

Abstract
This review aims to provide a snapshot of the actual state of knowledge on genetic variants of nuclear receptors (NR) involved in regulating important aspects of liver metabolism. It recapitulates recent evidence for the application of NR in genetic diagnosis of monogenic (“Mendelian”) liver disease and their use in clinical diagnosis. Genetic analysis of multifactorial liver diseases such as viral hepatitis or fatty liver disease identifies key players in disease predisposition and progression. Evidence from these analyses points towards a role of NR polymorphisms in common diseases, linking regulatory networks to complex and variable phenotypes. The new insights into NR variants also offer perspectives and cautionary advice for their use as handles towards diagnosis and treatment.
1. Introduction
Systematically, genetic analysis with regard to disease onset and progression can be separated into pre- and post-hoc examination of monogenic or polygenic diseases. Monogenic (“Mendelian”) diseases are caused by a single gene defect and follow relatively straightforward inheritance patterns. The most prominent of these disorders are rather rare, often severe, and characterized by early onset. Genetic testing for monogenic liver disease in symptomatic patients is based on known disease-associated gene variants, thereby confirming the genetic etiology and sometimes allowing prediction of disease progression [1].
In contrast, polygenic diseases such as fatty liver disease and gallstones result from combinations of multiple gene variants and environmental factors, all of which play a role in disease initiation and progression [2]. The assessment of predisposition towards polygenic disease is based on sequence analysis of known contributory genes and construction of “polygenic risk scores” from variants of these genes [1]. Still in its infancy, personal genome information might eventually be able to predict a variety of risks associated with an individual's lifestyle such as fatty food and alcohol consumption, as well as susceptibility to infectious diseases such as infection with hepatitis B or C virus.
2. Nuclear Receptors
Nuclear receptors (NRs) are a subclass of regulatory molecules that orchestrate gene transcription in response to the presence or absence of specific ligands. Due to these functional requirements, they are characterized by the presence of a ligand-binding and a DNA-binding domain. NRs represent a central point of interaction between environment and gene regulation. They are the “hinge” connecting endogenous and environmental stimuli, that is, ligands, with the cells' transcriptional response (Figure 1).
Figure 1.
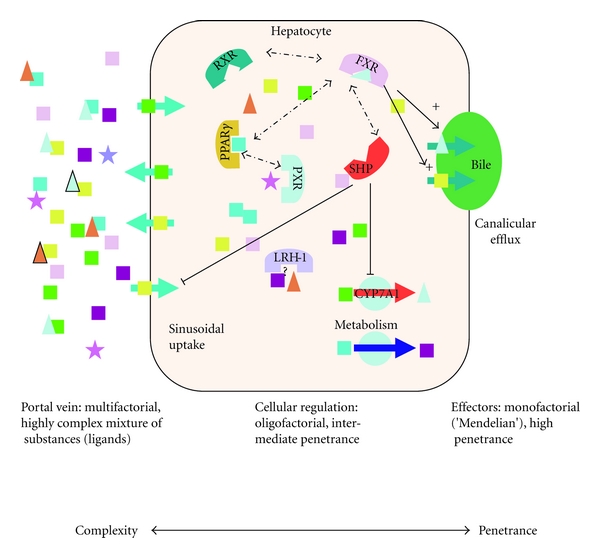
Schematic depiction of NR action in hepatocytes demonstrating a reduction in complexity and an increase in penetrance of genetic variants from the sinus to the canaliculus. Squares represent metabolic compounds such as triglycerides, cholesterol, fatty acids, and phospholipids; triangles represent bile salts; stars represent toxins; large semi-circles symbolise nuclear receptors, and circles stand for metabolic enzymes.
This position makes them a prime target for medical intervention by agonistic or antagonistic binding of synthetic compounds. However, the regulatory orchestra of molecules conducted by NR is highly complex and abounds with redundancy and crosstalk, hence any impact might be potentially difficult to predict as well as disappointingly diffuse. Even identical variations in a single NR can result in a wide variety of phenotypes due to genetic differences in the cofactors involved and higher order networks based on mutual regulatory interaction [3].
The core position in maintaining cellular equilibrium should render NR susceptible to the impact of natural sequence variation. On the other hand, it has been suggested that the phenotypic effects of NR gene variation are bound to be less severe than a variation in functionally defined effector molecules such as a single-substrate transmembrane transporter. The transcriptional regulation directed by NR is kept in tight check and fine-tuned by a set of co-regulators (co-activators or corepressors) [4, 5]. Consequently, relatively few congenital, “Mendelian” diseases have been identified to date, which are caused by genetic variations in NR. One example is maturity-onset diabetes of the young (MODY1), caused by mutations affecting the gene encoding the hepatic nuclear factor (HNF) 4-alpha (NR2A1) [6] (Table 1).
Table 1.
Single nucleotide polymorphisms (SNPs) associated with liver disease.
| Gene | SNP | rs number | Disease | OR (95% CI) | P-value | Cohort (controls) | Population | Reference |
|---|---|---|---|---|---|---|---|---|
| HNF4a (NR2A1) | Q268X | rs6093980 | MODY-1 | N/A | N/A | >360 | R-W pedigree | [6] |
| Y16X | N/A | N/A | N/A | 108 | UK | [9] | ||
| S34X | ||||||||
| R127W | ||||||||
| D206Y | ||||||||
| E276Q | ||||||||
| R303H I314F L332P |
||||||||
| M364R | ||||||||
| c. IVS5nt+1G>A | ||||||||
| c.IVS4nt-2A>G | ||||||||
| T(3;20) | ||||||||
| V393I | N/A | NIDDM | N/A | N/A | N/A | F-40 pedigree | [8] | |
| T130I | N/A | T2D | 1.26 (1.01–1.57) | 0.04 | 1,466 (4,520) | Danish | [10] | |
| V255M | N/A | Decreasing fasting serum C-peptide levels | 1.0 (0.28–3.65) | 1.0 | ||||
|
| ||||||||
| FXR (NR1H4) | −20,647T>G | N/A | Gallstones | 0.42 (0.17–1.01) | 0.053 | 77 (74) | Mexican | [34] |
| −1G>T | rs56163822 | 0.25 (0.07–0.95) | 0.042 | 75 (70) | ||||
| IVS7-31A>T | rs7138843 | 0.47 (0.22–1.01) | 0.053 | 77 (88) | ||||
| −1G>T | rs56163822 | ICP | 0.92 (0.35–2.44) | 0.96 | 342 (349) | British/Swedish | [35] | |
| M173T | N/A | 3.2 (1.1–11.2) | 0.02 | |||||
|
| ||||||||
| VDR (NR1I1) | c.1025-49G>T (ApaI) | rs7975232 | AIH | 0.72 (0.40–1.30) | 0.27 | 123 (214) | Caucasian | [22] |
| Intron 8 (BsmI) | rs1544410 | 0.63 (0.37–1.06) | 0.08 | |||||
| Exon 2 (FokI) | rs1073581 | 0.5 (0.28–0.92) | 0.02 | |||||
| I352I (TaqI) | rs731236 | 1.27 (0.69–2.33) | 0.43 | |||||
| c.1025-49G>T (ApaI) | rs7975232 | PBC | 1.85 (1.02–3.35) | 0.04 | 74 (214) | |||
| Intron 8 (BsmI) | rs1544410 | 2.1 (1.22–3.62) | 0.006 | |||||
| Exon 2 (FokI) | rs1073581 | 0.55 (0.27–1.12) | 0.09 | |||||
| I352I (TaqI) | rs731236 | 1.16 (0.56–2.39) | 0.69 | |||||
| c.1025-49G>T (ApaI) | rs7975232 | AIH | 0.82 (0.42–1.58) | 0,55 | 49 | Chinese | [20] | |
