

Abstract
Mitochondria can depolarize and trigger cell death through the opening of the mitochondrial permeability transition pore (MPTP). We recently showed that an increase in the long chain n3 polyunsaturated fatty acids (PUFA) docosahexaenoic acid (DHA; 22:6n3) and depletion of the n6 PUFA arachidonic acid (ARA; 20:4n6) in mitochondrial membranes is associated with a greater Ca2+ load required to induce MPTP opening. Here we manipulated mitochondrial phospholipid composition by supplementing the diet with DHA, ARA or combined DHA+ARA in rats for 10 weeks. There were no effects on cardiac function, or respiration of isolated mitochondria. Analysis of mitochondrial phospholipids showed DHA supplementation increased DHA and displaced ARA in mitochondrial membranes, while supplementation with ARA or DHA+ARA increased ARA and depleted linoleic acid (18:2n6). Phospholipid analysis revealed a similar pattern, particularly in cardiolipin. Tetralinoleoyl cardiolipin was depleted by 80% with ARA or DHA+ARA supplementation, with linoleic acid side chains replaced by ARA. Both the DHA and ARA groups had delayed Ca2+-induced MPTP opening, but the DHA+ARA group was similar to the control diet. In conclusion, alterations in mitochondria membrane phospholipid fatty acid composition caused by dietary DHA or ARA was associated with a greater cumulative Ca2+ load required to induced MPTP opening. Further, high levels of tetralinoleoyl cardiolipin were not essential for normal mitochondrial function if replaced with very-long chain n3 or n6 PUFAs.
Introduction
Cardiac muscle is densely packed with mitochondria, which are essential to support the high rate of ATP generation needed for contractile function. Mitochondria also are important for cell survival, as under conditions of stress they can depolarize and trigger cell death through the opening of the mitochondrial permeability transition pore (MPTP). The structure and regulation of the MPTP is not well understood [1], [2]. We recently found that increased long chain n3 polyunsaturated fatty acid (PUFA) and depletion of n6 PUFA in mitochondrial membrane phospholipids induced by high intake of docosahexaenoic acid (DHA; 20:6n3) were associated with resistance to Ca2+-induced MPTP opening [3]–[5]. Long chain PUFAs, specifically DHA and arachidonic acid (ARA; 20:4n6), are structurally distinguished from less unsaturated fatty acids such as oleic acid (18:1n9) or linoleic acid (18:2n6) by repeating double bonds that produces a highly flexible chain and a more fluid membrane [6]. DHA is the most unsaturated PUFA commonly found in mammals, followed by eicosapentaenoic acid (EPA; 20:5n3) and ARA. DHA supplementation has shown promise as a means to prevent and treat heart failure, which may be partially mediated by improvements in mitochondrial function [7]–[9].
ARA is depleted by the increase in membrane phospholipid DHA content induced by dietary DHA supplementation. This could be beneficial, as ARA is a precursor of inflammatory eicosanoids, and can also trigger MPTP opening when released from cell membranes by phospholipase A [10], [11]. Thus the greater Ca2+ load required to induce MPTP opening with DHA supplementation may occur secondary to lowering ARA in membrane phospholipids. If true, then an increase in ARA in mitochondrial membrane phospholipids above normal levels is predicted to increase MPTP opening.
Like other cardiac membranes, mitochondrial phospholipids are mainly comprised of phosphotidylethanolamine (PE) and phosphotidylcholine (PC), however they are unique in that they contain the tetra-acyl phospholipid cardiolipin (CL). CL comprises 15–20% of the mass of total mitochondrial phospholipid [12]. Depletion of CL, as seen in Barth syndrome patients who have an inherited defect in CL synthesis, results in severe mitochondrial dysfunction and cardiomyopathy [13]. Linoleic acid is the main fatty acyl moiety in CL, with 60–80% of CL being tetralinoleoyl CL (L4CL) in cardiac mitochondria in humans, dogs and rats [12], [14], [15]. It has been proposed that high levels of L4CL are essential for optimal mitochondrial function in the heart [12]. Evidence to the contrary comes from the mouse heart, where L4CL comprises only 22% of the total CL [14], and is replaced by CL species that contain DHA (53% of total CL) [14], [15]. A decrease in the total CL in cardiac mitochondria and less L4CL has been observed in acquired cardiac pathologies such as hypertension-induced hypertrophy and heart failure in rodents [12], but not dogs [16]. Importantly, there is also evidence that high intake of fish oil rich in DHA can increase CL in heart mitochondria [3], [17], [18], though this is not a consistent finding [5]. The effect of ARA intake on CL has not been reported, but presumably would increase ARA incorporation into all mitochondrial phospholipids and could alter mitochondrial function.
In the present investigation we used pharmacological levels of DHA and ARA (2.5% of energy intake [19], [20]), well above those consumed in food by humans, to manipulated cardiac mitochondrial phospholipid composition, and assessed the subsequent effects on respiratory function and susceptibility to MPTP opening in isolated cardiac mitochondria. We hypothesized that replacing linoleic acid with either DHA or ARA in mitochondrial membrane phospholipids would not adversely affect mitochondria respiratory function in the absence of stress, but that ARA would increase susceptibility to Ca2+-induced MPTP opening. We further hypothesized that dietary ARA supplementation would dramatically increase ARA in mitochondrial phospholipids, and specifically decrease L4CL and increase incorporation of ARA side chains of CL. Rats were fed diets supplemented with DHA, ARA or combined DHA+ARA at physiologically relevant doses. Cardiac contractile function was evaluated, and cardiac mitochondria were analyzed for susceptibility to MPTP opening, total phospholipid fatty acid composition, and individual molecular species within each phospholipid class by mass spectrometry.
Results
Morphometric data
All groups had similar weight gain and food consumption, and there were no significant differences in body, heart or liver mass between dietary treatments (Table 1). There was no effect of diet on LV dimensions as measured by echocardiography (Table 1).
