

Abstract
Major trauma results in a strong inflammatory response in injured tissue. This posttraumatic hyperinflammation has been implied in the adverse events leading to a breakdown of host defense mechanisms and ultimately to delayed organ failure. Ligands to peroxisome proliferator-activated receptors (PPARs) have recently been identified as potent modulators of inflammation in various acute and chronic inflammatory conditions. The main mechanism of action mediated by ligand binding to PPARs is the inhibition of the nuclear transcription factor NF-κB, leading to downregulation of downstream gene transcription, such as for genes encoding proinflammatory cytokines. Pharmacological PPAR agonists exert strong anti-inflammatory properties in various animal models of tissue injury, including central nervous system trauma, ischemia/reperfusion injury, sepsis, and shock. In addition, PPAR agonists have been shown to induce wound healing process after tissue trauma. The present review was designed to provide an up-to-date overview on the current understanding of the role of PPARs in the pathophysiology of the inflammatory response after major trauma. Therapeutic options for using recombinant PPAR agonists as pharmacological agents in the management of posttraumatic inflammation will be discussed.
1. Introduction
Severe trauma still represents the most frequent cause of death in people below the age of 40 years [1]. Despite research efforts and improved intensive care of patients with trauma, no causal protective therapy is currently available, and the clinical outcome of multiply injured patients is still poor. Following severe tissue injury, a series of inflammatory events is initiated that have to be tightly orchestrated to guarantee efficient tissue repair. A controlled posttraumatic inflammatory response consists of limited release of proinflammatory mediators and recruitment of immune cells that contribute to wound healing and restoration of organ function [2]. If trauma is severe or if additional “second hit” insults occur, this primary protective activation of the immune system may turn to an uncontrolled inflammatory response which might cause additional tissue damage and impair the outcome of trauma [3, 4]. In addition, early posttraumatic hypoxia and hypotension due to the interruption of the blood flow may induce ischemia and reperfusion injuries and increase the extent of harmful events [5].
Peroxisome proliferator-activated receptors (PPARs) are nuclear membrane-associated transcription factors which have recently been shown to possess profound anti-inflammatory functions in a broad field of injury-associated conditions including trauma of the central nervous system (CNS), ischemia/reperfusion injury, sepsis, and shock [6–8]. The present review outlines the current understanding of PPAR-mediated anti-inflammatory mechanisms and discusses both opportunities and limitations of PPAR ligands as potential treatment strategy in trauma.
2. Peroxisome Proliferator-Activated Receptors (PPARs)
PPARs are ligand-activated membrane-associated transcription factors belonging to the nuclear hormone receptor family. To date, three subtypes of PPARs have been identified in various species, PPARα, PPARβ/δ, and PPARγ, that exhibit differential tissue distributions and ligand specificities [9, 10]. Whereas PPARβ/δ is ubiquitously expressed in a wide range of cells, PPARα is found in tissues with high fatty acid catabolism such as brown adipose tissue, liver, heart, kidney, and skeletal muscle, and PPARγ is mainly expressed in the brown and white adipose tissue [10]. A wide range of natural and synthetic compounds can function as PPAR ligands. The natural agonists include fatty acids and fatty acid derivates, mainly eicosanoids, that can bind to and activate all three PPAR subtypes. With respect to the synthetic ligands, fibrates as lipid-lowering drugs function as PPARα agonists, and glitazones, a class of oral antidiabetic agents, have been described to bind to PPARγ [9, 11].
PPARs are activated by heterodimerization with the retinoid-X receptor (RXR) into biologically active transcription factors. PPAR-RXR heterodimers then bind to specific DNA sequences, known as peroxisome proliferator response elements (PPREs), in the promotor region of target genes, thereby acting as a transcriptional regulator [12]. In addition, PPARα and PPARγ are also capable of regulating gene expression independently of binding to PPREs. The activity of a number of transcription factors, for example NF-κB (nuclear factor-κB), AP-1 (activator protein-1), and STAT-1 (signal transducer and activator of transcription), can be inhibited by PPARs either via direct interaction or by competition for limited supplies of coactivators [11, 13]. This function is important in regard to the anti-inflammatory properties of PPARs since proinflammatory gene expression is mainly affected by this direct repression of transcription factors.
Since their first description, PPARs have been implicated in numerous biological processes and diseases. They play a central role in the regulation of glucose, lipid, and lipoprotein metabolism, and PPAR agonists are established drugs for treatment of diabetes (glitazones) and dyslipidemia (fibrates) [14]. Furthermore, angiogenesis, cellular differentiation and proliferation as well as apoptosis are mediated by PPARs, so that PPARs were suggested to contribute to tissue repair and cancer-related pathways [15, 16]. More recent studies could demonstrate an additional involvement of PPARs in senescence-related diseases, in the regulation of male and female fertility, and in cardiovascular conditions like atherosclerosis [17, 18].
In regard to the immune system, PPARs have been identified as crucial regulators of inflammatory gene expression. Various immune cells were shown to express PPARs including dendritic cells, monocytes, macrophages, B- and T-lymphocytes, and vascular endothelial cells [11]. At the site of inflammation, PPARβ/δ ligands reduce the expression of adhesion molecules by endothelial cells and decrease the release of cytokines and chemokines by macrophages. PPARα activation could be linked to inhibited production of proinflammatory cytokines from TH1 cells and increased release of anti-inflammatory cytokines from TH2 cells [19]. These numerous anti-inflammatory properties of PPARs have suggested a central role of PPAR activation in attenuating the inflammatory response after trauma and tissue injury.
3. The Role PPARs in Central Nervous System Trauma
Traumatic injury of the CNS remains a major health problem worldwide and represents one of the leading causes of death and persisting neurological impairment in industrialized countries [1, 20]. Brain damage after traumatic brain injury (TBI) is determined by a combination of primary and secondary insults [21, 22]. While the primary injury is induced by the mechanical impact to the skull and brain, secondary brain injury results from an uncontrolled neuroinflammatory response within the injured brain.
Recent experimental studies have indicated that activation of PPARs might represent a promising therapeutic strategy for counteracting this deleterious posttraumatic inflammation [23, 24]. PPAR expression has been described in intracerebral and spinal vascular endothelial cells, neurons, astrocytes, and oligodendrocytes [25, 26]. Interestingly, two recent studies could show an upregulation of cortical PPAR expression both in experimental models of CNS injury, and in the brain of head-injured patients [27–29].
The key role of PPARs in attenuating neuroinflammation has been examined in multiple models of stroke, ischemia/reperfusion injury and CNS trauma as well as in chronic brain disorders like Parkinson's and Alzheimer's disease [6, 30]. In these studies [30–32], PPAR activation was shown to induce neuroprotection through three distinct main mechanisms, as outlined schematically in Figure 1. First, the cerebral inflammatory response itself was modulated by PPAR-induced inhibition of macrophage/monocyte activation and proinflammatory cytokine release and suppressed upregulation of cellular adhesion molecules [33, 34]. Second, PPARs were able to modulate oxidative stress in the brain, a significant component enhancing the inflammatory cascade and secondary brain injury; activation of PPARs led to reduced production of reactive oxygen species and nitric oxide and increased the release of antioxidants around the injured tissue [35, 36]. Finally, PPARs have been shown to delay neuronal apoptosis and decrease lesion sizes by the inhibition of proapoptotic mediators [27, 37].
Figure 1.
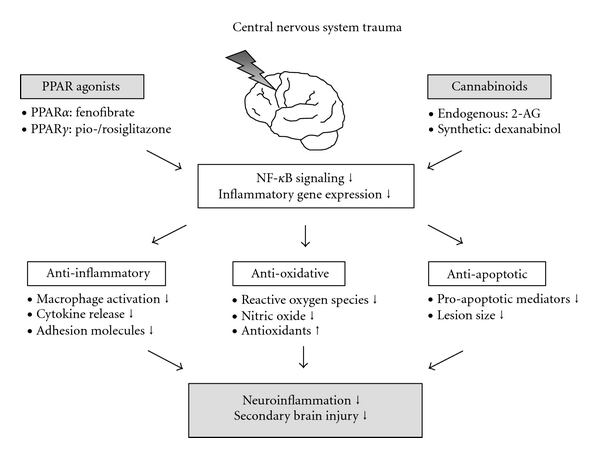
Overview of the anti-inflammatory and neuroprotective effects of selected PPAR agonists in central nervous system (CNS) injury, as exemplified in the setting of traumatic brain injury. See text for detailed explanations. Abbreviations: PPAR, peroxisome proliferator-activated receptor; 2-AG, 2-arachidonyl glycerol; NF-κB, nuclear factor-κB; CNS, central nervous system.
As mentioned above, PPARs act by repressing central inflammatory transcription factors like NF-κB or AP-1 [11]. This mechanism is likely to underlie the described anti-inflammatory effect of PPAR activation after CNS trauma and seems to be responsible for the reduced expression of several key downstream inflammatory genes in the injured CNS [32].
Genovese et al. have examined the effect of PPAR-α gene depletion in a mouse model of spinal cord injury: PPAR-α −/− mice showed significantly augmented injury parameters such as edema, neutrophil infiltration, and apoptosis [38]. Moreover, the therapeutic effect of glucocorticoids has been compared in PPAR-α −/− and wild-type mice [39]. Since the anti-inflammatory properties of glucocorticoids were markedly weakened in PPAR-α −/− mice, it was hypothesized that PPAR-α could contribute to the anti-inflammatory activity of glucocorticoids in CNS trauma [39].