| Intron 8 (BsmI) | rs1544410 | 1.44 (0.59–3.51) | 0.42 | |||||
| Exon 2 (FokI) | rs1073581 | 2.18 (1.07–4.43) | 0.019 | |||||
| I352I (TaqI) | rs731236 | 0.00 (0.00) | 0.28 | |||||
| c.1025-49G>T (ApaI) | rs7975232 | PBC | 0.90 (0.49–1.64) | 0.727 | 58 | |||
| Intron 8 (BsmI) | rs1544410 | 4.41 (1.29–15.02) | 0.01 | |||||
| Exon 2 (FokI) | rs1073581 | 1.30 (0.63–2.68) | 0.05 | |||||
| I352I (TaqI) | rs731236 | 0.00 (0.00) | 0.224 | |||||
| c.1025-49G>T (ApaI) | rs7975232 | PBC | 0.71 (0.47–1.08) | 0.133 | 195 (179) | Japanese | [21] | |
| Intron 8 (BsmI) | rs1544410 | 0.71 (0.44–1.16) | 0.179 | |||||
| I352I (TaqI) | rs731236 | 1.02 (1.00–1.04) | 0.109 | |||||
| c.1025-49G>T (ApaI) | rs7975232 | 1.02 (0.52–1.98) | 1.000 | 139 (156) | Italian | |||
| Intron 8 (BsmI) | rs1544410 | 0.33 (0.12–0.92) | 0.039 | |||||
| I352I (TaqI) | rs731236 | 0.94 (0.51–1.75) | 0.876 | |||||
| c.1025-49G>T (ApaI) | rs7975232 | HBV | 3.3 (1–11) | 0.05 | 214 (408) | [23] | ||
| c.1025-49G>T (ApaI) | rs7975232 | HCC | 0.852 (0.345–2.113) | n.s. | 80 (160) | Caucasian | [30] | |
| Intron 8 (BsmI) | rs1544410 | 1.711 (0.766–3.813) | n.s. | |||||
| Exon 2 (FokI) | rs1073581 | 1.338 (0.605–2.968) | n.s. | |||||
| I352I (TaqI) | rs731236 | 0.491 (0.212–1.141) | 0.09 | |||||
|
| ||||||||
| PPARγ (NR1C3) | P12A | rs1805192 | T2D | 0.78 (0.59–1.05) | 0.045 | 333 | Scandinavian | [40] |
| 1.37 | 0.04 | 2,126 (1,124) | French | [38] | ||||
| 0.12 (0.03–0.52) | 0.005 | 532 (386) | Asian Sikh | [39] | ||||
| C161T | rs121909245 | Obesity | 2.33 (1.03–5.29) | 0.042 | 292 (371) | Australian | [37] | |
| NAFLD | 4.606 (3.744–10.263) | 0.003 | 96 (96) | Chinese | [46] | |||
|
| ||||||||
| LXRα (NR1H3) | N/A | rs2167079 | HDL cholesterol level | N/A | 5.13 × 10−8 | 4763 | Northern Finland Birth cohort 1966 | [78] |
| rs7120118 | 3.57 × 10−8 | |||||||
| AR (NR3C4) | N/A | rs5031002 | LDL cholesterol level | N/A | 2.37 × 10−7 | |||
|
| ||||||||
| PXR (NR1I2) | Intronic | rs7643645 | NAFLD | 3.48 (1.25–10.62) | 0.008 | 188 | Argentine | [54] |
| rs2461823 | N/A | 0.039 | ||||||
| −25385 | rs3814055 | DILI | 3.37 (1.55–7.30) | 0.0023 | 51 (64) | European | [74] | |
Abbreviations: OR: odds ratio; N/A: not annotated; n.s.: not significant.
While the denominator of “monogenic disease” seems to imply a straightforward genotype-to-phenotype correlation, the reality in even those seemingly “simple” diseases is anything but simple: Tirona et al. [7] showed that HNF4A is critically involved in the PXR (NR1I2)- and CAR (NR1I3)-mediated transcriptional activation of cytochrome P450 (CYP) 3A4, hence involving two more NRs even in its basal regulatory function. Whereas Hani et al. [8] identified HNF4A mutations as being causative in maturity-onset diabetes of the young (MODY type 1, OMIM #125850) based on a nonsense mutation (p.Q268X) in an extended pedigree, many other seemingly functional variants have turned out to be either innocent bystanders or of relatively low penetrance. Pearson et al. [9] were able to show that functional HNF4A variants are associated with a considerable increase in birth weight and macrosomia, and a novel cause of neonatal hypoglycemia. Results from the investigation of 108 members of 15 families with MODY1 show how genotype-phenotype correlation is far from clearcut, with both described phenotypes being found only in 15–56% of mutation carriers. Ek et al. [10] examined the impact of two disease-associated variants (p.T130I and p.V255M, Table 1) on various aspects of HNF4-alpha function. Both variants showed decreased transactivation. Only the p.T130I polymorphism was associated with T2D, whereas the p.V255M variant was associated with a decrease in fasting serum C-peptide levels. Array analyses revealed that HNF4-alpha bound to the promoters of 12% of hepatocyte islet genes represented on a microarray and hence can be considered a “master regulator” of hepatocyte and beta-cell genes [11]. But even in a complex and occasionally ambiguous setting, where the detection of a functional variant does not necessarily predict disease phenotypes, genetic testing appears to be helpful, if only to identify at-risk relatives and motivate affected individuals towards lifestyle changes [12]. Evidently, there is no clear delineation between the “Mendelian” diseases and contribution towards complex phenotypes modulated by NR variants (Figure 1). A meta-analysis of polymorphisms in the promoter and along the entire coding region of the HNF4A gene and type 2 diabetes in 49.577 individuals revealed significant associations for more than one locus [13].
Nuclear transcription factors are known to undergo posttranslational modifications modulating their regulatory activity, which obviously makes the interpretation of genetic tests more difficult. Recent findings of epigenetic modification of the HNF4A promoter add an additional layer of uncertainty and environmental impact. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets [14]. Environmentally induced changes in promoter-enhancer interactions might represent a key epigenetic mechanism by which nutrition can influence NR signaling.
3. Candidate Receptor Studies
3.1. Vitamin D Receptor (VDR/NR1I1)
The human vitamin D receptor (VDR/NR1I1) has been in the focus of research for over a decade, a main reason being a wide spectrum of known effects of vitamin D deficiency. A second, more mundane reason might be the availability of four frequent variants (rs7975232, ApaI; rs1544410, BsmI; rs10735810, FokI; rs731236, TaqI) amenable to relatively quick and easy analysis using simple technology that has been available in every genetics laboratory, restriction fragment length polymorphism (RFLP) analysis. A Pubmed search (as of June 7, 2011) on “vitamin D receptor polymorphism” resulted in 1,200 articles, covering a wide-range of associated biochemical processes and diseases ranging from the more obvious bone density in various species at various ages [15, 16] to Parkinson disease [17] within the first 20 hits, ulcerative colitis [18], and inflammatory bowel disease [19]. Limiting the search to the liver results in a more manageable set of less than 30 publications, with a detectable focus on inflammatory and autoimmune liver diseases, in particular autoimmune hepatitis and primary biliary cirrhosis [20–22], but also hepatitis B virus (HBV) infection [23] (Table 1).
