

Abstract
Eosinophilic bronchitis is a common cause of chronic cough, which like asthma is characterized by sputum eosinophilia, but unlike asthma there is no variable airflow obstruction or airway hyperresponsiveness. Several studies suggest that prostaglandins may play an important role in orchestrating interactions between different cells in several inflammatory diseases such as asthma. PGE2 is important because of the multiplicity of its effects on immune response in respiratory diseases; however, respiratory system appears to be unique in that PGE2 has beneficial effects. We described that the difference in airway function observed in patients with eosinophilic bronchitis and asthma could be due to differences in PGE2 production. PGE2 present in induced sputum supernatant from NAEB patients decreases BSMC proliferation, probably due to simultaneous stimulation of EP2 and EP4 receptors with inhibitory activity. This protective effect of PGE2 may not only be the result of a direct action exerted on airway smooth-muscle proliferation but may also be attributable to the other anti-inflammatory actions.
1. Introduction
Asthma has consistently been reported as a major cause of chronic cough [1]. The development of noninvasive assessment of airway inflammation led to the identification of a condition that manifests chronic cough in individuals without the abnormalities of airway function that characterize asthma, but with sputum eosinophilia. This condition was named nonasthmatic eosinophilic bronchitis (NAEB) [2]. The reason for the absence of airway hyperresponsiveness in the NAEB remains unclear. Inflammation of the airways, with recruitment and activation of T lymphocytes, eosinophils, and mast cells and release of inflammatory mediators, plays an important role in the pathophysiology of asthma and NAEB. Among lipid mediators, PGE2 is a mediator thought to have an important role.
This paper shall summarize our current knowledge of the role of PGE2 in lung and in respiratory disease such as asthma and nonasthmatic eosinophilic bronchitis.
2. PGE2 Biosynthesis
Several studies suggest that prostaglandins may play an important role in orchestrating interactions between different cells in several inflammatory diseases such as asthma and rheumatoid arthritis.
Although the term prostaglandin was coined in the 1930s by Von Euler [3], and Bergstrom and Samuelsson defined the structure of two first prostaglandins in 1960 [4], the full structure of prostaglandins was not identified until 1965 by Orloff. Prostaglandins are arachidonic acid (AA) metabolites which result from enzymes with cyclooxygenase (COX) activity [5]. These metabolites are small lipidic molecules implicated in the regulation of many different processes in the organism. Their production begins with the liberation of AA from membrane phospholipids by phospholipase A2 in response to inflammatory stimuli [6]. AA is then transformed into prostaglandin H2 (PGH2) by COXs, which is the first step in eicosanoid biosynthesis. PGH2 is an unstable molecule that is transformed into several biologically active prostaglandins through specific enzymes with different tissue and cellular expression pattern [7].
Two isoforms of COX have been identified, and they are classified as COX-1 and COX-2. The main differences between them are their expression regulation and tissue distribution. In terms of expression, COX-1 is constitutively expressed in cells in which prostaglandins exert physiological functions, while COX-2 expression is enhanced after inflammatory stimuli [8], such as LPS, several proinflammatory cytokines (tumor necrosis factor-alpha, interleukin-1-beta), growth factors, or tumoral promoter as PMA [4, 9]. Both isoforms catalyze similar reactions although they are codified by different genes [10]. COX-1 is associated with immediate biosynthesis of prostaglandins (several minutes after stimulation) which develop homeostatic functions; COX-2 is linked to delayed biosynthesis of prostaglandins (several hours after stimulus) which exert pathological effects. Other difference is the cellular localization; thus, COX-1 is expressed in endoplasmic reticulo, whereas COX-2 is situated in perinuclear membrane [7, 11].
PGE2 is the most abundantly produced prostanoid in the body and has been shown to play an important role in regulating inflammatory processes. PGE2 production is largely dependent upon three enzymatic reactions: generation of arachidonic acid from membrane glycerophospholipids via phospholipase A2, conversion of AA to an unstable intermediate prostanoid (PGH2) by COXs, and metabolism of prostaglandin H2 to prostaglandin E2 via prostaglandin E synthase [12].
There are three enzymes that catalyze PGE2 generation starting from PGH2, namely, membrane-bound PGES (mPGES)-1 [13], mPGES-2 [14], and cytosolic PGE (cPGES) [15] which constitute two biosynthetic pathways to PGE2 secretion; on one hand, COX-1/cPGES, the pathway implicated in homeostasis and does not play an important role in PGE2 production [16], and on the other, COX-2/mPGES-1, the pathway associated with delayed response, inflammation, and pathology [17], and that is essential for basal production in some organs as brain, spleen, and stomach (at least in mice) [18].
Major cellular sources of PGE2 include epithelial cells, endothelial cells, airway smooth muscle, and monocytes/macrophages [19].
3. PGE2 Receptors
PGE2 exerts its effects by acting on a group of rhodopsin-type G-protein-coupled membrane receptors (GPCRs) termed E-prostanoid (EP) receptors. There are four GPCR subtypes: EP1, EP2, EP3, and EP4 [20, 21]. Expression regulation of the various subtypes of EP receptor by several agents, such as inflammatory stimuli, or even PGE2 itself, enables PGE2 to affect tissues in a very specific and diverse manner [22, 23]. These subtypes of EP receptor differ in the intracellular signaling [24]. The expression or a combination of receptors, each which may be differentially expressed in a number of tissues (i.e., airway smooth muscle, neurons, or immune effector cells), results in a specific physiological response. These receptors could be classified according to their intracellular signaling and second messenger. EP1 receptor activation leads to an increase in intracellular calcium, usually coupled to Gq protein, and its stimulation is linked to phospholipase C [25]. EP2 and EP4 receptors couple to Gs, and activation of these receptors results in stimulation of adenylyl cyclase and increases intracellular cAMP [26]. The major signaling pathway described for the EP3 receptor is mediated by Gαi and leads to a reduction in intracellular cAMP levels. However, several EP3 receptor isoforms generated by alternative splicing from the single EP3 receptor gene have been identified, and the intracellular signal may differ [27]. Some of these isoforms of the EP3 receptor coupled to multiple G proteins produced either inhibition of adenylate cyclase and calcium mobilization or stimulation of adenylate cyclase activity [26]. Thus, accumulation of cAMP promoted by EP2 and EP4 receptors is associated with the inhibition of effector cell functions; however, EP1 and some isoforms of the EP3 receptor that increase intracellular calcium could be linked to promotion of cellular activation [20].
In pathological conditions, the role of these receptors is determined by pattern expression, ligand affinity, and their differential coupling to transduction signaling pathways in which the cellular context of the receptor is very relevant.
4. PGE2 Effects on Respiratory System
PGE2 is almost ubiquitous in humans and evokes potent diverse actions. It regulates several functions in the major human systems, including the gastrointestinal, reproductive, neuroendocrine, and immune systems [28].
