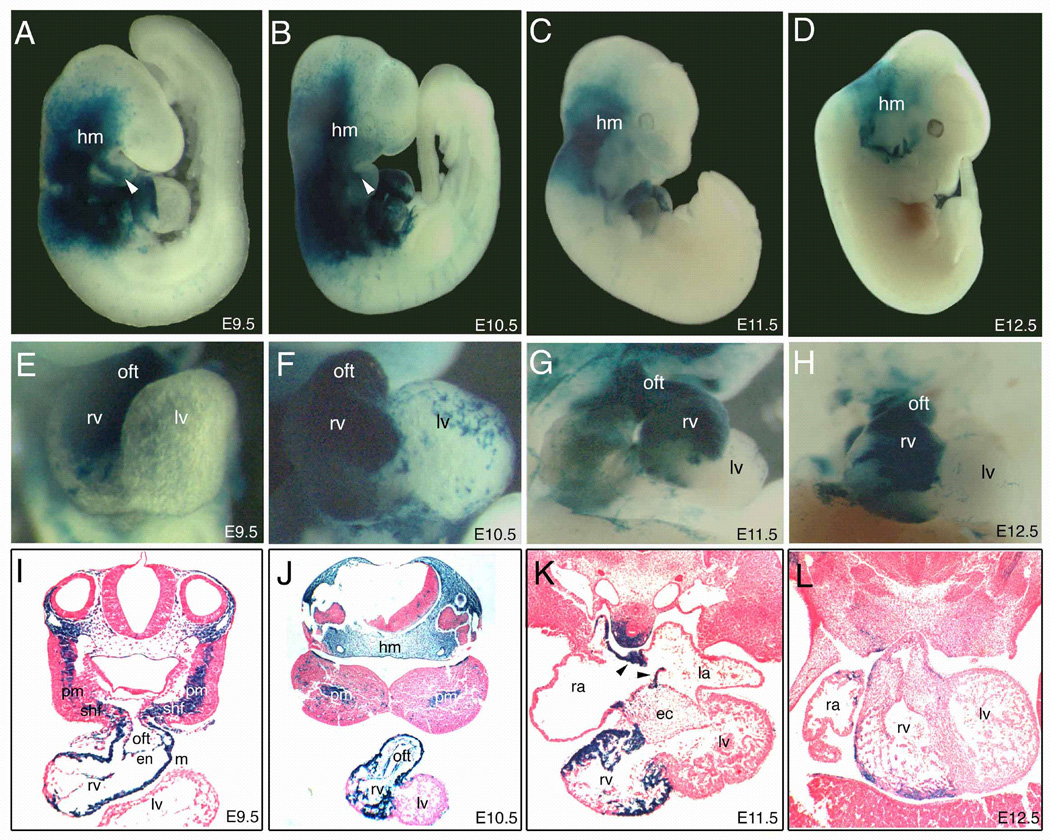
(A–D) Right lateral view of whole-mount lacZ staining of Tbx1-Cre/R26R embryos. The magnified view of each heart (E–H) and transverse sections of each embryo (I–L) are shown. Tbx1-expressing cells were observed at E9.5 (A,E,I), E10.5 (B,F,J), E11.5 (C,G,K), E12.5 (D,H,L). Tbx1-expressing cells were predominantly detectable in the outflow tract (oft) and the right ventricle (rv) at E9.5–10.5 (A, B, E, F, I, J), and relatively restricted in the rv at E11.5–12.5 (C, D, G, H, K, L). Note that blue cells appeared to be continuous from pharyngeal mesoderm (pm) and the secondary heart field (shf) to rv through the oft at E9.5 (I). Tbx1-expressing cells also contributed to the developing atrial septum (arrowheads in K). ec, endocardial cushion; en, endocardium; hm, head mesenchyme; la, left atrium; lv, left ventricle; m, myocardium; ra, right atrium.
