

Abstract
Metastasis to the central nervous system (CNS) remains a major cause of morbidity and mortality in patients with systemic cancers. Various crucial interactions between the brain environment and tumor cells take place during the development of the cancer at its new location. The rapid expansion in molecular biology and genetics has advanced our knowledge of the underlying mechanisms involved, from invasion to final colonization of new organ tissues. Understanding the various events occurring at each stage should enable targeted drug delivery and individualized treatments for patients, with better outcomes and fewer side effects. This paper summarizes the principal molecular and genetic mechanisms that underlie the development of brain metastasis (BrM).
1. Introduction
Brain metastases are the most frequently diagnosed intracranial neoplasms in adults, with an annual incidence estimated at 200,000 cases in the USA alone [1], an incidence 10 times greater than primary brain tumors [2]. Up to 20–40% of patients with adult systemic malignancies will develop brain metastases in the course of their disease; about 10–20% will be symptomatic [3, 4]. Improved treatment options for systemic disease, along with tools that permit less invasive screening, often when patients are asymptomatic, have increased patient survival, paradoxically escalating both its incidence and prevalence. A variety of systemic malignancies can metastasize to the central nervous system (CNS), although the majority of the lesions come from lung cancer (40–50%) followed by breast cancer (20–30%), melanoma (5–10%), lymphoma, and various other primary sites like the gastrointestinal tract (4–6%) and prostate [5, 6].
More than a century ago, Stephen Paget advanced his “seed and soil” hypothesis, which suggests that the occurrence of brain metastases is not random, but is secondary to certain tumor cells—“the seed”—having an attraction for the surrounding environment—“the soil” [7]. The hypothesis envisages three principles: first, that neoplasms are composed of heterogeneous subpopulations of cells, with different characteristics; second, that only a selectively “fit” subpopulation of cells will survive and multiply, invade, and migrate to other locations; finally, that colonization depends on tumor cell “seed” and host microenvironment “soil” interactions [8]. According to Ewing, circulatory patterns are responsible for the organ-specific spread between the primary tumor and their final destination [9]. Although complex, the metastatic process can be broadly divided into two main stages, the first being the migration of tumor cells from their primary tumor environment to various distant tissues and the second being the colonization of these tumor cells in their new location [10]. Underlying these two main stages are a number of cellular hallmarks taking place during the development and metastasis of human tumors [11]. The various molecular, genetic, and epigenetic changes that occur define the multistep dissemination process of the tumor, also known as the “metastatic cascade.”
Most BrMs occur in the cerebral hemispheres (80%), followed by the cerebellum (15%) and the brainstem (5%), corresponding to vascular distribution and tissue volumes [12]. BrMs are a major cause of morbidity and mortality, with clinical features of the metastasis corresponding to the location, causing focal neurological deficits, or presenting with nonspecific central nervous system features such as headache, cognitive impairment, and seizures [13]. The central nervous system (CNS) acts as a “safe haven,” generally beyond the reach of nearly all chemotherapeutic agents. The blood brain barrier (BBB) prevents the entry of most chemotherapeutic agents, and so the brain can act as a refuge for metastatic tumors [14]. The microenvironment of the CNS is exceptional in having a high chloride content, enabling tumors which prefer this environment, such as neuroepithelial tumors like small cell cancer of the lung and melanoma, to colonize, while potentially inhibiting invasion by other cancer cell types without this predilection [15]. Treatments targeting metastatic intracranial disease include surgery, whole-brain radiation therapy (WBRT), stereotactic radiosurgery (SRS), alone or in combination with various targeted agents, and generalized chemotherapy [16]. Following WBRT, survival ranges from anywhere between 4 and 6 months and can be as long as 24 months [17]. Various combinations of surgery, SRS, WBRT, and chemotherapy have been used to improve overall survival, obtain good clinical outcomes, and prevent recurrence of disease.
This paper will focus on metastatic brain tumors describing the hallmarks acquired in the metastatic cascade, which finally brings cancer cells to their “safe haven” in the CNS. The mechanisms through which cancer cells escape their primary focus of origin, invade adjacent tissues making their way into the microvasculature (intravasation), evade cell death, and make their way to a distant site (extravasation), finally proliferating and colonizing this new location, are outlined. With further understanding of the various molecular events that occur in metastasis, future-targeted therapies may lead to prevention or a slowdown in the development of BrM and more effective and less toxic therapy (ies).
2. The Metastatic Process
The ability of cancer cells to sever their link to the primary tumor site and commence the metastatic process begins once specific functions have been acquired by an appropriate subset of cancer cells. The multistep cascade can be grouped into two stages: migration, which includes intravasation, dissemination, and extravasation, and colonization (Figures 1(a) and 1(b)). We will review below the underlying pathobiology within each stage.