Paying attention to the above mentioned crucial role of PPARs in neuroprotection, pharmacological agonism to PPARs has been of particular interest to the neuroscience community in recent years (Figure 1) [33, 40]. Originally, PPAR-α ligands have been described as highly promising anti-inflammatory drugs (reviewed in [6]). In a rat model of traumatic brain injury, the PPAR-α agonist fenofibrate was revealed to significantly ameliorate pathophysiology after injury including an improvement of neurological scores and a reduction of brain edema [41]. In addition, fenofibrate showed antioxidant effects demonstrated by decrease of intracerebral markers of oxidative stress [36]. A study from the same group assessed a synergistical effect of a combination therapy with fenofibrate and simvastatin, a lipid-lowering drug [42]. The administration of these pharmacological compounds in combination after TBI exerted a more sustained neurological recovery than the monotherapies and might have important significance for the treatment of TBI [42].
More recently, studies have moved their focus on the therapeutic efficacy of agonists to PPAR-γ [24, 43]. Pioglitazone and rosiglitazone are both approved drugs for diabetes treatment and have been shown to bind to PPAR-γ [24]. Sauerbeck et al. hypothesized that pioglitazone would promote neuroprotection in a rat controlled cortical impact model of TBI where the drug was administered every 24 hours beginning 15 minutes after injury [44]. Treatment with pioglitazone resulted in significantly improved cognitive function, reduced lesion size, and prevented the activation of microglia, compared to the vehicle-treated animals [44, 45]. Similar beneficial effects could be demonstrated for rosiglitazone both in traumatic brain and spine injury [27, 34, 46, 47]. Taken together, PPAR-γ agonists have been shown to exert a wide range of anti-inflammatory, anti-oxidative, and anti-apoptotic effects and were like that able to counteract the main pathophysiological events occurring in the development of secondary CNS injury [27, 44, 47–49]. These observations could strengthen the idea that the field of application of PPAR-γ agonists could be expanded from antidiabetic drugs to therapeutic agents counteracting neuroinflammation and improving the outcome of patients with traumatic CNS injuries.
In addition to synthetic PPAR agonists, a protective role of cannabinoids on the sequelae of CNS trauma has been suggested in recent years [50–52]. The group of cannabinoids consists of endogenous ligands, such as 2-arachidonyl glycerol (2-AG), and synthetic drugs like dexanabinol (HU-211) [53]. After TBI in mice, elevated levels of 2-AG could be detected [54]. Administration of 2-AG to mice after experimental head injury suppressed the release of reactive oxygen species and proinflammatory cytokines and improved clinical recovery [54, 55]. Similar to that, the synthetic cannabinoid HU-211 revealed as a cerebroprotectant attenuating breakdown of the blood-brain barrier and production of cytokines [56, 57]. Recent studies have provided evidence that an increasing number of anti-inflammatory actions of cannabinoids are not mediated by the classical cannabinoid receptors but are induced by agonistic action of cannabinoids to PPARs [53, 58]. Oleylethanolamide that is structurally related to the endogenous cannabinoid anandamide was found to bind to PPAR-α, and Δ9-tetrahydrocannbinol (THC), the active ingredient of cannabis, has been described as a PPAR-γ ligand [59, 60]. Interestingly, the mechanism by which 2-AG suppresses interleukin-2 expression was linked to targeting PPAR-γ and was independent of cannabinoid receptors [58]. Due to the promising anti-inflammatory properties of cannabinoids, a randomized phase II and III clinical trial was initiated that aimed to test HU-211 in the treatment of brain trauma [52, 61, 62]. In the study patients treated with HU-211 achieved significantly decreased intracranial pressure, and a trend towards better neurological outcome was observed [61].
In summary, numerous attempts of targeting PPARs both by cannabinoids and by synthetic PPAR ligands have revealed the huge neuroprotective ability of PPAR agonism. Further studies will have to examine if administration of a single PPAR ligand or a combination of cannabinoids with synthetic PPAR agonists might have the potential to be introduced in the clinic of traumatic CNS injury.
4. PPARs in Ischemia/Reperfusion Injury
Ischemia/reperfusion (I/R) injury represents a challenging pathophysiological condition with serious clinical implications, in a broad field of conditions such as organ transplantation, compartment syndrome, myocardial infarction, stroke, and hemorrhagic, traumatic, or septic shock [5, 63]. Tissue ischemia together with subsequent reperfusion has been shown to trigger a whole cascade of inflammatory events that, if not counteracted in early stages, result in cell necrosis with irreversible tissue damage in affected organs [7]. Research efforts in recent years have provided increasing evidence that PPARs represent major regulators of this inflammatory response; PPAR activation could be shown to restrict inflammation and exert multiple beneficial effects against ischemia/reperfusion injury [7, 15]. Consequently, pharmacological agents targeting PPARs have been suggested as potential therapeutics for the treatment of I/R.
Similar as described for traumatic CNS injuries, a strong relationship between PPAR tissue expression and I/R injury could be demonstrated. In kidney I/R, PPARγ expression was strongly increased in endothelial cells, interstitial cells, and collecting ducts of the kidney peaking from 1.5 to 5 hours after reperfusion [64, 65]. Similar upregulation of PPARγ was detected in the cortical peri-infarct area after focal cerebral ischemia in rats [66]. Interestingly, Lee and colleagues have recently found in a model of transient cerebral ischemia that PPARγ-immunoreactivity in the hippocampus was colocalized with microglial cells indicating a high functional state of microglia in the ischemic brain [67].
In recent years, animal studies of I/R injury in various organs have revealed a crucial role of PPARs in reducing or even preventing tissue injury and organ dysfunction after ischemia and reperfusion [15, 68, 69]. Consequently, a wide variety of natural and synthetic PPAR agonists was tested in experimental I/R models and was shown to significantly improve the outcome of I/R injury [5, 70, 71]. The mechanisms of tissue protection by PPAR ligands have been thought to be multifactorial [7], since these agonists can interact with variable parameters of the IR-induced inflammatory cascade and inhibit multiple targets on the way to injury progression, as outlined in Figure 2. The proposed mechanisms of action include: (i) reduced expression of adhesion molecules like ICAM-1 and p-selectin on endothelial cells [72, 73], (ii) decreased vascular permeability with suppressed edema formation [74], (iii) inhibited release of proinflammatory mediators like cytokines and chemokines [68, 75], (iv) reduced activation of inflammatory cells like neutrophils [72, 76], (v) decreased formation of reactive oxygen species [77, 78], (vi) suppressed cell apoptosis and necrosis [79, 80], and (vii) inhibited platelet aggregation and thrombus formation [81]. Similar as described for CNS injuries, the majority of these anti-inflammatory effects is initiated by PPAR-induced suppression of transcription factors (mainly NF-κB) and subsequent inhibition of proinflammatory gene transcription (Figure 2) [35, 69, 70, 80].
Figure 2.
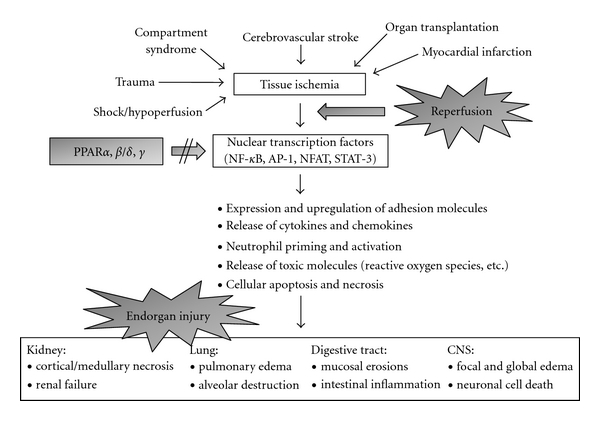
Schematic depiction of the inflammatory events occurring during the pathophysiology of ischemia/reperfusion (I/R) injury, and potential pharmacological effects of PPAR ligands, by inhibition of nuclear transcription factors. See text for detailed explanations. Abbreviations: PPAR, peroxisome proliferator-activated receptor; CNS, central nervous system; NF-κB, nuclear factor-κB; AP-1, activator protein-1; NFAT, nuclear factor of activated T cells; STAT-3, signal transducer and activator of transcription-3.
In addition to the mentioned general effects of PPAR activation on the inflammatory response in I/R (Figure 2), numerous tissue-specific impacts of PPAR agonists have been described in different organ systems (reviewed in [5, 15]).
4.1. Renal I/R
Renal ischemia is a major cause of acute renal failure that is complicated by the fact that subsequent reperfusion of hypoxic tissue may cause additional injury [82]. Agonists to all three PPAR isoforms, PPARα, PPARβ/δ, and PPARγ, significantly reduced tissue damage in mice subjected to kidney ischemia and reperfusion [77, 83, 84]. This renoprotection was reflected in attenuation of cortical and medullary necrosis, reduction of histological signs of renal damage, and finally in strongly increased renal function with lowered serum creatinine and urea nitrogen levels [69, 85, 86]. The mechanisms underlying these beneficial properties may consist in induction of fatty acid β-oxidation enzymes by PPARs in kidney tissue; transgenic mice expressing PPARα in the proximal tubule were shown to exert increased fatty acid oxidation and were protected from I/R-induced kidney failure [87, 88].
