

Summary
Background
Persistent inflammation has been proposed to contribute to various stages in the pathogenesis of cardiovascular disease. Interleukin-6 receptor (IL6R) signalling propagates downstream inflammation cascades. To assess whether this pathway is causally relevant to coronary heart disease, we studied a functional genetic variant known to affect IL6R signalling.
Methods
In a collaborative meta-analysis, we studied Asp358Ala (rs2228145) in IL6R in relation to a panel of conventional risk factors and inflammation biomarkers in 125 222 participants. We also compared the frequency of Asp358Ala in 51 441 patients with coronary heart disease and in 136 226 controls. To gain insight into possible mechanisms, we assessed Asp358Ala in relation to localised gene expression and to postlipopolysaccharide stimulation of interleukin 6.
Findings
The minor allele frequency of Asp358Ala was 39%. Asp358Ala was not associated with lipid concentrations, blood pressure, adiposity, dysglycaemia, or smoking (p value for association per minor allele ≥0·04 for each). By contrast, for every copy of 358Ala inherited, mean concentration of IL6R increased by 34·3% (95% CI 30·4–38·2) and of interleukin 6 by 14·6% (10·7–18·4), and mean concentration of C-reactive protein was reduced by 7·5% (5·9–9·1) and of fibrinogen by 1·0% (0·7–1·3). For every copy of 358Ala inherited, risk of coronary heart disease was reduced by 3·4% (1·8–5·0). Asp358Ala was not related to IL6R mRNA levels or interleukin-6 production in monocytes.
Interpretation
Large-scale human genetic and biomarker data are consistent with a causal association between IL6R-related pathways and coronary heart disease.
Funding
British Heart Foundation; UK Medical Research Council; UK National Institute of Health Research, Cambridge Biomedical Research Centre; BUPA Foundation.
Introduction
Although the hypothesis that persistent inflammation contributes to various stages in the pathogenesis of cardiovascular disease is supported by observational and experimental studies in human beings and animals,1,2 causality has not been established. Data from observational epidemiological studies have suggested that circulating concentrations of liver-derived inflammation biomarkers (eg, C-reactive protein, fibrinogen) are associated with subsequent risk of coronary heart disease.3–5 However, the likelihood of a direct causal role for these particular downstream inflammation biomarkers has been reduced because inherited DNA variations associated with them have not been linked to coronary heart disease.6–8 Clinical investigation of soluble concentrations of proximal regulators of inflammation cascades, such as the signalling of interleukin-6 receptor (IL6R) pathways, can be difficult because these factors are prone to fluctuation in the circulation.3 Consequently, to help to evaluate the relevance of proximal inflammatory mediators to coronary heart disease, studies of the genetic determinants of these factors might be informative, since such genetic variants are fixed at conception and are indicators of lifelong (rather than transient) inflammation status.6
In classic IL6R signalling, soluble interleukin 6 activates the membrane-bound receptor in hepatocytes and leucocytes, thereby initiating downstream proinflammatory cascades that increase hepatic production of C-reactive protein, fibrinogen, and other acute-phase reactants.9,10 Cell-based experiments have suggested that the Asp358Ala (rs2228145, formerly known as rs8192284) variant in IL6R might impair classic IL6R signalling (and hence dampen inflammation) by reducing membrane-bound IL6R levels.11–14 Although previous studies have reported associations between Asp358Ala (or variants closely linked with it) and circulating inflammation biomarkers,7,12,15–19 asthma,20 and some autoimmune diseases,21,22 associations of Asp358Ala with conventional cardiovascular risk factors or with risk of coronary heart disease have not been precisely characterised.7,23 To help to assess whether IL6R pathways are causally relevant to coronary heart disease, we analysed human genetic and biomarker data from more than 200 000 participants in a collaborative meta-analysis.
Methods
Study design
This meta-analysis had several interrelated components. First, we assessed associations of Asp358Ala (or two proxy genetic variants closely related to it) with several inflammation biomarkers and conventional cardiovascular risk factors in studies including up to 125 222 participants without a history of cardiovascular disease. Second, to gain insight into possible mechanisms, we undertook functional studies in monocytes and macrophages. Third, we assessed Asp358Ala in relation to risk of coronary heart disease in a meta-analysis of 51 441 patients with the disease and 136 226 controls to evaluate the previously suggested signal at this locus. Fourth, to compare the effect of Asp358Ala on liver-derived inflammation biomarkers with the corresponding physiological relationships of these soluble biomarkers with one another, we characterised the cross-sectional associations of circulating interleukin 6, C-reactive protein, and fibrinogen concentrations and their associations with incident coronary heart disease risk in up to 109 339 participants without a history of cardiovascular disease. Fifth, to compare the effects of Asp358Ala on concentrations of inflammation biomarkers and major lipids with those of tocilizumab, a monoclonal antibody that selectively inhibits IL6R, we analysed data for 4394 patients with inflammatory conditions enrolled in previous randomised placebo-controlled trials.
Contributing studies and data
For analyses of IL6R genotypes in relation to inflammation biomarkers, cardiovascular disease risk factors, and risk of coronary heart disease, we used systematic searches of the literature and registries of genetic studies to collate information from 46 studies with relevant summary-level data, as described in detail in the appendix pp 4–7, 20, 25. Studies provided data for Asp358Ala or, if this was not available, for either of two proxy variants highly correlated with rs2228145: rs4537545 or rs4129267 (r2=0·96 with each variant in white Europeans). Because of the high degree of linkage among these variants (appendix p 12), principal analyses were done for all three combined (sensitivity analyses assessed associations separately for each variant, where possible).
According to a uniform protocol, data were obtained from all studies, where available, for genotype frequencies by categorical disease outcome (separately for myocardial infarction cases, non-overlapping coronary stenosis cases, and healthy controls); criteria used to diagnose coronary heart disease; circulating concentrations of soluble IL6R, interleukin 6, C-reactive protein, fibrinogen, LDL cholesterol, HDL cholesterol, triglycerides, and fasting glucose; measurements of systolic blood pressure, body-mass index (BMI), and waist circumference; and information about smoking status and history of diabetes (appendix pp 4–7). Data for inflammation biomarkers and conventional risk factors were from participants with no known history of cardiovascular disease at time of survey. Data for coronary heart disease derived from participants in case-control studies included myocardial infarction or coronary stenosis (details provided in appendix p 6). Data from published studies were extracted in duplicate by two independent reviewers. All tabular data provided by study authors were cross-checked with registry data and queried with authors of papers, where necessary. Contributing prospective studies recorded coronary heart disease outcomes through linkage with hospital discharge records, mortality registers, and active follow-up.
To investigate whether observed associations of Asp358Ala might be due to effects on production of IL6R or interleukin 6 (rather than the shedding of membrane-bound IL6R into circulation), we assessed IL6R genotype in relation to gene expression in monocytes and macrophages derived from 363 patients with premature myocardial infarction and 395 healthy blood donors, and to postlipopolysaccharide stimulation of interleukin 6 in monocytes derived from 205 separate healthy blood donors (details provided in appendix pp 3, 26). We validated these results by accessing further data for circulating monocytes from 1490 healthy participants in the Gutenberg Heart Study,24 as well as further publicly available gene expression datasets that include liver, lymphoblastoid, and T cells.25–28 We did not undertake functional experiments to investigate whether Asp358Ala affects shedding of membrane-bound IL6R, because such work has been done previously.11–13 For analyses of cross-sectional correlations of inflammation biomarkers with one another and of associations of these biomarkers with incident coronary heart disease, we accessed individual-level data from participants without known cardiovascular disease at the baseline survey of 38 population-based long-term prospective studies (appendix pp 8, 21).4 Such data for soluble IL6R concentrations were not available. For analyses of tocilizumab, we did systematic searches of published randomised trials, as described in detail in appendix pp 22–24. We invited investigators and trial sponsors to provide tabular data for trial protocols, treatment regimens, and levels of inflammation and lipid biomarkers. Data were included from all relevant trials reporting both pre-trial and post-trial mean (SD) biomarker concentrations.