In contrast to the mouse liver, which showed no VDR expression [3], VDR was detected mainly in the nonparenchymal cells of rat liver, whereas hepatocytes expressed barely any VDR in murine livers [24]. Human hepatocytes express VDR, albeit at very low abundance [25]. One of the potential ligands of hepatic VDR in humans is the secondary bile acid lithocholic acid, resulting in a repression of bile salt synthesis by transcriptional repression of cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme in bile salt biosynthesis [26]. The effect is achieved by competing for promoter binding with HNF4-alpha. This example shows that results from animal models have to be treated with caution, and once again illustrates the complex interaction between NR-regulated pathways in human liver.
Association studies between gene variants and diseases provide signposts towards genes underlying functional effects, but do not elucidate how these effects are achieved. Investigations into the detailed effects of the respective polymorphisms in NR are harder to interpret than similar investigations in other molecules of clearer functional delineation [27]. Cell-type specific splicing events might modulate transcriptional activation or ligand binding and cause effects in a substrate-dependent manner. As an example, a functional effect of the 3′BsmI polymorphism in intron 8 of the VDR/NR1I1 gene was shown to have a modulatory function on epithelial cell proliferation when combined with the effects of calcium [28]. The FokI polymorphism of VDR/NR1I1 results in distinct translation initiation sites and was shown to have an effect on cell growth inhibition, possibly through estrogen receptor-α protein repression in a cancer cell line [29]. These pleiotropic and highly variable functions go some way towards explaining the miscellany of associations that have been detected for VDR polymorphisms, among others with the occurrence of hepatocellular carcinoma (HCC) in patients with liver cirrhosis, particularly in patients with an alcoholic etiology [30]. They also show why the idea of using NR as a handle towards personalized treatment of patients is not straightforward, due to the high number of unspecific side effects.
3.2. FXR: The Central Bile Salt Sensor
FXR/NR1H4 is the hepatic nuclear bile salt receptor, regulating bile salt synthesis and transport in hepatocytes, the central hub of cholesterol synthesis and conversion. Bile salts are direct FXR ligands and bind to the ligand binding domain of the molecule at low concentrations as dimers with the retinoid X receptor (RXR/NR2B1). Upon binding of the heterodimer, conformational change causes FXR activation. FXR also controls enterohepatic circulation through regulation of intestinal bile salt uptake via expression of the intestinal bile acid binding protein (I-BABP) in enterocytes. At the same time, FXR increases expression and release of fibroblast growth factor 19 (FGF19, mouse orthologue FGF15), which provides a feedback regulation loop from the intestine to the liver via association with β-klotho and activation of its dedicated receptor FGFR4 (Figure 2). In the liver, dimerization with RXR induces the expression of various genes involved in bile salt transport from the hepatocyte into the bile canaliculus such as the phosphatidylcholine floppase ABCB4 and the bile salt export pump ABCB11. Interaction of FXR with the short heterodimer partner (SHP/NR0B2) decreases bile salt synthesis by repression of CYP7A1 and CYP8B1. Hence, FXR occupies a key role and is a prime target for manipulating the balance of bile salts in multiple parts of the enterohepatic circulation. However, results from a pilot experiment assessing the metabolic impact of a synthetic FXR agonist indicate that caution is warranted. The administration of GW4064-induced obesity and diabetes in mice fed a high-fat diet and worsened the metabolic effects in liver and adipose tissue [31].
Figure 2.
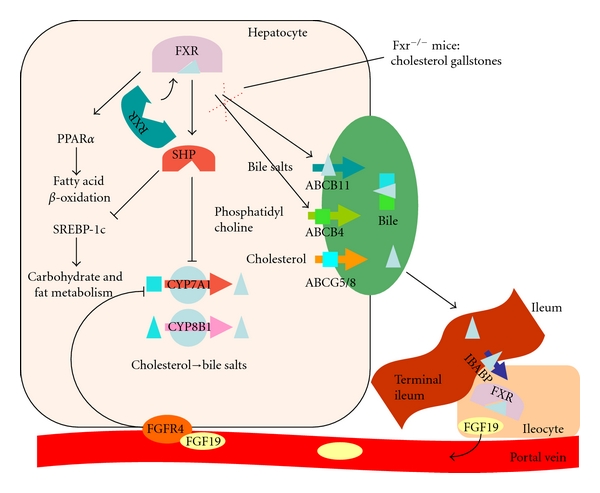
Schematic diagramme showing various aspects of FXR function as an example of complex nuclear receptor regulation and interaction. IBABP: intestinal bile acid binding protein, SREBP-1c: sterol regulatory element binding protein 1c, FGF: fibroblast growth factor, and FGFR: fibroblast growth factor receptor.
A potential role of FXR dysregulation in gallstone formation could be shown in the FXR deficient mouse. Due to the lack of positive feedback via FXR, the hepatocanalicular transporters ABCB4 and ABCB11 are not induced by bile salts (Figure 2) [32]. Under normal circumstances, biliary cholesterol is solubilized in mixed micelles, consisting of cholesterol, phosphatidylcholine, and bile salts. Lack of the latter two constituents causes supersaturation of cholesterol and the precipitation of crystals in the FXR-knockout mouse. When administered to gallstone-susceptible wild-type mice, the FXR agonist GW43456 reinstated the biliary balance of cholesterol, phospholipids, and bile salts by induction of hepatocanalicular transporter expression [32]. This makes FXR a potential target for the treatment of cholesterol gallstones.
3.2.1. FXR Variation and Functional Conservation
A survey of genetic variation in 13 NR that control the expression of drug metabolizing enzymes revealed an intriguing paucity of known functional variants in the coding region of FXR/NR1H4, comparable only in numbers to the androgen receptor (AR/NR3C4) and the aryl hydrocarbon receptor (AHR) [33]. This scarcity has been speculated to be indicative of considerable evolutionary selective pressures that conserve the functional domains in these receptors. However, compared to the both aforementioned receptors with low frequency of functional polymorphisms in the coding region, FXR revealed a relatively high number of base substitutions in the regulatory sequence. Thus, protein abundance of the molecule appears to be more variable than its conformation. Comparison of the frequency of variants in the sequences of drug metabolizing CYP enzymes with the frequency in essential enzymes in protein biosynthesis (ribosomal genes) and NR genes revealed similar patterns. A high level of variation in the regulatory sequence and a high conservation in coding areas of NR genes was juxtaposed by a reverse distribution in ribosomal genes [33]. No difference in variation frequency was observed in the noncoding, intronic areas.