It is in the area of inflammation that the actions of PGE2 are most diverse because of the many specialized cell types as well as the complex and sometimes seemingly opposing actions that make that PGE2 one of the most heterogeneous eicosanoids. This complexity reflects differences between endogenous formation and action versus pharmacological and exogenous addition of PGE2 in vivo and in vitro [29].
PGE2 acts like a pleiotropic prostaglandin with stimulating or inhibiting properties and could be classified as a regulatory element of immunity [30]. An example of this stimulating/inhibiting polarity is the opposite actions that PGE2 exerts in the cardiovascular and immune system, where PGE2 is able to enhance the inflammation caused by leukotrienes as well as inhibit the release of mediators and regulate monocyte macrophages and dendritic cells [31].
Although PGE2 acts in multiple systems, we will focus our attention on the role that this prostaglandin plays in the inflammatory process linked to respiratory system.
PGE2 is the major metabolite in the lower respiratory tract. In this system, epithelium and airway smooth muscle are the principal source of PGE2 [32], though fibroblast, alveolar macrophages, and pulmonary endothelial cells also produce it [33, 34]. PGE2 is important because of the multiplicity of its effects on immune response in respiratory diseases.
PGE2 is commonly presumed to be a proinflammatory mediator and has been implicated in several inflammatory disease conditions, including rheumatoid arthritis [35]; however, PGE2 has protective effects in different organs, and respiratory system appears to be one of them in that PGE2 has beneficial effects [26, 36–38].
During the 1970s, PGE2 was shown to protect against bronchoconstriction produced by ultrasonically nebulized distilled water [39] and exercise-induced asthma [40]. In the early 1990s, Pavord et al. [41] showed that inhaled PGE2 protected against bronchial hyperreactivity to sodium metabisulphite in which bronchoconstriction is thought to be neurally mediated. In this study, furosemide protected against bronchoconstrictor challenges in asthma, and this effect may be mediated through PGE2. Multiple subsequent studies have observed the bronchodilator effect of PGE2 in normal subjects [42] and patients with asthma and chronic bronchitis [43], showing that PGE2 attenuates bronchoconstriction, possibly inhibiting the release of the bronchoconstrictor mediators which are responsible for exercise bronchoconstriction. This prostanoid inhibits early and late allergen-induced bronchoconstriction, increasing the relaxation of airway smooth muscle and inhibiting the release of mast-cell mediators and the recruitment of inflammatory cells [34]. Moreover, PGE2 also decreases or inhibits the accompanying bronchial hyperresponsiveness to methacholine [36].
All these positive effects of PGE2 are mainly mediated through EP2 and EP4 receptors [44]. PGE2 can mediate bronchodilation via the EP2 receptor [45] and also anti-inflammatory effects via the EP2 and/or the EP4 receptor [46]. Also, EP2 plays an important role in aspirin-intolerant asthma because a reduction in released PGE2 and lower expression of its EP2 receptor provoked an increase in inflammatory process in the airways of these patients [47]. However, PGE2 also induces irritation of the upper airway, resulting in a reflex cough and enhancing the response to capsaicin. The coughing induced by this prostanoid is caused mainly, if not solely, by activation of the EP3 receptor, and in bronchoconstrictor effects are implicated by both EP1 and EP3 receptors [48].
5. PGE2 and Inflammatory Cells
Immune cells produce a variety of prostaglandins that have both proinflammatory and anti-inflammatory effects [49]. Eosinophils are one of predominant inflammatory cells in the lungs of asthmatic patients, and changes in the number and degree of lung eosinophils probably influence disease severity. Peacock and colleagues demonstrated that PGE2 suppresses eosinophil apoptosis, and this is likely mediated by interaction with the EP2 receptor subtype in eosinophils [50], by contrast, misoprostol (a PGE2 analogue) inhibits eosinophil survival in vitro [51]. Nevertheless, in recent years, all studies have shown a negative regulator role of PGE2 on eosinophils. This prostanoid directly controls eosinophils' migration on the cellular levels and is highly potent and efficacious. This effect is brought about mainly by EP2 receptor involving PI3K- and PKC-dependent pathways [52]. Furthermore, PGE2 not only attenuates eosinophil trafficking but also abolishes the production of reactive oxygen species, Ca2+ responses, and upregulation of adhesion molecules through the EP4 receptor [53]. EP4 does not seem to depend on activation of the adenylyl cyclase/PKA pathway. Its stimulation causes phosphorylation of extracellular signal-regulated kinases (ERKs) through a PI3K-dependent mechanism [53]. So, an alternative EP2/EP4 signaling pathway in which both PI3K and PKC activation are implicated has been postulated.
PGE2 also acts on T cells and alveolar macrophages. It is able to decrease proliferation of lymphocytes, subsequently decreasing the production of Th2 cytokines [54]. In another study, PGE2 produced an increase of IL-10 expression, an important regulatory cytokine [55]. In contrast, PGE2 positively controls the regulatory T cells (Treg) which are pivotal in suppressing immune responses and maintaining tolerance. PGE2 upregulates FOXP3 mRNA and protein expression and enhances FOXP3 promoter activity [56]. PGE2 also enhances Ig class switch to IgE in B cells [57].
PGE2 is a route through which airway epithelial cells (AECs) modulate specific cellular subtypes such as dendritic cells. Along these lines, Schmidt and colleagues [58] demonstrated that epithelial cells, through the constitutive secretion of PGE2, drive DCs to adopt an anti-inflammatory phenotype in an EP4 receptor-dependent manner coupled to cAMP production. As a result, PGE2-EP4 receptor signaling generates DCs with reduced proinflammatory properties (decreased production of TNF-α and enhancing IL-10 secretion), thereby limiting DC activation [58].
Also, in alveolar macrophages, it produces an increase of IL-10 production and a decrease of TNF-α levels generated by alveolar macrophages [59, 60].
6. PGE2 and Lung Structure
As mentioned above, the principal sources of PGE2 in airways are the epithelial, endothelial, fibroblast, and smooth muscle cells. The epithelium is the first barrier to protect against injury. Its cells create an anti-inflammatory microenvironment that modulates the phenotype of local antigen-presenting cells (APCs), regulating the activation of local professional immune cells. A chronic state of inflammation and wound healing exists in the lung as a part of asthma pathology. Moreover, fibroblasts actively migrate and proliferate, synthesize and secrete extracellular matrix components, and differentiate into myofibroblasts. In this process, PGE2 exerts significant negative regulatory functions by inhibiting fibroblast migration and chemotaxis with a dominant role of EP2 receptors in the cAMP-dependent inhibitory effects [61, 62], thereby decreasing fibroblast proliferation through Epac-1 and subsequent Rap1 activation (cAMP effectors) in a manner which is independent of ERK1/2 participation [63]; furthermore, PGE2 inhibits collagen expression by a PKA-mediated process which is linked to EP2 and EP4 receptors [63]. Impaired ex vivo COX-2 function and PGE2 synthesis that characterize fibroblast during the evolution of airway fibrosis may likewise promote fibrogenesis [64]. However, this inhibition of fibrosis may also occur through an inhibition of fibroblast PAR1 expression, by PAR2-mediated generation of PGE2, one of the protease-activated receptors coupled to G protein that possesses a unique mechanism of activation and induces COX activation in a variety of cell types [65]. PAR-1 signalling is a well-known profibrotic pathway [66], so inhibition of PAR1 expression diminished pulmonary fibrosis.