Table 1. Body mass, organ mass, and LV dimensions.
| Control | DHA | ARA | DHA+ARA | |
| Group size: | 15 | 14 | 15 | 15 |
| Terminal Body Mass (g) | 501±11 | 500±6 | 502±9 | 505±9 |
| LV Mass (g) | 0.895±0.021 | 0.917±0.017 | 0.902±0.020 | 0.938±0.024 |
| LV/Tibia length (g/cm) | 0.22±0.01 | 0.23±0.00 | 0.22±0.01 | 0. 23±0.01 |
| RV Mass (g) | 0.262±0.011 | 0.267±0.004 | 0.268±0.004 | 0.269±0.008 |
| Biatrial Mass (g) | 0.083±0.003 | 0.091±0.006 | 0.092±0.005 | 0.083±0.003 |
| Liver Mass (g) | 13.6±0.6 | 12.5±0.3 | 13.3±0.4 | 14.1±0.5 |
| Mitochondrial Yield (mg mito protein/g wet wt) | 14.57±0.69 | 14.17±0.64 | 14.42±0.86 | 15.52±1.00 |
| End-systolic diameter (mm) | 0.77±0.01 | 0.76±0.02 | 0.77±0.02 | 0.81±0.02 |
| End-diastolic diameter (mm) | 0.43±0.02 | 0.43±0.01 | 0.43±0.02 | 0.48±0.02 |
| Fractional shortening | 0.45±0.02 | 0.43±0.02 | 0.44±0.02 | 0.41±0.05 |
| Ejection fraction (%) | 83.0±1.4 | 81.3±1.5 | 81.9±1.8 | 79.2±1.4 |
| Malonaldehydes+Hydroxyalkenals (nmols/mg protein) | 0.56±0.04 | 0.58±0.04 | 0.53±0.03 | 0.68±0.04* † |
Data are the mean ± SEM. LV, left ventricle. RV, right ventricle.
p<0.05 vs CTRL.
p<0.05 vs ARA.
Mitochondrial Phospholipid Composition
The fatty acid composition of total mitochondrial phospholipids was dramatically altered by all 3 dietary interventions (Table 2, Figure 1). Both diets containing DHA raised mitochondrial phospholipid DHA content approximately 2-fold, while ARA supplementation led to a 70% reduction in DHA. Conversely, ARA supplementation increased membrane ARA by ∼50% while DHA supplementation decreased ARA by ∼50%. Combined DHA+ARA supplementation maintained ARA content at levels seen in the control diet group. Membrane EPA was increased from undetectable levels to approximately 1% by both the DHA and ARA diets, but was undetectable with the combined DHA+ARA diet. Oleate was only slightly affected by the different diets (Table 2). Most notably, the double bond index, a measure of membrane unsaturation, and the n3/n6 ratio were both dramatically decreased with ARA supplementation and preserved with the DHA+ARA diet.
Table 2. Mitochondrial phospholipid fatty acid composition expressed as molar percent of total phospholipid fatty acid.
| Fatty Acid | CTRL | DHA | ARA | DHA+ARA |
| Palimate (C16:0) | 16.0±0.4 | 17.7±1.0 | 14.8±0.6# | 16.8±0.7 |
| Stearate (C18:0) | 25.4±0.7 | 21.5±2.2 | 27.2±4.1 | 25.7±0.6 |
| Oleate (C18:1n9) | 4.1±0.3 | 3.3±0.3 | 3.6±0. | 2.7±0.1* |
| Vaccenic acid(C18:1n7) | 4.3±0.1 | 3.3±0.3* | 4.1±0.2# | 3.6±0.2 |
| Linoleic acid (C18:2n6) | 20.5±0.6 | 21.7±1.8 | 11.4±0.5# | 11.4±2.1# |
| Arachidonic acid (20:4n6) | 19.1±0.3 | 11.5±2.0* | 32.2±1.3* # | 17.2±1.8 |
| Eicosapentaenoic Acid (20:5n3) | BQL | 1.5±0.3* | 0.9±0.4* | BQL |
| Docosahexanoic acid (22:6n3) | 10.9±0.7 | 20.7±0.7* | 3.1±2.0* # | 21.4±0.9* † |
| Double Bond Index | 191±4.0 | 221±8 | 183±15 | 227±5* † |
| n3/n6 ratio | 0.27±0.01 | 0.73±0.04 | 0.15±0.03# | 0.73±0.04* † |
Data are means±SEM. CTRL, n = 7. DHA, n = 7. ARA, n = 5. DHA+ARA, n = 6.
p<0.05 vs CTRL.
p<0.05 vs DHA,
p<0.05 vs ARA; BQL, Below Quantifiable Limit.
Figure 1. Cardiac mitochondrial phospholipid fatty acid composition.
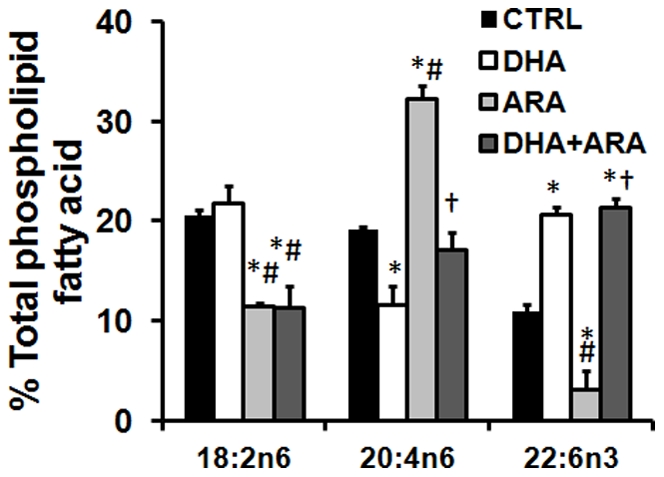
Values are expressed as percentage of total fatty acids. Data are mean±SEM. CTRL, n = 7. DHA, n = 7. ARA, n = 5. DHA+ARA, n = 6.