Using the Cre-mediated system we were able to trace the fate of a subset of cells that express Tbx1 at any stage using β-galactosidase staining, even if the gene is no longer being transcribed. During early cardiovascular development, lacZ positive cells were continuously detectable in the right ventricle and the cardiac outflow tract (Fig. 4A, E). In transverse sections at E9.5, blue cells appeared in a continuous stream from the pharyngeal mesoderm, encompassing the SHF, to the anterior portion of right ventricle through the outflow tract (Fig. 4I). In the outflow tract, both myocardial and endothelial layers were populated by lacZ expressing cells. At E10.5, lacZ positive cells were throughout the right ventricle (Fig. 4B, F, J), however they became restricted to a subset of right ventricular cells at E11.5–12.5 (Fig. 4G, H, K, L). The left ventricle had only spotty expression of lacZ outside of myocardial and endocardial layers at this stage (Fig. 4J–L). A small number of blue cells were also observed in the endocardial cushion, developing atrial wall and septum (Fig. 4K).
Outside of the heart, lacZ positive cells were widely detectable in head mesenchyme and pharyngeal arch at E9.5–10.5 (Fig. 4A, B), but were later restricted to a narrower domain consistent with contribution of Tbx1-expressing descendants to parts of the facial muscle and connective tissue (Fig. 4C, D).
At E17.5, lacZ positive cells predominantly accumulated in the conal area (infundibulum) of the right ventricle and the main trunk of the pulmonary artery (Fig. 5A, B). Only weak expression was detectable in the ascending aorta and its branching arteries (Fig. 5A, B). Although a previous study showed a subset of Tbx1 descendants in left ventricular myocardium (Brown et al. 2003), we could not detect blue cells there using our system at this stage. The ductus arteriosus was also excluded from lacZ expression (Fig. 5B). In transverse section, lacZ was predominantly expressed in the wall of the main pulmonary artery proximal to the bifurcation, and mesenchymal and endothelial tissue of the pulmonary valve (Fig. 5C). These results suggest that a subset of descendants of Tbx1-expressing cells from the SHF give rise to very specific domains of the outflow tract.
Fig. 5. The fate of Tbx1-expressing cells during late cardiac development.

(A) Frontal view of Tbx1-Cre/R26R embryonic heart at E17.5. (B) Left lateral view of the same heart after removing atria. (C) Transverse sections of the same heart at the pulmonary valve (pv) level. LacZ-positive cells were localized in the anterior portion (outflow tract) of the right ventricle (rv) and the main trunk of the pulmonary artery (mpa) (A, B). LacZ-positive cells were detectable in both endothelial and muscle layers of mpa as well as pv (C). Few blue cells were observed in the wall of aorta (ao in C). ra, right atrium; la, left atrium; da, ductus arteriosus.
Discussion
Fox proteins regulate Tbx1 during the SHF development through cardiac outflow tract enhancers in a dose-dependent fashion
In order to elucidate the regulation of Tbx1 in the SHF, we focused on the cardiac outflow tract-specific enhancer of Tbx1 in this study. Although we were previously able to identify an enhancer that was necessary and sufficient to direct Tbx1 expression in the pharyngeal endoderm and head mesenchyme (Yamagishi et al., 2003), a separate genomic region sufficient for outflow tract expression was not identified. Instead our results suggest that the outflow tract expression may be regulated through at least two enhancer regions. Interestingly, each of these regions contain a highly conserved consensus binding-site for Fox transcription factors: Fox site #1 that was previously reported to be sufficient for head mesenchyme and pharyngeal endoderm expression, and Fox site #2 that was identified in the present study. Our transgenic analyses revealed that Fox site #1 regulates the outflow tract expression of Tbx1 in conjunction with Fox site #2. Further, in vitro studies suggested that Foxa2, Foxc1 and Foxc2 proteins regulate the expression of Tbx1 through both Fox binding sites in a dose-dependent fashion. Together, our data indicate that Fox site #1 is indispensable, but not sufficient, for the outflow tract expression of Tbx1, and that the Fox site #2 plays a synergistic role in outflow tract expression with Fox site #1.
Because Fox proteins are inherently capable of initiating chromatin relaxation events (Cirillo et al., 2002), it is possible that the necessary Fox site facilitates access of other transcription factors to their essential cis-elements. Although we identified two Fox sites essential for expression in virtually all domains of Tbx1 transcription, we were unable to identify separable regulatory elements for expression within each domain. It is possible that other cis-elements around the Fox site or trans-acting factors are necessary to collaborate with the Fox site for expression in each specific domain.