PGE2 has complex effects on airway tone, and the existence of several E-prostanoid receptors, each one with different signalling characteristics, has provided a possible explanation for the seemingly contradictory actions of this lipid mediator. Potent relaxant effects on airway smooth muscle have been observed; however, human studies with aerosolized PGE2 have demonstrated inconsistent effects on airway tone, with most asthmatics showing a bronchodilator response but some developing profound bronchoconstriction requiring beta agonist rescue [36, 41, 43, 48].
7. Asthma and Nonasthmatic Eosinophilic Bronchitis
Asthma is a complex chronic disease of the airways that has been estimated to affect over 300 million people worldwide, and the burden is likely to rise in the coming decades [64, 67].
The inflammatory process in asthma is characterized by inflammatory cells, mainly eosinophils, mast cells, basophils, macrophages, and Th2 cells. These cells are involved in the development of another hallmark of asthma as airway hyperresponsiveness (AHR), reversible airway obstruction, cough, mucus secretion, and structural changes by releasing inflammatory mediators such as cytokines, chemokines, growth factors, and chemical and lipid mediators [68, 69]. The complex pathogenesis of asthma is contributed to by various cellular responses, based on the dysregulated interaction between innate and adaptative immune systems.
Chronic cough is a common reason for referral to a specialist, and asthma has consistently been reported among the most common causes of chronic cough, accounting for about 25% of such cases in adult nonsmokers [1, 70, 71]. The development of sputum induction has provided a safe noninvasive method of assessing airway inflammation. One of the most interesting early observations made using this method was the identification of a group of patients with sputum eosinophilia identical to that seen asthma, but with normal airway function. The physiological features of this condition were different from those of asthma, and Gibson and colleagues suggested that the new disease should be known as eosinophilic bronchitis [2]. The cough in patients with nonasthmatic eosinophilic bronchitis (NAEB) responds well to inhaled corticosteroids, though the same is not the case for cough in patients without sputum eosinophilia. NAEB is responsible for about 10% of cases of isolated chronic cough referred for specialist investigation [72].
The etiology of asthma and NAEB is usually unknown, although both can be associated with exposure to an occupational sensitizer or to common inhaled allergens [73–75]; thus, the triggers that cause eosinophilic bronchitis without asthma are similar to the triggers of eosinophilic bronchitis with asthma.
In patients with eosinophilic bronchitis, there is a clear dissociation between sputum eosinophilia and airway hyperresponsiveness. The pathophysiology of eosinophilic bronchitis and the reason for the absence of airway hyperresponsiveness in this disease remains unclear. One possible explanation is that the eosinophilic airway inflammation is less active than in asthma, with less release of effector mediators. Several studies using different techniques to assess airway inflammation have shown that the inflammatory component is very similar in patients with asthma and NAEB [76]. Indeed, asthma and eosinophilic bronchitis share many immunopathologic features including increased number of eosinophils and mast cells in the superficial airway. In addition to airway eosinophilia, both conditions are associated with reticular basement membrane thickening and similar number of subepithelial T lymphocytes, mast cells, and macrophages [76]. Eosinophilic bronchitis is a disease characterized by increased expression of Th2 cytokines (IL-4, IL-5, IL-10, and IL-13) [77, 78]. These data point to a dissociation between T-cell activation and the abnormalities in airway physiology that characterize asthma [79]. Such results suggest that Th2-mediated cytokines are closely linked to eosinophilic inflammation, though they are not necessarily associated with the physiologic hallmarks of the asthmatic phenotype.
One aspect of the inflammatory response that might be particularly important is the localization of mast cells in the airway wall, since they are present within the airway smooth muscle in asthma but not in NAEB [80]. Moreover, in subjects with eosinophilic bronchitis, CXCL8 and CXCL10 concentrations were elevated in airway secretions. These chemokines may play a key role in mast cell recruitment to the superficial airway in this condition [81]. Siddiqui and colleagues have demonstrated that mast cells are microlocalized within the airway smooth muscle bundle in asthma, which is associated with airway hyperresponsiveness [82]. Mast cells produce a variety of mediators that may interact with bronchial smooth muscle and subsequently become hyperresponsive to constrictive stimuli and proliferation [83].
8. PGE2 in Asthma and NAEB
We recently found that induced sputum prostaglandin E2 (PGE2) concentrations are strikingly increased in subjects with NAEB as compared with asthmatic and healthy subjects [78]. This study illustrates that, like asthma, there is active airway inflammation in eosinophilic bronchitis with release of different inflammatory mediators. These data suggest that the differences in airway function observed in subjects with NAEB and asthma may be due to differences in PGE2 production.
Brightling et al. [84] also found numerically higher levels of PGE2 in sputum from patients with eosinophilic bronchitis, though the differences were not statistically significant as compared with asthma patients. Recently, elevated sputum PGE2 concentrations have been found in all patients with chronic cough [73], although in this study cough variant asthma and eosinophilic bronchitis patients were included in the same group.
The differences in sputum PGE2 concentration between asthmatic and eosinophilic bronchitis patients may be the result of the implication of a different degradation kinetic or activity of enzymes in the synthesis and/or degradation of products of arachidonic acid metabolism. It has been recently shown that Th2 cytokines have specific effects on PGE synthase 1 and 15-PGDH enzymes in airway human epithelial cells decreasing PGE2 [85].
We postulate that PGE2 elevation in patients with eosinophilic bronchitis may have protective effects against the development of bronchial hyperresponsiveness. The fact that PGE2 and its analogue have a number of bronchoprotective and anti-inflammatory effects in vitro [37] or after inhalation [36], as well as on allergen-induced airway responses and airway inflammation in atopic asthma [38, 86], would support such a protective role. An imbalance in the ratio of bronchoconstrictor (LTC4) and bronchoprotective (PGE2) lipid mediators may have a role in the pathogenesis of eosinophilic bronchitis.