Phospholipid classes were not changed by diet to any significant extent, except for a decrease in CL and mono-lyso-CL (MLCL) with ARA and DHA+ARA supplementation (Table 3). There was also a small increase in PC with ARA supplementation. In contrast, analysis of side chain composition within each phospholipid class revealed some dramatic diet-induced changes in fatty acyl groups (Table S1, Figure 2, S1,S2,S3,S4,S5). Dietary supplementation with DHA increased DHA in PE, PI and PC, as assessed by mass spectrometry. The increase in DHA was determined by the increase in peak intensity at molecular masses that corresponded to the calculated theoretical mass based on probable side chains (Table S1). Similarly, ARA was increased by the ARA and DHA+ARA diets in PE, PG, PC, CL and MLCL.
Table 3. Composition of cardiac mitochondrial membrane by phospholipid class.
| Phospholipid Class (mol% of total) | CTRL | DHA | ARA | DHA+ARA |
| CL | 8.5±0.2 | 8.5±0.2 | 7.0±0.1* # | 7.2±0.1* # |
| MLCL | 0.5±0.0 | 0.5±0.0 | 1.0±0.1* # | 0.7±0.0* # † |
| PE | 47.7±0.5 | 48.2±0.6 | 46.1±0.5# | 46.9±0.5 |
| PI | 3.9±0.1 | 3.7±0.1 | 3.8±0.1 | 4.1±0.1 |
| PG | 0.6±0.0 | 0.6±0.1 | 0.7±0.1# | 0.6±0.0 |
| PC | 38.7±0.5 | 38.6±0.6 | 41.5±0.5* # | 40.5±0.6 |
Data are means±SEM. CTRL, n = 13. DHA, n = 12. ARA, n = 14. DHA+ARA, n = 14. CL, cardiolipin. MLCL, monolysocardiolipin. PE, phosphotidylethanolamine. PI, phosphotidylinositol. PG, phosphotidylglycine. PC, phosphotidylcholine.
p<0.05 vs CTRL.
p<0.05 vs DHA.
Figure 2. Representative mass spectra of cardiac mitochondrial CL species.

There was clear interaction between dietary DHA and ARA in their effect on phospholipid fatty acid composition, with DHA alone depleting ARA from CL, PE and PC (Figures 2 and 3, Table S1). Conversely, dietary ARA depleted DHA from PE and PC. Interestingly, the combination of DHA and ARA restored both DHA and ARA in PE and PC. Further, it gave a profile that was largely similar to the diet with ARA alone. This is illustrated in the mass spectrum (Figure 2 for CL, and Figures S1,S2,S3,S4,S5 for other phospholipids), which shows that treatment with ARA and DHA+ARA resulted in a profound shift in the profile, with an increase in peak intensities for higher molecular weight species. On the other hand, DHA had far less of an effect on the profile. As noted above, treatment with ARA or DHA+ARA, but not DHA alone, depleted linoleic acid content from total phospholipids by ∼50% (Figure 1), and the analysis of individual phospholipid species revealed that this occurred in CL, MLCL, PE PG and PC (Figure 3, Table S1).
Figure 3. Cardiac mitochondrial phospholipid species composition.

Values are expressed as percentage of total measured. Data are mean±SEM. CTRL, n = 14. DHA, n = 13. ARA, n = 13. DHA+ARA, n = 14. *p<0.05 vs CTRL. #p<0.05 vs DHA. †p<0.05 vs ARA.
Lipid Peroxidation
DHA+ARA supplementation significantly increased myocardial content of malonaldehydes and hydroxyalkenals compared to the control and ARA groups (p<0.05), and showed a strong trend toward being higher than the DHA group (p = 0.071).
Urine Thromboxane B2
TBX B2 is a metabolite of pro-inflammatory thromboxane A2, and is excreted by the kidneys. Both diets containing ARA increased TBX B2 excretion significantly by two- to three-fold compared to CTRL and DHA diets (29.0±2.9 and 22.8±2.6 pg/µmol creatine for CTRL and DHA groups, vs. 63.3±6.7 and 50.11±4.8 for ARA and DHA+ARA groups, respectively; p<0.05)
Mitochondrial Respiration
There was no effect of diet on State 3 or State 4 respiration, or on the respiratory control ratio (RCR) with any of the substrates used (Table 4).
Table 4. Mitochondrial Respiration.
| Control | DHA | ARA | DHA+ARA | |
| Pyruvate+Malate | ||||
| State 3 | 225±17 | 239±22 | 245±21 | 236±19 |
| State 4 (−oligomycin) | 51.6±2.0 | 51.3±3.1 | 47.3±2.6 | 49.7±3.7 |
| State 4 (+oligomycin) | 36.8±4.1 | 44.8±4.3 | 33.7±3.6 | 41.1±4.3 |
| RCR (−oligomycin) | 4.5±0.4 | 4.7±0.4 | 4.8±0.2 | 5.2±0.4 |
| RCR (+oligomycin) | 6.8±0.8 | 5.8±0.6 | 8.1±0.9 | 6.2±0.6 |
| Palmityl-CoA+Carnitine+Malate | ||||
| State 3 | 221±16 | 203±14 | 200±14 | 196±13 |
| State 4 (−oligomycin) | 45.6±2.9 | 53.4±5.0 | 46.0±2.6 | 46.6±2.9 |
| State 4 (+oligomycin) | 40.9±3.8 | 41.2±4.7 | 36.4±2.8 | 42.9±4.3 |
| RCR (−oligomycin) | 5.0±0.3 | 4.1±0.4 | 4.3±0.3 | 4.5±0.4 |
| RCR (+oligomycin) | 5.7±0.4 | 6.4±1.7 | 5.8±0.5 | 5.1±0.6 |
| Succinate+Rotenone | ||||
| State 3 | 392±16 | 398±19 | 360±22 | 393±25 |
| State 4 (−oligomycin) | 122.7±3.6 | 120.1±7.3 | 111.6±4.7 | 108.2±6.9 |
| State 4 (+oligomycin) | 107.8±4.9 | 123.9±7.8 | 100.0±8.3 | 123.6±7.0 |
| RCR (−oligomycin) | 3.2±0.1 | 3.4±0.2 | 3.7±0.2 | 3.2±0.2 |
| RCR (+oligomycin) | 3.7±0.1 | 3.3±0.1 | 3.8±0.3 | 3.2±0.1 |
Data are the mean ± SEM. CTRL, n = 14. DHA, n = 13. ARA, n = 13. DHA+ARA, n = 14. All Rates are expressed in ng atoms O·mg−1·min−1. The RCR, defined as the ratio of State 3 to State 4 respiration rate, was calculated from the State 4 rate with oligomycin.