Figure 1.

Schematics of the process of metastasis. (a) Formation of metastatic tumor cell lines at primary sites like breast, lung, and skin (melanoma) seen as the red nodes. Metastasis from these primary sites then spreads to the brain via the circulatory system (red arrows) and also to adjacent sites like the liver, bone, lung, and lymph nodes (black arrows). The inset shows the primary site of melanoma cells proliferating and migrating towards the vasculature, subsequently disseminating to secondary organ sites. (b) The metastatic tumor cells detach from the primary site and penetrate the adjacent parenchyma to reach the blood vessels. On reaching the vessels, the cells invade and enter the circulation (intravasation) and then disseminate within the vascular system (left half of figure). These cells eventually adhere to secondary sites “soil” to then extravasate out of the blood vessels and for colonies of metastatic cells (right half of figure).
2.1. Migration
2.1.1. Cellular Heterogeneity and Proliferation
The primary tumor consists of cancer cells which are genetically heterogeneous and have varying potentials to metastasize. These include the cell's ability to invade adjacent tissues, initiate (neo-) angiogenesis, disseminate, and adhere to new tissue substrates, while expressing an affinity for the CNS [3, 18]. Tumor cells have the ability to evade the structural organization present in normal tissues and cells. In spite of being exposed to various environmental pressures such as hypoxia and nutrient deprivation, low pH, poor blood supply, and immune and inflammatory mediators, a subset of tumor cells survive these pressures with the ability to metastasize to distant sites. Additionally, tumor cells are able to evade growth suppressors, which limit cell growth and proliferation, as well as circumvent inhibitors of cell proliferation such as cell cycle checkpoint and DNA damage control systems. Tumor cells can also resist apoptosis (programmed cell death) by the increased expression of antiapoptotic regulators (Bcl-2, Bcl-xL), survival signals (lgf 1/2), and downregulating proapoptotic factors (Bax, Bim, and Puma) [19]. The primary tumor cells have the ability to acquire genetic and epigenetic mutations such as DNA methylation and histone modification, allowing the fittest group of cells to survive [10, 20]. Emerging evidence also suggests that microRNA (miRNA) species interactions with pseudogenes may modify gene expression in cancer [21]. Various genetic mutations result in the ability of tumor cells to commence the proliferative process, and a number of genes associated with this process are listed in Table 1. Clonal expansion of these surviving fit cells leads to an acquisition of further changes, making subsequent cell lines progressively more carcinogenic (Figure 2).
Table 1.
Genes associated with increased metastatic potential.
| Genes | Cancer site (primary) | Role and implications | OMIM no. | Chromosome location |
|---|---|---|---|---|
| RHoC | Melanoma | Regulates remodeling of actin cytoskeleton during morphogenesis and motility. Important in tumor cell invasion | 165380 | 1p21-p13 |
|
| ||||
| LOX | Breast Head and neck cancer |
Increases invasiveness of hypoxic human cancer cells through cell matrix adhesion and focal adhesion kinase activity | 153455 | 5q23.1-q23.2 |
|
| ||||
| VEGF | Lung Breast Melanoma Colon |
Angiogenic growth factor Inhibition decreases brain metastasis formation; reduces blood vessel formation and cell proliferation; increases apoptosis |
192240 | 6p21.1 |
|
| ||||
| CSF1 | Breast Lung |
Stimulate macrophage proliferation and subsequent release of growth factors | 120420 | 1p13.3 |
|
| ||||
| ID1 | Breast Lung |
Involved in matrix remodeling, intracellular signaling, and angiogenesis | 600349 | 20q11.21 |
|
| ||||
| TWIST1 | Breast Gastric Rhabdomyosarcoma Melanoma Hepatocellular |
Causes loss of E-cadherin-mediated cell-cell adhesion, activates mesenchymal markers, and induces cell motility by promoting epithelial-mesenchymal transition | 601622 | 7p21.1 |
|
| ||||
| MET | Renal cell cancer | Affects a wide range of biological activity depending on the cell target, varying from mitogenesis, morphogenesis, and motogenesis | 164860 | 7q31.2 |
|
| ||||
| MMP-9 | Colorectal Breast Melanoma Chondrosarcoma |
Extracellular matrix degradation, tissue remodeling | 120361 | 20q13.12 |
|
| ||||
| NEDD9 | Melanoma | Acquisition of a metastatic potential | 602265 | 6p24.2 |
|
| ||||
| LEF1 | Lung | Transcriptional effecter—WNT pathway; predilection for brain metastasis Knockdown inhibits brain metastasis, decreases colony formation; in vitro decreases invasion |
153254 | 4q25 |
|
| ||||
| HOXB9 | Lung Breast |
Homeobox gene family; critical for embryonic segmentation and patterning. Also a TCF4 target Knockdown in vitro decreased invasion and colony formation; in vivo appears to inhibit brain metastasis |
142964 | 17q21.32 |
|
| ||||
| BMP4 | Lung Colorectal |
Plays an essential role in embryonic development and may be an essential component of the epithelial-mesenchymal transition | 112262 | 14q22.2 |
|
| ||||
| STAT3 | Melanoma | Cell signaling transcription factor Reduction suppresses brain metastasis; decreases angiogenesis in vivo and cellular invasion in vitro |
102582 | 17q21.2 |
Figure 2.