PGE2 can perform contrasting activities, which can thus lead to bronchodilation and act as anti-inflammatory or proinflammatory mediator substances in the lung [26]. Recently, several studies showed that the PGE2 protective actions were mediated in large part by the EP2 receptors [87, 88]. This protective action of PGE2 might not only result in a direct effect on airway smooth-muscle relaxation, but also in the inhibition of many inflammatory processes [89]. These data may explain why patients with eosinophilia have normal tests of variable airway obstruction and airway responsiveness and experience chronic cough due to high PGE2 levels [90].
The pathogenesis of eosinophilic bronchitis might be the opposite of that observed in aspirin-induced asthma. There is increasing evidence that, by inhibiting cyclooxygenase-1, the protective effect exerted by endogenous prostaglandin E2 on leukotriene generation by mast cells, eosinophils, and macrophages in the airways is removed in aspirin-induced asthma. In these patients with aspirin-induced bronchoconstriction, a low production of PGE2 has been observed, seemingly due to deficient COX-2 regulation and increased expression of leukotriene-C4 synthase [91].
9. Role of PGE2 in NAEB
Muscle hypertrophy and hyperplasia are characteristics of asthmatic airways, and this increased layer of bronchial smooth muscle contributes to AHR in asthmatic patients [92, 93]. We hypothesized that high PGE2 levels in the lower airways of NAEB patients constitute the essential regulatory mediator causing inhibition of bronchial smooth muscle cell proliferation (BSMC) and subsequent hyperresponsiveness [94]. Thus, we demonstrated that when BSMCs are cultured with induced sputum supernatant from NAEB patients, a strong inhibition of muscle cell proliferation takes place [94]. This inhibition was not due to the apoptotic effect of sputum supernatants or to differences in cytokine levels found in sputum. Formal proof of this finding was the addition of EP2 and EP4 antagonists to the culture recovery of the proliferation of smooth muscle cells. The lesser inhibition obtained from sputum of asthmatic patients may be explained by the absence or defectiveness of PGE2 production, as well as by differential EP2 and EP4 receptor expression from different pathologies. Similarly, Lundequist and colleagues recently described a role for PGE2 in protecting the pulmonary vasculature from remodeling dependent on more than one EP receptor [95].
An alternative explanation of the differences in airway function between asthma and NAEB is the different localization of mast cells within the airway wall. Siddiqui and colleagues have demonstrated that mast cells are microlocalized within the airway smooth muscle bundle in asthma, and this is associated with AHR [82]. Mast cells produce a variety of mediators that may interact with BSM and subsequently become hyperresponsive to constrictive stimuli and proliferation [83]. We hypothesize that both mechanisms may act synergistically (Figure 1). Recently, Duffy and colleagues reported that the engagement of the EP2 receptor closes the K+ channel KCa 3.1 in human lung mast cells and attenuates their migration [96]; thus, PGE2 present in sputum supernatant from NAEB patients could close the KCa 3.1 channel and inhibit mast cell migration to the airway wall and subsequently bring about microlocalization within BSMC. Furthermore, in the inhibition of BSM proliferation produced by PGE2, the K+ channel KCa 3.1 may be implicated since activated K+ channels regulate human airway smooth muscle proliferation [97].
Figure 1.
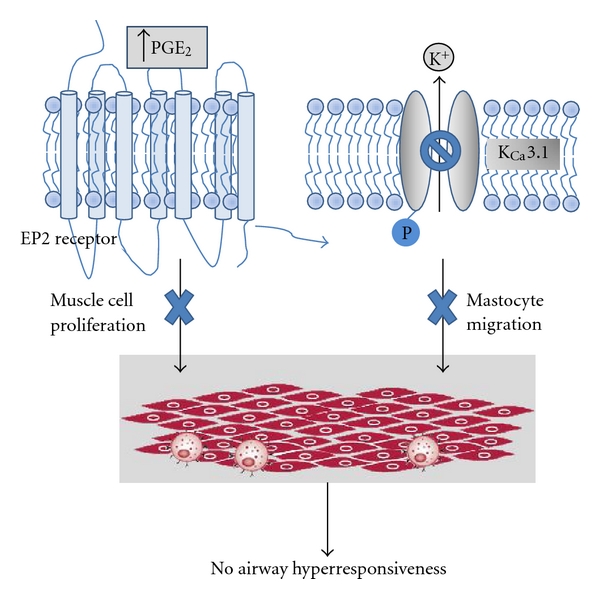
Hypothetical mechanisms through which PGE2 reduces the AHR and in NAEB. (a) The PGE2 decreases the smooth muscle proliferation producing a reduction of muscular hyperplasia, via EP2 and EP4 receptors; (b) The PGE2 closes the KCa 3.1 channel, preventing the migration of mastocytes by means of EP2. Both mechanisms will decrease or inhibit airway hyperresponsiveness, a relevant hallmark of asthma.
Airway smooth muscle cells are the major effector cells regulating bronchomotor tone in response to several mediators [98]. Some authors have reported that increased vascularity, reticular basement membrane thickening, and increased airway smooth muscle mass are features of both diseases [99, 100]. However, the same authors have recently reported that patients with asthma had airway wall thickening, as opposed to subjects with NAEB, who maintained airway patency without wall thickening [101]. In addition, AHR and altered airway geometry were found to be correlated in asthma patients. Maintained proximal airway patency in NAEB compared to the subjects with asthma may protect against the development of AHR. In line with this, Park et al. have reported that proximal airway wall thickening is not a feature of NAEB [102].
In conclusion, PGE2 present in induced sputum supernatant from NAEB patients decreases BSMC proliferation, probably due to simultaneous stimulation of EP2 and EP4 receptors with inhibitory activity. This protective effect of PGE2 may not only be the result of a direct action exerted on airway smooth-muscle proliferation but also may be attributable to the other anti-inflammatory actions. Thus, PGE2 agonist receptors may become a novel therapeutic approach for inflammatory respiratory diseases.
Acknowledgments
This study was supported by Fondo de Investigación Sanitaria—FIS [PI06/055 and PS09/00153]; CIBER de Enfermedades Respiratorias (CIBERES), a Carlos III Institute of Health Initiative. The authors recognize Oliver Shaw for his revision and editing in English.