3.2.2. FXR Variation in Complex Disease
Quantitative trait locus mapping in inbred mice identified the Nr1h4 gene encoding murine Fxr as a candidate gene for a gallstone susceptibility (lithogenic) locus (Lith7). Sequencing, genotyping, and haplotype analysis in humans revealed no more than three frequent haplotypes accounting for >95% of the variability. Kovacs et al. [34] described an association of a common risk haplotype NR1H4_1 (−20,647T; −1G; IVS7-31A) with gallstones in Mexican patients (OR = 2.1, P = 0.02) (Table 1). The association was inconsistent between different populations, pointing to a minor contributory role of FXR in overall gallstone susceptibility. Sequence variants of FXR have also been investigated in other pathological liver conditions, such as intrahepatic cholestasis of pregnancy (ICP). ICP is an interesting model disease as it illustrates the step up in complexity from monogenic diseases like severe familial cholestasis in children to complex cholestatic syndromes. The central role of FXR in balancing bile salt concentrations throughout the enterohepatic circulation makes it a good candidate for investigations into the causes of bile salt imbalances during pregnancy. Van Mil et al. [35] used an elegant and convincing experimental setup to prove the molecular impact of the few variants found in or near the transcribed sequence of FXR: They could show how two disease-associated single nucleotide polymorphisms (SNPs) at the methionine start codon or its immediate vicinity resulted in decreased translation. Expanding beyond the mere quantitative change, van Mil et al. [35] went on and proved that decreased translation diminished or abolished transactivation of FXR target genes in response to bile salt stimulation. A similar effect could be shown for the only nonconservative sequence variant that was found in both ICP patients and unaffected controls [35] (Table 1). Marzolini et al. [36] were able to confirm an effect of this variant in vivo by examining samples from a human liver bank. The expression levels of the FXR target genes short heterodimer partner (SHP/NR0B2) and organic anion transporting polypeptide 1B3 (OATP1B3) were reduced in livers harboring the rare [T] allele at position −1 of the FXR-coding sequence.
3.3. PPAR-Gamma and Diabetes
The peroxisome proliferator-activated receptor gamma (PPARG/NR1C3) is a fatty acid-activated member of the PPAR subfamily of NR. These receptors play important roles in lipid and glucose metabolism. Members of the family have been implicated in obesity-related metabolic diseases such as hyperlipidemia, insulin resistance, and coronary artery disease. Like FXR, PPARs form heterodimers with RXR, and these heterodimers regulate the transcription of various genes in liver. Polymorphisms in PPARG, particularly the proline-to-alanine substitution at aminoacid position 12, have been associated with diabetes, insulin levels, insulin sensitivity, body mass index, and dyslipidemia [37–40] (Table 1).
Disease prediction for population subgroups based on a combined “diabetes risk matrix” including PPARG p.P12A has been proposed to be informative and might, if accompanied by lifestyle intervention, prove a worthwhile path for prevention [41, 42]. Genome-wide association studies (GWAS) of type 2 diabetes in 2,335 Finns confirmed a contributory role of PPARG [43]. In a comprehensive example of complementarity between human and animal model research, Heikkinen et al. [44] were able to show that the p.P12A variant exerts its impact on various aspects of metabolism and human longevity in a diet-dependent manner. Hence, this prominent member of the NR family of molecules is a leading example of gene × environment interaction and “nature via nurture”.
3.4. PPAR-Gamma and NAFLD
Meirhaeghe et al. [45] described an association of a silent SNP in exon 6 of the PPARG gene (c.C161T) and the level of circulating leptin in obesity. Obese subjects carrying at least one [T] allele displayed higher plasma leptin levels than homozygous carriers of the common allele. The [T] allele was also associated with lower BMI at a given leptin level, indicating a complex interaction between PPAR-gamma and leptin signaling. These findings could be confirmed and extended by a study in 96 Chinese patients with nonalcoholic fatty liver disease (NAFLD), which reported an association of this variant with adiponectin levels and the development of NAFLD [46]. Zhou et al. [47] replicated this finding in an independent cohort, demonstrating that PPARG c.C161T and other polymorphisms are associated with the levels of tumor necrosis factor (TNF)-alpha, leptin, and adiponectin in NAFLD. When patients in Germany (NAFLD and AFLD, n = 363) [48] and Italy (NAFLD, n = 202) [49] were analyzed for the p.P12A variant, the results were less conclusive: German patients with fatty liver disease of either etiology were more likely to carry the rare minor allele, but no association was detected between p.P12A and the severity of steatosis, necroinflammation, or fibrosis.
3.5. Interactions between FXR and PPAR-Gamma
Bile salts are intimately entwined with lipid metabolism, and besides their well-known role in dietary lipid absorption and cholesterol homeostasis, evidence is accumulating that the body uses blood levels of bile salts as sensor for metabolic processes (reviewed comprehensively by Thomas et al. [50]). Hence, bile salts have been considered as metabolic signaling molecules. The decrease in energy expenditure following reduction of the bile salt pool by treatment of mice with the FXR-agonist GW4064 is proof of this concept, although the resulting weight gain and insulin resistance are not a desirable outcome [31]. The observation that PPAR-gamma might be induced by FXR in hepatic stellate cells underscores and highlights the complex interaction between bile salts, metabolism, inflammation, and fibrogenesis [51]. It also adds weight to the argument that NRs are involved in most aspects of metabolic regulation and liver cells' response to both internal and external stimuli. Further complexity is added to the spectrum of NR interactions in the liver by a recent report on the effect of GW4064 on the expression of the PPAR-gamma coactivator-1alpha (PGC1α/PPARGC1). The synthetic agonist GW4064 enhances expression of PGC1α and thus mitochondrial function, however enhanced expression of FXR increases PPARGC1 RNA not directly, but via protection from repression by the atypical corepressor SHP [52].
3.6. PXR and NAFLD
The pregnane X receptor (PXR/NR1I2) is known to be involved in the regulation of hepatic detoxification processes. Using PXR knockout and humanized mouse models, PXR was found to influence drug × drug interactions, hepatic steatosis, and the homeostasis of vitamin D, bile salts, and steroid hormones [53, 54]. Investigations into the genetic contribution of the PXR locus in 188 patients with NAFLD showed an association of two variants (rs7643645 and rs2461823) with several phenotypes of the disease, among others ALT levels [54] (Table 1). The combined analysis of both loci provided information with regards to disease progression. One of the associated SNPs (rs7643645) is localized within a potential binding site for HNF4-alpha, once again illustrating the interactions between NR pathways.
3.7. NR and Liver Cancer
Liver receptor homologue 1 (LRH-1, NR5A2) is an orphan member of the NR superfamily, that is, it has no known endogenous ligand. LRH-1 is involved in the regulation of genes that participate in steroid, bile salt, and cholesterol homeostasis [55]. Knockout of Nr5a2 in mice results in compromised intestinal lipid absorption as well as defective embryogenesis and cholesterol homeostasis [56, 57]. Application of dilauroyl-phosphatidylcholine, a specific LRH-1 agonist, increased bile salt levels and lowered hepatic triglyceride and serum glucose concentrations [58]. In mouse models of type 2 diabetes, the LRH-1 agonist also decreased hepatic steatosis and improved glucose homeostasis, pointing towards a new intervention target for the treatment of type 2 diabetes. LRH-1 has also been implicated in the growth of liver tumors via reversal of repression by SHP: In vitro methylation of the SHP promoter reversibly decreased transactivation and LRH-1 binding; overexpression of SHP inhibited HCC foci formation, arrested HCC tumor growth in xenografted nude mice, and increased the sensitivity of HCC cells to apoptotic stimuli [59].
3.8. NR and Viral Hepatitis
The replication of hepatitis C virus (HCV) is linked to lipid droplets, and a combined genomic/metabolomic analysis of HCV-infected HUH-7.5 cells by RNA sequencing, microarray, and proteomics revealed profound changes in, among others, PPAR signaling and PXR/RXR activation [60]. Viral replication efficiency has been linked to variations in cellular bile salt concentrations using the HCV replicon system [61]. Of note, variants of the human bile salt that export pump ABCB11 might be associated with sustained virological response [62] and progression towards liver cirrhosis [63]. In vitro experiments using HCV replicon-harboring cells have shown that the impact of bile salts on HCV replication might be through the action of FXR rather than a direct effect of bile salts themselves [64]. FXR antagonization by guggulsterone blocked the bile salt-mediated induction of HCV replication, and guggulsterone alone inhibited basal HCV replication by tenfold [64]. Hence, it seems feasible that HCV uses transcriptional activation via FXR. It would certainly be of interest to analyze the HCV-binding of natural FXR variants implicated in ICP [35]. We speculate that the increased susceptibility for cholestatic disease might be counterbalanced by decreased susceptibility to hepatotropic viruses.