Statistical analysis
We calculated estimates of association separately within each study before pooling across studies by fixed-effect meta-analysis (supplementary analyses used random-effects models), using methods described previously.29 Summary odds ratios (ORs) for coronary heart disease and differences in mean levels by IL6R genotype and per minor allele were calculated compared with the common homozygotes. Analyses were done with original units and then presented as percentage differences to enable comparisons across different biomarkers (calculated in reference to the weighted mean of each biomarker among common homozygotes). Cross-sectional associations among inflammation biomarkers were assessed with linear regression models to obtain means adjusted for age and sex. Biomarkers that were not normally distributed were analysed on a natural logarithmic scale. Hazard ratios for associations of biomarkers with incident coronary heart disease were calculated with Cox proportional hazards models (adjusting for conventional cardiovascular disease risk factors) and then pooled across studies, as previously described.4
We assessed the effect of tocilizumab intervention on inflammation and lipid biomarkers by calculating mean differences before and after intervention within each trial group, then pooling findings across studies. Heterogeneity was assessed by the I2 statistic,30 the Q statistic, and metaregression of prespecified groupings of study characteristics including study design, location, genetic variants used, and study size. Small-study effects were assessed with funnel plots and by comparison of pooled results from studies involving at least 500 coronary cases (or, for gene-intermediate phenotype investigations, at least 1000 participants) with those from smaller studies. We separately analysed data for Asp358Ala and coronary heart disease in the 16 studies (84 796 participants) that had previously reported on this association (the hypothesis-generating set7,19,23) and 24 additional studies (102 871 further participants) that provided an independent test of this hypothesis. A consequence of this focused hypothesis testing of one genetic variant with risk of coronary heart disease is that adjustment for multiple testing was not needed, by contrast with hypothesis-free genome-wide association studies exploring several hundred thousand variants. Analyses were done with Stata (version 11), two-sided p values, and 95% CIs.
Role of the funding source
The sponsors of the study have no role in study design, data analysis, data interpretation, or writing of the report. Nadeem Sarwar, Adam Butterworth, and John Danesh had full access to all the data and had final responsibility for the decision to submit for publication.
Results
We meta-analysed data from 46 genetic epidemiological studies (204 930 participants), 30 classical observational epidemiological studies (109 339 participants), nine randomised trials (4394 participants), and three explanatory functional studies (2372 participants), constituting 82 unique studies. Since more than 90% of these participants were of white European continental ancestry, there were insufficient data from non-Europeans to enable detailed comparisons with people of other continental ancestries.
In people without coronary heart disease, the overall minor allele frequency of Asp358Ala was 39%, and this frequency was similar across contributing studies (appendix pp 4–5). IL6R genotype was not related to concentrations of LDL or HDL cholesterol, triglyceride, fasting glucose, systolic blood pressure, BMI, waist circumference, smoking status, or history of diabetes (p value for association per minor allele ≥0·04 for each; table). By contrast, carriers of 358Ala had significantly higher mean concentrations of soluble IL6R and interleukin 6 (both p<10−13) and significantly lower mean concentrations of C-reactive protein and fibrinogen compared with non-carriers (both p<10−11). The results were consistent with a per-allele model of inheritance (figure 1). For every minor allele inherited, carriers had a 34·3% (95% CI 30·4–38·2) increase in mean concentration of soluble IL6R, a 14·6% (10·7–18·4) increase in mean interleukin 6, a 7·5% (5·9–9·1) reduction in mean C-reactive protein, and a 1·0% (0·7–1·3) reduction in mean fibrinogen (table).
Table.
Association of IL6R genotypes with levels of inflammation markers and conventional vascular risk factors
| Participants with available data | Percentage mean difference or odds ratio (95% CI) per minor allele | p value for association | |
|---|---|---|---|
| Inflammation markers | |||
| Soluble IL6R | 1645 | 34·3% (30·4 to 38·2) | 1·8×10−65 |
| Interleukin 6 | 27 185 | 14·6% (10·7 to 18·4) | 8·3×10−14 |
| C-reactive protein | 83 948 | −7·5% (−9·1 to −5·9) | 1·6×10−19 |
| Fibrinogen | 50 353 | −1·0% (−1·3 to −0·7) | 4·8×10−12 |
| Conventional vascular risk factors | |||
| LDL cholesterol | 80 343 | −0·03% (−0·3 to 0·3) | 0·83 |
| HDL cholesterol | 95 970 | 0·07% (−0·1 to 0·4) | 0·44 |
| Triglyceride | 94 473 | −0·3% (−0·9 to 0·3) | 0·28 |
| Fasting glucose | 51 109 | 0·02% (−0·2 to 0·3) | 0·87 |
| Systolic blood pressure | 99 577 | −0·1% (−0·3 to −0·01) | 0·04 |
| Body-mass index | 122 222 | 0·01% (−0·1 to 0·2) | 0·90 |
| Waist circumference | 81 492 | 0·2% (−0·01 to 0·3) | 0·06 |
| Ever vs never smokers | 54 523 vs 54 817 | 0·98 (0·96 to 1·01) | 0·07 |
| History vs no history of diabetes | 9722 vs 80 095 | 0·99 (0·96 to 1·03) | 0·62 |
Odds ratios are presented for smoking status and history of diabetes. Analyses were undertaken with standardised units of measurement for each marker. To enable comparison of the magnitude of associations across several different markers, associations are presented as percentage differences (calculated in reference to the weighted overall mean of each marker among the reference group). Details of the genotypes studied are provided in appendix p 12. IL6R=interleukin-6 receptor.
Figure 1.
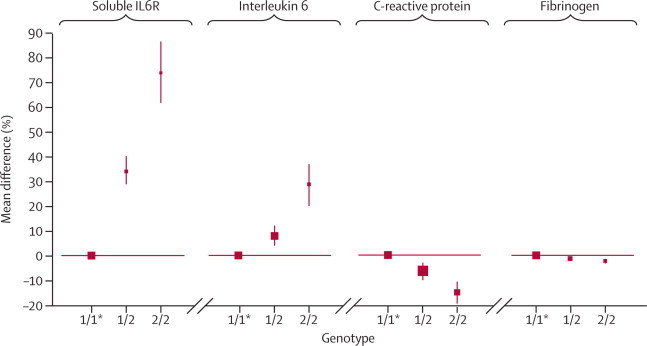
IL6R genotypes and circulating concentrations of inflammation markers
Analyses were undertaken with standardised units of measurement for each marker. To enable comparison of the magnitude of associations across several different markers, associations are presented as percentage differences (calculated in reference to the weighted overall mean of each marker among the reference group). The number of participants with information about each marker is shown in the table. Details of the genotypes studied are provided in appendix p 12. Error bars show 95% CI. IL6R=interleukin-6 receptor. *Reference group (represented by a square with an arbitrary fixed size).