References
- 1.Brightling CE. Cough due to asthma and nonasthmatic eosinophilic bronchitis. Lung. 2010;188:S13–S17. doi: 10.1007/s00408-009-9163-5. [DOI] [PubMed] [Google Scholar]
- 2.Gibson PG, Dolovich J, Denburg J, Ramsdale EH, Hargreave FE. Chronic cough: eosinophilic bronchitis without asthma. Lancet. 1989;1(8651):1346–1348. doi: 10.1016/s0140-6736(89)92801-8. [DOI] [PubMed] [Google Scholar]
- 3.Von Euler US. A depressor substance in the vesicular gland. Journal of Physiology. 1935;84:p. 21. [Google Scholar]
- 4.Clàiria J. Cyclooxygenase-2 biology. Current Pharmaceutical Design. 2003;9(27):2177–2190. doi: 10.2174/1381612033454054. [DOI] [PubMed] [Google Scholar]
- 5.Profita M, Sala A, Bonanno A, et al. Increased prostaglandin E2 concentrations and cyclooxygenase-2 expression in asthmatic subjects with sputum eosinophilia. Journal of Allergy and Clinical Immunology. 2003;112(4):709–716. doi: 10.1016/s0091-6749(03)01889-x. [DOI] [PubMed] [Google Scholar]
- 6.Harris SG, Padilla J, Koumas L, Ray D, Phipps RP. Prostaglandins as modulators of immunity. Trends in Immunology. 2002;23(3):144–150. doi: 10.1016/s1471-4906(01)02154-8. [DOI] [PubMed] [Google Scholar]
- 7.Ueno N, Takegoshi Y, Kamei D, Kudo I, Murakami M. Coupling between cyclooxygenases and terminal prostanoid synthases. Biochemical and Biophysical Research Communications. 2005;338(1):70–76. doi: 10.1016/j.bbrc.2005.08.152. [DOI] [PubMed] [Google Scholar]
- 8.Smith WL, Langenbach R. Why there are two cyclooxygenase isozymes. Journal of Clinical Investigation. 2001;107(12):1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gye YP, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. American Journal of Physiology. 2006;290(5):L797–L805. doi: 10.1152/ajplung.00513.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warner TD, Mitchell JA. Cyclooxygenases: new forms, new inhibitors, and lessons from the clinic. FASEB Journal. 2004;18(7):790–804. doi: 10.1096/fj.03-0645rev. [DOI] [PubMed] [Google Scholar]
- 11.Brock TG, Peters-Golden M. Activation and regulation of cellular eicosanoid biosynthesis. TheScientificWorldJournal. 2007;7:1273–1284. doi: 10.1100/tsw.2007.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henry PJ. The protease-activated receptor2 (PAR2)-prostaglandin E2-prostanoid EP receptor axis: a potential bronchoprotective unit in the respiratory tract? European Journal of Pharmacology. 2006;533(1–3):156–170. doi: 10.1016/j.ejphar.2005.12.051. [DOI] [PubMed] [Google Scholar]
- 13.Murakami M, Naraba H, Tanioka T, et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. Journal of Biological Chemistry. 2000;275(42):32783–32792. doi: 10.1074/jbc.M003505200. [DOI] [PubMed] [Google Scholar]
- 14.Tanikawa N, Ohmiya Y, Ohkubo H, et al. Identification and characterization of a novel type of membrane-associated prostaglandin E synthase. Biochemical and Biophysical Research Communications. 2002;291(4):884–889. doi: 10.1006/bbrc.2002.6531. [DOI] [PubMed] [Google Scholar]
- 15.Tanioka T, Nakatani Y, Semmyo N, Murakami M, Kudo I. Molecular identification of cytosolic prostaglandin E2 synthase that is functionally coupled with cyclooxygenase-1 in immediate prostaglandin E2 biosynthesis. Journal of Biological Chemistry. 2000;275(42):32775–32782. doi: 10.1074/jbc.M003504200. [DOI] [PubMed] [Google Scholar]
- 16.Lovgren AK, Kovarova M, Koller BH. cPGES/p23 is required for glucocorticoid receptor function and embryonic growth but not prostaglandin E2 synthesis. Molecular and Cellular Biology. 2007;27(12):4416–4430. doi: 10.1128/MCB.02314-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murakami M, Kudo I. Recent advances in molecular biology and physiology of the prostaglandin E2-biosynthetic pathway. Progress in Lipid Research. 2004;43(1):3–35. doi: 10.1016/s0163-7827(03)00037-7. [DOI] [PubMed] [Google Scholar]
- 18.Boulet L, Ouellet M, Bateman KP, et al. Deletion of microsomal prostaglandin E2 (PGE2) synthase-1 reduces inducible and basal PGE2 production and alters the gastric prostanoid profile. Journal of Biological Chemistry. 2004;279(22):23229–23237. doi: 10.1074/jbc.M400443200. [DOI] [PubMed] [Google Scholar]
- 19.Ying S, O’Connor BJ, Meng Q, et al. Expression of prostaglandin E2 receptor subtypes on cells in sputum from patients with asthma and controls: effect of allergen inhalational challenge. Journal of Allergy and Clinical Immunology. 2004;114(6):1309–1316. doi: 10.1016/j.jaci.2004.08.034. [DOI] [PubMed] [Google Scholar]
- 20.Tilley SL, Coffman TM, Koller BH. Mixed messages: modulation of inflammation and immune responses by prostaglandins and thromboxanes. Journal of Clinical Investigation. 2001;108(1):15–23. doi: 10.1172/JCI13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annual Review of Pharmacology and Toxicology. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 22.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiological Reviews. 1999;79(4):1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 23.Narumiya S. Prostanoid receptors. Structure, function, and distribution. Annals of the New York Academy of Sciences. 1994;744:126–138. doi: 10.1111/j.1749-6632.1994.tb52729.x. [DOI] [PubMed] [Google Scholar]
- 24.Norel X, Walch L, Labat C, Gascard JP, Dulmet E, Brink C. Prostanoid receptors involved in the relaxation of human bronchial preparations. British Journal of Pharmacology. 1999;126(4):867–872. doi: 10.1038/sj.bjp.0702392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watabe A, Sugimoto Y, Honda A, et al. Cloning and expression of cDNA for a mouse EP1 subtype of prostaglandin E receptor. Journal of Biological Chemistry. 1993;268(27):20175–20178. [PubMed] [Google Scholar]
- 26.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. The lung as a privileged site for the beneficial actions of PGE2 . Trends in Immunology. 2004;25(1):40–46. doi: 10.1016/j.it.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen M, Solle M, Audoly LP, et al. Receptors and signaling mechanisms required for prostaglandin E2-mediated regulation of mast cell degranulation and IL-6 production. Journal of Immunology. 2002;169(8):4586–4593. doi: 10.4049/jimmunol.169.8.4586. [DOI] [PubMed] [Google Scholar]