Ca2+ Retention Capacity
Mitochondria from rats supplemented with DHA or ARA alone had significantly enhanced Ca2+ retention capacity compared to CTRL animals, as reflected by significantly lower extramitochondrial [Ca2+] for a given cumulative Ca2+ load with all substrates except palmitoylcarnitine+malate (Figure 4). Surprising, dietary supplementation with the combination of DHA+ARA resulted in greater sensitivity to Ca2+ MPTP opening, as reflected in a higher extramitochondrial [Ca2+] with all substrates when compared to either CTRL, DHA or ARA with pyruvate+malate as a substrate (Figure 4).
Figure 4. Effect of diet on Ca2+ retention capacity in the presence of different respiratory substrates.

Data are mean±SEM. CTRL, n = 9. DHA, n = 9. ARA, n = 9. DHA+ARA, n = 10.
To assess the possible role of Ca2+-dependent and –independent phospholipase A2 in Ca2+ induced MPTP opening, we assessed the effects of addition of aristolochic acid and bromoenolactone, inhibitors of phospholipase A2, on the relationship between cumulated Ca2+ load and extramitochondrial [Ca2+]. This shifted the relationship to the right in mitochondrial from the CTRL and DHA+ARA groups, but not in the DHA or ARA groups (Figure 5).
Figure 5. Effect of phospholipase A2 inhibition on the Ca2+ retention capacity.

Data are mean ±SEM. CTRL, n = 9. DHA, n = 9. ARA, n = 9. DHA+ARA, n = 10. CTRL vehicle vs CTRL inhibitors p<0.006. DHA+ARA vehicle vs DHA+ARA inhibitors p<0.01.
Mitochondrial Swelling
The decrease in absorbance at 520 nm following the addition of Ca2+ to isolated mitochondria was used as a measure of MPTP opening. DHA supplementation attenuated the in response to 2 µmols Ca2+/mg mitochondrial protein compared to CTRL, ARA and DHA+ARA groups (Figure 6). Both DHA and ARA supplementation attenuated the decrease in absorbance in response to 4 µM µmols Ca2+/mg mitochondrial protein compared to CTRL.
Figure 6. Effect of diet on mitochondrial Ca2+ induced swelling.

Average decrease in absorbance 900 seconds after Ca2+ addition. Data are mean ± SEM. CTRL, n = 9. DHA, n = 8. ARA, n = 9. DHA+ARA, n = 9. * p<0.05 vs CTRL.
Discussion
Here we present the novel observation that a large increase in either DHA or ARA in mitochondrial membrane phospholipids is associated with a significant increase in the mitochondrial capacity for Ca2+ retention, an index of MPTP opening. Further, we showed that L4CL is not essential for normal mitochondrial function if replaced with very-long chain n3 or n6 PUFA. On the other hand, the extreme increase in the sum of ARA and DHA accompanied by depletion of linoleic acid that occurred with the DHA+ARA diet was associated with increased susceptibility to MPTP opening, and suggests that the combination of elevated DHA and ARA with low linoleic acid in membrane phospholipids is detrimental.
Our data do not support our initial hypothesis that the beneficial effect of DHA on MPTP opening is due to decreased membrane phospholipid ARA content. ARA has been identified as an MPTP inducer [21] and increased free ARA in cells can result in necrosis or apoptosis [10], [22], [23]. For example, inhibition of mitochondrial Ca2+-independent phospholipase A2γ, which is predicted to lower free ARA, reduced Ca2+-induced MPTP opening, mitochondrial phospholipid loss and reduced infarct size following I/R [23]. Similarly, Penzo and colleagues demonstrated that ARA released by Ca2+-dependent cytosolic phospholipase A2 activation triggers MPTP opening and cell death [11]. We found that dual inhibition of Ca2+-dependent and independent phospholipase A2 enzymes with a combination of aristolochic acid and bromoenolactone, respectively, resulted in delayed Ca2+-induced MPTP in the mitochondria of control-fed rats. Interestingly, it also provided a protective effect in the DHA+ARA group and altered the calcium sensitivity back to control levels. Both control and DHA+ARA groups, with phospholipase inhibition, are not significantly different from either DHA or ARA groups. This supports the conclusions made by previous studies in favor of a role for ARA release by phospholipases in inducing MPTP opening. However, we also saw that ARA supplementation does not induce MPTP opening as this hypothesis would predict, but rather delays it with no added benefit of phospholipase inhibition. Similarly, in the DHA group phospholipase inhibition did not provide additional resistance in MPTP opening. Thus, it is unlikely that modulation of free ARA explains the downward shift in the relationship between cumulative Ca2+ load and extramitochondrial [Ca2+] induced MPTP opening seen with ARA or DHA supplementation.
Although ARA supplementation provided a protective effect on MPTP opening, it also increased thromboxane production. This increase in inflammatory mediators also was seen in the combined DHA+ARA diet. The potential for ARA to modulate inflammation has been described by others [24]. Prostaglandin E2 synthesis can be increased by dietary supplementation with ARA, and this is modulated by other saturated fatty acids [25]–[27]. However, given the potential for ARA to increase inflammation, it is necessary to reconsider its use as a dietary supplement to prevent MPTP, especially in diseases that have an inflammatory component to their pathology, such as heart failure.