Differential regulation of the pre-C and pregenomic promoters of HBV by members of the NR superfamily (HNF4-alpha and PPAR-gamma) has been known for some time [65]. Ramière et al. [66] were able to show that FXR-RXR-heterodimers bind to two motifs on the HBV enhancer II and core promoter regions, which are characterized by high homology to the consensus inverted repeat FXR response elements [66]. The tight connection between hepatotropic viruses, liver nutrition, and metabolism by means of NR is intriguing and might reveal new therapeutic targets or even dietary recommendations to optimize the efficacy of antiviral treatment for HBV or HCV infected patients.
3.9. NR and Drug-Induced Liver Injury (DILI)
While diet-induced metabolic overload, alcohol and HCV are the most common insults to the liver, drug-induced injury (DILI) is gaining in prominence due to increasing age and multimorbidity of the general population. The likelihood of medication-induced liver damage increases substantially beyond the threshold of 50 mg per day cumulative ingestion [67]. The involvement of the bile salt transporter system in estrogen-induced cholestasis has been observed for a long time in patients with ICP [35, 68, 69]. Functional variants of ABCB11 are known to be associated with cholestasis induced by oral contraceptives and other drugs [70], as reviewed elsewhere [71].
The role of gene polymorphisms in predisposition towards DILI has been reviewed comprehensively [72, 73]. Suffice to say that next to drug-metabolising enzymes, drug transporters and genes for the immune response to injury, variants in the NR genes PXR [74] and CAR [58, 75, 76] are the most prominent contributors, for example, towards acetaminophen (APAP) toxicity. Evidence comes from knockout or knockdown of these genes, conferring resistance to the toxic effects of APAP.
4. The Search for New Targets
4.1. NR and Metabolic Traits
Identifying and quantifying genes associated with metabolic traits is one of the prime challenges when devising means to deal with the epidemics of diabetes and metabolic syndrome. This information might be used as cost-efficient leverage to identify and motivate at-risk individuals towards lifestyle changes, but to date there is no evidence for clinical impact [77]. A long standing question with regard to the usefulness of genome wide association studies (GWAS) has been how to detect the impact of other than high-frequency low-risk variants. While the setup of GWAS is a case-control scenario in an otherwise unselected population, more in-depth information is expected from longitudinal studies on large cohorts of individuals with defined but limited genetic heterogeneity. A combination of results from both approaches might be required: since GWAS needs to accommodate higher levels of heterogeneity, and hence a greater variety of factors that influence the trait under examination, only factors that reach the threshold across the total population are identified. Studies on smaller cohorts, possibly with less genetic variety due to a relatively small common founder population, enable researchers to identify the impact of comparatively rare variants that have been enriched in the respective population due to a lack of admixture, while contributory genetic factors that are not present in the founder population might be missed. In fact, using large cohorts with relatively small founder populations have enabled researchers to identify metabolic risk factors by GWAS and association mapping. Typing 4,763 individuals from the Northern Finland birth cohort 1966 for 329,091 SNPs identified 21 genomic loci that were associated with metabolic traits such as HDL and LDL cholesterol levels [78]. Besides the “usual suspects” such as a frequent polymorphism in the NR1H3 gene encoding LXR-alpha, which affects HDL cholesterol level with an effect size of only 4% but shows a risk allele frequency of 42%, the study also detected some higher impact rare alleles. A variation in the gene for the androgen receptor AR (NR3C4) exerted a strong influence on LDL cholesterol levels, with a considerable effect size of 30% set aside a risk allele frequency of only 2% [78] (Table 1).
4.2. Complex NR Genetics
At our present state of knowledge, the variation of transcript abundance is probably a more important mechanism underlying disease susceptibility than structural protein alterations by nonsynonymous SNPs [79]. The view of health as a balanced state or equilibrium of interrelated gene expressions holds a lot of attraction for liver homeostasis, with diseases representing “network perturbations” [80]. This view also explains why transcription factors are more likely to be detected in GWAS studies of quantitative metabolic traits, since they might be master regulators of multiple other genes. Hence, NR variants impact not just on their own expression but can rather, via multiple effectors, exert stronger effects on a complex phenotype.
A GWAS with more than 2.5 million SNPs in 19,840 individuals, with replication in up to 20,632 individuals, identified more than 30 loci that had an impact on serum LDL cholesterol, HDL cholesterol, or triglyceride levels. A rare variant (risk allele frequency 3%) in HNF4A (p.T130I) was identified as a new contributory factor for HDL cholesterol levels, with an effect size of 19% [81].
4.3. Metabolic Traits as Quantitative Trait Loci (QTLs)
Technical progress, particularly the combination of increased speed and decreased cost of genotyping and expression quantification, is ushering in systematic approaches to metabolic traits. Once sufficiently large numbers of individuals from any population (humans, animals, or plants) have been genotyped for markers covering the entire genome, statistics can be used to calculate the likelihood of correlation between the inheritance of genotypes and the expression values of quantifiable phenotypes, including the abundance of transcripts assayed by expression arrays. This methodology, denoted as “expression quantitative trait loci (eQTL) mapping” can be used to unravel gene regulation by association and identify regulatory networks involving multiple loci. To extend the information from these large datasets, Schadt et al. [82] proposed mathematical models and tools to infer relationships between genes and groups of genes as well as between gene expression and disease phenotypes. Liu et al. [83] used data from the mouse phenome database covering 173 mouse phenotypes to map 937 quantitative trait loci in silico. Phenotypes examined included various metabolic traits such as fat mass at different diets, cholesterol, and triglyceride levels under normal and atherogenic diet. Ten of the QTL regions identified in this study contained candidate genes that had previously been characterized and shown to cause metabolic phenotypes in agreement with the trait used for mapping, serving as a proof of principle for the application of this methodology.
A note of caution is obviously due when transferring data and results from mouse models into the human genomic context. Transcriptional regulation, particularly organ-specific mechanisms and binding sites, has diverged significantly between man and mouse, probably more so than NR function [84]. Nevertheless, results from animal models, particularly knockouts, still provide valuable clues towards unexpected mechanisms of disease [85, 86].
4.4. Combining the Power of GWAS and eQTL
The identification and functional characterization of regulatory SNPs have encouraged the use of eQTL data for the interpretation of GWAS results. These genome-wide scans using anonymous SNP markers usually detect associations of disease with polymorphic markers, often in regions without known candidate genes [87]. For an example we refer to Kathiresan et al. [81], who identified 30 loci that contributed towards the regulation of three dyslipidemia traits. Increasing knowledge about transregulatory effects by eQTL might help to pinpoint functional mechanisms for these disease-associated variants. eQTL data enables us to identify a mechanism by which a SNP controls expression of a remote locus, hence causing predisposition to disease by allelic variation across long genomic distances [88].