- 28.Serhan CN, Levy B. Success of prostaglandin E2 in structure-function is a challenge for structure-based therapeutics. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):8609–8611. doi: 10.1073/pnas.1733589100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trebino CE, Stock JL, Gibbons CP, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phipps RP, Stein SH, Roper RL. A new view of prostaglandin E regulation of the immune response. Immunology Today. 1991;12(10):349–352. doi: 10.1016/0167-5699(91)90064-Z. [DOI] [PubMed] [Google Scholar]
- 31.Rocca B, FitzGerald GA. Cyclooxygenases and prostaglandins: shaping up the immune response. International Immunopharmacology. 2002;2(5):603–630. doi: 10.1016/s1567-5769(01)00204-1. [DOI] [PubMed] [Google Scholar]
- 32.Ozaki T, Rennard SI, Crystal RG. Cyclooxygenase metabolites are compartmentalized in the human lower respiratory tract. Journal of Applied Physiology. 1987;62(1):219–222. doi: 10.1152/jappl.1987.62.1.219. [DOI] [PubMed] [Google Scholar]
- 33.Widdicombe JH, Ueki IF, Emery D, Margolskee D, Yergey J, Nadel JA. Release of cyclooxygenase products from primary cultures of tracheal epithelia of dog and human. American Journal of Physiology. 1989;257(6):L361–L365. doi: 10.1152/ajplung.1989.257.6.L361. [DOI] [PubMed] [Google Scholar]
- 34.Sheller JR, Mitchell D, Meyrick B, Oates J, Breyer R. EP2 receptor mediates bronchodilation by PGE2 in mice. Journal of Applied Physiology. 2000;88(6):2214–2218. doi: 10.1152/jappl.2000.88.6.2214. [DOI] [PubMed] [Google Scholar]
- 35.McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. Journal of Clinical Investigation. 2002;110(5):651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavord ID, Wong CS, Williams J, Tattersfield AE. Effect of inhaled prostaglandin E2 on allergen-induced asthma. American Review of Respiratory Disease. 1993;148(1):87–90. doi: 10.1164/ajrccm/148.1.87. [DOI] [PubMed] [Google Scholar]
- 37.Pavord ID, Tattersfield AE. Bronchoprotective role for endogenous prostaglandin E2 . Lancet. 1995;345(8947):436–438. doi: 10.1016/s0140-6736(95)90409-3. [DOI] [PubMed] [Google Scholar]
- 38.Gauvreau GM, Watson RM, O’Byrne PM. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. American Journal of Respiratory and Critical Care Medicine. 1999;159(1):31–36. doi: 10.1164/ajrccm.159.1.9804030. [DOI] [PubMed] [Google Scholar]
- 39.Pasargiklian M, Bianco S, Allegra L. Clinical, functional and pathogenetic aspects of bronchial reactivity to prostaglandins F2alpha, E1, and E2 . Advances in prostaglandin and thromboxane research. 1976;1:461–475. [PubMed] [Google Scholar]
- 40.Pasargiklian M, Bianco S, Allegra L. Aspects of bronchial reactivity to prostaglandins and aspirin in asthmatic patients. Respiration. 1977;34(2):79–91. [PubMed] [Google Scholar]
- 41.Pavord ID, Wisniewski A, Mathur R, Wahedna I, Knox AJ, Tattersfield AE. Effect of inhaled prostaglandin E2 on bronchial reactivity to sodium metabisulphite and methacholine in patients with asthma. Thorax. 1991;46(9):633–637. doi: 10.1136/thx.46.9.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Costello JF, Dunlop LS, Gardiner PJ. Characteristics of prostaglandin induced cough in man. British Journal of Clinical Pharmacology. 1985;20(4):355–359. doi: 10.1111/j.1365-2125.1985.tb05077.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melillo E, Woolley KL, Manning PJ, Watson RM, O’Byrne PM. Effect of inhaled PGE2 on exercise-induced bronchoconstriction in asthmatic subjects. American Journal of Respiratory and Critical Care Medicine. 1994;149(5):1138–1141. doi: 10.1164/ajrccm.149.5.8173753. [DOI] [PubMed] [Google Scholar]
- 44.Maher SA, Belvisi MG. Prostanoids and the cough reflex. Lung. 2010;188:S9–S12. doi: 10.1007/s00408-009-9190-2. [DOI] [PubMed] [Google Scholar]
- 45.Kay LJ, Yeo WW, Peachell PT. Prostaglandin E2 activates EP2 receptors to inhibit human lung mast cell degranulation. British Journal of Pharmacology. 2006;147(7):707–713. doi: 10.1038/sj.bjp.0706664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takayama K, García-Cardeña G, Sukhova GK, Comander J, Gimbrone MA, Libby P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. Journal of Biological Chemistry. 2002;277(46):44147–44154. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- 47.Roca-Ferrer J, Garcia-Garcia FJ, Pereda J, et al. Reduced expression of COXs and production of prostaglandin E2 in patients with nasal polyps with or without aspirin-intolerant asthma. Journal of Allergy and Clinical Immunology. 2011;128(1):66–72. doi: 10.1016/j.jaci.2011.01.065. [DOI] [PubMed] [Google Scholar]
- 48.Tilley SL, Hartney JM, Erikson CJ, et al. Receptors and pathways mediating the effects of prostaglandin E2 on airway tone. American Journal of Physiology - Lung Cellular and Molecular Physiology. 2003;284(4):L599–L606. doi: 10.1152/ajplung.00324.2002. [DOI] [PubMed] [Google Scholar]
- 49.Sousa AR, Pfister R, Christie PE, et al. Enhanced expression of cyclo-oxygenase isoenzyme 2 (COX-2) in asthmatic airways and its cellular distribution in aspirin-sensitive asthma. Thorax. 1997;52(11):940–945. doi: 10.1136/thx.52.11.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peacock CD, Misso NLA, Watkins DN, Thompson PJ. PGE2 and dibutyryl cyclic adenosine monophosphate prolong eosinophil survival in vitro. Journal of Allergy and Clinical Immunology. 1999;104(1):153–162. doi: 10.1016/s0091-6749(99)70127-2. [DOI] [PubMed] [Google Scholar]
- 51.Alam R, Dejarnatt A, Stafford S, Forsythe PA, Kumar D, Grant JA. Selective inhibition of the cutaneous late but not immediate allergic response to antigens by misoprostol, a PGE analog: results of a double-blind, placebo-controlled randomized study. American Review of Respiratory Disease. 1993;148(4):1066–1070. doi: 10.1164/ajrccm/148.4_Pt_1.1066. [DOI] [PubMed] [Google Scholar]
- 52.Sturm EM, Schratl P, Schuligoi R, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. Journal of Immunology. 2008;181(10):7273–7283. doi: 10.4049/jimmunol.181.10.7273. [DOI] [PubMed] [Google Scholar]