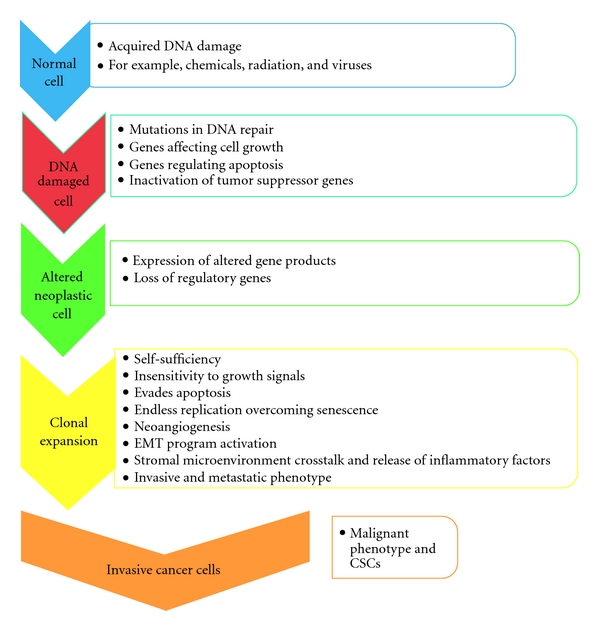
Schematic of the metastatic cascade. Cascade of events taking place at the primary site during oncogenesis, illustrating the steps creating the neoplastic cell line followed by clonal expansion and survival of the fittest cells, becoming the invasive and metastatic phenotype.
Observations within the primary tumor mass have revealed the presence of heterogeneous cell lines including cancer stem cells (CSCs), partially differentiated progenitor cells, and fully differentiated end-stage cells; these appear to recapitulate the same hierarchal patterns in normal tissue types but in an uncontrolled manner [22]. Present evidence suggests that these CSCs may be the primary drivers of the enhanced malignant potential of primary tumors, giving origin to their aggressive phenotypes with the ability to degrade the extracellular matrix (ECM), invade blood vessels and lymph nodes, migrate, extravasate, colonize, and renew themselves at their new locations [23, 24]. These CSCs can reside in clusters or niches, at two or more locations within the primary tumor cell mass [23, 24]. Thus, the key role a CSC plays in the metastatic cascade cannot be overstated, due to its ability to initiate tumor proliferation and “self-renew” itself at alternative tissue locations. Other observations reveal that, in addition to the abilities discussed, they are also motile and invasive and are resilient to the apoptotic process [25].
2.1.2. Epithelial-Mesenchymal Transition (EMT)
The epithelial-mesenchymal transition (EMT), which is currently at the forefront of investigation by numerous groups, describes a temporary, reversible phenomenon wherein cells can dedifferentiate, migrate to a distant focus, and then redifferentiate back to their original cell, forming a new structure [26]. Signals activating the EMT can be intrinsic, such as gene mutations, and extrinsic, such as growth factor signaling. Transdifferentiation appears to be initiated by release of certain EMT-inducing transcription factors (EMT-TFs) that transform epithelial cells into mesenchymal derivatives, giving these cells the capacity to invade, resist apoptosis, and disseminate [11, 27]. Transforming growth factor β (TGFβ) [28], hepatocyte growth factor (HGF) [29], epidermal growth factor (EGF), insulin-like growth factor (IGF) [30], fibroblast growth factor (FGF), and members of the Notch signaling family [31] play a role in inducing the EMT pathway. More recent evidence indicates that the EMT program enables non-CSCs to derive characteristics of the CSC state, which enables them to invade and disseminate from the primary tumor to a distant, metastatic focus [32]. Some of these traits include the ability to loosen adherent junctions, express matrix-degrading enzymes, resist apoptosis, and to undergo morphological conversion. Using the EMT program, cancer cells can, transiently or for longer time frames, activate themselves and acquire attributes critical to survival and dissemination. To activate the EMT, a certain amount of crosstalk has to exist between the tumor cells and adjacent stromal cells, which are done by various EMT-TFs and signals from within the adjacent tumor stroma [11, 32].