Intriguingly, mutation of Fox site #2 in transgenic embryos in conjunction with the 200 bp fragment including intact Fox site #1 (Fox site #2 mutation in construct 2), abolished all expression of lacZ in vivo (data not shown), suggesting that a putative repressor may exist within the 200 bp proximal enhancer surrounding Fox site #2. Although we have been unable to identify the repressive cis-element, we did find a conserved binding site for Runt domain transcription factors in this region. Runt factors have been reported to act as transcriptional silencers in the development of T lymphocytes (Taniuchi et.al., 2002). Our preliminary date indicates that this site is not sufficient for repression and we are currently searching for additional repressor elements flanking Fox site #2.
Of numerous Fox proteins, Foxc1 and Foxc2, and Foxh1 are known to be involved in the cardiac outflow tract development. Foxa2 is expressed in the pharyngeal mesoderm, which contributes to the SHF (Kume et al., 1998, 2001; Hu et al., 2004; von Both et al., 2004). Although early embryonic demise of Foxa2-null mutants precludes studying the requirement for Foxa2 in outflow tract development (Weinstein et al., 1994), we previously demonstrated a reinforcing autoregulatory loop involving Foxa2 and Tbx1 in the pharyngeal mesoderm involving the SHF (Hu et al., 2004). Foxc1 or Foxc2 null mice and compound heterozygous mice for Foxc1 and Foxc2 display aortic arch defects reminiscent of Tbx1 heterozygous mice (Kume et al., 1998, 2001). Interestingly, they also display reduction in the size of the outflow tract and right ventricle (Kume T, personal communication), suggesting a dose-dependent regulatory mechanism of Foxc1 and Foxc2 in the SHF development as well as aortic arch formation. Furthermore, embryos homozygous-null for both Foxc1 and Foxc2 display absence of the outflow tract and right ventricle, as well as downregulation of Tbx1, Fgf8 and Fgf10 (Kume T, personal communication). These in vivo data support our conclusion that Fox proteins regulate Tbx1 during the SHF development through the outflow tact enhancers in a dose-dependent fashion.
In contrast, there has been no evidence of genetic interaction between Foxh1 and Tbx1 although Foxh1 plays an important role in the SHF development. A recent study demonstrated that Foxh1 could form a transcriptional complex with Nkx2.5 and regulate Mef2c expression in the SHF and outflow tract, and that Foxh1-null mice displayed defects of the outflow tract (von Both et al., 2004). However, the consensus binding sequence for Foxh1 is different from the one for Foxa2, Foxc1 and Foxc2, suggesting that Foxh1 may not directly regulate outflow tract development through the Tbx1 enhancer. We speculate that a Foxa2/c1/c2-Tbx1 pathway may be parallel or independent to the Foxh1/Nkx2.5-Mef2c pathway in regulation of the SHF.
The fate of Tbx1-expressing cells during cardiac development
Cre-mediated lacZ expression directed by a 1.1 kb region containing Fox site #1 alone recapitulated the endogenous expression of Tbx1 in all domains including the outflow tract at E9.5 even though the 1.1 kb hsp68-lacZ reporter system marked only head mesenchyme and pharyngeal endoderm (Yamagishi et al., 2003). This inconsistency may be explained by high copy numbers of integrated transgene or high transcriptional efficiency of the nls-Cre reporter system. Our experiment using the hsp68-lacZ reporter system with five tandem repeats of Fox site #1 may support the possibility that Fox site #1 has weak enhancer activity in the outflow tract, in addition to its other domains of activity. Another possibility is that sequences flanking Fox site #1 in the context of the 1.1 kb enhancer involve positive regulatory elements and those cis-elements may amplify the function of Fox enhancer in the outflow tract in this system.
In either case, the Cre-mediated transgenic system was useful to analyze the fate of a subset of SHF-derived cells where Tbx1 was expressed. Cell fate analyses using Cre driven by the Fox site #1 enhancer suggested that Tbx1-expressing cells derived from the SHF contribute to development of the right ventricular myocardium as well as the outflow tract at E9.5–10.5. At this developmental stage, the Tbx1-espressing cells appeared to be the primary source of cells that form the right ventricle as well as the outflow tract. Later at E11.5–12.5, the right ventricle consisted of a mixture of lacZ positive and negative cells, and finally Tbx1-expressing cells became restricted to the right ventricular outflow tract (infundibulum), main pulmonary trunk and pulmonary valves. It is of note that only a few Tbx1-expressing cells were detectable in the developing aortic arch, although aortic arch anomalies are one of the common defects in 22q11DS and mice lacking Tbx1 (reviewed in Yamagishi and Srivastava, 2003). Recently it was suggested that aortic arch anomalies occur independent of the outflow tract defect based on mice lacking Tbx1 only in the domain of Nkx2.5 expression (Xu et al., 2004). Taken together, cells derived from the SHF may not directly contribute to aortic arch formation, but Tbx1 may play a role in patterning of the aortic arch in a non-cell autonomous fashion.