- 53.Luschnig-Schratl P, Sturm EM, Konya V, et al. EP4 receptor stimulation down-regulates human eosinophil function. Cellular and Molecular Life Sciences. 2011;68(21):3573–3587. doi: 10.1007/s00018-011-0642-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jarvinen L, Badri L, Wettlaufer S, et al. Lung resident mesenchymal stem cells isolated from human lung allografts inhibit T cell proliferation via a soluble mediator. Journal of Immunology. 2008;181(6):4389–4396. doi: 10.4049/jimmunol.181.6.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benbernou N, Esnault S, Shin HCK, Fekkar H, Guenounou M. Differential regulation of IFN-γ IL-10 and inducible nitric oxide synthase in human T cells by cyclic AMP-dependent signal transduction pathway. Immunology. 1997;91(3):361–368. doi: 10.1046/j.1365-2567.1997.00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baratelli F, Lin Y, Zhu L, et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. Journal of Immunology. 2005;175(3):1483–1490. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 57.Roper RL, Brown DM, Phipps RP. Prostaglandin E2 promotes B lymphocyte Ig isotype switching to IgE. Journal of Immunology. 1995;154(1):162–170. [PubMed] [Google Scholar]
- 58.Schmidt LM, Belvisi MG, Bode KA, et al. Bronchial epithelial cell-derived prostaglandin E2 dampens the reactivity of dendritic cells. Journal of Immunology. 2011;186(4):2095–2105. doi: 10.4049/jimmunol.1002414. [DOI] [PubMed] [Google Scholar]
- 59.Ménard G, Turmel V, Bissonnette EY. Serotonin modulates the cytokine network in the lung: involvement of prostaglandin E2 . Clinical and Experimental Immunology. 2007;150(2):340–348. doi: 10.1111/j.1365-2249.2007.03492.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ratcliffe MJ, Walding A, Shelton PA, Flaherty A, Dougall IG. Activation of E-prostanoid4 and E-prostanoid2 receptors inhibits TNF-α release from human alveolar macrophages. European Respiratory Journal. 2007;29(5):986–994. doi: 10.1183/09031936.00131606. [DOI] [PubMed] [Google Scholar]
- 61.Li YJ, Wang XQ, Sato T, et al. Prostaglandin E2 inhibits human lung fibroblast chemotaxis through disparate actions on different E-prostanoid receptors. American Journal of Respiratory Cell and Molecular Biology. 2010;44(1):99–107. doi: 10.1165/rcmb.2009-0163OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang S, Wettlaufer SH, Hogaboam C, Aronoff DM, Peters-Golden M. Prostaglandin E2 inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. American Journal of Physiology. 2007;292(2):L405–L413. doi: 10.1152/ajplung.00232.2006. [DOI] [PubMed] [Google Scholar]
- 63.Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. American Journal of Respiratory Cell and Molecular Biology. 2008;39(4):482–489. doi: 10.1165/rcmb.2008-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stumm CL, Wettlaufer SH, Jancar S, Peters-Golden M. Airway remodeling in murine asthma correlates with a defect in PGE2 synthesis by lung fibroblasts. American Journal of Physiology. 2011;301(5):L636–L644. doi: 10.1152/ajplung.00158.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peters T, Henry PJ. Protease-activated receptors and prostaglandins in inflammatory lung disease. British Journal of Pharmacology. 2009;158(4):1017–1033. doi: 10.1111/j.1476-5381.2009.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Howell DC, Laurent GJ, Chambers RC. Role of thrombin and its major cellular receptor, protease-activated receptor-1, in pulmonary fibrosis. Biochemical Society Transactions. 2002;30(2):211–216. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- 67.Brightling CE, Gupta S, Hollins F, Sutcliffe A, Amrani Y. Immunopathogenesis of severe asthma. Current Pharmaceutical Design. 2011;17(7):667–673. doi: 10.2174/138161211795429028. [DOI] [PubMed] [Google Scholar]
- 68.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma: from bronchoconstriction to airways inflammation and remodeling. American Journal of Respiratory and Critical Care Medicine. 2000;161(5):1720–1745. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 69.Jeffery PK, Wardlaw AJ, Nelson FC, Collins JV, Kay AB. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. American Review of Respiratory Disease. 1989;140(6):1745–1753. doi: 10.1164/ajrccm/140.6.1745. [DOI] [PubMed] [Google Scholar]
- 70.Irwin RS, Madison JM. The persistently troublesome cough. American Journal of Respiratory and Critical Care Medicine. 2002;165(11):1469–1474. doi: 10.1164/rccm.2110097. [DOI] [PubMed] [Google Scholar]
- 71.McGarvey LPA, Heaney LG, Lawson JT, et al. Evaluation and outcome of patients with chronic non-productive cough using a comprehensive diagnostic protocol. Thorax. 1998;53(9):738–743. doi: 10.1136/thx.53.9.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brightling CE, Ward R, Goh KL, Wardlaw AJ, Pavord ID. Eosinophilic bronchitis is an important cause of chronic cough. American Journal of Respiratory and Critical Care Medicine. 1999;160(2):406–410. doi: 10.1164/ajrccm.160.2.9810100. [DOI] [PubMed] [Google Scholar]
- 73.Birring SS, Parker D, Brightling CE, Bradding P, Wardlaw AJ, Pavord ID. Induced sputum inflammatory mediator concentrations in chronic cough. American Journal of Respiratory and Critical Care Medicine. 2004;169(1):15–19. doi: 10.1164/rccm.200308-1092OC. [DOI] [PubMed] [Google Scholar]
- 74.Gibson PG, Fujimura M, Niimi A. Eosinophilic bronchitis: clinical manifestations and implications for treatment. Thorax. 2002;57(2):178–182. doi: 10.1136/thorax.57.2.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lemière C, Efthimiadis A, Hargreave FE. Occupational eosinophilic bronchitis without asthma: an unknown occupational airway disease. Journal of Allergy and Clinical Immunology. 1997;100(6):852–853. doi: 10.1016/s0091-6749(97)70286-0. [DOI] [PubMed] [Google Scholar]
- 76.Gibson PG, Zlatic K, Scott J, Sewell W, Woolley K, Saltos N. Chronic cough resembles asthma with IL-5 and granulocyte-macrophage colony-stimulating factor gene expression in bronchoalveolar cells. Journal of Allergy and Clinical Immunology. 1998;101(3):320–326. doi: 10.1016/S0091-6749(98)70242-8. [DOI] [PubMed] [Google Scholar]
- 77.Brightling CE, Symon FA, Birring SS, Bradding P, Pavord ID, Wardlaw AJ. TH2 cytokine expression in bronchoalveolar lavage fluid T lymphocytes and bronchial submucosa is a feature of asthma and eosinophilic bronchitis. Journal of Allergy and Clinical Immunology. 2002;110(6):899–905. doi: 10.1067/mai.2002.129698. [DOI] [PubMed] [Google Scholar]
