

Abstract
More than 50% of children with severe 46,XY disorders of sex development (DSD) do not have a definitive etiological diagnosis. Besides gonadal dysgenesis, defects in androgen biosynthesis, and abnormalities in androgen sensitivity, the Mastermind-like domain containing 1 (MAMLD1) gene, which was identified as critical for the development of male genitalia, may be implicated. The present study investigated whether MAMLD1 is implicated in cases of severe 46,XY DSD and whether routine sequencing of MAMLD1 should be performed in these patients.
Seventy children with severe non-syndromic 46,XY DSD of unknown etiology were studied. One hundred and fifty healthy individuals were included as controls. Direct sequencing of the MAMLD1, AR, SRD5A2 and NR5A1 genes was performed. The transactivation function of the variant MAMLD1 proteins was quantified by the luciferase method.
Two new mutations were identified: p.S143X (c.428C>A) in a patient with scrotal hypospadias with microphallus and p.P384L (c.1151C>T) in a patient with penile hypospadias with microphallus. The in vitro functional study confirmed no residual transactivating function of the p.S143X mutant and a significantly reduced transactivation function of the p.P384L protein (p = 0.0032). The p.P359S, p.N662S and p.H347Q variants are also reported with particularly high frequency of the p.359T- p.662G haplotype in the DSD patients.
Severe undervirilization in XY newborns can reveal mutations of MAMLD1. MAMLD1 should be routinely sequenced in these patients with otherwise normal AR, SRD5A2 and NR5A1genes.
Introduction
The disorders of sex development (DSD) comprise a variety of anomalies defined by congenital conditions in which chromosomal, gonadal, or anatomical sex is atypical. The prevalence of the 46,XY disorders of sex development (46,XY DSD) is difficult to determine with accuracy because of the heterogeneity in the clinical presentation and the etiologies. The estimated incidence of severe 46,XY DSD with uncertain sex is 2.2 per 10,000 births [1], and for a minor form of 46,XY DSD with isolated and non-severe hypospadias, the incidence is estimated at 1 in 250–400 births [2]. Two independent surveillance systems in the United States, the nationwide Birth Defects Monitoring Program (BDMP) and the Metropolitan Atlanta Congenital Defects Program (MACDP), reported a near doubling in the hypospadias rate in comparison with the immediately preceding decades [3]. Although recent studies have questioned this reported rise and provide conflicting data [4], [5], the elucidation of the pathophysiology of these genital malformations remains challenging.
The etiologies of 46,XY DSD are usually gonadal dysgenesis (defect in SRY and downstream genes such as SOX9, WT1, NR5A1 [6], [7], etc.), defects in androgen biosynthesis and, more frequently, abnormalities in androgen sensitivity. Unfortunately, more than 50% of children with severe 46,XY DSD presenting with uncertain sex do not have a definitive clinical diagnosis [8]. For instance, an AR gene defect is identified in less than 10% of the cases [9].
In addition to these well classified causes, a recent candidate gene was identified as critical for the development of male genitalia: the Mastermind-like domain containing 1 (MAMLD1) gene (formerly CXorf6). This gene was discovered during studies to find the gene responsible for X-linked myotubular myopathy, MTM1, which maps to proximal Xq28 [10]: MAMLD1 was observed to be deleted in patients with both the myopathy and external genital malformations [10], [11], [12]. Polymorphisms of MAMLD1 have been reported in patients with isolated hypospadias, the less severe form of 46,XY DSD, but these variants usually do not affect the transactivation of the protein [13], [14]. Conversely, severe 46,XY DSD with uncertain sex has been sparsely studied. To date, only one study has focused on these patients: Fukami et al. identified three nonsense mutations in four individuals from a group of 166 patients [15]. The aim of the present study was to determine whether MAMLD1 is frequently implicated in newborns and children with severe 46,XY DSD with uncertain sex and whether MAMLD1 should be routinely sequenced in these patients.