Associations between Asp358Ala and each of interleukin 6, C-reactive protein, and fibrinogen concentration did not vary by the specific variant assessed (ie, lead single nucleotide polymorphism or proxy variant) or the study design used, nor was there evidence of small-study effects (appendix p 13). Data were insufficient for detailed analyses by ethnicity, although results remained unchanged when restricted to studies of people of predominantly white European descent (appendix p 9) and did not significantly differ when results were grouped by geographical location. There were too few data to enable such subanalyses for soluble IL6R. Asp358Ala was not associated with IL6R mRNA levels in monocytes or macrophages (appendix p 10), nor was any significant association seen in the range of tissues in publicly available gene expression datasets. Furthermore, Asp358Ala was not associated with interleukin-6 production by monocytes after lipopolysaccharide stimulation (appendix p 14).
Risk of coronary heart disease was lower in carriers of 358Ala compared with non-carriers. Again, there was evidence of a per-allele model of inheritance (figure 2). For every minor allele inherited, carriers had a 3·4% (1·8–5·0) reduction in risk of coronary heart disease. Results were similar in the 24 hypothesis-testing studies that did not contribute to previous analyses (4·2%, 2·1–6·3), thereby providing independent confirmation of this association. Findings did not vary by the variant assessed, the type of coronary outcome recorded, or study design used, nor was there evidence of small-study effects (appendix p 15). Results did not differ according to study location and were similar in analyses restricted to populations of white European descent.
Figure 2.
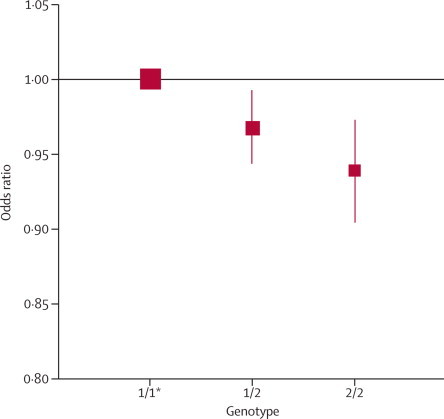
IL6R genotypes and risk of coronary heart disease
Data are shown for 51 441 cases and 136 226 controls. The odds ratio per minor allele was 0·966 (95% CI 0·950–0·982, p=4·5×10−5). Details of the genotypes studied are provided in appendix p 12. Error bars show 95% CI. *Reference group (represented by a square with in arbitrary fixed size).
Among people with no history of cardiovascular disease at the time of blood sampling, circulating concentrations of interleukin 6, C-reactive protein, and fibrinogen were each positively associated with one another (figure 3A). The age-adjusted and sex-adjusted partial correlation coefficient was 0·49 (95% CI 0·46–0·52) between loge interleukin 6 and loge C-reactive protein, 0·35 (0·31–0·39) between loge interleukin 6 and fibrinogen, and 0·48 (0·45–0·50) between loge C-reactive protein and fibrinogen. There were roughly log-linear associations between baseline concentrations of each of interleukin 6, C-reactive protein, and fibrinogen and subsequent risk of coronary heart disease (figure 3B).
Figure 3.
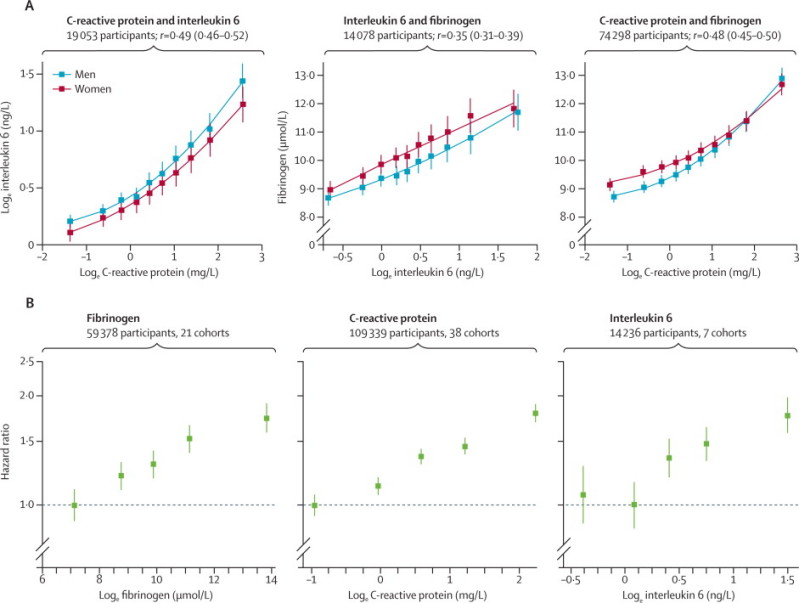
(A) Cross-sectional associations between interleukin 6, C-reactive protein, and fibrinogen concentrations in the general population and (B) associations of interleukin 6, C-reactive protein, and fibrinogen concentrations with incident coronary heart disease
Adjusted for age, sex, smoking, body-mass index, diabetes status, systolic blood pressure, and total cholesterol. Studies included in these analyses had information on at least two of the biomarkers studied and had recorded at least ten incident coronary heart disease events during follow-up. Error bars show 95% CI.
Nine randomised trials of tocilizumab, involving 4394 patients with rheumatoid arthritis or Crohn's disease, were included in our analyses (appendix p 11). Original data were provided by investigators for five of these trials (4087 patients) and published data were abstracted for four trials (307 patients). Five of the nine trials also involved methotrexate use. In analyses of lipids, comparing post-treatment with pre-treatment levels, tocilizumab intervention resulted in increased concentrations of total, LDL, and HDL cholesterol and triglycerides (appendix p 16), as well as increased concentrations of proximal inflammatory mediators (soluble IL6R and interleukin 6) and decreased concentrations of downstream mediators (C-reactive protein and fibrinogen; appendix p 17). For some biomarker endpoints (eg, interleukin 6, IL6R, LDL cholesterol), there was substantial heterogeneity among the results of the contributing trials (appendix pp 18–19). However, most of the statistical scatter was due to only one or two of the trials, the omission of which did not affect the overall results.
Discussion
Interest in the inflammation hypothesis of cardiovascular disease has intensified since the launch of phase 3 trials of various anti-inflammatory agents in the secondary prevention of cardiovascular disease, including two trials of darapladib (an inhibitor of lipoprotein-associated phospholipase A2),31 a trial of canakinumab (a monoclonal antibody to interleukin 1β),32 and a trial of low-dose methotrexate.33 Although this hypothesis has received some support from previous human genetic studies that implicate loci of potential relevance to inflammation, including CXCL12,34 ADAMTS7,35 SH2B3,34 and IL5,23 in coronary heart disease, such studies have not provided specific insight into inflammation and coronary heart disease because the relevant genes (or causal alleles) in these loci still await identification.
By contrast, the present study has focused on a specific functional allele known to affect IL6R signalling. This meta-analysis has provided large-scale human genetic and biomarker evidence that is consistent with a causal association between IL6R-related pathways and coronary heart disease (panel). First, our results have shown that Asp358Ala—a common functional variant in IL6R—is unrelated to a panel of conventional cardiovascular disease risk factors, suggesting that any effect of Asp358Ala on risk of coronary heart disease is unlikely to be mediated by conventional risk factors. Second, we noted that carriage of 358Ala was associated, in a dose-dependent manner, with decreased concentrations of C-reactive protein and fibrinogen (two different liver-derived proteins that sensitively reflect inflammation status), suggesting that 358Ala dampens the systemic inflammatory response.22 Third, our results show that 358Ala is significantly related to decreased risk of coronary heart disease in the same dose-dependent manner. In aggregate, therefore, these results support the inflammation hypothesis in coronary heart disease and encourage exploration of modulation of IL6R pathways as a means to prevent coronary heart disease.