2.1.3. Interactions with Tumor Stroma
Progression in cancer also involves activating a number of cells in the adjacent stroma via paracrine signaling [33]. These stromal cells, including endothelial cells, pericytes, fibroblasts, and leukocytes, provide a number of protumorigenic factors which sustain tumor growth. The two prominent cell types are the cancer-associated fibroblasts (CAFs) and the pericytes [34]. These cells produce growth factors, hormones, and cytokines that promote tumor proliferation. CAFs are known to express high amounts of TGFβ, HGF, EGF, FGF, canonical Wnt families, and cytokines like stromal-derived factor- (SDF-) 1α (CXL12) and interleukin-6 (IL-6) [35]. Invasion of cancer cells can be enhanced by stromal macrophages which supply matrix-degrading enzymes such as matrix metalloproteinases and cysteine cathepsin protease [36]. Experimental tumor models suggest that cancer cells release factors such as CSF-1, which stimulates macrophages in the tumor microenvironment, with the subsequent release of EGF, which promotes proliferation of the tumor mass [37]. In addition, CAFs are activated by various inflammatory mediators and induced to produce increased quantities of VEGF, FGF-2, among other cytokines and growth factors, which recruits endothelial progenitor cells thereby promoting angiogenesis [38, 39]. This dynamic stromal environment further stresses the tumor cells, potentially enhancing additional genomic instability, and heterogeneity and epigenetic dysregulation [40].
2.1.4. Local Invasion
Once the phenotypically aggressive clone has developed, spread of the tumor consists of a series of two sequential steps: namely, invasion of the extracellular matrix (ECM), with penetration into the vasculature and hematogenous dissemination to the CNS. Tumor expansion causes adjacent ECM compression and modifies lymphatic and blood vessel flow, eventually leading to basement membrane (BM) thinning. Combined with the various molecular and cellular events, this leads to eventual tumor metastasis.
To reach the circulation, tumor cells must penetrate the BM, traverse the extracellular connective tissue matrix (ECM) tissue, and then breach the vascular basement membrane (VBM) to enter the circulation. The process is dependent on a number of protein complexes that regulate cellular interactions and proteolytic enzymes, with degradation of the ECM, which permits extravasation.
2.1.5. E-Cadherin-Catenin Complex (ECCC), Integrins, and Other Molecules
The E-cadherin-catenin molecular complex is essential to maintain a normal and tumoral cytoarchitecture. It is a necessary mediator of cell-cell adhesion that, among other functions, determines the polarity of normal (and tumor) cells and their organization into tissues [41]. Cadherin molecules are integral cell membrane glycoproteins that interact in a homophilic manner with one another. They have a stable extracellular fragment and possess a cytoplasmic undercoat protein of one or more proteins called catenins. In the process of tumor metastasis, tumor clones become discohesive, fail to adhere to one another, and develop a more disordered cytoarchitecture, which allows these cells to separate from the tumor mass. E-cadherin maintains cell adhesion by anchoring its cytoplasmic domain to actin cytoskeleton via α-catenin and β-catenin. Infiltrating malignancies have mutations in the genes for α- and β-catenins and E-cadherin, thus decreasing the expression of this complex. This has been correlated with invasion, metastasis, and an unfavorable prognosis. Furthermore, DNA hypermethylation of the promoter region of E-cadherin can diminish or silence its expression, thereby disturbing ECCC function, and is a common event in many metastatic cancers [42]. N-cadherin is another molecule connected to the cellular cytoskeleton via α-catenin and β-catenin in a manner similar to E-cadherin. One of the hallmarks of the EMT described above is a cadherin switch, with loss of epithelial E-cadherin and gain of mesenchymal N-cadherin functions. This induces loss of epithelial cellular affinity, while at the same time increasing the affinity of cells for the mesenchymal cells like fibroblasts. Gain-of-function mutations in N-cadherin also trigger increased migration and invasion in tumors [43].