Materials and Methods
Patients and controls
Two hundred and twenty individuals were included in this study. Seventy children presented with non-syndromic 46,XY DSD of unknown etiology. According to the Quigley classification [16], 8 patients exhibited a stage 2 phenotype; 32 patients, stage 3; 20 patients, stage 4; 5 patients, stage 5; and 5 patients, stage 6. One hundred and fifty healthy individuals were included as controls. Controls were chosen among patients without urinary, genital, or endocrine disease, or any other congenital malformation. For instance, patients with acute appendicitis or operated on for circumcision without phimosis were included. This study was approved by the Institutional Review Board (CPP-Montpellier, ID RCB No. 2008-A00781-54). Written consent was obtained from the parents, carers or guardians on behalf of the participating minors. When a mutation was identified, other family members were examined if possible. The patients and controls were Caucasian.
DNA extraction
DNA was extracted from peripheral blood using a QIAamp DNA blood minikit (Qiagen, Courtaboeuf, France).
Mutational analysis of MAMLD1
Direct sequencing of MAMLD1 coding exons and their flanking splice sites was performed in all patients and controls using primers as previously described [17]. The 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) was used. Sequencing reactions were repeated twice with at least two different PCR products. The DNA sequences were compared with the sequences of normal controls and the reference genomes from the ensembl.org database (Ensembl: ENSG00000013619) and the genebank database (MIM: 300120, NCBI Gene ID: 10046). It is notable that the number of the cDNA and amino acids has been changed recently because of the recognition of a novel MAMLD1 start codon. This report describes MAMLD1 cDNA and amino acids according to the new system.
Molecular analysis of androgen sensitivity
A molecular analysis of the androgen receptor (AR) and 5 alpha reductase type 2 (SRD5A2) genes was performed in all patients. Exons 1–8 of the AR gene were amplified by PCR using sets of primers and reactions previously described [18]. Molecular analysis of the SRD5A2 gene (exons 1–5) was performed as previously reported [19]. PCRs were verified for correct length on agarose gel, purified with Qiaquick PCR columns (Qiagen), and sequenced with the ABI Prism Big Dye terminator sequencing kit. NR5A1was sequenced in 46,XY DSD children with low plasma testosterone as previously published [6], [20].
Homology study
Ensembl.org detected the putative homologs of the human MAMLD1 gene and alignments were made with the ClustalW software at http://www.ebi.ac.uk/Tools/msa/clustalw2/.
Structure prediction
The potential impact of variants was first predicted using X in-silico tools for secondary structure, tertiary structure and prediction of the consequences of amino acid changes.
The secondary structure for wildtype and variants was predicted using JPred software [21] (http://www.compbio.dundee.ac.uk/www-jpred/). The relative accessibility of amino acids was studied with Netsurf software [22] (http://www.cbs.dtu.dk/services/NetSurfP/). The three-dimensional structure was predicted by the Protein Homology/analogY Recognition Engine (PhyreEngine) from the Structural Bioinformatics Group, Imperial College, London, at http:www.sbg.bio.ic.ac.uk/phyrew/. This tool can detect remote homologous proteins with similar tertiary structures, based on multiple sequence profiles with structure-based profiles [23].
The functional consequences of amino acid changes were predicted using four algorithms. Polyphen (Harvard, USA) [24], [25], Panther [26], Sift (University of British Columbia) [27] and SNP-3D (University of Maryland) [28] were used, respectively, at http://genetics.bwh.harvard.edu/pph/, http://www.pantherdb.org/tools/csnpScoreForm.jsp., http://sift.jcvi.org/, and http://www.snps3d.org/modules.php?name=Search&op=advanced%20search. These algorithms are based on the alignment of orthologous and/or paralogous protein sequences and/or structural constraints.
Transactivation analysis of MAMLD1
The transactivation function of the variant MAMLD1 proteins was analyzed by the luciferase method [29]. We used the previously reported luciferase reporter vector containing the promoter sequence of mouse hairy/enhancer of split 3 (Hes3) (–2,715∼+261 bp) [30] and expression vectors containing cDNAs for wildtype MAMLD1, p.S143X and p.P384L [29]. Mouse Leydig tumor (MLTC1) cells (ATCC, CRL-2065) seeded in 12-well dishes (0.5–1.0×105 cells/well) were transiently transfected using Lipofectamine 2000 (Invitrogen) with 0.6 µg of luciferase reporter vector and 0.6 µg of expression vector for wildtype or variant MAMLD1, together with 20 ng of pRL-CMV vector (Promega) used as an internal control. As a control for the expression vectors, an empty counterpart vector was transfected. Luciferase assays performed with a Lumat LB9507 (Berthold) 48 hours after transfection were repeated three times.