4.2. Pulmonary I/R
I/R injury of the lung still occurs in 20% of patients after lung transplantation and remains the main cause of death during the first month after transplantation [89]. Application of the synthetic PPARγ ligand pioglitazone or the natural PPARγ agonist 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) before ischemia could attenuate lung I/R injury in rats [70, 74]. A recent study of Okada et al. indicated that PPARγ activation suppresses activation of the zinc finger transcription factor early growth response gene-1 (Egr-1) that has a crucial role in the inflammatory response in ischemic vessels [70]. Thus, as a consequence of PPARγ activation, the induction of Egr-1 target genes such as interleukin-1β is prevented, IR-associated leukostasis is decreased, and overall survival is improved [70].
4.3. Gastrointestinal I/R
Intestinal and gastric I/R injuries are serious clinical conditions resulting from abdominal aneurism, acute mesenteric ischemia, small bowel transplantation, or shock [90]. In rodent models of intestinal I/R, all three isotypes of PPAR agonists showed profound anti-inflammatory, anti-oxidative and anti-apoptotic effects that were associated with a decreased I/R-induced mortality rate [68, 72, 80]. Similar to that, pioglitazone and rosiglitazone suppressed gastric mucosal erosion and damage in gastric I/R rats [78, 91]. Additionally, beneficial effects of early enteral nutrition after gut I/R could be linked to PPAR induction. The nutrition component glutamine was reported to exert gut protection by activation of PPARγ [92, 93].
4.4. Ischemic Brain Injury
Ischemic cerebrovascular disease represents the third leading cause of death and is one of the major causes of neurological dysfunction and disability [94]. Various studies have suggested that PPAR agonists may prevent or decrease the severity of both focal and global ischemia [66, 95]. In humans, stroke incidence was reduced when men with coronary heart disease and low HDL and LDL cholesterol values were treated with the fibrate and PPARα agonist gemfibrozil [96]. Application of PPARα, PPARβ/δ, and PPARγ ligands in transient ischemic brain injury of rodents resulted in significantly attenuated neuronal damage and reduced infarction volume, increased cerebral blood flow, and improved neurological outcome parameters [71, 97–99]. This neuroprotection was observed when animals were treated preventively before ischemia, at the time of cerebral infarction, or shortly after that with a time window of efficacy of two hours after ischemia [73, 76, 100]. In contrast to transient ischemia, PPARγ activation failed to decrease infarction volume when the blood flow was interrupted permanently without subsequent reperfusion [101–103]. These findings support evidence that the neuroprotective role of PPARγ is specific to events occurring during reperfusion.
Overall, various studies provide evidence that ligands to PPARs cause a substantial reduction of I/R injury in diverse organs by interfering with multiple targets of the I/R-induced inflammatory cascade.
5. PPARs in Sepsis and Shock
Shock and sepsis are serious complications of severely ill patients in intensive care units that can ultimately lead to multiple organ failure and death. There is now increasing evidence that the immune response following severe trauma is divided into two phases [4]. The early hyperinflammatory state is characterized by an overactivation of the innate immune system with increased priming of neutrophils and extensive release of inflammatory mediators like cytokines or reactive oxygen species. Often this first period is followed by a second immunosuppressive phase associated with the attenuation of adaptive immunity and decreased T cell function [3, 8].
The role of PPAR activation in modulating this posttraumatic immune response is ambiguous and has been described as “double-edged sword” [8]. Numerous preclinical studies have supported central beneficial effects of PPAR activation in the first proinflammatory period [104–107]. On the other hand, PPARs have been shown to exert proapoptotic and desensitizing characteristics on inflammatory cells what might increase the susceptibility of the trauma patient to secondary infections during the immunosuppressive phase [108, 109].
The protective properties of PPAR agonists have been examined in multiple animal models of sepsis and shock (Table 1) (reviewed in [8, 12]). Using intraperitoneal LPS injection as a model for endotoxic shock in mice, Kaplan et al. could demonstrate that endotoxin-induced inflammation was associated with reduced expression of PPARγ and with activation of NF-κB in the lung [104]. Treatment with the natural PPARγ ligand 15d-PGJ2 significantly improved survival rate and attenuated inflammation signs by repressing activation of NF-κB and enhancing the expression of cytoprotective heat shock protein in the lung [104]. Similar to that biomarkers of liver and kidney injury and inflammatory cytokines were suppressed when PPARγ agonists were applied before induction of endotoxic shock [110].
Table 1.
Selected publications on experimental studies testing the anti-inflammatory effects of PPAR ligands in various models of sepsis and shock.
| Pathological condition | PPAR isotype/ligand | Ligand-induced effects | Affected signaling pathway | Reference |
|---|---|---|---|---|
| Endotoxic shock | PPARα/fenofibrate | Decrease in coagulation activation (monocyte tissue factor expression), protection against endothelial dysfunction | Not examined | Wiel et al. [107] |
| PPARγ/rosiglitazone | Suppression of biomarkers for liver and kidney injury and of cytokines, inhibition of heart rate increase | Not examined | Wu et al. [110] | |
| PPARγ/15d-PGJ2 | Improvement of survival rate, reduction of adhesion molecule expression, and of neutrophil infiltration in tissues | NF-κB, HSP (heat shock protein) 70 | Kaplan et al. [104] | |
|
| ||||
| Hemorrhagic shock | PPARγ/ciglitazone | Amelioration of mean arterial pressure, reduction of plasma cytokine levels, decrease of apoptosis in lung and liver | NF-κB | Chima et al. [112] |
| Caspase-3, PI3/Akt | Chima et al. [113], Zingarelli et al. [105] | |||
| PPARγ/15d-PGJ2 | Attenuation of renal dysfunction and of liver, lung, and intestine injury | Not examined | Abdelrahman et al. [111] | |
|
| ||||
| Polymicrobial sepsis/septic shock | PPARβ/δ | Decrease in cytokine release, attenuation of organ dysfunction, reduced expression of inducible nitric oxide synthase | Akt, GSK-3β, ERK1/2, STAT-3, NF-κB | Kapoor et al. [106] |
| Zingarelli et al. [115] | ||||
| PPARγ/ciglitazone and 15d-PGJ2 | Amelioration of hypotension and survival, decreased inflammatory signs in lung, colon, and liver | NF-κB, AP-1 | Zingarelli et al. [114] | |
|
| ||||
| Multiple organ failure | PPARβ/δ | Reduction of peritoneal exsudate formation and of neutrophil infiltration, attenuation of multiple organ dysfunction syndrome | Not examined | Galuppo et al. [117] |
| PPARγ/rosiglitazone | Attenuation of peritoneal exsudation and of organ injury and dysfunction | Not examined | Cuzzocrea et al. [116] | |
In hemorrhagic shock in rats, the PPARγ agonists 15d-PGJ2 and ciglitazone ameliorated mean arterial pressure, blunted neutrophil activation, and abolished dysfunction of kidney, liver, lung, and intestine [111, 112]. These effects were mediated through inhibition of the NF-κB pathway. Moreover, two recent studies have found that hemorrhaghic shock-induced apoptosis in the liver and the lung can be decreased by ciglitazone treatment [105, 113]. Herein, attenuation of lung apoptosis was associated with significant reduction in caspase-3 activity and increased phosphorylation of the prosurvival kinase Akt [113].
In a rodent model of polymicrobial sepsis, both PPARγ and PPARβ/δ activation limited the extent of organ dysfunction caused by cecal ligation and puncture (CLP) [106, 114]. PPARβ/δ-deficient mice suffered exaggerated lethality when subjected to CLP and exhibited severe lung injury and higher levels of TNFα [106, 115]. Application of a PPARβ/δ agonist significantly improved survival in polymicrobial sepsis by a mechanism that might involve activation of Akt, inhibition of the MAPK-ERK1/2-signaling pathway, and subsequent suppression of NF-κB activity [106].
Multiple organ failure as a final consequence of severe shock and sepsis can be induced in mice by intraperitoneal administration of zymosan. When animals were treated with a PPARγ or PPARβ/δ agonist after zymosan injection, peritoneal exude formation and neutrophil infiltration were reduced, and lung, liver and pancreatic injury, and renal dysfunction were attenuated [116, 117].
Besides of the classical PPAR agonists, endogenous factors have recently been shown to exert immunomodulatory properties by activating PPARs in different shock models. C-peptide is a 31-amino acid peptide cleaved from insulin during insulin synthesis that has been considered to have minimal biological activity [118]. However, in vitro and in vivo studies have now reported that C-peptide may stimulate PPARγ and thus modulate the inflammatory response in ischemia/reperfusion and shock [119–121]. A study of Vish et al. could demonstrate that treatment with C-peptide improved survival rates and reduced plasma levels of cytokines when mice were subjected to endotoxic shock [121]. C-peptide also upregulated nuclear expression of PPARγ and reduced phosphorylation of ERK-1 and -2 [121]. Moreover, in a model of hemorrhagic shock in rats, hypotension and lung inflammation were significantly ameliorated after C-peptide infusion what was associated with reduced expression of AP-1 and NF-κB and activation of PPARγ [122].
Similarly, the vasoactive hormone adrenomedullin has been shown to be beneficial in sepsis by abrogating the progression to irreversible shock. In addition to its vasodilatory function, adrenomedullin decreases cytokines in the circulation of septic animals what synergistically protects animals from dying of sepsis [123]. These latter anti-inflammatory effects seem to be mediated by mechanisms involving intracellular cAMP increase, followed by upregulation of PPARγ and subsequent suppression of cytokine release [124].