5. Bringing It Together: Future Perspectives
Technical advances increase our knowledge regarding the biochemical and gene-regulatory mechanisms underlying metabolic diseases. Transgenic animals inform us about gene function, eQTL studies in human samples and model systems provide us with information about genetic loci that are associated with the inheritance of multiple metabolic parameters, and GWAS in genetically well-characterized cohorts yield candidate genes for metabolic disturbances at ever increasing resolution and depth. Considering all this amassed knowledge, our understanding of disease pathobiology is improving constantly. Considering the reality of how little lifestyle modification is achieved by risk information, as illustrated by antismoking campaigns, it remains a challenging task to employ this knowledge to combat the epidemics of the metabolic syndrome and its associated burden of disease.
Acknowledgment
Experimental work of the authors related to this paper has been supported by Deutsche Forschungsgemeinschaft (SFB/TRR 57 TP 01, DFG LA997/5-1, DFG LA997/6-1).
References
- 1.Krawczyk M, Müllenbach R, Weber SN, Zimmer V, Lammert F. Genome-wide association studies and genetic risk assessment of liver diseases. Nature Reviews Gastroenterology and Hepatology. 2010;7(12):669–681. doi: 10.1038/nrgastro.2010.170. [DOI] [PubMed] [Google Scholar]
- 2.Korstanje R, Paigen B. From QTL to gene: the harvest begins. Nature Genetics. 2002;31(3):235–236. doi: 10.1038/ng0702-235. [DOI] [PubMed] [Google Scholar]
- 3.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Viswakarma N, Jia Y, Bai L, Vluggens A, Borensztajn J, Xu J. Coactivators in PPAR-regulated gene expression. PPAR Research. 2010;2010:21 pages. doi: 10.1155/2010/250126. Article ID 250126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lonard DM, Lanz RB, O'Malley BW. Nuclear receptor coregulators and human disease. Endocrine Reviews. 2007;28(5):575–587. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 6.Yamagata K, Furuta H, Oda N, et al. Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1) Nature. 1996;384(6608):458–460. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 7.Tirona RG, Lee W, Leake BF, et al. The orphan nuclear receptor HNF4α determines PXR- and CAR-mediated xenobiotic induction of CYP3A4. Nature Medicine. 2003;9(2):220–224. doi: 10.1038/nm815. [DOI] [PubMed] [Google Scholar]
- 8.Hani EH, Suaud L, Boutin P, et al. A missense mutation in hepatocyte nuclear factor-4α, resulting in a reduced transactivation activity, in human late-onset non-insulin-dependent diabetes mellitus. Journal of Clinical Investigation. 1998;101(3):521–526. doi: 10.1172/JCI1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pearson ER, Boj SF, Steele AM, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Medicine. 2007;4(4, article e118) doi: 10.1371/journal.pmed.0040118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ek J, Rose CS, Jensen DP, et al. The functional Thr130Ile and Val255Met polymorphisms of the hepatocyte nuclear factor-4α (HNF4A): gene associations with type 2 diabetes or altered β-cell function among danes. Journal of Clinical Endocrinology and Metabolism. 2005;90(5):3054–3059. doi: 10.1210/jc.2004-2159. [DOI] [PubMed] [Google Scholar]
- 11.Odom DT, Zizlsperger N, Gordon DB, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303(5662):1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naylor R, Philipson LH. Who should have genetic testing for MODY? Clinical Endocrinology. [DOI] [PubMed]
- 13.Sookoian S, Gemma C, Pirola CJ. Influence of hepatocyte nuclear factor 4α (HNF4α) gene variants on the risk of type 2 diabetes: a meta-analysis in 49,577 individuals. Molecular Genetics and Metabolism. 2010;99(1):80–89. doi: 10.1016/j.ymgme.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Sandovici I, Smith NH, Nitert MD, et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(13):5449–5454. doi: 10.1073/pnas.1019007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoldemir T, Yavuz DG, Anik G, Verimli N, Erenus M. Vitamin D receptor gene polymorphisms in a group of postmenopausal Turkish women: association with bone mineral density. Climacteric. 2011;14(3):384–391. doi: 10.3109/13697137.2010.550973. [DOI] [PubMed] [Google Scholar]
- 16.Swamy GK, Garrett ME, Miranda ML, Ashley-Koch AE. Maternal vitamin D receptor genetic variation contributes to infant birthweight among black mothers. American Journal of Medical Genetics, Part A. 2011;155(6):1264–1271. doi: 10.1002/ajmg.a.33583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler MW, Burt A, Edwards TL, et al. Vitamin D receptor gene as a candidate gene for Parkinson disease. Annals of Human Genetics. 2011;75(2):201–210. doi: 10.1111/j.1469-1809.2010.00631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pei FH, Wang YJ, Gao SL, et al. Vitamin D receptor gene polymorphism and ulcerative colitis susceptibility in Han Chinese. Journal of Digestive Diseases. 2011;12(2):90–98. doi: 10.1111/j.1751-2980.2011.00483.x. [DOI] [PubMed] [Google Scholar]
- 19.Bentley RW, Keown D, Merriman TR, et al. Vitamin D receptor gene polymorphism associated with inflammatory bowel disease in New Zealand males. Alimentary Pharmacology and Therapeutics. 2011;33(7):855–856. doi: 10.1111/j.1365-2036.2011.04588.x. [DOI] [PubMed] [Google Scholar]
- 20.Fan L, Tu X, Zhu Y, et al. Genetic association of vitamin D receptor polymorphisms with autoimmune hepatitis and primary biliary cirrhosis in the Chinese. Journal of Gastroenterology and Hepatology. 2005;20(2):249–255. doi: 10.1111/j.1440-1746.2005.03532.x. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka A, Nezu S, Uegaki S, et al. Vitamin D receptor polymorphisms are associated with increased susceptibility to primary biliary cirrhosis in Japanese and Italian populations. Journal of Hepatology. 2009;50(6):1202–1209. doi: 10.1016/j.jhep.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Vogel A, Strassburg CP, Manns MP. Genetic association of vitamin D receptor polymorphisms with primary biliary cirrhosis and autoimmune hepatitis. Hepatology. 2002;35(1):126–131. doi: 10.1053/jhep.2002.30084. [DOI] [PubMed] [Google Scholar]
- 23.Suneetha PV, Sarin SK, Goyal A, Kumar GT, Shukla DK, Hissar S. Association between vitamin D receptor, CCR5, TNF-α and TNF-β gene polymorphisms and HBV infection and severity of liver disease. Journal of Hepatology. 2006;44(5):856–863. doi: 10.1016/j.jhep.2006.01.028. [DOI] [PubMed] [Google Scholar]
- 24.Gascon-Barré M, Demers C, Mirshahi A, Néron S, Zalzal S, Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology. 2003;37(5):1034–1042. doi: 10.1053/jhep.2003.50176. [DOI] [PubMed] [Google Scholar]
- 25.Han S, Chiang JY. Mechanism of vitamin D receptor inhibition of cholesterol 7α-hydroxylase gene transcription in human hepatocytes. Drug Metabolism and Disposition. 2009;37(3):469–478. doi: 10.1124/dmd.108.025155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han S, Li T, Ellis E, Strom S, Chiang JY. A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Molecular Endocrinology. 2010;24(6):1151–1164. doi: 10.1210/me.2009-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byrne JA, Strautnieks SS, Ihrke G, et al. Missense mutations and single nucleotide polymorphisms in ABCB11 impair bile salt export pump processing and function or disrupt pre-messenger RNA splicing. Hepatology. 2009;49(2):553–567. doi: 10.1002/hep.22683. [DOI] [PubMed] [Google Scholar]