Panel. Research in context.
Systematic review
The inflammation hypothesis in coronary heart disease has received potential support from human genetic studies that implicate several loci of potential relevance to inflammation in coronary heart disease. However, because the relevant genes (or causal variants) in these loci await identification, they do not provide specific insight. By contrast, the present study has focused on a specific functional genetic variant (Asp358Ala; rs2228145) known to affect interleukin-6 receptor (IL6R) signalling. Previous studies7,23 of Asp358Ala, inflammation biomarkers, and coronary heart disease have been insufficiently powerful or insufficiently detailed, or both, to enable causal evaluation.
In our collaborative meta-analysis, we studied Asp358Ala in IL6R in relation to a panel of conventional risk factors and inflammation biomarkers in 125 222 participants. We also compared the frequency of Asp358Ala in 51 441 patients with coronary heart disease and in 136 226 controls. These studies were identified by computerised searches of electronic databases (eg, PubMed), registries of genetic association studies in coronary heart disease, and discussion with investigators. The studies included met quality criteria in relation to methods for population sampling, genotyping, biomarker assessment, and coronary heart disease outcome definition.
Interpretation
358Ala carriers have substantially raised soluble interleukin 6 and IL6R concentrations and reduced C-reactive protein and fibrinogen concentrations, which suggests that 358Ala dampens the systemic inflammatory response. 358Ala carriers also have reduced risk of coronary heart disease. These findings are consistent with a causal role of IL6R-related pathways in coronary heart disease, and they support the inflammation hypothesis in this disease.
Our study has confirmed previous reports of positive associations between circulating interleukin-6 concentration and subsequent risk of coronary heart disease.3,36 That 358Ala, which is inversely related to coronary heart disease, is strongly related with higher concentrations of both soluble IL6R and interleukin 6 in a dose-dependent manner might, therefore, seem paradoxical. However, this apparent paradox might be resolved by previous suggestions that 358Ala impairs classical IL6R signalling by reducing membrane-bound IL6R levels, either through increased receptor shedding or alternative splicing, rather than by changes in production of IL6R or interleukin 6.11–14 Such mechanisms could account for the accumulation of both soluble IL6R and interleukin 6 in the circulation of people who carry 358Ala. It would also be consistent with two findings in our study. First, expression analyses showed that Asp358Ala was not associated with interleukin-6 production in several different tissues or in monocytes after lipopolysaccharide stimulation. Second, whereas we reported positive interrelations among circulating concentrations of interleukin 6, C-reactive protein, and fibrinogen with one another in the general population, carriage of 358Ala was associated with increased interleukin 6 but decreased C-reactive protein and fibrinogen concentrations. Nevertheless, further mechanistic studies are needed to improve understanding of these findings. For example, investigation of 358Ala in relation to other inflammation-related phenotypes (and its potential effects on gp130-mediated trans-signalling) to further understand its pleiotropic immunomodulatory effects would be of particular interest in view of the recently reported association of 358Ala with an increased risk of asthma.20
Pharmacological agents such as tocilizumab, an antihuman monoclonal antibody that competitively inhibits IL6R, reduce circulating concentrations of downstream inflammation biomarkers and produce clinical benefits in rheumatoid arthritis and in other inflammatory conditions.37,38 There have also been suggestions that such agents improve endothelial function in arthritis patients and reduce atherosclerosis in mice.39,40 However, whether these agents affect risk of coronary heart disease is not yet known. Our meta-analysis confirms that tocilizumab use in patients with inflammatory conditions increases LDL and HDL cholesterol and triglyceride concentrations. It is uncertain whether such lipid changes reflect improvements in underlying chronic inflammation with tocilizumab, mechanism-based effects of IL6R modification, or tocilizumab-specific effects.41–43 Hence, the US Food and Drug Administration has mandated further study of tocilizumab and cardiovascular disease risk factors in postmarketing clinical trials.44 Increases in LDL cholesterol with tocilizumab might be avoidable, or at least attenuated, with concomitant statin use.45,46 However, as shown by our meta-analysis, carriage of 358Ala is unrelated to concentrations of major lipids, suggesting that interleukin-6 modulation might not inherently increase these lipids. Further work is needed to understand why the effect of tocilizumab on major lipids differs from that of Asp358Ala, despite directionally consistent effects on inflammation biomarkers.
The generalisability and validity of our findings have been improved by our analysis of genetic and biomarker data for more than 200 000 participants. Our observation of an association between a single functional variant in IL6R and coronary heart disease has been supported by a recent replication study of a genome-wide association scan.47 By analogy with the fairly small effects of common variants in LDLR on coronary heart disease still being compatible with the major benefits of statins in coronary heart disease,48 the effect of 358Ala on coronary heart disease does not necessarily restrict the scope for potential therapeutic effect of IL6R modification in coronary heart disease.49 Future studies will seek to study Asp358Ala (or lower frequency variants with more pronounced effects) in relation to additional traits, non-coronary heart disease outcomes, and expression studies involving multiple cell types. Although our study involves the principle of mendelian randomisation, we did not do an instrumental variables analysis for two reasons: first, the relevant intermediate phenotype of the IL6R pathway remains uncertain because Asp358Ala is associated with changes in several inflammation biomarkers; second, our study was limited to aggregated (rather than individual-level) genetic data.
In conclusion, our data support a causal association between IL6R-related pathways and coronary heart disease.
Correspondence to: Dr Nadeem Sarwar or Dr Adam S Butterworth, Department of Public Health and Primary Care, University of Cambridge, Strangeways Research Laboratory, Cambridge CB1 8RN, UK
Acknowledgments
Acknowledgments
The coordinating centre was supported by the British Heart Foundation (RG/08/014), the UK Medical Research Council, the UK National Institute of Health Research, Cambridge Biomedical Research Centre, and a specific grant from the BUPA Foundation. Various sources have supported recruitment, follow-up, and laboratory measurements in the studies contributing to this report. Investigators of several of these studies have contributed to a list naming some of these funding sources. Sekar Kathiresan, Kenneth G C Smith, and John Todd commented helpfully on an earlier version of this report.
Contributors
Nadeem Sarwar, Adam Butterworth, and John Danesh drafted the report. Nadeem Sarwar, Adam Butterworth, Pei Gao, Daniel Freitag, Donal Gorman, Peter Willeit, John Gregson, Danish Saleheen, Stephen Kaptoge, and Emanuele Di Angelantonio undertook literature searches and analysed data. Augusto Rendon, Christopher Nelson, and Peter Braund also analysed data. All investigators shared data and had opportunities to contribute to the interpretation of the results and critical revision of the report. All members of the writing committee provided critical revisions. All members of the coordinating centre contributed to the collection, harmonisation, analysis, and interpretation of the data. The data management team undertook data collation and harmonisation.