Integrins are another family of major adhesion and signaling receptor proteins linking the ECM to the cellular actin cytoskeletal structure called focal adhesions and play an important role in mediating cell migration and invasion [44]. They trigger a variety of signal transduction pathways and regulate cytoskeletal organization, specific gene expression, control of growth, and apoptosis. Animal models of human nonsmall cell lung cancer (NSCLC) have shown that blocking α 3 β 1 integrin significantly decreases brain metastasis [45]. Additionally, Carbonell et al. have shown that blocking the β 1 integrin subunit prevents adhesion to the VBM and attenuates the development of metastasis [46]. Integrins induce the release of a key mediator in signaling known as focal adhesion kinase (FAK). FAK is a ubiquitously expressed nonreceptor cytoplasmic tyrosine kinase, thought to play a key role in migration and proliferation, by providing abnormal signals for survival, EMT, invasion, and angiogenesis [47]. FAK may also play an important role in the regulation of CSCs. Dephosphorylation and inhibition of FAK at the Y397 locus via the activated Ras (rat sarcoma) oncogene promote tumor migration by facilitating focal adhesion at the leading edge of tumor cells [15, 48].
The ability of tumor cells to escape the primary site is dependent on their ability to remodel the ECM. This remodeling occurs by breaking down or degrading the ECM via proteolytic enzymes, thus creating a pathway for invasion. The advancing edge of tumor cells posses the ability to carry out this proteolytic activity by releasing signals that promote cell proliferation and angiogenesis in the metastatic cascade. Neurotrophins (NTs) promote brain invasion by enhancing the production of heparinase, which is an ECM proteolytic enzyme. Heparinase is a β-d-glucuronidase that cleaves the heparin sulfate chain of the ECM. It is the prominent heparin sulfate degradative enzyme [49] and is known to destroy both the ECM and the BBB [3]. Evidence suggests the presence of NTs at the tumor-brain interface in melanomas, and reports have suggested a role for the p75 NT receptor in brain metastasis [50].
Matrix metalloproteinases (MMPs) are members of a family of zinc-dependent endopeptidases that function at physiological pH and help remodeling human connective tissue at low levels. About 25 human family members have been identified, and they have been grouped according to their substrate on which they act, namely, collagenases, stromelysins, matrilysins, and gelatinases [51]. They also play a critical role in the EMT and tumor microenvironment [52]. Cytokines and inducers present on the surface of tumor cells in the ECM regulate their expression. Once these MMPs are induced and stimulated, they aid in breakdown of type I collagen, fibronectin, and laminin in the ECM [53] and enhance tumor cell migration. MMP activity correlates with invasiveness, metastasis, and poor prognosis [54]. In one study of brain metastasis, MMP-2 was identified in all metastases regardless of site of origin. Moreover, MMP-2 activity correlated inversely with survival [55]. In a murine tumor model, the incidence of brain metastasis was reduced by 75% when compared to the wild type following the use of tissue inhibitor of metalloprotease1 (TIMP-1), which suggests that inhibitors of MMPs suppress BrMs [56].
The urokinase-type plasminogen activator (uPA) system consists of uPA, its receptor (uPAR), and plasminogen. The uPA binds to the receptor uPA-R (CD87), the activity of which is regulated by the action of plasminogen activator inhibitor type 1 and 2 (PAI-1/2) on the cell membrane and causes urokinase to convert plasminogen to plasmin. The proteolytic activity of plasmin then degrades components of the ECM including fibrin, fibronectin, proteoglycans, and laminin. Further, plasmin activates other proteolytic enzymes with resultant local invasion and migration [57]. As far back as 1994, researchers have found that there is a high level of uPA in metastatic tumors, that uPA correlates with necrosis and edema, and that there is an inverse correlation with a tumor's levels of uPA and survival [58]. Additionally, high levels of uPA and absent tissue plasminogen activator (tPA) correlate with aggressiveness and decreased survival [58].
More recent evidence describes the role of “invadopodia,” which are three-dimensional protrusive processes, compared to the two-dimensional lamellipodia and filopodia, in metastatic invasion [59]. Invadopodia appear to share a number of structural and functional features with filopodia, but spatially focus proteolytic secretion, remodeling the ECM matrix and establishing tracts supporting subsequent invasion [60]. Integrins play a major role in organizing the components, triggering the formation of invadopodia. α 3 β 1 activation promotes Src-dependent tyrosine phosphorylation of p190RhoGAP, via RhoGTPases family, which activates invadopodia and invasion [61]. Integrins also appear to focus proteolytic activity to the region of these processes, as in melanoma cells, where collagen-induced α 3 β 1 association with the serine protease Seprase (surface-expressed protease) enhances the activity of matrix-degrading enzymes focally at the invadopodia [62]. Numerous cancer cell lines such as melanoma, breast cancer, glioma, and head and neck cancer have shown the presence of invadopodia. A number of other molecules, such as EGF, HGF, or TGF-β, can induce their formation as well [63]. The release of tumor-released chemokines such as CSF-1 and PIGF attract tumor-associated macrophages (TAM) to the microenvironment, which in turn release multiple factors stimulating invadopodia [64]. In addition, a family of proteins called aquaporins may also facilitate migration. Aquaporin-dependent tumor angiogenesis and metastases enhance water transport in the lamellipodia of migrating cells [65]. Studies on brain-specific breast metastasis reveal that increased expression of KCNMA1, a gene encoding for a big conductance type potassium channel (BKCa) that is upregulated in breast cancer, leads to greater invasiveness and transendothelial migration [66].