Statistical methods
Haplotype frequencies were compared between cases and controls using the χ2 test and the Fisher test on SPSS 16.0 software. The odds ratio (OR) was also considered with the logit confidence intervals method: 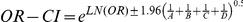 . Hapmap and ensembl.org were used to exclude linkage disequilibrium. Regarding the transactivation analysis of MAMLD1, the results are expressed using the mean and SD, and statistical significance was determined by the t-test.
. Hapmap and ensembl.org were used to exclude linkage disequilibrium. Regarding the transactivation analysis of MAMLD1, the results are expressed using the mean and SD, and statistical significance was determined by the t-test.
Results
Mutations of MAMLD1 and functional analyses
Among the 70 newborns and children with 46,XY DSD, two new mutations were identified in two unrelated patients: p.S143X (c.428C>A) and p.P384L (c.1151C>T) (Fig. 1). The clinical and genetic data are summarized in Table 1. None of these mutations was noted in the control group. The sequences of the AR, SRD5A2 and NR5A1 genes were normal in these patients.
Figure 1. Electrochromatograms and pedigrees of the three patients with MAMLD1 mutations.

The black squares indicate patients with posterior hypospadias. All mutant sequences were controlled by wildtype (WT) DNA. Regarding case 1's family, only the members III-3 and II-4 were genotyped, as the other members in the pedigree declined genetic testing.
Table 1. Clinical and hormonal data of patients with mutated MAMLD1.
| Patient | Case 1 | Case 2 |
| MAMLD1 mutation | pS143X | pP384L |
| Previous medical history | None | Maternal diabetes |
| Genital phenotype | ||
| Urethral meatus | Scrotal | Penile posterior |
| Age at exam (yr,mo) | 0,0 | 0,0 |
| Microphallus | Yes, 20 mm | Yes, 20 mm with cuvature |
| Testis position | Intra-scrotal | Intra-scrotal |
| Testis size (normal = 1–2 ml) | Normal | Normal |
| Scrotal appearance | Ventral transposition, Bifid Scrotum | Bifid Scrotum |
| Renal and urinary tract structure | Normal | Normal |
| Extragenital phenotype | Normal | Normal |
| Growth | ||
| Birth height, cm (SDS) | 51 (+0) | 50.5 (+0) |
| Birth weight, Kg (SDS) | 3.540 (+0) | 3.750 (+0.5) |
| Serum hormone level | ||
| Time of measurment (yr,mo) | 0,0 | 0,3 |
| Testosterone (ng/ml) (1–3 ng/ml) | 1.78 | <0.07 |
| LH (UI/l) (1–12 UI/l) | 10 | 0.3 |
| FSH (UI/l) (1–10 UI/l) | 0.8 | 0.8 |
| AMH | 336 ng/ml | 19 ng/ml* |
| Inhibin | NA | <15 ng/ml* |
SD: standard deviation. ND: not determined. NA: not available. DHT: dihydrotestosterone. DHEA: dihydroepiandrsosterone. Parentheses indicate the standard deviation for height and weight and the normal range for hormone serum levels. Testes of 1–2 ml can be regarded as normal, as recently reported by Shibata et al. [34].
It is notable that anti-mullerian hormone and inhibin were lowered in one case. MAMLD1 is indeed reported to be expressed in Sertoli cells, as well [15].
a- The p.S143X mutation was predicted to cause a short and truncated protein. The in silico prediction showed profoundly modified amino acid accessibility and 3D structure. Relative surface accessibility and absolute surface accessibility of the last amino acid changed from 0.248 to 0.834 and from 29.124 to 97.721, respectively. PhyreEngine predicted the loss of any functional site without a residual consensus sequence (no homologous sequence over 5% through whole genome) (Fig. 2). The in vitro functional study confirmed no residual transactivating function of the mutant (Fig. 3). Interestingly, a maternal uncle and a maternal cousin of the index case both exhibited severe hypospadias (not available for genetic testing). The mother was indeed heterozygous for the mutation (Fig. 1).