Writing committee
Nadeem Sarwar* PhD, University of Cambridge, UK; Adam S Butterworth* PhD, University of Cambridge, UK; Daniel F Freitag MPhil, University of Cambridge, UK; John Gregson MPhil, University of Cambridge, UK; Peter Willeit MD, University of Cambridge, UK; Donal N Gorman MPhil, University of Cambridge, UK; Pei Gao PhD, University of Cambridge, UK; Danish Saleheen MD, University of Cambridge, UK; Augusto Rendon PhD, University of Cambridge and MRC Biostatistics Unit, UK; Christopher P Nelson PhD, University of Leicester, UK; Peter S Braund PhD, University of Leicester, UK; Alistair S Hall PhD, University of Leeds, UK; Daniel I Chasman PhD, Brigham and Women's Hospital and Harvard Medical School, USA; Anne Tybjærg-Hansen MD, Copenhagen University Hospital, University of Copenhagen, Denmark; John C Chambers PhD, Imperial College London, UK; Emelia J Benjamin MD, Boston University School of Medicine and Public Health, and National Heart, Lung, and Blood Institute's Framingham Heart Study, USA; Paul W Franks PhD, Department of Clinical Sciences, Genetic and Molecular Epidemiology Unit, Skåne University Hospital, Lund University, Malmö, Sweden, Department of Nutrition, Harvard School of Public Health, Boston, MA, USA, and Department of Public Health and Clinical Medicine, Section for Medicine, Umeå University, Umeå, Sweden; Robert Clarke MD, University of Oxford, UK; Arthur A M Wilde MD, Academic Medical Center, University of Amsterdam, Netherlands; Mieke D Trip MD, Academic Medical Center, University of Amsterdam, Netherlands; Maristella Steri PhD, IRGB-CNR, Italy; Jacqueline C M Witteman PhD, Erasmus Medical Center, Netherlands; Lu Qi MD, Harvard School of Public Health, Brigham and Women's Hospital, USA; C Ellen van der Schoot MD, Sanquin Research, Netherlands; Ulf de Faire MD, Karolinska Institutet, Sweden; Jeanette Erdmann PhD, University of Lübeck, Germany; Heather M Stringham PhD, University of Michigan, Ann Arbor, USA; Wolfgang Koenig MD, University of Ulm, Germany; Daniel J Rader MD, University of Pennsylvania, USA; David Melzer PhD, Peninsula College of Medicine and Dentistry, University of Exeter, UK; David Reich PhD, Harvard Medical School, USA; Bruce M Psaty MD, University of Washington, USA; Marcus E Kleber PhD, LURIC Study nonprofit LLC, Freiburg, Germany; Demosthenes B Panagiotakos MD, Harokopio University of Athens, Greece; Johann Willeit MD, Medical University Innsbruck, Austria; Patrik Wennberg MD, Umeå University, Sweden; Mark Woodward PhD, University of Sydney, Australia; Svetlana Adamovic PhD, Sahlgrenska University Hospital, Sweden; Eric B Rimm ScD, Harvard University, USA; Tom W Meade FRS, London School of Hygiene and Tropical Medicine, UK; Richard F Gillum MD, Center for Disease Control and Prevention, USA; Jonathan A Shaffer PhD, Columbia University Medical Center, USA; Albert Hofman MD, Erasmus Medical Center, Netherlands; Altan Onat MD, Istanbul University, Turkey; Johan Sundström MD, Uppsala University, Sweden; Sylvia Wassertheil-Smoller PhD, Albert Einstein College of Medicine, USA; Dan Mellström MD, Sahlgrenska University Hospital, Sweden; John Gallacher PhD, Cardiff University, UK; Mary Cushman MD, University of Vermont, USA; Russell P Tracy PhD, University of Vermont, USA; Jussi Kauhanen MD, University of Eastern Finland, Finland; Magnus Karlsson MD, Lund University, Sweden; Jukka T Salonen MD, Metabolic Analytical Services Inc, Finland; Lars Wilhelmsen MD, University of Gothenburg, Sweden; Philippe Amouyel MD, Institut Pasteur de Lille, France; Bernard Cantin MD, Hôpital Laval, Canada; Lyle G Best MD, Missouri Breaks Industries Research Inc, Timber Lake, USA; Yoav Ben-Shlomo PhD, University of Bristol, UK; JoAnn E Manson MD, Harvard Medical School, USA; George Davey-Smith MD, University of Bristol, UK; Paul I W de Bakker PhD, Brigham and Women's Hospital, USA; Christopher J O'Donnell MD, Harvard Medical School, USA; James F Wilson DPhil, University of Edinburgh, UK; Anthony G Wilson PhD, University of Sheffield, UK; Themistocles L Assimes MD, Stanford University School of Medicine, USA; John-Olov Jansson MD, Gothenburg University, Sweden; Claes Ohlsson MD, Sahlgrenska University Hospital, Gothenburg, Sweden; Åsa Tivesten MD, Sahlgrenska University Hospital, Gothenburg, Sweden; Östen Ljunggren MD, University of Uppsala, Sweden; Muredach P Reilly MB, University of Pennsylvania, USA; Anders Hamsten FRCP, Karolinska Institutet, Stockholm, Sweden; Erik Ingelsson MD, Karolinska Institutet, Sweden; Francois Cambien MD, INSERM, France; Joseph Hung FRACP, University of Western Australia, Australia; G Neil Thomas PhD, University of Birmingham, UK; Michael Boehnke PhD, University of Michigan, USA; Heribert Schunkert MD, University of Lübeck, Germany; Folkert W Asselbergs MD, University Medical Centre Utrecht, Netherlands; John J P Kastelein MD, Academic Medical Center, University of Amsterdam, Netherlands; Vilmundur Gudnason MD, Icelandic Heart Association and University of Iceland, Iceland; Veikko Salomaa MD, National Institute for Health and Welfare, Finland; Tamara B Harris MD, US National Institute on Aging, USA; Jaspal S Kooner FRCP, National Heart and Lung Institute, Imperial College London, UK; Kristine H Allin MD, Copenhagen University Hospital, University of Copenhagen, Denmark; Børge G Nordestgaard MD, Copenhagen University Hospital, University of Copenhagen, Denmark; Jemma C Hopewell PhD, University of Oxford, UK; Alison H Goodall PhD, University of Leicester, UK; Cardiogenics Consortium; Paul M Ridker MD, Brigham and Women's Hospital and Harvard Medical School, USA; Hilma Hólm MD, deCODE genetics, Reykjavik, Iceland; Hugh Watkins FMedSci, University of Oxford, UK; Willem H Ouwehand MD, University of Cambridge, UK; Nilesh J Samani FMedSci, University of Leicester, UK; Stephen Kaptoge PhD, University of Cambridge, UK; Emanuele Di Angelantonio MD, University of Cambridge, UK; Olivier Harari PhD, Hoffmann-La Roche, Switzerland; John Danesh FRCP, University of Cambridge, UK. *Denotes equal contribution.