2.1.6. Genetic Alterations
Several known tumor suppressor genes (TSGs) that function at the level of escape and migration/intravasation are worth exploring and are enumerated in Table 2. The best known of these is the KiSS1 gene on chromosome 1. KiSS1 encodes metastin, which is a ligand of the orphan G protein couples receptor hOT7T175. Lee et al. [67] have found that the forced expression of KiSS1 suppressed both melanoma and breast metastasis. Other authors have found an inverse correlation between KiSS1 expression and melanoma progression [68].
Table 2.
Representative metastasis and invasion suppressor genes.
| Gene | Cancer/metastatic tumor | Function(s) of protein | OMIM no. | Chromosome Location |
|---|---|---|---|---|
| NM23 | Breast, colon, melanoma | A histidine kinase. Nm23 phosphorylates KSR and can lead to decreased ERK 1/2 activation. appears to play a role in normal development and differentiation | 156490 | 17q21.3 |
|
| ||||
| MKK4 | Breast, ovarian, and prostate | A mitogen-activated protein kinase (MAPKK) that phosphorylates p38 and Jun (JNK) kinases | 601335 | 17p11.2 |
|
| ||||
| BRMS1 | Breast, melanoma | Functions in gap-junction communication | 606259 | 11q13.1-q13.2 |
|
| ||||
| KiSS1 | Breast, melanoma | A G-protein coupled receptor ligand, also known as metastin. | 603286 | 1q32 |
|
| ||||
| KAI1 (CD82) | Bladder, breast, lung, pancreas, and prostate | Interact with beta-catenin-reptin and histone deacetylases. It may desensitize EGFR activity, also known as kangai | 600623 | 11p11.2 |
|
| ||||
| CD44 | Breast, colon, lung, melanoma, prostate | An integral cell membrane glycoprotein that affects cell adhesion. Decreased expression due in part to hypermethylation | 107269 | 11pter-p13 |
|
| ||||
| CRSP3 | Melanoma | A transcriptional coactivator that may work through the enhancer binding factor Sp1 | 605042 | |
|
| ||||
| RHOGDI2 | Bladder, breast, colon, kidney, liver, lung, and prostate | Regulates function of Rho and Rac, GTP-binding proteins of the Ras superfamily | 11p11.2 | |
|
| ||||
| VDUP1 | Melanoma | A differentiation factor via thioredoxin inhibition | 606599 | 1q21 |
|
| ||||
| PTEN/MMAC1 | Breast, colon, endometrial, germ cell, kidney, lung, melanoma, and thyroid | A homologue of cytoskeletal tension, leading to invasion and metastasis through interaction with actin filaments at focal adhesions | 601728 | 10q23.31 |
|
| ||||
| VHL | Renal cell, pheochromocytoma, and hemangioblastoma | Encodes protein products playing an essential role in microtubule stability, orientation, tumor suppression, cilia formation, signaling of cytokines, and extracellular matrix assembly | 608537 | 3p25.3 |
|
| ||||
| TIMP2 | Melanoma | Protease inhibitor plays a role in preventing excessive ECM disruption | 188825 | 17q25.3 |
|
| ||||
| SMAD4 | Pancreatic cancer, colorectal, and prostate | Transcription factor, pivotal role in signal transduction of TGFβ | 600993 | 18q21.2 |
|
| ||||
| RRM1 | Lung | Cell cycle regulator | 180410 | 11p15.4 |
|
| ||||
| PTPN11 | Lung, colon, thyroid, and melanoma | Regulates tyrosine phosphatase, proliferation, differentiation, motility, and apoptosis of cells | 176876 | 12q24.1 |
|
| ||||
| CDH1 | Gastric, breast | Cellular adherens junctional protein | 192090 | 16q22.1 |
|
| ||||
| CASP8 | Gastric, breast, lung, and PNETs | Apoptotic cascade via aspartate-specific cysteine proteases | 601763 | 2q33 |
Definitions: EGFR: epidermal growth factor; ERK: extracellular signal-regulated kinase; JNK: Jun-terminal kinase; KSR: kinase suppressor of Ras. OMIM no.: Online Mendelian Inheritance in Man Identification number (http://www.ncbi.nlm.nih.gov/), which provides detailed information and references for these genes, their protein products, and potential functions.