Figure 2. Tertiary structure prediction of the wildtype protein (left column) and with the mutants.

3D structure was predicted at Protein Homology/analogY Recognition Engine (PhyreEngine) from the Structural Bioinformatics Group, Imperial College, London, at http:www.sbg.bio.ic.ac.uk/phyre~/. The plain arrows show the changes in the shape of the protein between the wildtype and p.P384L.
Figure 3. Transactivation function of the variants of the MAMLD1 protein analyzed by the luciferase method.
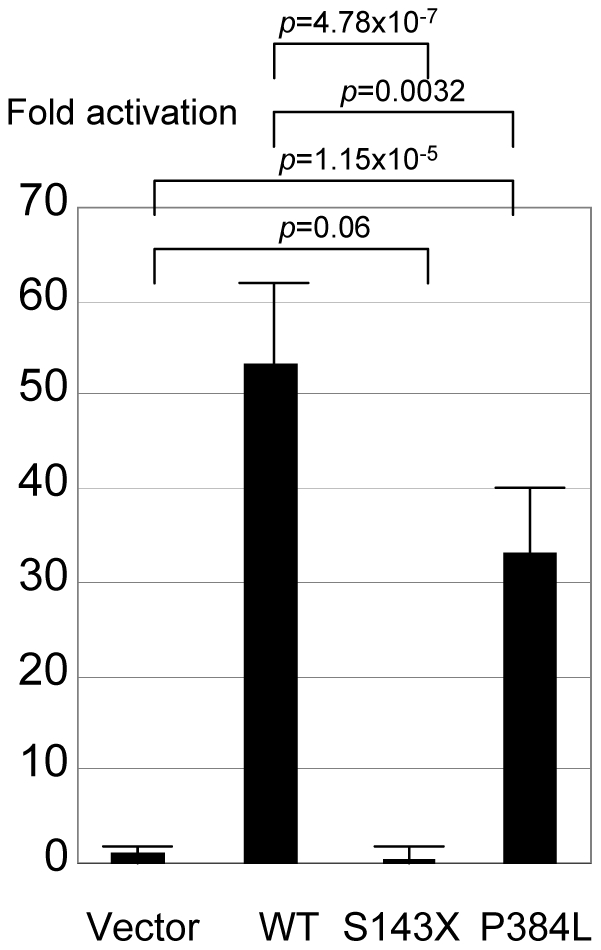
The activity is evaluated for pHes3-luc vector.
b- The p.P384L mutation was found in a patient with posterior penile hypospadias and microphallus. No cryptrochidism was noted. The secondary structure was predicted to be changed in the next four amino acids. The relative and absolute accessibilities of the amino acid were modified from 0.27 to 0.35 and from 39.07 to 65.25, respectively. The 3D structure prediction of the mutated protein was significantly changed (Fig. 2). All four in silico algorithms predicted affected protein function (Table 2) with a conserved amino acid throughout species (Table 3). Functional studies confirmed the significantly reduced transactivation function of the p.P384L protein with 60% residual activity when compared with the wildtype protein, p = 0.0032 (Fig. 3).
Table 2. Prediction of affected protein function using four algorithms.
| Algorithm | pP384L |
| Polyphen | Probably damaging |
| score = 0.961 (sensitivity: 0.71; specificity: 0.93) | |
| Sift | Affect protein function |
| Sift score = 0.04 | |
| Panther | Probability of deleterious effect = 0.42 |
| (subPSEC score = −2.7) | |
| SNPS3D | Deleterious |
| (svm score = −1.75) |
References and online access are indicated in the text. Mathematical calculation of the significance of each score is available online.