Members of the IL6R Genetics Consortium and the Emerging Risk Factors Collaboration
ADVANCE: Themistocles L Assimes, Thomas Quertermous, Alan S Go, Mark A Hlatky, Joshua W Knowles; AGES: Vilmundur Gudnason, Albert V Smith; ATTICA: Demosthenes B Panagiotakos, Christina Chrysohoou, Christos Pitsavos, Christodoulos Stefanadis; BHF-FHS: Christopher P Nelson, Peter S Braund, Nilesh J Samani, Alistair S Hall, Anthony J Balmforth, John R Thompson; BLOODOMICS: Augusto Rendon, Arthur A M Wilde, Mieke D Trip, C Ellen van der Schoot, John J P Kastelein, Willem H Ouwehand, Suthesh Sivapalaratnam, Stephani Maiwald, Hanneke Basart, Mahdi Motazacker, Jonas S S G de Jong, Lucas R C Dekker, Michael Tanck, Connie R Bezzina; BRHS: Peter H Whincup, Richard W Morris, S Goya Wannamethee; BRUN: Johann Willeit, Stefan Kiechl; CAPS: John Gallacher, John W G Yarnell, Gordon Lowe, Ann Rumley; CARDIOGENICS: Nilesh J Samani, Alison H Goodall, Francois Cambien (see appendix p 26 for acknowledgments); CHS: Mary Cushman, Kenneth J Mukamal (see http://www.chs-nhlbi.org for acknowledgments); COPEN/CIHDS: Børge G Nordestgaard, Anne Tybjærg-Hansen, Kristine H Allin; COROGENE: Veikko Salomaa, Aki S Havulinna, Marja-Liisa Lokki, Markku S Nieminen, Samuli Ripatti, Juha Sinisalo; CUDAS/CUPID: Joseph Hung, Brendan M McQuillan, John P Beilby, Peter L Thompson; DECODE: Hilma Hólm, Gu mar Thorleifsson, Gumundur Thorgeirsson, Unnur Thorsteinsdóttir, Kari Stefansson; DILGOM: Veikko Salomaa, Antti Jula, Satu Männistö, Markus Perola, Emmi Tikkanen; EPICNL: Folkert W Asselbergs, Jolanda M A Boer, N Charlotte Onland-Moret, Yvonne T van der Schouw, W M Monique Verschuren, Paul I W de Bakker; FIA: Patrik Wennberg, Jan-Håkan Jansson; FINRISK92,97: Veikko Salomaa; FLETCHER: Mark Woodward; FRAM: Emelia J Benjamin, Josée Dupuis, João D Fontes, Xiaoyan Yin, Christopher J O'Donnell; FUSION: Michael Boehnke, Heather M Stringham, Jaakko Tuomilehto; GerMIFS1 / GerMIFS2: Heribert Schunkert, Jeanette Erdmann, Inke R Koenig, Janja Nahrstaedt, Christina Loley, Klaus Stark, Christina Willenborg, Christian Hengstenberg, Stefan Schreiber, Michael Preuss; GLACIER: Paul W Franks, Inês Barroso, Göran Hallmans, Dmitry Shungin; Guangzhou Biobank Cohort Study: G Neil Thomas, Kar Keung Cheng, Tai Hing Lam, Chao Chiang Jiang; HEALTH: David Reich, Tamara B Harris; HPFS: Eric B Rimm, Jennifer Pai; HPS: Jemma C Hopewell, Rory Collins, Sarah Parish, Jane Armitage; HUNT: Heather M Stringham, Anne Jackson, Kristian Hveem; HVHS: Bruce M Psaty, Kerri L Wiggins, Susan R Heckbert, Nicholas L Smith, Joshua C Bis; inCHIANTI: David Melzer, Luigi Ferrucci, Jack M Guralnik, Stefania Bandinelli, Andrew B Singleton; KIHD: Jussi Kauhanen, Jukka T Salonen, Tomi-Pekka Tuomainen, Sudhir Kurl; LEADER: Tom W Meade; LOLIPOP: John C Chambers, Jaspal S Kooner, Weihua Zhang, Angad S Kooner, Debashis Das; LURIC: Marcus E Kleber, Winfried März, Hubert Scharnagl, Bernhard O Böhm, Bernhard R Winkelmann; MESA: Mary Cushman, Russell P Tracy, Aaron R Folsom, Bruce M Psaty, Steven J Shea (see http://www.mesa-nhlbi.org for acknowledgments); METSIM: Heather M Stringham, Markku Laakso, Johanna Kuusisto; MOGERAUG: Wolfgang Koenig, Jens Baumert, Barbara Thorand, Thomas Illig, Christa Meisinger; MOSWEGOT: Lars Wilhelmsen, Annika Rosengren; MrOS: John-Olov Jansson, Svetlana Adamovic, Magnus K Karlsson, Östen Ljunggren, Dan Mellström, Claes Ohlsson, Åsa Tivesten; NHANESIII: Richard F Gillum; NHS: Eric B Rimm, JoAnn E Manson, Lu Qi, Frank B Hu, Susan E Hankinson; NSHS: Jonathan A Shaffer, Karina W Davidson; ORCADES: James F Wilson, Ross Fraser, Sarah Wild, Harry Campbell; PENNCATH: Daniel J Rader, Muredach P Reilly, Atif Qasim, Liming Qu, Mingyao Li; PIVUS: Erik Ingelsson, Lars Lind, Johan Sundström, Ann-Christine Syvänen; PRIME: Philippe Amouyel, Dominique Arveiler; PROCARDIS: Robert Clarke, Hugh Watkins, Martin Farrall, Jemma C Hopewell, John F Peden; PROMIS: Danish Saleheen, Panos Deloukas, Nasir Sheikh, Asif Rasheed, John Danesh; QUEBEC: Bernard Cantin, Gilles R Dagenais; ROTT: Jacqueline C M Witteman, Albert Hofman, Abbas Dehghan, Cornelia M van Duijn, Andre G Uitterlinden; SARDINIA: Maristella Steri, Goncalo R Abecasis, Francesco Cucca, Serena Sanna, Manuela Uda, David Schlessinger; SCARF: Anders Hamsten, Maria Sabater-Lleal, Angela Silveira; SHEEP: Ulf de Faire, Bruna Gigante; SHS: Lyle G Best, Barbara V Howard; SPEED: George Davey-Smith, Yoav Ben-Shlomo; TARFS: Altan Onat; ULSAM: Erik Ingelsson, Johan Sundström, Lars Lind, Samar Basu, Ann-Christine Syvänen; WGHS: Paul M Ridker, Daniel I Chasman, Lynda M Rose; WHI-HaBPS: Sylvia Wassertheil-Smoller; WHS: Paul M Ridker, Julie Buring.