- 28.Holt PR, Arber N, Halmos B, et al. Colonic epithelial cell proliferation decreases with increasing levels of serum 25-hydroxy vitamin D. Cancer Epidemiology Biomarkers and Prevention. 2002;11(1):113–119. [PubMed] [Google Scholar]
- 29.Alimirah F, Peng X, Murillo G, Mehta RG. Functional significance of vitamin D receptor Foki polymorphism in human breast cancer cells. PLoS ONE. 2011;6(1, article e16024) doi: 10.1371/journal.pone.0016024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Falleti E, Bitetto D, Fabris C, et al. Vitamin D receptor gene polymorphisms and hepatocellular carcinoma in alcoholic cirrhosis. World Journal of Gastroenterology. 2010;16(24):3016–3024. doi: 10.3748/wjg.v16.i24.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watanabe M, Horai Y, Houten SM, et al. Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure. Journal of Biological Chemistry. 2011;286(30):26913–26920. doi: 10.1074/jbc.M111.248203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moschetta A, Bookout AL, Mangelsdorf DJ. Prevention of cholesterol gallstone disease by FXR agonists in a mouse model. Nature Medicine. 2004;10(12):1352–1358. doi: 10.1038/nm1138. [DOI] [PubMed] [Google Scholar]
- 33.Okey AB, Boutros PC, Harper PA. Polymorphisms of human nuclear receptors that control expression of drug-metabolizing enzymes. Pharmacogenetics and Genomics. 2005;15(6):371–379. doi: 10.1097/01213011-200506000-00001. [DOI] [PubMed] [Google Scholar]
- 34.Kovacs P, Kress R, Rocha J, et al. Variation of the gene encoding the nuclear bile salt receptor FXR and gallstone susceptibility in mice and humans. Journal of Hepatology. 2008;48(1):116–124. doi: 10.1016/j.jhep.2007.07.027. [DOI] [PubMed] [Google Scholar]
- 35.van Mil SWC, Milona A, Dixon PH, et al. Functional variants of the central bile acid sensor FXR identified in intrahepatic cholestasis of pregnancy. Gastroenterology. 2007;133(2):507–516. doi: 10.1053/j.gastro.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Marzolini C, Tirona RG, Gervasini G, et al. A common polymorphism in the bile acid receptor farnesoid X receptor is associated with decreased hepatic target gene expression. Molecular Endocrinology. 2007;21(8):1769–1780. doi: 10.1210/me.2007-0025. [DOI] [PubMed] [Google Scholar]
- 37.Swarbrick MM, Chapman CM, McQuillan BM, Hung J, Thompson PL, Beilby JP. A Pro12Ala polymorphism in the human peroxisome proliferator-activated receptor-γ2 is associated with combined hyperlipidaemia in obesity. European Journal of Endocrinology. 2001;144(3):277–282. doi: 10.1530/eje.0.1440277. [DOI] [PubMed] [Google Scholar]
- 38.Ghoussaini M, Meyre D, Lobbens S, et al. Implication of the Pro12Ala polymorphism of the PPAR-gamma 2 gene in type 2 diabetes and obesity in the French population. BMC Medical Genetics. 2005;6, article 11 doi: 10.1186/1471-2350-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sanghera DK, Ortega L, Han S, et al. Impact of nine common type 2 diabetes risk polymorphisms in Asian Indian Sikhs: PPARG2 (Pro12Ala), IGF2BP2, TCF7L2 and FTO variants confer a significant risk. BMC Medical Genetics. 2008;9, article 59 doi: 10.1186/1471-2350-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Altshuler D, Hirschhorn JN, Klannemark M, et al. The common PPARγ Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics. 2000;26(1):76–80. doi: 10.1038/79216. [DOI] [PubMed] [Google Scholar]
- 41.Weedon MN, McCarthy MI, Hitman G, et al. Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Medicine. 2006;3(10, article e374) doi: 10.1371/journal.pmed.0030374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316(5829):1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in finns detects multiple susceptibility variants. Science. 2007;316(5829):1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heikkinen S, Argmann C, Feige JN, et al. The Pro12Ala PPARγ2 variant determines metabolism at the gene-environment interface. Cell Metabolism. 2009;9(1):88–98. doi: 10.1016/j.cmet.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 45.Meirhaeghe A, Fajas L, Helbecque N, et al. A genetic polymorphism of the peroxisome proliferator-activated receptor γ gene influences plasma leptin levels in obese humans. Human Molecular Genetics. 1998;7(3):435–440. doi: 10.1093/hmg/7.3.435. [DOI] [PubMed] [Google Scholar]
- 46.Hui Y, Yu-yuan L, Yu-qiang N, et al. Effect of peroxisome proliferator-activated receptors-γ and co-activator-1α genetic polymorphisms on plasma adiponectin levels and susceptibility of non-alcoholic fatty liver disease in Chinese people. Liver International. 2008;28(3):385–392. doi: 10.1111/j.1478-3231.2007.01623.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhou YJ, Li YY, Nie YQ, et al. Influence of polygenetic polymorphisms on the susceptibility to non-alcoholic fatty liver disease of Chinese people. Journal of Gastroenterology and Hepatology. 2010;25(4):772–777. doi: 10.1111/j.1440-1746.2009.06144.x. [DOI] [PubMed] [Google Scholar]
- 48.Rey JW, Noetel A, Hardt A, et al. Pro12Ala polymorphism of the peroxisome proliferator-activated receptor gamma2 in patients with fatty liver diseases. World Journal of Gastroenterology. 2010;16(46):5830–5837. doi: 10.3748/wjg.v16.i46.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dongiovanni P, Rametta R, Fracanzani AL, et al. Lack of association between peroxisome proliferator-activated receptors alpha and gamma2 polymorphisms and progressive liver damage in patients with non-alcoholic fatty liver disease: a case control study. BMC Gastroenterology. 2010;10, article 102 doi: 10.1186/1471-230X-10-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nature Reviews Drug Discovery. 2008;7(8):678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 51.Renga B, Mencarelli A, Migliorati M, et al. SHP-dependent and -independent induction of peroxisome proliferator- activated receptor-γ by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflammation Research. 2011;60(6):577–587. doi: 10.1007/s00011-010-0306-1. [DOI] [PubMed] [Google Scholar]
- 52.Dwivedi SK, Singh N, Kumari R, et al. Bile acid receptor agonist GW4064 regulates PPARγ coactivator-1α expression through estrogen receptor-related receptor α . Molecular Endocrinology. 2011;25(6):922–932. doi: 10.1210/me.2010-0512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma X, Idle JR, Gonzalez FJ. The pregnane X receptor: from bench to bedside. Expert Opinion on Drug Metabolism and Toxicology. 2008;4(7):895–908. doi: 10.1517/17425255.4.7.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sookoian S, Castaño GO, Burgueño AL, Gianotti TF, Rosselli MS, Pirola CJ. The nuclear receptor PXR gene variants are associated with liver injury in nonalcoholic fatty liver disease. Pharmacogenetics and Genomics. 2010;20(1):1–8. doi: 10.1097/FPC.0b013e328333a1dd. [DOI] [PubMed] [Google Scholar]
- 55.Mataki C, Magnier BC, Houten SM, et al. Compromised intestinal lipid absorption in mice with a liver-specific deficiency of liver receptor homolog 1. Molecular and Cellular Biology. 2007;27(23):8330–8339. doi: 10.1128/MCB.00852-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paré JF, Malenfant D, Courtemanche C, et al. The fetoprotein transcription factor (FTF) gene is essential to embryogenesis and cholesterol homeostasis and is regulated by a DR4 element. Journal of Biological Chemistry. 2004;279(20):21206–21216. doi: 10.1074/jbc.M401523200. [DOI] [PubMed] [Google Scholar]