KAI1 (CD82), a TSG on chromosome 11p11.2, regulates adhesion, migration, growth, and differentiation of tumor cell lines. KAI1 expression is inversely correlated with prostate cancer progression [69] as well as breast [26, 27] and melanoma metastasis [28]. Additionally, KAI1 is known to be associated with the epidermal growth factor receptor (EGFR), discussed later in this paper, and is thought to affect the Rho GTPase pathway [29] resulting in suppression of lamellipodia formation and migration [30].
Hypermethylation of the TSG Drg1 inhibits both liver metastasis and colorectal carcinoma invasion [70]. Conversely, overexpression of Drg1 has been linked to resistance to irinotecan chemotherapy [71]. Finally, in a murine model of breast cancer metastasis, the Notch signaling pathway was found to be activated via increased Jag2 mRNA levels, thereby, creating a cell line that was both more migratory and more invasive in collagen assays. Additionally, inactivation of the Notch pathway significantly decreased tumor cell migratory and invasive activity [72]. In addition to the suppressor genes responsible for invasion and metastasis, there are a number of promoter genes responsible for invasion and metastasis as well, a few of which are enumerated in Table 3. Genetic activation or inactivation of promoter/suppressor genes in human cancer can be the result of mutations, deletions, loss of heterozygosity, multiplication, and translocation [73]. The same genes that are responsible for normal cellular functioning, signaling, signal transduction, modulating, and mediating cellular response are frequently the genes that enhance invasion and metastasis when altered by genetic or epigenetic dysfunction [74, 75].
Table 3.
Representative metastasis and invasion promoter genes.
| Gene | Cancer/metastatic tumor | Function(s) of protein | OMIM no. | Chromosome location |
|---|---|---|---|---|
| ERBB2 (HER2) | Breast | Receptor tyrosine kinase, critical component of IL6, and cytokine signaling | 164870 | 17q21.1 |
|
| ||||
| TIAM1 | Lymphomas, renal cell cancer, colon, prostate, and breast | Activates Rho-like GTPase Rac1, Tiam1Rac1 signaling which affects invasion in numerous ways | 600687 | 21q22.1 |
|
| ||||
| SRC | Colorectal, breast, melanoma, and lung | Critical role in cellular signal transduction pathways, regulating cell division, motility, adhesion, angiogenesis, and survival | 190090 | 20q12-q13 |
|
| ||||
| S100A4 | Colorectal Breast Gastric cancers |
Increases endothelial cell motility and induces angiogenesis, increases invasive properties through deregulation of the extracellular matrix | 114210 | 1q21 |
|
| ||||
| MTA1 | Breast Ovary Lung Gastrointestinal Colorectal |
Nucleosome remodeling and deacetylating (NuRD) complex serves multiple functions in cellular signaling, chromosome remodeling and transcription processes, that are important in the progression, invasion, and growth of metastatic epithelial cells | 603526 | 14q32.3 |
|
| ||||
| KRAS | Pancreatic Lung Colorectal |
Encode GDP/GTP-binding proteins involved in signal transduction during cellular proliferation, differentiation, and senescence | 190070 | 12p12.1 |
|
| ||||
| HRAS | Bladder Renal Thyroid |
Small GTPase growth promoting factor | 190020 | 11p15.5 |
These changes within the primary tumor microenvironment give rise to an “active seed” ready to implant itself in a fertile environmental “soil” (Figure 3). These cellular modifications enable the next steps of migration, namely, dissemination and extravasation.
Figure 3.
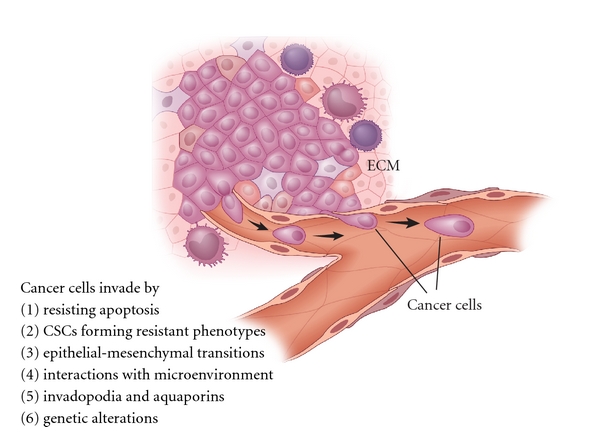
Invasion and migration. Subsets of cancer cells at the primary site develop an invasive phenotype; survive environmental pressures such as hypoxia and nutrient deprivation, low pH, poor blood supply, immune, and inflammatory mediators, gaining the ability to metastasize to distant sites. These cancer cells can evade growth suppressors and circumvent inhibitors of cell proliferation to intravasate and disseminate to various other sites.