The combined DHA+ARA diet did not provide the protective effect of the diets containing DHA or ARA alone, and was detrimental to MPTP opening and increased inflammatory markers. The very high dose of PUFA provided by this diet increases membrane unsaturation to very high levels. Taking into account the high susceptibility of the highly unsaturated fatty acids to undergo peroxidation, and their proximity in the mitochondria to an ample source of reactive oxygen species, the DHA+ARA diet may increase MPTP susceptibility due to increased lipid peroxidation. This is further supported by the increase in malonaldehydes and hydroxyalkenals seen in the combined diet. Thus, the optimal dose of PUFAs might be below the threshold where increased lipid peroxidation outweighs the benefits provided by increasing membrane unsaturation. A less likely possibility is that there is negative interaction between DHA and ARA that sensitizes the MPTP. To fully exclude this type of effect, future studies should compare DHA alone and ARA alone to DHA+ARA at an equivalent dose of total PUFA.
We did not observe any differences in mitochondrial respiration with DHA supplementation with any of the substrates tested. We had previously shown that DHA supplementation decreases state 4 respiration with pyruvate+malate as substrates [3]. A subsequent study failed to replicate this finding [4], and again, we saw no difference in state 4 respiration in this study. The larger sample size used here allows for more confidence in concluding that DHA does not affect non-phosphorylating respiration in the healthy mitochondria compared to the CTRL diet. This, however, may not apply to mitochondria that have been stressed. Given DHA's protective effect in heart failure [7]–[9], we expect that DHA supplementation would ameliorate respiratory defects associated with heart disease.
There are limitations to the present investigation that need to be addressed. First, we assess only subsarcolemmal mitochondria, and not the whole population of cardiac mitochondria that includes the subpopulation of cardiac mitochondrial found amongst the fibrils (“interfibrillar mitochondria”). Isolation of total cardiac mitochondria requires incubation with a protease (trypsin) to disrupt the fibrils and release interfibrillar mitochondria, however we did not want to risk potential damage to mitochondrial proteins by this procedure. The effects protease digestion on mitochondrial Ca2+ uptake or MPTP opening is not known, thus we did not wish to introduce this potential confounding variable. We have previously found that there are no differences in membrane phospholipid fatty acid composition between interfibrillar and subsarcolemmal cardiac mitochondria in rats fed a standard low n-3 PUFA lab chow, or in the changes in mitochondrial membrane phospholipids caused by long term dietary supplementation with DHA or eicosapentaenoic acid (EPA) [4], [5]. This suggests that the changes we observed in subsarcolemmal mitochondria in the present study would very likely be duplicated in interfibrillar mitochondria. A second important limitation is that lack of measurement of the initial mitochondrial [Ca2+], as dietary supplementation with DHA or ARA could affect the residual Ca2+ in the mitochondrial matrix following isolation. Decreased initial matrix [Ca2+] could increase mitochondrial Ca2+ uptake capacity and give the impression of delayed PTP opening in response to successive Ca2+ additions. Our main conclusion regarding mitochondrial Ca2+ retention is that dietary supplementation with docosahexaenoic acid or arachidonic acid are associated with a greater exogenous Ca2+ load required to induce MPTP opening. We know of no rationale to suggest there would be differences in the initial [Ca2+] among treatment groups, nevertheless there may be differences that could affect the capacity for Ca2+ retention. Clearly future studies should assess this variable. Lastly, we did not assess the effects of cyclosporin A on Ca2+-induced MPTP opening in the present investigation. In a previous study we found that acute ex vivo treatment with cyclosporine A delayed Ca2+-induced MPTP opening, on rats fed a normal diet or with supplementation with DHA+EPA or EPA, but not DHA alone [4], [5]. Future studies should compare the effects of ARA and DHA supplementation on the delay in MPTP opening caused by cyclosporine A.
In summary, dietary supplementation with DHA or ARA caused dramatic alterations in mitochondria membrane phospholipid fatty acid composition, and delayed Ca2+-induced MPTP opening to approximately the same extent when compared to the standard control diet. This suggests that the effect of DHA on MPTP is most likely due to membrane unsaturation rather than depletion of ARA. Furthermore, DHA+ARA supplementation showed no added benefit, but rather sensitized MPTP to opening, suggesting that a very large dose of long chain PUFA can be detrimental. Finally, we found that high levels of L4CL were not essential for normal mitochondrial function if replaced by very-long chain n3 or n6 PUFAs.
Methods
Experimental Design
The animal protocol was conducted according to the Guideline for the Care and Use of Laboratory Animals (NIH publication 85-23) and was approved by the University of Maryland School of Medicine Institutional Animal Care and Use Committee (Protocol number 1009011). In accordance with this document, every effort was made to prevent pain to the animal, and to aleviate pain with anesthetic when it could potentially occur. Investigators were blinded to treatment when measurements were performed. The animals were maintained on a reverse 12-h light–dark cycle and all procedures were performed in the fed state between 3 and 6 h from the start of the dark phase. Male Wistar rats weighing 190–200 g were fed a standard low fat diet (CTRL) or a modified standard diet containing DHA or ARA at 2.5% of total caloric intake, which corresponds to a human intake of approximately 5.5 g/day (calculated assuming an energy intake of 2000 kcal/day and 9 kcal/g of fat) or a combined DHA+ARA diet at 5% of total caloric intake. After 10 weeks of dietary treatment, rats were anesthetized with isoflurane, urine was collected from the bladder, and hearts were harvested for biochemical analysis and mitochondrial isolation. Urine was analyzed for creatinine and thromboxane metabolites. Cardiac mitochondria were analyzed for respiration, Ca2+ retention capacity, Ca2+ induced swelling and membrane phospholipid composition as described below.