In contrast to the multiple beneficial properties of PPAR agonism, recent studies have revealed additional effects of PPAR activation on immune cells that might compromise the role of these cells in host defense. Neutrophils represent the central cellular component in the sepsis-induced innate immune response and were previously shown to express PPARγ [125]. Treatment of isolated neutrophils with PPARγ agonists resulted in a significant reduction of neutrophil chemotactic activity in vitro [109]. When sepsis was induced in mice, chemotaxis of neutrophils was suppressed compared to healthy mice, but treatment with a PPARγ antagonist restored chemotactic activity to control levels [109]. Since neutrophil expression was increased in septic patients and mice, the authors of the study suggested that the inhibited migration of neutrophils during sepsis might occur as a result of PPARγ activation.
Similar to neutrophils, upregulation of PPARγ was observed in T cells of septic patients [108]. These cells responded with apoptosis when they were exposed to PPARγ agonists [108]. In an animal model of sepsis, sepsis-induced T cell depletion was abrogated after application of a PPARγ antagonist [126]. Consequently, a pivotal involvement of PPARγ in T cell apoptosis was hypothesized what might contribute to lymphocyte loss and breakdown of defense mechanisms during sepsis.
Taken together, there is increasing evidence that PPAR activation has multiple anti-inflammatory properties in shock, sepsis, and multiple organ failure. However, PPARγ agonism was also found to contribute to function loss of neutrophils and depletion of lymphocytes what might be deleterious in the immunosuppressive phase of sepsis. This fact has to be taken in consideration when a potential therapeutical use of PPAR agonists in the treatment of shock and sepsis is discussed.
6. The Role of PPARs in Wound Healing Processes
After skin trauma and injury, wound healing is a life-saving process during which the wound bed is covered with a new protective epithelium. This healing process is mainly divided into three phases. The initial inflammatory stage is followed by proliferation and migration of keratinocytes with reepithelialization of the wound [19]. In parallel, dermal repair involves activation and proliferation of fibroblasts and angiogenesis (remodeling phase) what finally results in wound closure.
While PPAR expression progressively disappears from the interfollicular epidermis after birth, PPARα and PPARβ/δ are reactivated in keratinocytes at the wound edges of damaged skin [127]. Hereby, the upregulation of PPARα is transient and correlates with the early inflammatory phase of wound healing. A study of Michalik et al. could demonstrate that PPARα −/− mice exert a transient delay of wound healing during the first 4 days after injury coinciding with increased PPARα expression during the inflammatory stage [127, 128]. Furthermore, both the recruitment of neutrophils and monocytes was impaired in PPARα −/− mice. In contrast to PPARα, PPARβ/δ expression persists throughout the entire repair process and is correlated to keratinocyte proliferation, adhesion, and migration in order to reepithelialize the wounded area. Consistent to that, wound healing of PPARβ +/− mice was delayed during the whole repair process, and final wound closure was postponed by two to three days [127].
As outlined in Figure 3, the expression of PPARβ/δ during wound healing is characterized by a specific time pattern and is tightly regulated by signaling cascades, providing both stimulatory and negative feedback mechanisms [18, 129]. Upon injury, proinflammatory cytokines like TNFα activate the stress-associated protein kinase pathway and induce AP-1 binding to the PPARβ/δ promotor, leading to the stimulation of PPARβ/δ expression [130]. In the healing process, PPARβ/δ represses apoptosis cascades through transcriptional upregulation of integrin-linked kinase (ILK) and 3-phosphoinositide-dependent kinase-1 (PDK), and consequent Akt-1 activation [131]. The resulting resistance to cell death helps to maintain a sufficient number of viable keratinocytes for reepithelialization. Moreover, PPARβ/δ is implicated in keratinocyte adhesion and migration of keratinocytes, two key processes during reepithelialization [131, 132]. After the inflammatory and early reepithelialization phase, during which PPARβ/δ expression is maximal, PPARβ/δ is progressively reduced in the epithelium. The responsible antagonization process is initiated by TGFβ-1/Smad3-mediated inhibition of AP-1 binding to the PPARβ/δ promotor in the late reepithelialization/remodeling phase, resulting in downregulation of the PPARβ/δ gene [133, 134]. Taken together, the healing process represents a delicate balance between early proinflammatory signals triggering PPARβ/δ expression and negative feedback pathways at later stages of wound healing. Since both processes temporally overlap, an intensive crosstalk between the two signaling pathways is suggested that contribute to fine-tuning of wound closure [133].
Figure 3.
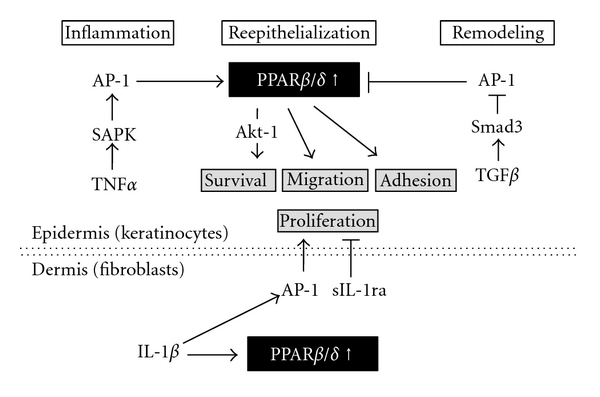
Role of PPARβ/δ expression in the wound healing processes after tissue injury. See text for detailed explanations. Abbreviations: PPAR, peroxisome proliferator-activated receptor; TNFα, tumor necrosis factor α; SAPK, stress-associated protein kinase; AP-1, activator protein-1; PPAR, peroxisome proliferator-activated receptor; TGFβ, transforming growth factor β; IL-1β, interleukin-1 β; sIL-1ra, soluble interleukin-1 receptor antagonist.
In addition to the described anti-apoptotic properties, PPARβ/δ also influences keratinocyte differentiation and proliferation by to-date unknown mechanism. Chong et al. could recently demonstrate that PPARβ/δ-dependent interplay between the epidermis and dermis is essential for controlling epidermal proliferation (Figure 3) [18, 135]. They found that interleukin-1 (IL-1) produced by keratinocytes stimulates AP-1 transcription factor in dermal fibroblasts and consequently increases the production of mitogenic factors that enhance keratinocyte proliferation. In parallel, IL-1 activates PPARβ/δ expression in fibroblasts, which increases the production of the secreted IL-1 receptor antagonist (sIL-1ra), a protein inhibiting IL-1 signaling. As a consequence, the IL-1-induced production of mitogenic factors by fibroblasts is reduced, and keratinocyte proliferation is decreased when wound closure comes to an end [135]. This study provides evidence that PPARβ/δ participates in regulating epidermal proliferation via a paracrine mechanism.
In summary, efficiency in the wound healing process is guaranteed both by interactions within the epidermis as well as by epithelial-mesenchymal crosstalk. Thus, keratinocyte differentiation and survival are provided in the early healing process and, at the same time, exaggerated proliferation and hypertrophic scarring are avoided in later stages. It has been suggested that these insights into the coordination of wound healing might contribute to develop better treatment strategies for chronic wound disorders [18].
7. Conclusion
In summary, various studies have provided evidence that PPARs have a substantial impact on reducing the extent of post-injury inflammation after major trauma, mainly by the suppression of the central transcription factor NF-κB. These anti-inflammatory properties could be supported by the use of synthetic and natural PPAR ligands in multiple animal models of CNS trauma, I/R injury, and shock. Furthermore, PPAR activation was found to be responsible for the balance between differentiation, cell survival, and apoptosis during wound healing. Synthetic PPAR ligands are readily available from clinical trials in diabetes and hyperlipidemia. The basic science studies described in this review article provide a strong rationale for testing these pharmacological compounds for their anti-inflammatory potential in the clinical setting of posttraumatic hyperinflammation in the near future.