Brown et al. generated transgenic mice expressing Cre recombinase under control of a 7.6 kb fragment subcloned from a region upstream of Tbx1 and investigated the fate of Tbx1-expressing cells by crossing with R26R mice (Brown et al., 2004). Their 7.6 kb fragment contained both Fox site #1 and #2 presented in our study, and directed expression of Tbx1 similar to the enhancers described here. However, unlike our results, Brown et al. found Tbx1-expressing descendents in the left ventricular myocardium, and ascending and descending aorta. LacZ positive cells in this study may represent a subset of those described by Brown et al., since the 7.6 kb enhancer previously reported includes the whole 1.1 kb enhancer we described earlier (Yamagishi et al., 2003). Interestingly, placement of tamoxifen-inducible Cre into the Tbx1 genomic locus using homologous recombination demonstrated Cre activity only in the context of pharyngeal arch and the cardiac outflow tract (Xu et al., 2004). The distribution and strength of lacZ expression, under control of the genomic Tbx1 locus may most accurately reflect the fate of endogenous Tbx1 expressing cells and is more restricted than that described by Brown et al. Our slightly broader lacZ distribution may result from high copy numbers of integrated transgene or high transcriptional efficiency in our system. In any case, a number of studies, including that of Xu et al., suggest that cells derived from the SHF contribute to outflow tract and right ventricle formation, but not abundantly to the left ventricle (Waldo et al., 2001; Mjaatvedt CH et al., 2001; Kelly et al., 2001; Dodou et al., 2004; Cai et al., 2003, Xu et al., 2004).
Clinical implications of Tbx1 regulation
Despite intensive efforts to find TBX1 mutations in patients with the 22q11DS phenotype who have no chromosomal deletion, only two missense mutations and a frameshift mutation have been identified (Yagi et al., 2003). Although the frameshift mutation deleted the C-terminus of TBX1 which contains a nuclear localization signal conserved across species (Stoller et al., 2005), the functional significance of other two mutations remains unknown. It is possible that some patients with 22q11DS phenotype but no chromosomal deletion may have mutations in the cis-regulatory region of Tbx1. Since the phenotype of 22q11DS is highly variable in spite of the relatively uniform chromosomal deletion, allelic polymorphism in these regions or genes encoding trans-regulatory factors may be associated with the phenotypic variability of 22q11DS. For example, mutations of FOXC2, an upstream regulator of TBX1, cause lymphedema-distichiasis syndrome (OMIM#153400), which is occasionally associated with congenital heart defects such as tetralogy of Fallot, ventricular septal defect or patent ductus arteriosus (Brice et al., 2002). Further mutation analyses of the cis- and trans-regulators of TBX1 in patients with cardiac outflow tract defects should ultimately reveal their significance in human disease.
The precise molecular mechanisms for normal outflow tract development remain uncertain, although it is clear that disruption of the process results in a variety of outflow tract defects ranging from tetralogy of Fallot to persistent truncus arteriosus. The anatomical defects in tetralogy of Fallot are believed to result from incomplete rotation of the outflow tract during septation. Malrotation of the outflow tract results in misalignment of the outlet and trabecular septum and consequent overriding of the aorta above the malaligned ventricular septum. Contribution of neural crest cells is believed to be essential for proper rotation and septation of the outflow tract. Alternatively, hypoplasia and underdevelopment of the pulmonary infundibulum may also be responsible for the infundibular obstruction and malalignment of the outlet septum (Siwik et al., 2001). Our data suggest that Tbx1-expressing descendents representing a subset of cells derived from the SHF contribute predominantly to the pulmonary infundibulum. Developmental defects of this subset of cardiac progenitor cells may cause hypoplasia of the pulmonary infundibulum, resulting in tetralogy of Fallot. If severe hypoplasia or absence of this subset of cells occurs, the main pulmonary trunk might be missing and the pulmonary arteries would originate from the resultant single common vessel arising from the heart. This anatomy is reminiscent of persistent truncus arteriosus and supports the observation that tetralogy of Fallot and persistent truncus arteriosus occur in 22q11DS. Further study utilizing our transgenic system may provide new insights into the pulmonary infundibulum’s contribution to outflow tract development and pathogenesis of outflow tract defects.