Table 3. Homology study showed that this amino acid was highly conserved through species for the c.1041C>A and c.1151C>T mutations.
| Patient | MSSNTLSGSTLRGSLNALLSSMTSSSNAAL |
| Human-MAMLD1 | MSSNTLSGSTLRGSPNALLSSMTSSSNAAL |
| Pig | MSSSSLPGSTLHGSPGALLSSGAPSSSSAL |
| Horse | MSSSNLPGSTLQGSPNALLSSMVSGSSAAL |
| Chimpanzee | MSSNTLSGSTLRGSPNALLSSMTSSSNAAL |
| Mouse | MSSSSLSGSAVQSSPNALLSSMAPSSNASL |
| Rabbit | MAPHSLPGSSLQGSPNALLSSMAPNSSGAL |
| Dog | MASSNLPGSSFQASPNALLASMASASSAGL |
| Cat | MASGNLPGSAFQGSPNALLASMASGSSAAL |
Polymorphisms of MAMLD1
We identified three polymorphisms of MAMLD1 in our series: p.P359S (c.1075C>T, rs41313406), p.N662S (c.1985A>G, rs2073043) and p.H347Q (c.1041C>A, rs62641609). Regarding the p.P359S and p.N662S polymorphisms, 14 patients exhibited double polymorphisms (S-S haplotype) and five had the p.N359S polymorphism. The phenotypes of the patients with the S-S haplotype were as follows: penile posterior hypospadias and cryptorchidism in three cases, hypospadias and microphallus in five cases (anterior n = 1, penile posterior n = 2 and scrotal hypospadias n = 2), and cryptorchidism and microphallus in six cases (bilateral cryptorchidism n = 5, unilateral cryptorchidism n = 1). Using hapmap and ensembl.org, no linkage disequilibrium was found for these two variants. In previous studies, we and others found that the S-S haplotype was present in only 6/150 controls (4.0%) and 23/360 controls (6.4%) [13], [14]. By combining the published series for controls (matched patients and controls), we determined that the incidence of the S-S haplotype was higher in the DSD patients (20%, n = 70 vs. 6%, n = 510, p = 0.0003) (OR = 3.86, CI from 1.94 to 7.70, p = 0.05). Haplotypes and their relative frequencies in each group of patients are summarized in Table 4.
Table 4. Incidence of exonic polymorphisms p.P359S and p.N662S, and relative haplotypes in normal controls and 46,XY DSD patients.
| Haplotype 359–662 | Patients, n = 70 | Controls, n = 510 | Fisher, p value | OR | OR confidence interval (p = 0.05) |
| p.359C- p.662A | 72.9% (n = 51) | 90.6% (n = 462) | p = 0.0001 | 0.28 | 0.15–0.51 |
| p.359T- p.662A | 0% | 1.5% (n = 8) | p = 0.60 | 0.42 | 0.02–7.35 |
| p.359C- p.662G | 7.1% (n = 5) | 0.8% (n = 9) | p = 0.02 | 4.28 | 1.39–13.17 |
| p.359T- p.662G (S-S polymorphism) | 20% (n = 14) | 6% (n = 31) | p = 0.0003 | 3.86 | 1.94–7.70 |
Controls are combined with the published series (matched for ethnicity of patients and controls) [13] [14]. The χ-square test was performed. When combining all patients with the p.662G polymorphism whatever the p.359 allele, this p.662G was significantly more frequent in 46,XY DSD patients: 27.1% (n = 19) vs. 6.8% (n = 40), p = 0.0001.
The p.H347Q variant, previously reported as a polymorphism especially in sub-Saharan populations (rs62641609, http://www.ensembl.org/Homo_sapiens/Variation/Summary?r=X:149638386-149639386v=rs62641609vdb=variationvf=16740729), was identified in a patient with posterior hypospadias and microphallus (25 mm length at birth).
Discussion
MAMLD1 is a good candidate to explore in patients with unexplained 46,XY DSD, as it has been shown to be expressed in fetal Leydig cells around the critical period for sex development [15]. The transient knockdown of MAMLD1 mRNA expression results in significantly reduced testosterone production in mouse Leydig tumor cells [29]. MAMLD1 is further coexpressed with steroidogenic factor (NR5A1), which regulates the transcription of genes involved in sex development, and an NR5A1target site was found within the MAMLD1 gene [29], [31]. MAMLD1 thus seems to have an important role in modulating testosterone production during sex development and is involved in the 46,XY disorders of sex development [32].