mar Thorleifsson, Gumundur Thorgeirsson, Unnur Thorsteinsdóttir, Kari Stefansson; DILGOM: Veikko Salomaa, Antti Jula, Satu Männistö, Markus Perola, Emmi Tikkanen; EPICNL: Folkert W Asselbergs, Jolanda M A Boer, N Charlotte Onland-Moret, Yvonne T van der Schouw, W M Monique Verschuren, Paul I W de Bakker; FIA: Patrik Wennberg, Jan-Håkan Jansson; FINRISK92,97: Veikko Salomaa; FLETCHER: Mark Woodward; FRAM: Emelia J Benjamin, Josée Dupuis, João D Fontes, Xiaoyan Yin, Christopher J O'Donnell; FUSION: Michael Boehnke, Heather M Stringham, Jaakko Tuomilehto; GerMIFS1 / GerMIFS2: Heribert Schunkert, Jeanette Erdmann, Inke R Koenig, Janja Nahrstaedt, Christina Loley, Klaus Stark, Christina Willenborg, Christian Hengstenberg, Stefan Schreiber, Michael Preuss; GLACIER: Paul W Franks, Inês Barroso, Göran Hallmans, Dmitry Shungin; Guangzhou Biobank Cohort Study: G Neil Thomas, Kar Keung Cheng, Tai Hing Lam, Chao Chiang Jiang; HEALTH: David Reich, Tamara B Harris; HPFS: Eric B Rimm, Jennifer Pai; HPS: Jemma C Hopewell, Rory Collins, Sarah Parish, Jane Armitage; HUNT: Heather M Stringham, Anne Jackson, Kristian Hveem; HVHS: Bruce M Psaty, Kerri L Wiggins, Susan R Heckbert, Nicholas L Smith, Joshua C Bis; inCHIANTI: David Melzer, Luigi Ferrucci, Jack M Guralnik, Stefania Bandinelli, Andrew B Singleton; KIHD: Jussi Kauhanen, Jukka T Salonen, Tomi-Pekka Tuomainen, Sudhir Kurl; LEADER: Tom W Meade; LOLIPOP: John C Chambers, Jaspal S Kooner, Weihua Zhang, Angad S Kooner, Debashis Das; LURIC: Marcus E Kleber, Winfried März, Hubert Scharnagl, Bernhard O Böhm, Bernhard R Winkelmann; MESA: Mary Cushman, Russell P Tracy, Aaron R Folsom, Bruce M Psaty, Steven J Shea (see http://www.mesa-nhlbi.org for acknowledgments); METSIM: Heather M Stringham, Markku Laakso, Johanna Kuusisto; MOGERAUG: Wolfgang Koenig, Jens Baumert, Barbara Thorand, Thomas Illig, Christa Meisinger; MOSWEGOT: Lars Wilhelmsen, Annika Rosengren; MrOS: John-Olov Jansson, Svetlana Adamovic, Magnus K Karlsson, Östen Ljunggren, Dan Mellström, Claes Ohlsson, Åsa Tivesten; NHANESIII: Richard F Gillum; NHS: Eric B Rimm, JoAnn E Manson, Lu Qi, Frank B Hu, Susan E Hankinson; NSHS: Jonathan A Shaffer, Karina W Davidson; ORCADES: James F Wilson, Ross Fraser, Sarah Wild, Harry Campbell; PENNCATH: Daniel J Rader, Muredach P Reilly, Atif Qasim, Liming Qu, Mingyao Li; PIVUS: Erik Ingelsson, Lars Lind, Johan Sundström, Ann-Christine Syvänen; PRIME: Philippe Amouyel, Dominique Arveiler; PROCARDIS: Robert Clarke, Hugh Watkins, Martin Farrall, Jemma C Hopewell, John F Peden; PROMIS: Danish Saleheen, Panos Deloukas, Nasir Sheikh, Asif Rasheed, John Danesh; QUEBEC: Bernard Cantin, Gilles R Dagenais; ROTT: Jacqueline C M Witteman, Albert Hofman, Abbas Dehghan, Cornelia M van Duijn, Andre G Uitterlinden; SARDINIA: Maristella Steri, Goncalo R Abecasis, Francesco Cucca, Serena Sanna, Manuela Uda, David Schlessinger; SCARF: Anders Hamsten, Maria Sabater-Lleal, Angela Silveira; SHEEP: Ulf de Faire, Bruna Gigante; SHS: Lyle G Best, Barbara V Howard; SPEED: George Davey-Smith, Yoav Ben-Shlomo; TARFS: Altan Onat; ULSAM: Erik Ingelsson, Johan Sundström, Lars Lind, Samar Basu, Ann-Christine Syvänen; WGHS: Paul M Ridker, Daniel I Chasman, Lynda M Rose; WHI-HaBPS: Sylvia Wassertheil-Smoller; WHS: Paul M Ridker, Julie Buring.
Data management team
Matthew Walker, Sarah Watson.
Coordinating centre
Adam S Butterworth, Emanuele Di Angelantonio, Daniel F Freitag, Pei Gao, Donal N Gorman, John Gregson, Stephen Kaptoge, Lisa Pennells, Nadeem Sarwar, Danish Saleheen, Simon G Thompson, Matthew Walker, Sarah Watson, Peter Willeit, Angela M Wood, David Wormser, John Danesh (principal investigator).
Conflicts of interest
Since April, 2011, Nadeem Sarwar has been a full-time employee of Pfizer Inc. Olivier Harari is an employee of Roche Products. Hilma Holm is an employee of deCODE genetics. Bruce Psaty serves on a data safety and monitoring board for a clinical trial of a device funded by the manufacturer Zoll. JoAnn Manson is listed as a co-inventor on a pending patent held by Brigham and Women's Hospital, Harvard Medical School, that relates to inflammatory markers in diabetes prediction.
Paul M Ridker is listed as a co-inventor on patents held by Brigham and Women's Hospital that relate to the use of inflammatory markers in cardiovascular disease, and that have been licensed to AstraZeneca and Siemens. John Danesh has received research funding from the British Heart Foundation, BUPA Foundation, Denka, diaDexus, European Union, European Research Council, Evelyn Trust, Fogarty International Centre, GlaxoSmithKline, Medical Research Council, Merck Sharp and Dohme, National Heart, Lung and Blood Institute, National Institute of Neurological Disorders and Stroke, National Institute for Health Research, Novartis, Pfizer, Roche, Wellcome Trust, and UK Biobank, and has served on advisory boards for Merck, Pfizer, and Novartis, for which he has received compensation. All other members of the writing committee declare that they have no conflicts of interest.
Supplementary Material
References
- 1.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 3.Danesh J, Kaptoge S, Mann AG. Long-term interleukin-6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med. 2008;5:e78. doi: 10.1371/journal.pmed.0050078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Emerging Risk Factors Collaboration C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fibrinogen Studies Collaboration Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294:1799–1809. doi: 10.1001/jama.294.14.1799. [DOI] [PubMed] [Google Scholar]
- 6.C Reactive Protein Coronary Heart Disease Genetics Collaboration (CCGC) Association between C reactive protein and coronary heart disease: mendelian randomisation analysis based on individual participant data. BMJ. 2011;342:d548. doi: 10.1136/bmj.d548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott P, Chambers JC, Zhang W. Genetic loci associated with C-reactive protein levels and risk of coronary heart disease. JAMA. 2009;302:37–48. doi: 10.1001/jama.2009.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keavney B, Danesh J, Parish S. Fibrinogen and coronary heart disease: test of causality by ‘Mendelian randomization’. Int J Epidemiol. 2006;35:935–943. doi: 10.1093/ije/dyl114. [DOI] [PubMed] [Google Scholar]
- 9.Abeywardena MY, Leifert WR, Warnes KE, Varghese JN, Head RJ. Cardiovascular biology of interleukin-6. Curr Pharm Des. 2009;15:1809–1821. doi: 10.2174/138161209788186290. [DOI] [PubMed] [Google Scholar]
- 10.Schuett H, Luchtefeld M, Grothusen C, Grote K, Schieffer B. How much is too much? Interleukin-6 and its signalling in atherosclerosis. Thromb Haemost. 2009;102:215–222. doi: 10.1160/TH09-05-0297. [DOI] [PubMed] [Google Scholar]
- 11.Müllberg J, Oberthür W, Lottspeich F. The soluble human IL-6 receptor. Mutational characterization of the proteolytic cleavage site. J Immunol. 1994;152:4958–4968. [PubMed] [Google Scholar]
- 12.Reich D, Patterson N, Ramesh V, the Health, Aging and Body Composition (Health ABC) Study Admixture mapping of an allele affecting interleukin 6 soluble receptor and interleukin 6 levels. Am J Hum Genet. 2007;80:716–726. doi: 10.1086/513206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lamas JR, Rodriguez-Rodriguez L, Tornero-Esteban P. Mechanisms underlying the generation of soluble IL-6 soluble receptor (sIL-6R) in rheumatoid arthritis: mRNA alternative splicing and proteolytic cleavage as independent contributors. Arthritis Rheum. 2011;63(suppl 10):383. [Google Scholar]