References
- 1.Kauvar DS, Wade CE. The epidemiology and modern management of traumatic hemorrhage: US and international perspectives. Critical Care. 2005;9(supplement 5):S1–S9. doi: 10.1186/cc3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianchi ME, Manfredi AA. Dangers in and out. Science. 2009;323(5922):1683–1684. doi: 10.1126/science.1172794. [DOI] [PubMed] [Google Scholar]
- 3.Stahel PF, Smith WR, Moore EE. Role of biological modifiers regulating the immune response after trauma. Injury. 2007;38(12):1409–1422. doi: 10.1016/j.injury.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 4.Keel M, Trentz O. Pathophysiology of polytrauma. Injury. 2005;36(6):691–709. doi: 10.1016/j.injury.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 5.Cuzzocrea S. Peroxisome proliferator-activated receptors gamma ligands and ischemia and reperfusion injury. Vascular Pharmacology. 2004;41(6):187–195. doi: 10.1016/j.vph.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Stahel PF, Smith WR, Bruchis J, Rabb CH. Peroxisome proliferator-activated receptors: “Key” regulators of neuroinflammation after traumatic brain injury. PPAR Research. 2008;2008 doi: 10.1155/2008/538141. Article ID 538141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giaginis C, Tsourouflis G, Theocharis S. Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands: novel pharmacological agents in the treatment of ischemia reperfusion injury. Current Molecular Medicine. 2008;8(6):562–579. doi: 10.2174/156652408785748022. [DOI] [PubMed] [Google Scholar]
- 8.Von Knethen A, Soller M, Brüne B. Peroxisome proliferator-activated receptor γ (PPARγ) and sepsis. Archivum Immunologiae et Therapiae Experimentalis. 2007;55(1):19–25. doi: 10.1007/s00005-007-0005-y. [DOI] [PubMed] [Google Scholar]
- 9.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. Journal of Medicinal Chemistry. 2000;43(4):527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 10.Staels B, Fruchart JC. Therapeutic roles of peroxisome proliferator-activated receptor agonists. Diabetes. 2005;54(8):2460–2470. doi: 10.2337/diabetes.54.8.2460. [DOI] [PubMed] [Google Scholar]
- 11.Daynes RA, Jones DC. Emerging roles of PPARs in inflammation and immunity. Nature Reviews Immunology. 2002;2(10):748–759. doi: 10.1038/nri912. [DOI] [PubMed] [Google Scholar]
- 12.Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR-γ ligands in ischemia-reperfusion injury, inflammation and shock. Cardiovascular Research. 2005;65(4):772–781. doi: 10.1016/j.cardiores.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 13.van Neerven S, Mey J. RAR/RXR and PPAR/RXR signaling in spinal cord injury. PPAR Research. 2007;2007 doi: 10.1155/2007/29275. Article ID 29275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocrine Reviews. 1999;20(5):649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 15.Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. Journal of Clinical Investigation. 2006;116(3):598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youssef J, Badr M. Peroxisome proliferator-activated receptors and cancer: challenges and opportunities. British Journal of Pharmacology. 2011;164(1):68–82. doi: 10.1111/j.1476-5381.2011.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plutzky J. The PPAR-RXR transcriptional complex in the vasculature: energy in the balance. Circulation Research. 2011;108(8):1002–1016. doi: 10.1161/CIRCRESAHA.110.226860. [DOI] [PubMed] [Google Scholar]
- 18.Montagner A, Rando G, Degueurce G, Leuenberger N, Michalik L, Wahli W. New insights into the role of PPARs. Prostaglandins Leukotrienes and Essential Fatty Acids. 2011;85(5):235–243. doi: 10.1016/j.plefa.2011.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Yessoufou A, Wahli W. Multifaceted roles of peroxisome proliferator-activated receptors (PPARs) at the cellular and whole organism levels. Swiss Medical Weekly. 2010;140:4–10. doi: 10.4414/smw.2010.13071. Article ID w13071. [DOI] [PubMed] [Google Scholar]
- 20.McMillan TM, Teasdale GM. Death rate is increased for at least 7 years after head injury: a prospective study. Brain. 2007;130, part 10:2520–2527. doi: 10.1093/brain/awm185. [DOI] [PubMed] [Google Scholar]
- 21.Schmidt OI, Heyde CE, Ertel W, Stahel PF. Closed head injury—an inflammatory disease? Brain Research Reviews. 2005;48(2):388–399. doi: 10.1016/j.brainresrev.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 22.Gaetz M. The neurophysiology of brain injury. Clinical Neurophysiology. 2004;115(1):4–18. doi: 10.1016/s1388-2457(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 23.Bright JJ, Kanakasabai S, Chearwae W, Chakraborty S. PPAR regulation of inflammatory signaling in CNS diseases. PPAR Research. 2008;2008 doi: 10.1155/2008/658520. Article ID 658520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kapadia R, Yi JH, Vemuganti R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Frontiers in Bioscience. 2008;13(5):1813–1826. doi: 10.2741/2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cullingford TE, Bhakoo K, Peuchen S, Dolphin CT, Patel R, Clark JB. Distribution of mRNAs encoding the peroxisome proliferator-activated receptor α, β, and γ and the retinoid X receptor α, β, and γ in rat central nervous system. Journal of Neurochemistry. 1998;70(4):1366–1375. doi: 10.1046/j.1471-4159.1998.70041366.x. [DOI] [PubMed] [Google Scholar]
- 26.Moreno S, Farioli-vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid X receptors in the adult rat CNS. Neuroscience. 2004;123(1):131–145. doi: 10.1016/j.neuroscience.2003.08.064. [DOI] [PubMed] [Google Scholar]
- 27.Yi JH, Park SW, Brooks N, Lang BT, Vemuganti R. PPARγ agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Research. 2008;1244:164–172. doi: 10.1016/j.brainres.2008.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Almad A, McTigue DM. Chronic expression of PPAR-δ by oligodendrocyte lineage cells in the injured rat spinal cord. Journal of Comparative Neurology. 2010;518(6):785–799. doi: 10.1002/cne.22242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng D, Zhang Y, Chen G. Cortical expression of peroxisome proliferator-activated receptor-α after human brain contusion. Journal of International Medical Research. 2008;36(4):783–791. doi: 10.1177/147323000803600421. [DOI] [PubMed] [Google Scholar]
- 30.Bordet R, Gelé P, Duriez P, Fruchart JC. PPARs: a new target for neuroprotection. Journal of Neurology, Neurosurgery and Psychiatry. 2006;77(3):285–286. doi: 10.1136/jnnp.2005.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qi L, Jacob A, Wang P, Wu R. Peroxisome proliferator activated receptor-γ and traumatic brain injury. International Journal of Clinical and Experimental Medicine. 2010;3(4):283–292. [PMC free article] [PubMed] [Google Scholar]
- 32.Bordet R, Ouk T, Petrault O, et al. PPAR: a new pharmacological target for neuroprotection in stroke and neurodegenerative diseases. Biochemical Society Transactions. 2006;34, part 6:1341–1346. doi: 10.1042/BST0341341. [DOI] [PubMed] [Google Scholar]
- 33.Bernando A, Minghetti L. PPAR-γ agonists as regulators of microglial activation and brain inflammation. Current Pharmaceutical Design. 2006;12(1):93–109. doi: 10.2174/138161206780574579. [DOI] [PubMed] [Google Scholar]
- 34.Hyong A, Jadhav V, Lee S, et al. Rosiglitazone, a PPAR gamma agonist, attenuates inflammation after surgical brain injury in rodents. Brain Research. 2008;1215(C):218–224. doi: 10.1016/j.brainres.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Collino M, Aragno M, Mastrocola R, et al. Modulation of the oxidative stress and inflammatory response by PPAR-γ agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. European Journal of Pharmacology. 2006;530(1-2):70–80. doi: 10.1016/j.ejphar.2005.11.049. [DOI] [PubMed] [Google Scholar]
- 36.Chen XR, Besson VC, Palmier B, Garcia Y, Plotkine M, Marchand-Leroux C. Neurological recovery-promoting, anti-inflammatory, and anti-oxidative effects afforded by fenofibrate, a PPAR alpha agonist, in traumatic brain injury. Journal of Neurotrauma. 2007;24(7):1119–1131. doi: 10.1089/neu.2006.0216. [DOI] [PubMed] [Google Scholar]
- 37.Zhao X, Zhang Y, Strong R, Grotta JC, Aronowski J. 15d-Prostaglandin J2 activates peroxisome proliferator-activated receptor-γ, promotes expression of catalase, and reduces inflammation, behavioral dysfunction, and neuronal loss after intracerebral hemorrhage in rats. Journal of Cerebral Blood Flow and Metabolism. 2006;26(6):811–820. doi: 10.1038/sj.jcbfm.9600233. [DOI] [PubMed] [Google Scholar]