Acknowledgement
We thank K. Kaestner for the Foxa2 plasmid; T. Kume for Foxc1/2 plasmids and sharing unpublished observation; J. Wrana for the Foxh1 plasmid. This work was supported by grants from the NHLBI/NIH, March of Dimes Birth Defects Foundation, and American Heart Association to D.S. H.Y. was supported by grants from Pfizer Funds for Development and Growth, and grants from the Japanese Ministry of Education and Science.
References
- Brice G, Mansour S, Bell R, Collin JR, Child AH, Brady AF, Sarfarazi M, Burnand KG, Jeffery S, Mortimer P, Murday VA. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet. 2002;39:478–438. doi: 10.1136/jmg.39.7.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CB, Wenning JM, LU MM, Epstein DJ, Meyers EN, Epstein JA. Cre-mediated excision of Fgf8 in the Tbx1 expression domain reveals a critical role for Fgf8 in cardiovascular development in the mouse. Dev Biol. 2003;267:190–202. doi: 10.1016/j.ydbio.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Cai C-L, Liang X, Shi Y, Chu P-H, Pfaff SL, Chen J, Evans S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev Cell. 2003;5:877–889. doi: 10.1016/s1534-5807(03)00363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Zaret KS. An early developmental transcription factor complex that is more stable on nucleosome core particles than on free DNA. Mol Cell. 1999;4:961–969. doi: 10.1016/s1097-2765(00)80225-7. [DOI] [PubMed] [Google Scholar]
- Dodou E, Verzi MP, Anderson JP, Xu S-M, Black BL. Mef2c is a direct transcriptional target of ISL1 and GATA factors in the anterior heart field during mouse embryonic development. Development. 2004;131:3931–3942. doi: 10.1242/dev.01256. [DOI] [PubMed] [Google Scholar]
- Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- Hu T, Yamagishi H, Maeda J, McAnally J, Yamagishi C, Srivastava D. Tbx1 regulates fibroblast growth factors in the anterior heart field through a reinforcing autoregulatory loop involving forkhead transcription factors. Development. 2004;131:5491–5502. doi: 10.1242/dev.01399. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nature Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Kelly RG, Brown NA, Buckingham ME. The arterial pole of the mouse heart forms from Fgf10-expressing cells in pharyngeal mesoderm. Dev Cell. 2001;1:435–440. doi: 10.1016/s1534-5807(01)00040-5. [DOI] [PubMed] [Google Scholar]
- Kume T, Deng KY, Winfrey V, Gould DB, Walter MA, Hogan BL. The forkhead/winged helix gene Mf1 is disrupted in the pleiotropic mouse mutation congenital hydrocephalus. Cell. 1998;93:985–996. doi: 10.1016/s0092-8674(00)81204-0. [DOI] [PubMed] [Google Scholar]
- Kume T, Jiang H, Topczewska JM, Hogan BL. The murine winged helix transcription factors, Foxc1 and Foxc2, are both required for cardiovascular development and somitogenesis. Genes Dev. 2001;15:2470–2482. doi: 10.1101/gad.907301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Shioi T, Kasahara H, Jobe SM, Wiese RJ, Markham BE, Izumo S. The cardiac tissue-restricted homeobox protein Csx/Nkx2.5 physically associates with the zinc finger protein GATA4 and cooperatively activates atrial natriuretiv factor gene expression. Mol Cell Biol. 1998;18:3120–3129. doi: 10.1128/mcb.18.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, von Both I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodeling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Vitelli F, Su Hong, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, Bradley A, Baldini A. Tbx1 haploinsufficiency in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, Tokooya K, St. Jore B, Lopez M, Pandita RK, Lia M, Carrion D, Xu H, Schorie H, Kobler JB, Scambler P, Wynshaw-Boris A, Skoultchi AI, Morrow BE, Kucheriapati R. TBX1 is responsible for cardiovascular defects in vero-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Mjaatvedt CH, Nakaoka T, Moreno-Rodriguez R, Norris RA, Ken MJ, Eisenberg CA, Turner D, Markwald RR. The outflow tract of the heart is recruited from a novel heart-forming field. Dev Biol. 2001;238:97–109. doi: 10.1006/dbio.2001.0409. [DOI] [PubMed] [Google Scholar]