- 14.Stephens OW, Zhang Q, Qu P. An intermediate-risk multiple myeloma subgroup is defined by sIL-6r: levels synergistically increase with incidence of SNP rs2228145 and 1q21 amplification. Blood. 2012;119:503–512. doi: 10.1182/blood-2011-07-367052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridker PM, Pare G, Parker A. Loci related to metabolic-syndrome pathways including LEPR, HNF1A, IL6R, and GCKR associate with plasma C-reactive protein: the Women's Genome Health Study. Am J Hum Genet. 2008;82:1185–1192. doi: 10.1016/j.ajhg.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wassel CL, Lange LA, Keating BJ. Association of genomic loci from a cardiovascular gene SNP array with fibrinogen levels in European Americans and African-Americans from six cohort studies: the Candidate Gene Association Resource (CARe) Blood. 2011;117:268–275. doi: 10.1182/blood-2010-06-289546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Melzer D, Perry JR, Hernandez D. A genome-wide association study identifies protein quantitative trait loci (pQTLs) PLoS Genet. 2008;4:e1000072. doi: 10.1371/journal.pgen.1000072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Danik JS, Paré G, Chasman DI. Novel loci, including those related to Crohn disease, psoriasis, and inflammation, identified in a genome-wide association study of fibrinogen in 17 686 women: the Women's Genome Health Study. Circ Cardiovasc Genet. 2009;2:134–141. doi: 10.1161/CIRCGENETICS.108.825273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dehghan A, Dupuis J, Barbalic M. Meta-analysis of genome-wide association studies in >80 000 subjects identifies multiple loci for C-reactive protein levels. Circulation. 2011;123:731–738. doi: 10.1161/CIRCULATIONAHA.110.948570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferreira MA, Matheson MC, Duffy DL, the Australian Asthma Genetics Consortium Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. Lancet. 2011;378:1006–1014. doi: 10.1016/S0140-6736(11)60874-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eleftherohorinou H, Hoggart CJ, Wright VJ, Levin M, Coin LJ. Pathway-driven gene stability selection of two rheumatoid arthritis GWAS identifies and validates new susceptibility genes in receptor mediated signalling pathways. Hum Mol Genet. 2011;20:3494–3506. doi: 10.1093/hmg/ddr248. [DOI] [PubMed] [Google Scholar]
- 22.Marinou I, Walters K, Winfield J, Bax DE, Wilson AG. A gain of function polymorphism in the interleukin 6 receptor influences RA susceptibility. Ann Rheum Dis. 2010;69:1191–1194. doi: 10.1136/ard.2008.100644. [DOI] [PubMed] [Google Scholar]
- 23.The IBC 50K CAD Consortium Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. 2011;7:e1002260. doi: 10.1371/journal.pgen.1002260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeller T, Wild P, Szymczak S. Genetics and beyond—the transcriptome of human monocytes and disease susceptibility. PLoS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nica AC, Parts L, Glass D, the MuTHER Consortium The architecture of gene regulatory variation across multiple human tissues: the MuTHER study. PLoS Genet. 2011;7:e1002003. doi: 10.1371/journal.pgen.1002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dimas AS, Deutsch S, Stranger BE. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009;325:1246–1250. doi: 10.1126/science.1174148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schadt EE, Molony C, Chudin E. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stranger BE, Nica AC, Forrest MS. Population genomics of human gene expression. Nat Genet. 2007;39:1217–1224. doi: 10.1038/ng2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010;375:1634–1639. doi: 10.1016/S0140-6736(10)60545-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lp-PLA2 Studies Collaboration Lipoprotein-associated phospholipase A2 and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–1544. doi: 10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 33.Ridker PM. Testing the inflammatory hypothesis of atherothrombosis: scientific rationale for the cardiovascular inflammation reduction trial (CIRT) J Thromb Haemost. 2009;7(suppl 1):332–339. doi: 10.1111/j.1538-7836.2009.03404.x. [DOI] [PubMed] [Google Scholar]
- 34.Schunkert H, König IR, Kathiresan S, the Cardiogenics Consortium, and the CARDIoGRAM Consortium Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. 2011;43:333–338. doi: 10.1038/ng.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reilly MP, Li M, He J, the Myocardial Infarction Genetics Consortium, and the Wellcome Trust Case Control Consortium Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome-wide association studies. Lancet. 2011;377:383–392. doi: 10.1016/S0140-6736(10)61996-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation. 2000;101:1767–1772. doi: 10.1161/01.cir.101.15.1767. [DOI] [PubMed] [Google Scholar]
- 37.Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376:1094–1108. doi: 10.1016/S0140-6736(10)60826-4. [DOI] [PubMed] [Google Scholar]
- 38.Furst DE, Keystone EC, Braun J. Updated consensus statement on biological agents for the treatment of rheumatic diseases, 2010. Ann Rheum Dis. 2011;70(suppl 1):i2–36. doi: 10.1136/ard.2010.146852. [DOI] [PubMed] [Google Scholar]
- 39.Protogerou AD, Zampeli E, Fragiadaki K. A pilot study of endothelial dysfunction and aortic stiffness after interleukin-6 receptor inhibition in rheumatoid arthritis. Atherosclerosis. 2011;219:734–736. doi: 10.1016/j.atherosclerosis.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 40.Schuett H, Oestreich R, Waetzig GH. Transsignaling of interleukin-6 crucially contributes to atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:281–290. doi: 10.1161/ATVBAHA.111.229435. [DOI] [PubMed] [Google Scholar]
- 41.Bannwarth B, Richez C. Clinical safety of tocilizumab in rheumatoid arthritis. Expert Opin Drug Saf. 2011;10:123–131. doi: 10.1517/14740338.2011.537256. [DOI] [PubMed] [Google Scholar]
- 42.Choy E, Sattar N. Interpreting lipid levels in the context of high-grade inflammatory states with a focus on rheumatoid arthritis: a challenge to conventional cardiovascular risk actions. Ann Rheum Dis. 2009;68:460–469. doi: 10.1136/ard.2008.101964. [DOI] [PubMed] [Google Scholar]
- 43.Nurmohamed MT. Atherogenic lipid profiles and its management in patients with rheumatoid arthritis. Vasc Health Risk Manag. 2007;3:845–852. [PMC free article] [PubMed] [Google Scholar]
- 44.US Food and Drug Administration FDA approves new drug for rheumatoid arthritis. 2011. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm197108.htm (accessed Feb 6, 2012).
- 45.Genovese MC, Smolen JS, Emery P. Analysis of low-density lipoprotein and adverse events in rheumatoid arthritis patients treated with tocilizumab plus statins: data from five phase III clinical trials. Rheumatology (Oxford) 2009;48:I81. (abstr 175). [Google Scholar]
- 46.Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease-drug-drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2011;89:735–740. doi: 10.1038/clpt.2011.35. [DOI] [PubMed] [Google Scholar]
- 47.Kanoni S, Willenborg C, Thompson J, et al. Multiple novel loci conferring risk to coronary artery disease identified in a study of 63,253 cases and 126,820 controls. ICHG/ASHG Scientific Meeting; Montreal, Canada; Oct 11–15, 2011. Abstr 75.
- 48.Linsel-Nitschke P, Götz A, Erdmann J, the Wellcome Trust Case Control Consortium (WTCCC), and the Cardiogenics Consortium Lifelong reduction of LDL-cholesterol related to a common variant in the LDL-receptor gene decreases the risk of coronary artery disease—a Mendelian Randomisation study. PLoS One. 2008;3:e2986. doi: 10.1371/journal.pone.0002986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McCarthy MI. Genomics, type 2 diabetes, and obesity. N Engl J Med. 2010;363:2339–2350. doi: 10.1056/NEJMra0906948. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.