- 38.Genovese T, Mazzon E, Di Paola R, et al. Role of endogenous ligands for the peroxisome proliferators activated receptors alpha in the secondary damage in experimental spinal cord trauma. Experimental Neurology. 2005;194(1):267–278. doi: 10.1016/j.expneurol.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 39.Genovese T, Esposito E, Mazzon E, et al. PPAR-α modulate the anti-inflammatory effect of glucocorticoids in the secondary damage in experimental spinal cord trauma. Pharmacological Research. 2009;59(5):338–350. doi: 10.1016/j.phrs.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 40.Esposito E, Cuzzocrea S. Targeting the peroxisome proliferator-activated receptors (PPARs) in spinal cord injury. Expert Opinion on Therapeutic Targets. 2011;15(8):943–959. doi: 10.1517/14728222.2011.581231. [DOI] [PubMed] [Google Scholar]
- 41.Besson VC, Chen XR, Plotkine M, Marchand-Verrecchia C. Fenofibrate, a peroxisome proliferator-activated receptor α agonist, exerts neuroprotective effects in traumatic brain injury. Neuroscience Letters. 2005;388(1):7–12. doi: 10.1016/j.neulet.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 42.Xiao RC, Besson VC, Beziaud T, Plotkine M, Marchand-Leroux C. Combination therapy with fenofibrate, a peroxisome proliferator-activated receptor α agonist, and simvastatin, a 3-hydroxy-3-methylglutaryl-coenzyme a reductase inhibitor, on experimental traumatic brain injury. Journal of Pharmacology and Experimental Therapeutics. 2008;326(3):966–974. doi: 10.1124/jpet.108.140368. [DOI] [PubMed] [Google Scholar]
- 43.McTigue DM. Potential therapeutic targets for PPARγ after spinal cord injury. PPAR Research. 2008;2008 doi: 10.1155/2008/517162. Article ID 517162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sauerbeck A, Gao J, Readnower R, et al. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Experimental Neurology. 2011;227(1):128–135. doi: 10.1016/j.expneurol.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Semple BD, Noble-Haeusslein LJ. Broad-spectrum neuroprotection against traumatic brain injury by agonism of peroxisome proliferator-activated receptors. Experimental Neurology. 2011;229(2):195–197. doi: 10.1016/j.expneurol.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Q, Hu W, Meng B, Tang T. PPARγ agonist rosiglitazone is neuroprotective after traumatic spinal cord injury via anti-inflammatory in adult rats. Neurological Research. 2010;32(8):852–859. doi: 10.1179/016164110X12556180206112. [DOI] [PubMed] [Google Scholar]
- 47.Park SW, Yi JH, Miranpuri G, et al. Thiazolidinedione class of peroxisome proliferator-activated receptor γ agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. Journal of Pharmacology and Experimental Therapeutics. 2007;320(3):1002–1012. doi: 10.1124/jpet.106.113472. [DOI] [PubMed] [Google Scholar]
- 48.McTigue DM, Tripathi R, Wei P, Lash AT. The PPAR gamma agonist Pioglitazone improves anatomical and locomotor recovery after rodent spinal cord injury. Experimental Neurology. 2007;205(2):396–406. doi: 10.1016/j.expneurol.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Q, Huang C, Tang T, Shi Q, Yang H. Comparative neuroprotective effects of methylprednisolone and rosiglitazone, a peroxisome proliferator-activated receptor-γ following spinal cord injury. Neurosciences. 2011;16(1):46–52. [PubMed] [Google Scholar]
- 50.Mechoulam R, Panikashvili D, Shohami E. Cannabinoids and brain injury: therapeutic implications. Trends in Molecular Medicine. 2002;8(2):58–61. doi: 10.1016/s1471-4914(02)02276-1. [DOI] [PubMed] [Google Scholar]
- 51.Mechoulam R, Shohami E. Endocannabinoids and traumatic brain injury. Molecular Neurobiology. 2007;36(1):68–74. doi: 10.1007/s12035-007-8008-6. [DOI] [PubMed] [Google Scholar]
- 52.Bayir H, Clark RSB, Kochanek PM. Promising strategies to minimize secondary brain injury after head trauma. Critical Care Medicine. 2003;31(1):S112–S117. doi: 10.1097/00003246-200301001-00016. [DOI] [PubMed] [Google Scholar]
- 53.Sun Y, Bennett A. Cannabinoids: a new group of agonists of PPARs. PPAR Research. 2007;2007 doi: 10.1155/2007/23513. Article ID 23513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Panikashvili D, Simeonidou C, Ben-Shabat S, et al. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413(6855):527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- 55.Gallily R, Breuer A, Mechoulam R. 2-Arachidonylglycerol, an endogenous cannabinoid, inhibits tumor necrosis factor-α production in murine macrophages, and in mice. European Journal of Pharmacology. 2000;406(1):R5–R7. doi: 10.1016/s0014-2999(00)00653-1. [DOI] [PubMed] [Google Scholar]
- 56.Shohami E, Novikov M, Mechoulam R. A nonpsychotropic cannabinoid, HU-211, has cerebroprotective effects after closed head injury in the rat. Journal of Neurotrauma. 1993;10(2):109–119. doi: 10.1089/neu.1993.10.109. [DOI] [PubMed] [Google Scholar]
- 57.Shohami E, Gallily R, Mechoulam R, Bass R, Ben-Hur T. Cytokine production in the brain following closed head injury: dexanabinol (HU-211) is a novel TNF-α inhibitor and an effective neuroprotectant. Journal of Neuroimmunology. 1997;72(2):169–177. doi: 10.1016/s0165-5728(96)00181-6. [DOI] [PubMed] [Google Scholar]
- 58.Rockwell CE, Snider NT, Thompson JT, Vanden Heuvel JP, Kaminski NE. Interleukin-2 suppression by 2-arachidonyl glycerol is mediated through peroxisome proliferator-activated receptor γ independently of cannabinoid receptors 1 and 2. Molecular Pharmacology. 2006;70(1):101–111. doi: 10.1124/mol.105.019117. [DOI] [PubMed] [Google Scholar]
- 59.Fu J, Gaetani S, Oveisi F, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α . Nature. 2003;425(6953):90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 60.O'Sullivan SE, Tarling EJ, Bennett AJ, Kendall DA, Randall MD. Novel time-dependent vascular actions of Δ9- tetrahydrocannabinol mediated by peroxisome proliferator-activated receptor gamma. Biochemical and Biophysical Research Communications. 2005;337(3):824–831. doi: 10.1016/j.bbrc.2005.09.121. [DOI] [PubMed] [Google Scholar]
- 61.Knoller N, Levi L, Shoshan I, et al. Dexanabinol (HU-211) in the treatment of severe closed head injury: a randomized, placebo-controlled, phase II clinical trial. Critical Care Medicine. 2002;30(3):548–554. doi: 10.1097/00003246-200203000-00009. [DOI] [PubMed] [Google Scholar]
- 62.Narayan RK, Michel ME, Ansell B, et al. Clinical trials in head injury. Journal of Neurotrauma. 2002;19(5):503–557. doi: 10.1089/089771502753754037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heyndrickx GR. Early reperfusion phenomena. Seminars in Cardiothoracic and Vascular Anesthesia. 2006;10(3):236–241. doi: 10.1177/1089253206291137. [DOI] [PubMed] [Google Scholar]
- 64.Matsuyama M, Yoshimura R, Kawahito Y, et al. Relationship between peroxisome proliferator-activated receptor-γ and renal ischemia-reperfusion injury. Molecular Medicine Reports. 2008;1(4):499–503. [PubMed] [Google Scholar]
- 65.Yoshimura R, Matsuyama M, Segawa Y, et al. Study of peroxisome proliferator-activated receptor (PPAR)-γ in renal ischemia-reperfusion injury. Transplantation Proceedings. 2004;36(7):1946–1948. doi: 10.1016/j.transproceed.2004.08.039. [DOI] [PubMed] [Google Scholar]
- 66.Zhao Y, Patzer A, Herdegen T, Gohlke P, Culman J. Activation of cerebral peroxisome proliferator-activated receptors gamma promotes neuroprotection by attenuation of neuronal cyclooxygenase-2 overexpression after focal cerebral ischemia in rats. FASEB Journal. 2006;20(8):1162–1175. doi: 10.1096/fj.05-5007com. [DOI] [PubMed] [Google Scholar]
- 67.Lee CH, Park OK, Yoo KY, et al. The role of peroxisome proliferator-activated receptor γ, and effects of its agonist, rosiglitazone, on transient cerebral ischemic damage. Journal of the Neurological Sciences. 2011;300(1-2):120–129. doi: 10.1016/j.jns.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 68.Cuzzocrea S, Pisano B, Dugo L, et al. Rosiglitazone and 15-deoxy-Δ12,14-prostaglandin J 2, ligands of the peroxisome proliferator-activated receptor-γ (PPAR-γ), reduce ischaemia/reperfusion injury of the gut. British Journal of Pharmacology. 2003;140(2):366–376. doi: 10.1038/sj.bjp.0705419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chatterjee PK, Patel NSA, Cuzzocrea S, et al. The cyclopentenone prostaglandin 15-deoxy-Δ12,14- prostaglandin J2 ameliorates ischemic acute renal failure. Cardiovascular Research. 2004;61(3):630–643. doi: 10.1016/j.cardiores.2003.10.024. [DOI] [PubMed] [Google Scholar]
- 70.Okada M, Yan SF, Pinsky DJ. Peroxisome proliferator-activated receptor-γ (PPAR-γ) activation suppresses ischemic induction of Egr-1 and its inflammatory gene targets. FASEB Journal. 2002;16(14):1861–1868. doi: 10.1096/fj.02-0503com. [DOI] [PubMed] [Google Scholar]
- 71.Guo Q, Wang G, Namura S. Fenofibrate improves cerebral blood flow after middle cerebral artery occlusion in mice. Journal of Cerebral Blood Flow and Metabolism. 2010;30(1):70–78. doi: 10.1038/jcbfm.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cuzzocrea S, Di Paola R, Mazzon E, Genovese T, Muià C, Caputi AP. WY 14643, a potent exogenous PPAR-α ligand, reduces intestinal injury associated with splanchnic artery occlusion shock. Shock. 2004;22(4):340–346. doi: 10.1097/01.shk.0000136704.26372.2d. [DOI] [PubMed] [Google Scholar]