- Poelmann RE, Mikawa T, Gittenberger-de Groot AC. Neural crest cells in outflow tract septation of the embryonic chicken heart: Differentiation and apoptosis. Dev Dyn. 1998;212:373–384. doi: 10.1002/(SICI)1097-0177(199807)212:3<373::AID-AJA5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: New fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott V, Clark AR, Docherty K. The gel retardation assay. In: Harwood AJ, editor. Methods in molecular biology. Vol. 31: Protocolas for gene analysis. Totowa N: Human Press; 1994. pp. 339–347. [DOI] [PubMed] [Google Scholar]
- Siwik ES, Patel CR, Zahka KG. Tetralogy of Fallot. In: Allen HD, Gutgesell HP, Clark EB, Driscoll DJ, editors. Moss and Adams’ Heart Disease in Infants, Children, and Adolescents including the Fetus and Young Adult. Philadelphia: Lippincott: Williams & Wilkins; 2001. pp. 880–902. [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srivastava D, Olson EN. A genetic blueprint of heart development. Nature. 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- Stoller JZ, Epstein JA. Identification of a novel nuclear localization signal in Tbx1 that is deleted in DiGeorge syndrome patients harboring the 1223delC mutation. Hum Mol Genet. 2005;14:885–892. doi: 10.1093/hmg/ddi081. [DOI] [PubMed] [Google Scholar]
- Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae S-C, Komori T, Ito Y, Littman DR. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- Verzi MP, McCulley DJ, De Val S, Dodou E, Black BL. The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev Biol. 2005 doi: 10.1016/j.ydbio.2005.08.041. (in press) [DOI] [PubMed] [Google Scholar]
- von Both I, Silvestri C, Erdemir T, Lickert H, Walls JR, Henkelman RM, Rossant J, Harvey RP, Attisano L, Wrana JL. Foxh1 is essential for development of the anterior heart field. Dev Cell. 2004;7:331–345. doi: 10.1016/j.devcel.2004.07.023. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Kumiski DH, Wallis KT, Stadt HA, Hutson MR, Platt DH, Kirby ML. Conotruncal myocardium arises from a secondary heart field. Development. 2001;128:3179–3188. doi: 10.1242/dev.128.16.3179. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Jafri A, Fisher SA. Apoptosis is required for the proper formation of the ventriculo-arterial connections. Dev Biol. 2001;240:274–288. doi: 10.1006/dbio.2001.0466. [DOI] [PubMed] [Google Scholar]
- Weinstein DC, Ruiz i Altaba A, Chen WS, Hoodless P, Prezioso VR, Jessell TM, Darnel JE., Jr The winged-helix transcription factor HNF-3β is required for notochord development in the mouse embryo. Cell. 1994;78:575–588. doi: 10.1016/0092-8674(94)90523-1. [DOI] [PubMed] [Google Scholar]
- Xu H, Morishima M, Wylie JN, Schwartz RJ, Bruneau BG, Lindsay EA, Baldini A. Tbx1 has a dual role in the morphogenesis of the cardiac outflow tract. Development. 2004;131:3217–3227. doi: 10.1242/dev.01174. [DOI] [PubMed] [Google Scholar]
- Yagi H, Furutani Y, Hamada H, Sasaki T, Azakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, Kamatani N, Momma K, Takao A, Nakazawa M, Shimizu N, Matsuoka R. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. doi: 10.1016/s0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yamagishi H. The 22q11.2 deletion syndrome. Keio J Med. 2002;51:77–88. doi: 10.2302/kjm.51.77. [DOI] [PubMed] [Google Scholar]
- Yamagishi H, Maeda J, Hu T, McAnally J, Conway SJ, Kume T, Meyers EN, Yamagishi C, Srivastava D. Tbx1 is regulated by tissue-specific frokhead proteins through a commom Sonic Hedgehog-responsive enhancer. Genes Dev. 2003;17:269–281. doi: 10.1101/gad.1048903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi H, Srivastava D. Unraveling the genetic and developmental mysteries of 22q11 deletion syndrome. Treneds Mol Med. 2003;9:383–383. doi: 10.1016/s1471-4914(03)00141-2. [DOI] [PubMed] [Google Scholar]