Diets
All diets were custom-manufactured (Research Diets Inc., New Brunswick, NJ), and had 66% of total energy from carbohydrate (54% of total energy from cornstarch and 12% from maltodextrin), 20% protein (casein supplemented with l-cystine) and 14% energy from fat (see Table 5 for fatty acid composition). In the CTRL diet, the fat was made up of 71.5% cocoa butter, 17.1% soybean oil, 7.2% palm oil, 2.8% safflower oil and 1.4% linseed oil. The DHA diet contained 5.75% of total energy from DHASCO (Martek Inc, Columbia, MD, USA) that was comprised of 43.6% DHA by, in place of part of the cocoa butter. The DHASCO oil was free from ARA. The ARA diet had 6.25% of energy from ARASCO (Martek Inc, Columbia, MD, USA) that was comprised of 40.1% ARA by mass partially replaced cocoa. The ARASCO oil was free from DHA. The DHA+ARA diet had both the DHA and ARA enriched oils added at 5.75 and 6.25% of energy respectively. DHASCO and ARASCO oils were purified from algae and contained ascorbyl palmitate (250 ppm) and tocopherols (250 ppm) to prevent lipid oxidation, which was less than 0.5 meq/kg at the time of manufacture of the diet. All diets were supplemented with the same amount of vitamins (Vitamin Mix V10001, 10 g/kg), minerals (Mineral Mix S10026, 10 g/kg), cellulose (50 g/kg) and choline (2 g/kg). It is important to note that the levels of DHA and ARA used in this study are equivalent to ∼5.5 g/day in humans (calculated assuming an energy intake of 2000 kcals/day). Tthus the dose of DHA and ARA use in this study are pharmacological, and are far above what is typically consumed in food (∼0.0.5 to ∼0.7 g/day of each) [19], [20].
Table 5. Fatty acid compositions of the rodent diets expressed as the molar percent of total fatty acids in the diet.
| Fatty Acid | Diet | |||
| CRTL | DHA | ARA | DHA+ARA | |
| C12:0 | 3.4 | 1.5 | 3.4 | 1.5 |
| C14:0 | 1.1 | 3.6 | 1.6 | 4.1 |
| C16:0 | 21.7 | 14.6 | 16.2 | 9.5 |
| C16:1 | 0.2 | 1.4 | 0.1 | 1.4 |
| C18:0 | 25.8 | 14.2 | 16.1 | 5.3 |
| C18:1n-9 | 30.3 | 28.8 | 23.4 | 21.9 |
| C18:2n6 | 13.9 | 14.7 | 15.2 | 14.1 |
| C18:3n3 | 2.2 | 2.2 | 2.3 | 2.4 |
| C20:4n6 | - | - | 18.3 | 18.4 |
| C20:5n3 | - | 0.3 | - | 0.3 |
| C22:6n3 | - | 17.8 | - | 18.3 |
| Total saturated | 52 | 34 | 37 | 20 |
| Total mono-sat | 30 | 30 | 23 | 23 |
| Total n3 | 2.2 | 20 | 2.3 | 21 |
| Total n6 | 14 | 15 | 33 | 32 |
| n6/n3 | 6.3 | 0.7 | 15 | 1.5 |
| PUFA/saturated | 0.3 | 1.0 | 1.3 | 2.6 |
All diets had 14% of total energy from fat, 20% from protein and 66% from carbohydrates.
Echocardiography
Since dietary lipids can effect cardiac chamber size and contractile function, we assessed LV dimensions in anesthetized rats using a high resolution small animal imaging system (model Vevo 770, with transducer model RMV 716, VisualSonics Inc., Toronto, Canada), as previously described in detail [5]. Briefly, rats were anesthetized with isoflurane by mask, placed supine on a heated platform, and 2-dimensional cine loops and guided M-mode frames were acquired from the short and long axis. Measurements were made off line using software resident on the system, with the investigator blind to the treatment.
Metabolic and Biochemical Parameters
Lipid peroxidation was measured in whole tissue homogenate using a commercially available kit reacting to malonaldehydes and 4-hydroxyalkenals (Oxford Biomedical Research, MI, USA). Urine creatinine and thromboxane B2 were measured using commercially available kits (Cayman Chemicals, MI, USA)
Mitochondrial Preparation
Subsarcolemmal mitochondria were isolated as previously described [3], [26]. LV tissue (approximately 500 mg) was thoroughly rinsed and cut immediately following excision of the heart and placed in Ca2+ free ice-cold modified Chappel-Perry buffer (100 mM KCl, 50 mM MOPS, 5 mM MgSO4, 1 mM Na2ATP, 1 mM EGTA, 2 mg/ml BSA). LV tissue was dissected, rinsed again, blotted dry, then minced in ice-cold buffer. Prior to homogenization, the buffer was replaced twice to remove blood contamination. The tissue was homogenized in 1∶10 (wt/vol) Chappel-Perry buffer, and subsarcolemmal mitochondria were then isolated and purified by differential centrifugation. Mitochondrial protein concentration was measured by the Lowry method using bovine serum albumin (BSA) as a standard.
Membrane Lipid Composition
Cardiac phospholipid fatty acid composition was assessed in isolated cardiac mitochondria homogenates by gas chromatography coupled with mass spectrometry according to a modification of the transesterification method as previously described [28]. Individual phospholipid species were quantified after extraction using the method of Christiansen in the presence of internal standard [29]. The resultant extract was subjected to silica gel chromatography to harvest phospholipids [30]. The phospholipid classes were separated on a normal-phase HPLC coupled to a LCQ Deca (Thermo Scientific) ion trap mass spectrometer. Quantitation was performed by interpolation to standard curves for each class of phospholipids. (Kim J. & Hoppel C.L., manuscript submitted). Mass spectrometric data was adjusted for natural abundance using predicted molecular composition from the RCM Lipid Calculator (http://pharmacology.ucdenver.edu/lipidcalc/Default.aspx) and the GENEBIO SmileMS Low Precision Isotope Distribution Calculator (http://research.smilems.com/molecule-tk/moleculeUtils/isotoperatio).
Mitochondrial respiration
Mitochondrial oxygen consumption was measured using a Clark-type oxygen electrode (Qubit Systems, Ontario, Canada). Mitochondria (0.25 mg protein) were suspended in respiration buffer (0.5 ml; 100 mM KCl, 50 mM MOPS, 5 mM KH2PO4 1 mM EGTA, and 0.5 mg fatty acid-free BSA) at pH 7.4 and 37°C [31]. State 3 (ADP-stimulated) and state 4 (non-phosphorylating) respiration were measured with 3 different sets of substrates: pyruvate+malate (10 and 5 mM, respectively) and palmityl-CoA+carnitine+malate (40 µM, 5 mM and 5 mM, respectively) to assess respiration through complex I–IV, and succinate+rotenone (10 mM and 7.5 µM, respectively) to assess respiration through complex II–IV of the ETC exclusively. Oligomycin was also used to inhibit mitochondrial ATP synthase and assess state 4 respiration.