- 73.Deplanque D, Gelé P, Pétrault O, et al. Peroxisome proliferator-activated receptor-α activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. Journal of Neuroscience. 2003;23(15):6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ito K, Shimada J, Kato D, et al. Protective effects of preischemic treatment with pioglitazone, a peroxisome proliferator-activated receptor-γ ligand, on lung ischemia-reperfusion injury in rats. European Journal of Cardio-thoracic Surgery. 2004;25(4):530–536. doi: 10.1016/j.ejcts.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 75.Honda T, Kaikita K, Tsujita K, et al. Pioglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates myocardial ischemia-reperfusion injury in mice with metabolic disorders. Journal of Molecular and Cellular Cardiology. 2008;44(5):915–926. doi: 10.1016/j.yjmcc.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 76.Luo Y, Yin W, Signore AP, et al. Neuroprotection against focal ischemic brain injury by the peroxisome proliferator-activated receptor-γ agonist rosiglitazone. Journal of Neurochemistry. 2006;97(2):435–448. doi: 10.1111/j.1471-4159.2006.03758.x. [DOI] [PubMed] [Google Scholar]
- 77.Sivarajah A, Chatterjee PK, Patel NSA, et al. Agonists of peroxisome-proliferator activated receptor-gamma reduce renal ischemia/reperfusion injury. American Journal of Nephrology. 2003;23(4):267–276. doi: 10.1159/000072088. [DOI] [PubMed] [Google Scholar]
- 78.Villegas I, Martín AR, Toma W, Alarcón de la Lastra C. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, protects against gastric ischemia-reperfusion damage in rats: role of oxygen free radicals generation. European Journal of Pharmacology. 2004;505(1–3):195–203. doi: 10.1016/j.ejphar.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 79.Doi S, Masaki T, Arakawa T, et al. Protective effects of peroxisome proliferator-activated receptor γ ligand on apoptosis and hepatocyte growth factor induction in renal ischemia-reperfusion injury. Transplantation. 2007;84(2):207–213. doi: 10.1097/01.tp.0000269614.21367.3f. [DOI] [PubMed] [Google Scholar]
- 80.Di Paola R, Esposito E, Mazzon E, Paterniti I, Galuppo M, Cuzzocrea S. GW0742, a selective PPAR-β/δ agonist, contributes to the resolution of inflammation after gut ischemia/reperfusion injury. Journal of Leukocyte Biology. 2010;88(2):291–301. doi: 10.1189/jlb.0110053. [DOI] [PubMed] [Google Scholar]
- 81.Li D, Chen K, Sinha N, et al. The effects of PPAR-γ ligand pioglitazone on platelet aggregation and arterial thrombus formation. Cardiovascular Research. 2005;65(4):907–912. doi: 10.1016/j.cardiores.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 82.Paller MS. The cell biology of reperfusion injury in the kidney. Journal of Investigative Medicine. 1994;42(4):632–639. [PubMed] [Google Scholar]
- 83.Patel NSA, di Paola R, Mazzon E, Britti D, Thiemermann C, Cuzzocrea S. Peroxisome proliferator-activated receptor-α contributes to the resolution of inflammation after renal ischemia/reperfusion injury. Journal of Pharmacology and Experimental Therapeutics. 2009;328(2):635–643. doi: 10.1124/jpet.108.146191. [DOI] [PubMed] [Google Scholar]
- 84.Collino M, Benetti E, Miglio G, et al. Peroxisome proliferator-activated receptor β/δ agonism protects the kidney against ischemia/reperfusion injury in diabetic rats. Free Radical Biology and Medicine. 2011;50(2):345–353. doi: 10.1016/j.freeradbiomed.2010.10.710. [DOI] [PubMed] [Google Scholar]
- 85.Letavernier E, Perez J, Joye E, et al. Peroxisome proliferator-activated receptor β/δ exerts a strong protection from ischemic acute renal failure. Journal of the American Society of Nephrology. 2005;16(8):2395–2402. doi: 10.1681/ASN.2004090802. [DOI] [PubMed] [Google Scholar]
- 86.Chen HH, Chen TW, Lin H. Prostacyclin-induced peroxisome proliferator-activated receptor-α translocation attenuates NF-κB and TNF-α activation after renal ischemia-reperfusion injury. American Journal of Physiology—Renal Physiology. 2009;297(4):F1109–F1118. doi: 10.1152/ajprenal.00057.2009. [DOI] [PubMed] [Google Scholar]
- 87.Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARα-modulated enzymes protect during acute renal failure. American Journal of Physiology—Renal Physiology. 2000;278(4):F667–F675. doi: 10.1152/ajprenal.2000.278.4.F667. [DOI] [PubMed] [Google Scholar]
- 88.Li S, Nagothu KK, Desai V, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-α in mice confers protection during acute kidney injury. Kidney International. 2009;76(10):1049–1062. doi: 10.1038/ki.2009.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.King RC, Binns OAR, Rodriguez F, et al. Reperfusion injury significantly impacts clinical outcome after pulmonary transplantation. Annals of Thoracic Surgery. 2000;69(6):1681–1685. doi: 10.1016/s0003-4975(00)01425-9. [DOI] [PubMed] [Google Scholar]
- 90.Cappell MS. Intestinal (mesenteric) vasculopathy II. Ischemic colitis and chronic mesenteric ischemia. Gastroenterology Clinics of North America. 1998;27(4):827–860. doi: 10.1016/s0889-8553(05)70034-0. [DOI] [PubMed] [Google Scholar]
- 91.Ichikawa H, Naito Y, Takagi T, Tomatsuri N, Yoshida N, Yoshikawa T. A specific peroxisome proliferator-induced receptor-γ (PPAR-γ) ligand, pioglitazone, ameliorates gastric mucosal damage induced by ischemia and reperfusion in rats. Redox Report. 2002;7(5):343–346. doi: 10.1179/135100002125000956. [DOI] [PubMed] [Google Scholar]
- 92.Sato N, Moore FA, Kone BC, et al. Differential induction of PPAR-γ by luminal glutamine and iNOS by luminal arginine in the rodent postischemic small bowel. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2006;290(4):G616–G623. doi: 10.1152/ajpgi.00248.2005. [DOI] [PubMed] [Google Scholar]
- 93.Ban K, Kozar RA. Enteral glutamine: a novel mediator of PPARγ in the postischemic gut. Journal of Leukocyte Biology. 2008;84(3):595–599. doi: 10.1189/jlb.1107764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hankey GJ, Warlow CP. Treatment and secondary prevention of stroke: evidence, costs, and effects on individuals and populations. Lancet. 1999;354(9188):1457–1463. doi: 10.1016/S0140-6736(99)04407-4. [DOI] [PubMed] [Google Scholar]
- 95.Fatehi-Hassanabad Z, Tasker RA. Peroxisome proliferator-activated receptor-γ (PPAR-γ) activation confers functional neuroprotection in global ischemia. Neurotoxicity Research. 2011;19(3):462–471. doi: 10.1007/s12640-010-9201-3. [DOI] [PubMed] [Google Scholar]
- 96.Rubins HB, Davenport J, Babikian V, et al. Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol the veterans affairs HDL intervention trial (VA-HIT) Circulation. 2001;103(23):2828–2833. doi: 10.1161/01.cir.103.23.2828. [DOI] [PubMed] [Google Scholar]
- 97.Iwashita A, Muramatsu Y, Yamazaki T, et al. Neuroprotective efficacy of the peroxisome proliferator-activated receptor δ-selective agonists in vitro and in vivo. Journal of Pharmacology and Experimental Therapeutics. 2007;320(3):1087–1096. doi: 10.1124/jpet.106.115758. [DOI] [PubMed] [Google Scholar]
- 98.Victor NA, Wanderi EW, Gamboa J, et al. Altered PPARγ expression and activation after transient focal ischemia in rats. European Journal of Neuroscience. 2006;24(6):1653–1663. doi: 10.1111/j.1460-9568.2006.05037.x. [DOI] [PubMed] [Google Scholar]
- 99.Lin TN, Cheung WM, Wu JS, et al. 15d-prostaglandin J2 protects brain from ischemia-reperfusion injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26(3):481–487. doi: 10.1161/01.ATV.0000201933.53964.5b. [DOI] [PubMed] [Google Scholar]
- 100.Sundararajan S, Gamboa JL, Victor NA, Wanderi EW, Lust WD, Landreth GE. Peroxisome proliferator-activated receptor-γ ligands reduce inflammation and infarction size in transient focal ischemia. Neuroscience. 2005;130(3):685–696. doi: 10.1016/j.neuroscience.2004.10.021. [DOI] [PubMed] [Google Scholar]
- 101.Shimazu T, Inoue I, Araki N, et al. A peroxisome proliferator-activated receptor-γ agonist reduces infarct size in transient but not in permanent ischemia. Stroke. 2005;36(2):353–359. doi: 10.1161/01.STR.0000152271.21943.a2. [DOI] [PubMed] [Google Scholar]
- 102.Gamboa J, Blankenship DA, Niemi JP, et al. Extension of the neuroprotective time window for thiazolidinediones in ischemic stroke is dependent on time of reperfusion. Neuroscience. 2010;170(3):846–857. doi: 10.1016/j.neuroscience.2010.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kasahara Y, Taguchi A, Uno H, et al. Telmisartan suppresses cerebral injury in a murine model of transient focal ischemia. Brain Research. 2010;1340(C):70–80. doi: 10.1016/j.brainres.2010.03.101. [DOI] [PubMed] [Google Scholar]
- 104.Kaplan JM, Cook JA, Hake PW, O’Connor M, Burroughs TJ, Zingarelli B. 15-Deoxy-Δ12,14-prostaglandin J2 (15D-PGJ 2), a peroxisome proliferator activated receptor γ ligand, reduces tissue leukosequestration and mortality in endotoxic shock. Shock. 2005;24(1):59–65. doi: 10.1097/01.shk.0000167108.88376.f2. [DOI] [PubMed] [Google Scholar]