Regarding the minor forms of 46,XY DSD with isolated and non-severe hypospadias, mutational studies of MAMLD1 have identified several polymorphisms in this gene. We reported the following variants in patients with isolated hypospadias: p.P359S, p.V505A, p.N662S and p.604ins3Q [13], [17], all of which were recently confirmed as polymorphisms [14]. The p.Q602K mutation was also found in one patient with posterior hypospadias and was predicted to affect the splicing process. An association between isolated hypospadias and the rare haplotype p.P359S-p.N662S is also suspected [13], [14].
Regarding severe 46,XY DSD with uncertain sex, only one published paper to date has reported three MAMLD1 mutations (p.E124X, p.Q197X and p.R653X) [15]. It is precisely in this situation of severe genital malformation that the diagnosis of the causative mechanism is of clinical interest for medical treatment (hormone substitution, pubertal follow-up). In order to determine whether this report was an exceptional observation or of practical clinical interest, we screened 70 patients with severe 46,XY DSD of unknown origin. We identified two new mutations of MAMLD1 in patients with severe hypospadias and microphallus (1 stop codon and 1 missense mutation). These mutations were associated with a severe phenotype, and reduced (p.P384L) or abolished (p.S143X) transactivation function was found in two cases. 46,XY DSD with normal AR, SRD5A2 and NR5A1gene sequences can thus reveal a mutation of MAMLD1. This finding suggests a new diagnostic investigation for these patients and may be helpful in genetic counselling if a mutation is identified. It also provides new insight into the pathophysiology of DSD. Indeed, in the family of the child bearing the p.S143X mutation, the mother was heterozygous and two other males on the maternal side of the family exhibited a consistent phenotype. Unfortunately, the family declined any further investigation.
The mechanisms by which these mutations with reduced transactivation induce DSD are still under investigation. As noted above, several studies have provided strong evidence of MAMLD1 implication in fetal sex development through modulation of testosterone production at the time of sex differentiation. The plasma testosterone measured in one of our cases was indeed lowered but it was normal in the other one, as previously reported in patients with nonsense mutations [15]. Plasma testosterone evaluation is thus not systematically helpful in orienting the diagnosis of DSD since mutations of the genes implicated in testosterone production - such as MAMLD1 and NR5A1 - have been reported in 46,XY DSD patients with normal plasma testosterone. These findings, along with the absence of correlation between the in vitro functional analysis and the biological and clinical phenotype, suggest that the genital malformation is primarily related to a transient prenatal testicular (Leydig cell) dysfunction and the resulting compromised testosterone production around the critical period of sex differentiation [33]. In the postnatal period, the mouse homolog of MAMLD1 was indeed reported to be weakly expressed in the testis at one week of age and the expression was faint thereafter.
We also report a high incidence of the rare haplotype p.P359S-p.N662S in our series. The p.P359S (which was designated p.P286S in the previous report) variant was first reported in a patient with hypospadias but it was absent in his brother and nephew with the same phenotype [15]. The p.N662S (which was designated p.P589S in the previous report) variant was found in hypospadiac patients but was also reported in a normal population, although with low incidence [15]. We and others have found that the S-S haplotype is associated with a minor form of DSD, i.e., isolated hypospadias [14], but the in vitro functional study of the p.P359S-p.N662S MAMLD1 variant was inconclusive with unchanged transactivation function [13]. In the present study, we show that the combination of these alleles was present in as much as 15% of patients with severe 46,XY DSD. This is significantly higher than in the controls [combining the series, 15% (n = 70) vs. 10.7% (n = 510), p = 0.0003]. Again, a transient testosterone production failure during prenatal development may have contributed to the undervirilization of the external genitalia, but how this haplotype can be present in normal, mild and severe phenotypes remains to be elucidated.
Severe undervirilization in XY newborns can reveal mutations of MAMLD1. MAMLD1 should be routinely sequenced in these patients with otherwise normal AR, SRD5A2 and NR5A1 genes.