- 78.Sastre B, Fernández-Nieto M, Mollá R, et al. Increased prostaglandin E2 levels in the airway of patients with eosinophilic bronchitis. Allergy. 2008;63(1):58–66. doi: 10.1111/j.1398-9995.2007.01515.x. [DOI] [PubMed] [Google Scholar]
- 79.Brightling CE, Symon FA, Birring SS, Bradding P, Wardlaw AJ, Pavord ID. Comparison of airway immunopathology of eosinophilic bronchitis and asthma. Thorax. 2003;58(6):528–532. doi: 10.1136/thorax.58.6.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brightling CE, Pavord ID. Location, location, location: microlocalisation of inflammatory cells and airway dysfunction. Thorax. 2004;59(9):734–735. doi: 10.1136/thx.2004.026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woodman L, Sutcliffe A, Kaur D, et al. Chemokine concentrations and mast cell chemotactic activity in BAL fluid in patients with eosinophilic bronchitis and asthma, and in normal control subjects. Chest. 2006;130(2):371–378. doi: 10.1378/chest.130.2.371. [DOI] [PubMed] [Google Scholar]
- 82.Siddiqui S, Hollins F, Saha S, Brightling CE. Inflammatory cell microlocalisation and airway dysfunction: cause and effect? European Respiratory Journal. 2007;30(6):1043–1056. doi: 10.1183/09031936.00162506. [DOI] [PubMed] [Google Scholar]
- 83.Robinson DS. The role of the mast cell in asthma: induction of airway hyperresponsiveness by interaction with smooth muscle? Journal of Allergy and Clinical Immunology. 2004;114(1):58–65. doi: 10.1016/j.jaci.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 84.Brightling CE, Ward R, Woltmann G, et al. Induced sputum inflammatory mediator concentrations in eosinophilic bronchitis and asthma. American Journal of Respiratory and Critical Care Medicine. 2000;162(3):878–882. doi: 10.1164/ajrccm.162.3.9909064. [DOI] [PubMed] [Google Scholar]
- 85.Trudeau J, Hu H, Chibana K, Chu HW, Westcott JY, Wenzel SE. Selective downregulation of prostaglandin E2-related pathways by the TH2 cytokine IL-13. Journal of Allergy and Clinical Immunology. 2006;117(6):1446–1454. doi: 10.1016/j.jaci.2006.01.049. [DOI] [PubMed] [Google Scholar]
- 86.Pavord ID, Ward R, Woltmann G, Wardlaw AJ, Sheller JR, Dworski R. Induced sputum eicosanoid concentrations in asthma. American Journal of Respiratory and Critical Care Medicine. 1999;160(6):1905–1909. doi: 10.1164/ajrccm.160.6.9903114. [DOI] [PubMed] [Google Scholar]
- 87.Feng C, Beller EM, Bagga S, Boyce JA. Human mast cells express multiple EP receptors for prostaglandin E 2 that differentially modulate activation responses. Blood. 2006;107(8):3243–3250. doi: 10.1182/blood-2005-07-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hartney JM, Coggins KG, Tilley SL, et al. Prostaglandin E2 protects lower airways against bronchoconstriction. American Journal of Physiology. 2006;290(1):L105–L113. doi: 10.1152/ajplung.00221.2005. [DOI] [PubMed] [Google Scholar]
- 89.Tanaka H, Kanako S, Abe S. Prostaglandin E2 receptor selective agonists E-prostanoid 2 and E-prostanoid 4 may have therapeutic effects on ovalbumin-induced bronchoconstriction. Chest. 2005;128(5):3717–3723. doi: 10.1378/chest.128.5.3717. [DOI] [PubMed] [Google Scholar]
- 90.Coleridge HM, Coleridge JCG, Ginzel KH. Stimulation of ’irritant’ receptors and afferent C fibres in the lungs by prostaglandins. Nature. 1976;264(5585):451–453. doi: 10.1038/264451a0. [DOI] [PubMed] [Google Scholar]
- 91.Picado C. Aspirin-intolerant asthma: role of cyclo-oxygenase enzymes. Allergy. 2002;57(72):58–60. doi: 10.1034/j.1398-9995.57.s72.14.x. [DOI] [PubMed] [Google Scholar]
- 92.Jeffery PK. Remodeling and inflammation of bronchi in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1(3):176–183. doi: 10.1513/pats.200402-009MS. [DOI] [PubMed] [Google Scholar]
- 93.Johnson PRA, Roth M, Tamm M, et al. Airway smooth muscle cell proliferation is increased in asthma. American Journal of Respiratory and Critical Care Medicine. 2001;164(3):474–477. doi: 10.1164/ajrccm.164.3.2010109. [DOI] [PubMed] [Google Scholar]
- 94.Sastre B, Fernández-Nieto M, López E, et al. PGE2 decreases muscle cell proliferation in patients with non-asthmatic eosinophilic bronchitis. Prostaglandins and Other Lipid Mediators. 2011;95(1-4):11–18. doi: 10.1016/j.prostaglandins.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 95.Lundequist A, Nallamshetty SN, Xing W, et al. Prostaglandin E2 exerts homeostatic regulation of pulmonary vascular remodeling in allergic airway inflammation. Journal of Immunology. 2010;184(1):433–441. doi: 10.4049/jimmunol.0902835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Duffy SM, Cruse G, Cockerill SL, Brightling CE, Bradding P. Engagement of the EP2 prostanoid receptor closes the K+ channel KCa3.1 in human lung mast cells and attenuates their migration. European Journal of Immunology. 2008;38(9):2548–2556. doi: 10.1002/eji.200738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shepherd MC, Duffy SM, Harris T, et al. KCa3.1 Ca2+-activated K+ channels regulate human airway smooth muscle proliferation. American Journal of Respiratory Cell and Molecular Biology. 2007;37(5):525–531. doi: 10.1165/rcmb.2006-0358OC. [DOI] [PubMed] [Google Scholar]
- 98.Hirst SJ. Regulation of airway smooth muscle cell immunomodulatory function: Role in asthma. Respiratory Physiology and Neurobiology. 2003;137(2-3):309–326. doi: 10.1016/s1569-9048(03)00155-1. [DOI] [PubMed] [Google Scholar]
- 99.Siddiqui S, Sutcliffe A, Shikotra A, et al. Vascular remodeling is a feature of asthma and nonasthmatic eosinophilic bronchitis. Journal of Allergy and Clinical Immunology. 2007;120(4):813–819. doi: 10.1016/j.jaci.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 100.Siddiqui S, Mistry V, Doe C, et al. Airway hyperresponsiveness is dissociated from airway wall structural remodeling. Journal of Allergy and Clinical Immunology. 2008;122(2):335–341.e3. doi: 10.1016/j.jaci.2008.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Siddiqui S, Gupta S, Cruse G, et al. Airway wall geometry in asthma and nonasthmatic eosinophilic bronchitis. Allergy. 2009;64(6):951–958. doi: 10.1111/j.1398-9995.2009.01951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Park SW, Park JS, Lee YM, et al. Differences in radiological/HRCT findings in eosinophilic bronchitis and asthma: Implication for bronchial responsiveness. Thorax. 2006;61(1):41–47. doi: 10.1136/thx.2005.044420. [DOI] [PMC free article] [PubMed] [Google Scholar]