- 105.Zingarelli B, Chima R, O'Connor M, Piraino G, Denenberg A, Hake PW. Liver apoptosis is age dependent and is reduced by activation of peroxisome proliferator-activated receptor-γ in hemorrhagic shock. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2010;298(1):G133–G141. doi: 10.1152/ajpgi.00262.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kapoor A, Shintani Y, Collino M, et al. Protective role of peroxisome proliferator-activated receptor-β/δ in septic shock. American Journal of Respiratory and Critical Care Medicine. 2010;182(12):1506–1515. doi: 10.1164/rccm.201002-0240OC. [DOI] [PubMed] [Google Scholar]
- 107.Wiel E, Lebuffe G, Robin E, et al. Pretreatment with peroxysome proliferator-activated receptor α agonist fenofibrate protects endothelium in rabbit Escherichia coli endotoxin-induced shock. Intensive Care Medicine. 2005;31(9):1269–1279. doi: 10.1007/s00134-005-2730-1. [DOI] [PubMed] [Google Scholar]
- 108.Soller M, Tautenhahn A, Brüne B, et al. Peroxisome proliferator-activated receptor γ contributes to T lymphocyte apoptosis during sepsis. Journal of Leukocyte Biology. 2006;79(1):235–243. doi: 10.1189/jlb.0205058. [DOI] [PubMed] [Google Scholar]
- 109.Reddy RC, Narala VR, Keshamouni VG, Milam JE, Newstead MW, Standiford TJ. Sepsis-induced inhibition of neutrophil chemotaxis is mediated by activation of peroxisome proliferator-activated receptor-γ . Blood. 2008;112(10):4250–4258. doi: 10.1182/blood-2007-12-128967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wu WT, Lee CC, Lee CJ, Subeq YM, Lee RP, Hsu BG. Rosiglitazone ameliorates endotoxin-induced organ damage in conscious rats. Biological Research for Nursing. 2011;13(1):38–43. doi: 10.1177/1099800409353358. [DOI] [PubMed] [Google Scholar]
- 111.Abdelrahman M, Collin M, Thiemermann C. The peroxisome proliferator-activated receptor-γ ligand 15-deoxy Δ12,14 prostaglandin J2 reduces the organ injury in hemorrhagic shock. Shock. 2004;22(6):555–561. doi: 10.1097/01.shk.0000144132.13900.24. [DOI] [PubMed] [Google Scholar]
- 112.Chima RS, Hake PW, Piraino G, Mangeshkar P, Denenberg A, Zingarelli B. Ciglitazone ameliorates lung inflammation by modulating the inhibitor κb protein kinase/nuclear factor-κB pathway after hemorrhagic shock. Critical Care Medicine. 2008;36(10):2849–2857. doi: 10.1097/ccm.0b013e318187810e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chima RS, Hake PW, Piraino G, Mangeshkar P, O’Connor M, Zingarelli B. Ciglitazone, a novel inhibitor of lung apoptosis following hemorrhagic shock. International Journal of Clinical and Experimental Medicine. 2010;3(1):1–9. [PMC free article] [PubMed] [Google Scholar]
- 114.Zingarelli B, Sheehan M, Hake PW, O’Connor M, Denenberg A, Cook JA. Peroxisome proliferator activator receptor-γ ligands, 15-deoxy-Δ12,14-prostaglandin J2 and ciglitazone, reduce systemic inflammation in polymicrobial sepsis by modulation of signal transduction pathways. Journal of Immunology. 2003;171(12):6827–6837. doi: 10.4049/jimmunol.171.12.6827. [DOI] [PubMed] [Google Scholar]
- 115.Zingarelli B, Piraino G, Hake PW, et al. Peroxisome proliferator-activated receptor δ regulates inflammation via NF-κB signaling in polymicrobial sepsis. American Journal of Pathology. 2010;177(4):1834–1847. doi: 10.2353/ajpath.2010.091010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cuzzocrea S, Pisano B, Dugo L, et al. Rosiglitazone, a ligand of the peroxisome proliferator-activated receptor-γ, reduces the development of nonseptic shock induced by zymosan in mice. Critical Care Medicine. 2004;32(2):457–466. doi: 10.1097/01.CCM.0000109446.38675.61. [DOI] [PubMed] [Google Scholar]
- 117.Galuppo M, Di Paola R, Mazzon E, et al. Role of PPAR-δ in the development of zymosan-induced multiple organ failure: an experiment mice study. Journal of Inflammation. 2010;7, article 12 doi: 10.1186/1476-9255-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Steiner DF, Cunningham D, Spigelman L, Aten B. Insulin biosynthesis: evidence for a precursor. Science. 1967;157(3789):697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- 119.Al-Rasheed NM, Chanai RS, Baines RJ, Willars GB, Brunskill NJ. Ligand-independent activation of peroxisome proliferator-activated receptor-γ by insulin and C-peptide in kidney proximal tubular cells: dependent on phosphatidylinositol 3-kinase activity. Journal of Biological Chemistry. 2004;279(48):49747–49754. doi: 10.1074/jbc.M408268200. [DOI] [PubMed] [Google Scholar]
- 120.Young LH, Ikeda Y, Scalia R, Lefer AM. C-peptide exerts cardioprotective effects in myocardial ischemia-reperfusion. American Journal of Physiology—Heart and Circulatory Physiology. 2000;279(4):H1453–H1459. doi: 10.1152/ajpheart.2000.279.4.H1453. [DOI] [PubMed] [Google Scholar]
- 121.Vish MG, Mangeshkar P, Piraino G, et al. Proinsulin c-peptide exerts beneficial effects in endotoxic shock in mice. Critical Care Medicine. 2007;35(5):1348–1355. doi: 10.1097/01.CCM.0000260245.61343.B3. [DOI] [PubMed] [Google Scholar]
- 122.Chima RS, LaMontagne T, Piraino G, Hake PW, Denenberg A, Zingarelli B. C-peptide, a novel inhibitor of lung inflammation following hemorrhagic shock. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2011;300(5):L730–L739. doi: 10.1152/ajplung.00308.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wu R, Zhou M, Wang P. Adrenomedullin and adrenomedullin binding protein-1 downregulate TNF-α in macrophage cell line and rat Kupffer cells. Regulatory Peptides. 2003;112(1–3):19–26. doi: 10.1016/s0167-0115(03)00018-1. [DOI] [PubMed] [Google Scholar]
- 124.Miksa M, Wu R, Cui X, et al. Vasoactive hormone adrenomedullin and its binding protein: anti-inflammatory effects by up-regulating peroxisome proliferator-activated receptor-γ . Journal of Immunology. 2007;179(9):6263–6272. doi: 10.4049/jimmunol.179.9.6263. [DOI] [PubMed] [Google Scholar]
- 125.Greene ME, Blumberg B, McBride OW, et al. isolation of the human peroxisome proliferator activated receptor gamma cDNA: expression in hematopoietic cells and chromosomal mapping. Gene Expression. 1995;4(4-5):281–299. [PMC free article] [PubMed] [Google Scholar]
- 126.Schmidt MV, Paulus P, Kuhn AM, et al. Peroxisome proliferator-activated receptor g-induced T cell apoptosis reduces survival during polymicrobial sepsis. American Journal of Respiratory and Critical Care Medicine. 2011;184(1):64–74. doi: 10.1164/rccm.201010-1585OC. [DOI] [PubMed] [Google Scholar]
- 127.Michalik L, Desvergne B, Tan NS, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)α and PPARβ mutant mice. Journal of Cell Biology. 2001;154(4):799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dubrac S, Schmuth M. PPAR-alpha in cutaneous inflammation. Dermato-Endocrinology. 2011;3(1):23–26. doi: 10.4161/derm.3.1.14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Icre G, Wahli W, Michalik L. Functions of the peroxisome proliferator-activated receptor (PPAR) α and β in skin homeostasis, epithelial repair, and morphogenesis. Journal of Investigative Dermatology Symposium Proceedings. 2006;11(1):30–35. doi: 10.1038/sj.jidsymp.5650007. [DOI] [PubMed] [Google Scholar]
- 130.Tan NS, Michalik L, Noy N, et al. Critical roles of PPARβ/δ in keratinocyte response to inflammation. Genes and Development. 2001;15(24):3263–3277. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Di-Po N, Tan NS, Michalik L, Wahli W, Desvergne B. Antiapoptotic role of PPARβ in keratinocytes via transcriptional control of the Akt1 signaling pathway. Molecular Cell. 2002;10(4):721–733. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 132.Nguan ST, Icre G, Montagner A, Bordier-Ten Heggeler B, Wahli W, Michalik L. The nuclear hormone receptor peroxisome proliferator-activated receptor β/δ potentiates cell chemotactism, polarization, and migration. Molecular and Cellular Biology. 2007;27(20):7161–7175. doi: 10.1128/MCB.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tan NS, Michalik L, Di-Poï N, et al. Essential role of Smad3 in the inhibition of inflammation-induced PPARβ/δ expression. EMBO Journal. 2004;23(21):4211–4221. doi: 10.1038/sj.emboj.7600437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tan NS, Michalik L, Desvergne B, Wahli W. Genetic- or transforming growth factor-β1-induced changes in epidermal peroxisome proliferator-activated receptor β/δ expression dictate wound repair kinetics. Journal of Biological Chemistry. 2005;280(18):18163–18170. doi: 10.1074/jbc.M412829200. [DOI] [PubMed] [Google Scholar]
- 135.Chong HC, Tan MJ, Philippe V, et al. Regulation of epithelial-mesenchymal IL-1 signaling by PPARβ/δ is essential for skin homeostasis and wound healing. Journal of Cell Biology. 2009;184(6):817–831. doi: 10.1083/jcb.200809028. [DOI] [PMC free article] [PubMed] [Google Scholar]