Acknowledgments
We would like to thank Dr Béroud (Laboratoire de Génétique Chromosomique, Institut Universitaire de Recherche Clinique, Université de Montpellier 1, France) for his great help in the statistical study of haplotypes.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This study was funded by a Programme Hospitalier de Recherch Clinique Inter-Régional (PHRC number UF 8270) provided by CHU de Montpellier and by a grant from Fondation pour la Recherche Médicale FRM110309. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Thyen U, Lanz K, Holterhus PM, Hiort O. Epidemiology and initial management of ambiguous genitalia at birth in Germany. Horm Res. 2006;66:195–203. doi: 10.1159/000094782. [DOI] [PubMed] [Google Scholar]
- 2.Nelson P. Epidemiology of Hypospadias. Dialogues in Pediatric Urology. 2007;28:2–3. [Google Scholar]
- 3.Paulozzi LJ, Erickson JD, Jackson RJ. Hypospadias trends in two US surveillance systems. Pediatrics. 1997;100:831–834. doi: 10.1542/peds.100.5.831. [DOI] [PubMed] [Google Scholar]
- 4.Martinez-Frias ML, Prieto D, Prieto L, Bermejo E, Rodriguez-Pinilla E, et al. Secular decreasing trend of the frequency of hypospadias among newborn male infants in Spain. Birth Defects Res A Clin Mol Teratol. 2004;70:75–81. doi: 10.1002/bdra.10149. [DOI] [PubMed] [Google Scholar]
- 5.Fisch H, Hyun G, Hensle TW. Rising hypospadias rates: disproving a myth. J Pediatr Urol. 2010;6:37–39. doi: 10.1016/j.jpurol.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 6.Lin L, Philibert P, Ferraz-de-Souza B, Kelberman D, Homfray T, et al. Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab. 2007;92:991–999. doi: 10.1210/jc.2006-1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coutant R, Mallet D, Lahlou N, Bouhours-Nouet N, Guichet A, et al. Heterozygous mutation of steroidogenic factor-1 in 46,XY subjects may mimic partial androgen insensitivity syndrome. J Clin Endocrinol Metab. 2007;92:2868–2873. doi: 10.1210/jc.2007-0024. [DOI] [PubMed] [Google Scholar]
- 8.Morel Y, Rey R, Teinturier C, Nicolino M, Michel-Calemard L, et al. Aetiological diagnosis of male sex ambiguity: a collaborative study. Eur J Pediatr. 2002;161:49–59. doi: 10.1007/s00431-001-0854-z. [DOI] [PubMed] [Google Scholar]
- 9.Choi J, Kim G, Seo E, KS K, Kim S, et al. Molecular analysis of the AR and SRD5A2 genes in patients with 46,XY disorders of sex development. J Pediatr Endocrinol Metab. 2008;21:545–553. [PubMed] [Google Scholar]
- 10.Laporte J, Kioschis P, Hu LJ, Kretz C, Carlsson B, et al. Cloning and characterization of an alternatively spliced gene in proximal Xq28 deleted in two patients with intersexual genitalia and myotubular myopathy. Genomics. 1997;41:458–462. doi: 10.1006/geno.1997.4662. [DOI] [PubMed] [Google Scholar]
- 11.Bartsch O, Kress W, Wagner A, Seemanova E. The novel contiguous gene syndrome of myotubular myopathy (MTM1), male hypogenitalism and deletion in Xq28:report of the first familial case. Cytogenet Cell Genet. 1999;85:310–314. doi: 10.1159/000015284. [DOI] [PubMed] [Google Scholar]
- 12.Hu LJ, Laporte J, Kress W, Kioschis P, Siebenhaar R, et al. Deletions in Xq28 in two boys with myotubular myopathy and abnormal genital development define a new contiguous gene syndrome in a 430 kb region. Hum Mol Genet. 1996;5:139–143. doi: 10.1093/hmg/5.1.139. [DOI] [PubMed] [Google Scholar]
- 13.Kalfa N, Cassorla F, Audran F, Oulad Abdennabi I, Philibert P, et al. Polymorphisms of MAMLD1 gene in hypospadias. J Pediatr Urol. 2011;7:585–591. doi: 10.1016/j.jpurol.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 14.Chen Y, Thai HT, Lundin J, Lagerstedt-Robinson K, Zhao S, et al. Mutational study of the MAMLD1-gene in hypospadias. Eur J Med Genet. 2010;53:122–126. doi: 10.1016/j.ejmg.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Fukami M, Wada Y, Miyabayashi K, Nishino I, Hasegawa T, et al. CXorf6 is a causative gene for hypospadias. Nat Genet. 2006;38:1369–1371. doi: 10.1038/ng1900. [DOI] [PubMed] [Google Scholar]