Mitochondrial Ca2+ Retention Capacity
Mitochondrial Ca2+ retention was assessed using a 96-well fluorescence plate reader (FLUOstar Optima, BMG Labtech, Germany) as previously described [3]. 25 µg of mitochondria per well were resuspended in 200 µL of the same buffer used above, but with varying substrates and inhibitors; either glutamate+malate (10 and 5 mM, respectively), pyruvate+malate (10 and 5 mM, respectively), palmitoylcarnitine+malate (40 µM and 5 mM, respectively) or succinate (10 mM) with rotenone (7.5 µM), and glutamate+malate with phospholipase A2 inhibitors bromoenolactone (5 µM) and aristolochic acid (5 µM). Extramitochondrial Ca2+ was monitored using 1 µM Calcium Green 5N and fluorescence was measured at 485 nm and 538 nm for excitation and emission wavelengths respectively. Automated additions of 25 nmoles Ca2+/mg mitochondrial protein were performed at regular 7 minute intervals and fluorescence measured every 17 seconds for 160 min at 37°C.
Ca2+-Induced Mitochondrial Swelling
Light scattering, an index of Ca2+-induced mitochondrial swelling, was monitored using a 96 well spectrophotometric plate reader (SpectraMax, Molecular Devices, USA) [3]. Briefly, 25 µg of mitochondria were resuspended in 200 µl of the same buffer as used for the Ca2+ retention capacity assay. Baseline absorbance at 540 nm was read at 7 second intervals for 2 min, then either 50 or 100 nmoles of Ca2+ was rapidly added to the wells and the absorbance was read for 15 min at 37°C.
Statistical Analyses
Mean values are presented ± SEM, and the level of significance was set at p<0.05. Differences among dietary treatment groups were assessed using a One-Way ANOVA with Holm-Sidak post hoc test. Data sets that were not normally distributed were analyzed with a Kruskal-Wallis One-Way ANOVA on Ranks, with a Dunn's post hoc test. The response of isolated mitochondria to progressive additions of Ca2+ were assessed by comparing the extramitochondrial Ca2+ concentrations using a two way repeated measures ANOVA followed by a Holm-Sidak post hoc test for main effects and interactions.
Supporting Information
Representative spectra for mono-lyso-cardiolipin (MLCL).
(TIF)
Representative spectra for phosphotidylethanolamine (PE).
(TIF)
Representative spectra for phosphotidylcholine (PC).
(TIF)
Representative spectra for phosphotidylinositol (PI).
(TIF)
Representative spectra for phosphotidylglycerol (PG).
(TIF)
Phospholipid composition of mitochondrial membranes.
(DOCX)
Acknowledgments
We would like to thank Dr. Liron Boyman for his technical expertise in calcium calibration and measurements.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by National Institutes of Health grants HL074237, HL091307 and HL101434. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Ricchelli F, Sileikyte J, Bernardi P. Shedding light on the mitochondrial permeability transition. Biochim Biophys Acta. 2011;1807:482–490. doi: 10.1016/j.bbabio.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Halestrap AP. A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem Soc Trans. 2010;38:841–860. doi: 10.1042/BST0380841. [DOI] [PubMed] [Google Scholar]
- 3.Khairallah RJ, Sparagna GC, Khanna N, O'shea KM, Hecker PA, et al. Dietary supplementation with docosahexaenoic acid, but not eicosapentaenoic acid, dramatically alters cardiac mitochondrial phospholipid fatty acid composition and prevents permeability transition. Biochim Biophys Acta. 2010;1797:1555–1562. doi: 10.1016/j.bbabio.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khairallah RJ, O'shea KM, Brown BH, Khanna N, Des Rosiers C, et al. Treatment with docosahexaenoic acid, but not eicosapentaenoic acid, delays Ca2+-induced mitochondria permeability transition in normal and hypertrophied myocardium. J Pharmacol Exp Ther. 2010;335:155–162. doi: 10.1124/jpet.110.170605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'shea KM, Khairallah RJ, Sparagna GC, Xu W, Hecker PA, et al. Dietary omega-3 fatty acids alter cardiac mitochondrial phospholipid composition and delay Ca2+-induced permeability transition. J Mol Cell Cardiol. 2009;47:819–827. doi: 10.1016/j.yjmcc.2009.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitz G, Ecker J. The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res. 2008;47:147–155. doi: 10.1016/j.plipres.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 7.Duda MK, O'shea KM, Tintinu A, Xu W, Khairallah RJ, et al. Fish oil, but not flaxseed oil, decreases inflammation and prevents pressure overload-induced cardiac dysfunction. Cardiovasc Res. 2009;81:319–327. doi: 10.1093/cvr/cvn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nodari S, Triggiani M, Campia U, Manerba A, Milesi G, et al. Effects of n-3 Polyunsaturated Fatty Acids on Left Ventricular Function and Functional Capacity in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol. 2011;57:870–879. doi: 10.1016/j.jacc.2010.11.017. [DOI] [PubMed] [Google Scholar]
- 9.Gissi-Hf I. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1223–1230. doi: 10.1016/S0140-6736(08)61239-8. [DOI] [PubMed] [Google Scholar]
- 10.Kinsey GR, McHowat J, Patrick KS, Schnellmann RG. Role of Ca2+-independent phospholipase A2gamma in Ca2+-induced mitochondrial permeability transition. J Pharmacol Exp Ther. 2007;321:707–715. doi: 10.1124/jpet.107.119545. [DOI] [PubMed] [Google Scholar]