- 57.Gu P, Goodwin B, Chung AC, et al. Orphan nuclear receptor LRH-1 is required to maintain Oct4 expression at the epiblast stage of embryonic development. Molecular and Cellular Biology. 2005;25(9):3492–3505. doi: 10.1128/MCB.25.9.3492-3505.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee JM, Lee YK, Mamrosh JL, et al. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474(7352):506–510. doi: 10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He N, Park K, Zhang Y, Huang J, Lu S, Wang L. Epigenetic inhibition of nuclear receptor small heterodimer partner is associated with and regulates hepatocellular carcinoma growth. Gastroenterology. 2008;134(3):793–802. doi: 10.1053/j.gastro.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 60.Woodhouse SD, Narayan R, Latham S, et al. Transcriptome sequencing, microarray, and proteomic analyses reveal cellular and metabolic impact of hepatitis C virus infection in vitro. Hepatology. 2010;52(2):443–453. doi: 10.1002/hep.23733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chang KO, George DW. Bile acids promote the expression of hepatitis C virus in replicon-harboring cells. Journal of Virology. 2007;81(18):9633–9640. doi: 10.1128/JVI.00795-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iwata R, Stieger B, Mertens JC, et al. The role of bile acid retention and a common polymorphism in the ABCB11 gene as host factors affecting antiviral treatment response in chronic hepatitis C. Journal of Viral Hepatitis. 2011;18(11):768–778. doi: 10.1111/j.1365-2893.2010.01363.x. [DOI] [PubMed] [Google Scholar]
- 63.Iwata R, Baur K, Stieger B, et al. A common polymorphism in the ABCB11 gene is associated with advanced fibrosis in hepatitis C but not in non-alcoholic fatty liver disease. Clinical Science. 2011;120(7):287–296. doi: 10.1042/CS20100246. [DOI] [PubMed] [Google Scholar]
- 64.Scholtes C, Diaz O, Icard V, et al. Enhancement of genotype 1 hepatitis C virus replication by bile acids through FXR. Journal of Hepatology. 2008;48(2):192–199. doi: 10.1016/j.jhep.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 65.Yu X, Mertz JE. Differential regulation of the pre-C and pregenomic promoters of human hepatitis B virus by members of the nuclear receptor superfamily. Journal of Virology. 1997;71(12):9366–9374. doi: 10.1128/jvi.71.12.9366-9374.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramière C, Scholtès C, Diaz O, et al. Transactivation of the hepatitis B virus core promoter by the nuclear receptor FXRα . Journal of Virology. 2008;82(21):10832–10840. doi: 10.1128/JVI.00883-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2008;47(6):2003–2009. doi: 10.1002/hep.22272. [DOI] [PubMed] [Google Scholar]
- 68.Jacquemin E, Cresteil D, Manouvrier S, Boute O, Hadchouel M. Heterozygous non-sense mutation of the MDR3 gene in familial intrahepatic cholestasis of pregnancy. Lancet. 1999;353(9148):210–211. doi: 10.1016/S0140-6736(05)77221-4. [DOI] [PubMed] [Google Scholar]
- 69.Dixon PH, Weerasekera N, Linton KJ, et al. Heterozygous MDR3 missense mutation associated with intrahepatic cholestasis of pregnancy: evidence for a defect in protein trafficking. Human Molecular Genetics. 2000;9(8):1209–1217. doi: 10.1093/hmg/9.8.1209. [DOI] [PubMed] [Google Scholar]
- 70.Meier Y, Zodan T, Lang C, et al. Increased susceptibility for intrahepatic cholestasis of pregnancy and contraceptive-induced cholestasis in carriers of the 1331T > C polymorphism in the bile salt export pump. World Journal of Gastroenterology. 2008;14(1):38–45. doi: 10.3748/wjg.14.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stieger B, Geier A. Genetic variations of bile salt transporters as predisposing factors for drug-induced cholestasis, intrahepatic cholestasis of pregnancy and therapeutic response of viral hepatitis. Expert Opinion on Drug Metabolism and Toxicology. 2011;7(4):411–425. doi: 10.1517/17425255.2011.557067. [DOI] [PubMed] [Google Scholar]
- 72.Wagner M, Zollner G, Trauner M. Nuclear receptors in liver disease. Hepatology. 2011;53(3):1023–1034. doi: 10.1002/hep.24148. [DOI] [PubMed] [Google Scholar]
- 73.Russmann S, Jetter A, Kullak-Ublick GA. Pharmacogenetics of drug-induced liver injury. Hepatology. 2010;52(2):748–761. doi: 10.1002/hep.23720. [DOI] [PubMed] [Google Scholar]
- 74.Andrews E, Armstrong M, Tugwood J, et al. A role for the pregnane X receptor in flucloxacillin-induced liver injury. Hepatology. 2010;51(5):1656–1664. doi: 10.1002/hep.23549. [DOI] [PubMed] [Google Scholar]
- 75.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. Journal of Biological Chemistry. 2004;279(47):49517–49522. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 76.Amador-Noguez D, Dean A, Huang W, Setchell K, Moore D, Darlington G. Alterations in xenobiotic metabolism in the long-lived Little mice. Aging Cell. 2007;6(4):453–470. doi: 10.1111/j.1474-9726.2007.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haga SB. Ethical issues of predictive genetic testing for diabetes. Journal of Diabetes Science and Technology. 2009;3(4):781–788. doi: 10.1177/193229680900300427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sabatti C, Service SK, Hartikainen AL, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nature Genetics. 2009;41(1):35–46. doi: 10.1038/ng.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M. Mapping complex disease traits with global gene expression. Nature Reviews Genetics. 2009;10(3):184–194. doi: 10.1038/nrg2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.del Sol A, Balling R, Hood L, Galas D. Diseases as network perturbations. Current Opinion in Biotechnology. 2010;21(4):566–571. doi: 10.1016/j.copbio.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 81.Kathiresan S, Willer CJ, Peloso GM, et al. Common variants at 30 loci contribute to polygenic dyslipidemia. Nature Genetics. 2009;41(1):56–65. doi: 10.1038/ng.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schadt EE, Lamb J, Yang X, et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nature Genetics. 2005;37(7):710–717. doi: 10.1038/ng1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu P, Vikis H, Lu Y, Wang D, You M. Large-scale in silico mapping of complex quantitative traits in inbred mice. PLoS ONE. 2007;2(7, article e651) doi: 10.1371/journal.pone.0000651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Odom DT, Dowell RD, Jacobsen ES, et al. Tissue-specific transcriptional regulation has diverged significantly between human and mouse. Nature Genetics. 2007;39(6):730–732. doi: 10.1038/ng2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Das SK, Eder S, Schauer S, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333(6039):233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 86.Skarnes WC, Rosen B, West AP, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474(7351):337–342. doi: 10.1038/nature10163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber SN, Gressner OA, Hall R, Grünhage F, Lammert F. Genetic determinants in hepatic fibrosis: from experimental models to fibrogenic gene signatures in humans. Clinics in Liver Disease. 2008;12(4):747–757. doi: 10.1016/j.cld.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 88.Libioulle C, Louis E, Hansoul S, et al. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS genetics. 2007;3(4, article e58) doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]