- 16.Quigley CA, French FS. Androgen insensitivity syndromes. Curr Ther Endocrinol Metab. 1994;5:342–351. [PubMed] [Google Scholar]
- 17.Kalfa N, Liu B, Ophir K, Audran F, Wang MH, et al. Mutations of CXorf6 are associated with a range of severities of hypospadias. Eur J Endocrinol. 2008;159:453–458. doi: 10.1530/EJE-08-0085. [DOI] [PubMed] [Google Scholar]
- 18.Philibert P, Audran F, Pienkowski C, Morange I, Kohler B, et al. Complete androgen insensitivity syndrome is frequently due to premature stop codons in exon 1 of the androgen receptor gene: an international collaborative report of 13 new mutations. Fertil Steril. 2010;94:472–476. doi: 10.1016/j.fertnstert.2009.03.057. [DOI] [PubMed] [Google Scholar]
- 19.Maimoun L, Philibert P, Cammas B, Audran F, Bouchard P, et al. Phenotypical, Biological, and Molecular Heterogeneity of 5{alpha}-Reductase Deficiency: An Extensive International Experience of 55 Patients. J Clin Endocrinol Metab. 2010;96:296–307. doi: 10.1210/jc.2010-1024. [DOI] [PubMed] [Google Scholar]
- 20.Philibert P, Leprieur E, Zenaty D, Thibaud E, Polak M, et al. Steroidogenic factor-1 (SF-1) gene mutation as a frequent cause of primary amenorrhea in 46,XY female adolescents with low testosterone concentration. Reprod Biol Endocrinol. 2010;8:28. doi: 10.1186/1477-7827-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct Biol. 2009;9:51. doi: 10.1186/1472-6807-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 24.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomas PD, Kejariwal A. Coding single-nucleotide polymorphisms associated with complex vs. Mendelian disease: evolutionary evidence for differences in molecular effects. Proc Natl Acad Sci U S A. 2004;101:15398–15403. doi: 10.1073/pnas.0404380101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, et al. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38:D204–210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 28.Yue P, Melamud E, Moult J. SNPs3D: candidate gene and SNP selection for association studies. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukami M, Wada Y, Okada M, Kato F, Katsumata N, et al. Mastermind-like domain containing 1 (MAMLD1 or CXorf6) transactivates the Hes3 promoter, augments testosterone production, and contains the SF1 target sequence. J Biol Chem. 2008;29:5525–5532. doi: 10.1074/jbc.M703289200. [DOI] [PubMed] [Google Scholar]
- 30.Nishimura M, Isaka F, Ishibashi M, Tomita K, Tsuda H, et al. Structure, chromosomal locus, and promoter of mouse Hes2 gene, a homologue of Drosophila hairy and Enhancer of split. Genomics. 1998;49:69–75. doi: 10.1006/geno.1998.5213. [DOI] [PubMed] [Google Scholar]
- 31.Sadovsky Y, Dorn C. Function of steroidogenic factor 1 during development and differentiation of the reproductive system. Rev Reprod. 2000;5:136–142. doi: 10.1530/ror.0.0050136. [DOI] [PubMed] [Google Scholar]
- 32.Ogata T, Laporte J, Fukami M. MAMLD1 (CXorf6): a new gene involved in hypospadias. Horm Res. 2009;71:245–252. doi: 10.1159/000208797. [DOI] [PubMed] [Google Scholar]
- 33.Welsh M, MacLeod DJ, Walker M, Smith LB, Sharpe RM. Critical androgen-sensitive periods of rat penis and clitoris development. Int J Androl. 2010;33:e144–152. doi: 10.1111/j.1365-2605.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shibata Y, Kojima Y, Mizuno K, Nakane A, Kato T, et al. Optimal cutoff value of contralateral testicular size for prediction of absent testis in Japanese boys with nonpalpable testis. Urology. 76:78–81. doi: 10.1016/j.urology.2010.02.043. [DOI] [PubMed] [Google Scholar]