- 11.Penzo D, Petronilli V, Angelin A, Cusan C, Colonna R, et al. Arachidonic acid released by phospholipase A2 activation triggers Ca2+-dependent apoptosis through the mitochondrial pathway. J Biol Chem. 2004;279:25219–25225. doi: 10.1074/jbc.M310381200. [DOI] [PubMed] [Google Scholar]
- 12.Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol. 2009;53:290–301. doi: 10.1097/FJC.0b013e31819b5461. [DOI] [PubMed] [Google Scholar]
- 13.Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, et al. Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann Neurol. 2002;51:634–637. doi: 10.1002/ana.10176. [DOI] [PubMed] [Google Scholar]
- 14.Minkler PE, Hoppel CL. Separation and characterization of cardiolipin molecular species by reverse-phase ion pair high-performance liquid chromatography-mass spectrometry. J Lipid Res. 2010;51:856–865. doi: 10.1194/jlr.D002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, et al. Phospholipid abnormalities in children with Barth syndrome. J Am Coll Cardiol. 2003;42:1994–1999. doi: 10.1016/j.jacc.2003.06.015. [DOI] [PubMed] [Google Scholar]
- 16.Rosca M, Minkler P, Hoppel CL. Cardiac mitochondria in heart failure: Normal cardiolipin profile and increased threonine phosphorylation of complex IV. Biochim Biophys Acta. 2011;1807:1373–1382. doi: 10.1016/j.bbabio.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 17.Pepe S, Tsuchiya N, Lakatta EG, Hansford RG. PUFA and aging modulate cardiac mitochondrial membrane lipid composition and Ca2+ activation of PDH. Am J Physiol. 1999;276:H149–H158. doi: 10.1152/ajpheart.1999.276.1.H149. [DOI] [PubMed] [Google Scholar]
- 18.McMillin JB, Bick RJ, Benedict CR. Influence of dietary fish oil on mitochondrial function and response to ischemia. Am J Physiol. 1992;263:H1479–H1485. doi: 10.1152/ajpheart.1992.263.5.H1479. [DOI] [PubMed] [Google Scholar]
- 19.Kawabata T, Hirota S, Hirayama T, Adachi N, Hagiwara C, et al. Age-related changes of dietary intake and blood eicosapentaenoic acid, docosahexaenoic acid, and arachidonic acid levels in Japanese men and women. Prostaglandins Leukot Essent Fatty Acids. 2011;84:131–137. doi: 10.1016/j.plefa.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 20.Farina EK, Kiel DP, Roubenoff R, Schaefer EJ, Cupples LA, et al. Dietary intakes of arachidonic acid and alpha-linolenic acid are associated with reduced risk of hip fracture in older adults. J Nutr. 2011;141:1146–1153. doi: 10.3945/jn.110.133728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scorrano L, Penzo D, Petronilli V, Pagano F, Bernardi P. Arachidonic acid causes cell death through the mitochondrial permeability transition. Implications for tumor necrosis factor-alpha aopototic signaling. J Biol Chem. 2001;276:12035–12040. doi: 10.1074/jbc.M010603200. [DOI] [PubMed] [Google Scholar]
- 22.Pompeia C, Lima T, Curi R. Arachidonic acid cytotoxicity: can arachidonic acid be a physiological mediator of cell death? Cell Biochem Funct. 2003;21:97–104. doi: 10.1002/cbf.1012. [DOI] [PubMed] [Google Scholar]
- 23.Williams SD, Gottlieb RA. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem J. 2002;362:23–32. doi: 10.1042/0264-6021:3620023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo GL. Dietary n-6 and n-3 polyunsaturated fatty acids: from biochemistry to clinical implications in cardiovascular prevention. Biochem Pharmacol. 2009;77:937–946. doi: 10.1016/j.bcp.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 25.Seyberth HW, Oelz O, Kennedy T, Sweetman BJ, Danon A, et al. Increased arachidonate in lipids after administration to man: effects on prostaglandin biosynthesis. Clin Pharmacol Ther. 1975;18:521–529. doi: 10.1002/cpt1975185part1521. [DOI] [PubMed] [Google Scholar]
- 26.Abeywardena MY, McLennan PL, Charnock JS. Differential effects of dietary fish oil on myocardial prostaglandin I2 and thromboxane A2 production. Am J Physiol. 1991;260:H379–H385. doi: 10.1152/ajpheart.1991.260.2.H379. [DOI] [PubMed] [Google Scholar]
- 27.Abeywardena MY, McLennan PL, Charnock JS. Changes in myocardial eicosanoid production following long-term dietary lipid supplementation in rats. Am J Clin Nutr. 1991;53:1039S–1041S. doi: 10.1093/ajcn/53.4.1039S. [DOI] [PubMed] [Google Scholar]
- 28.Gelinas R, Thompson-Legault J, Bouchard B, Daneault C, Mansour A, et al. Prolonged QT interval and lipid alterations beyond {beta}-oxidation in very long-chain acyl-CoA dehydrogenase null mouse hearts. Am J Physiol Heart Circ Physiol. 2011;301:H813–H823. doi: 10.1152/ajpheart.01275.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Christiansen K. Lipid extraction procedure for in vitro studies of glyceride synthesis with labeled fatty acids. Anal Biochem. 1975;66:93–99. doi: 10.1016/0003-2697(75)90728-9. [DOI] [PubMed] [Google Scholar]
- 30.Ingalls ST, Kriaris MS, Xu Y, DeWulf DW, Tserng KY, et al. Method for isolation of non-esterified fatty acids and several other classes of plasma lipids by column chromatography on silica gel. J Chromatogr. 1993;619:9–19. doi: 10.1016/0378-4347(93)80441-6. [DOI] [PubMed] [Google Scholar]
- 31.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Representative spectra for mono-lyso-cardiolipin (MLCL).
(TIF)
Representative spectra for phosphotidylethanolamine (PE).
(TIF)
Representative spectra for phosphotidylcholine (PC).
(TIF)
Representative spectra for phosphotidylinositol (PI).
(TIF)
Representative spectra for phosphotidylglycerol (PG).
(TIF)
Phospholipid composition of mitochondrial membranes.
(DOCX)