2.1.7. Dissemination
Once a cancer cell has breached its microenvironment and arrived at the vasculature (brain metastasis) or lymphatic system (other sites), the tumor cell must survive its exposure to high shear forces and varied stress patterns. Tumor cells respond by reenforcing their cytoskeleton and increasing the ability to adhere to the vascular wall [76]. More recent experimental evidence suggests shear induces a paradoxical enhancement of adhesion to the VBM via activation of Src [77] and FAK phosphorylation seen in colon cancer cell lines [78]. On adhering to endothelium of target tissue, the tumor cells behave like macrophages, creating pseudopodia, and penetrating the cell-cell junctions, driven by dynamic remodeling of the cellular cytoskeleton [60]. There are a subset of circulating tumor cells which maintain their physical plasticity and, although much larger in diameter (20–30 μ) than lung capillaries (~8 μ), can survive the sieving action of lung capillaries. These cells can be found either growing as clumps in the lung or colonizing other organ sites [10]. Cancer cells in circulation appear to attract platelets because of their expressed surface tissue proteins, and these protect the cells from the immune system [79]. Once these mobile cancer cells get lodged in a secondary organ tissue site, there are two pathways for colonization. One is mediated by cellular diapedesis, extravasation, and proliferation of the tumor cell mass, whereas the other consists of accumulation of tumor cells within the site of obstruction in the foreign tissue vascular bed, wherein they proliferate, prior to their rupture into the adjacent stroma where they begin to grow [80].
2.2. Colonization
2.2.1. Organ-Specific Infiltration
Subsequent to intravasation and dissemination, special mechanisms are necessary to extravasate and colonize secondary sites. The metastatic deposits occur in certain organ tissues because of the influence of hematogenous dynamics, for example, colon cancer metastasis preferentially metastasizing to the liver because of mesenteric circulation and large vascular sinusoids [81]. The overexpression of the cell adhesion molecule, metadherin, in breast cancer makes it easier for tumor cells to target and adhere to endothelial lining in the lung parenchyma [82], making it possible for these endothelial-adhesive interactions to enhance the possibility of brain metastasis. Although the exact causes of preferential metastatic sites have not been clearly elucidated, one theory states that direct neurotropic interactions with yet undiscovered brain homing mechanisms result in BrM. “Vascular co-option,” a term put forward by Carbonell et al., describes the ability of metastatic cells to grow along the preexisting vessels much before overt secondaries are detected. Once adherent to the VBM, tumor cells can extravasate into the parenchyma, the VBM thus being the “soil” for BrM (Figure 4) [46]. Saito et al. demonstrated that the pia-glial membrane along the external surface of blood vessels serves as a scaffold for the angiocentric spread of metastatic cells [83].
Figure 4.

Colonization of metastatic tumor cells in the brain. Overexpression of the adhesion molecules makes it easy for tumor cells to target and adhere to endothelial lining in the parenchyma, making it possible for these endothelial-adhesive interactions to enhance the possibility of brain metastasis. Direct neurotropic interactions with brain homing mechanisms result in BrM. “Vascular co-option” is the ability of metastatic cells to grow along the preexisting vessels, and once adherent to the VBM, tumor cells can extravasate into the parenchyma, the VBM thus being the “soil” for BrM.
In a mouse model of CNS metastasis, tumor cells function like macrophages within the vasculature and during extravasation, expressing CD11b, Iba1, F4/80, CD68, CD45, and CXCR, which are proteins normally expressed specifically by macrophages [84]. The ability of tumor cells to mimic macrophages may enable them to evade the immune system while in the vasculature.
2.2.2. The BBB, Function of the Brain Microenvironment, and Brain Metastasis
Passage of tumor cells across the BBB occurs via mechanisms which have not yet been delineated fully. Recently, three proteins that mediate breast metastasis to the brain and lungs have been described, namely, cyclooxygenase 2 or COX2 (also known as PTGS2), EGFR, ligand and heparin binding epidermal growth factor (HBEGF). These proteins facilitate extravasation through nonfenestrated blood vessels and enhance colonization [85]. Other molecules targeting organ specific colonization may also be expressed by the cancer cell [86]. These molecules include ezrin (an intracellular protein necessary for the survival of osteosarcoma cells in the lung) and serine-threonine kinase 11 (STK11, or LKB1, a metastasis suppressor which regulates NEDD9 in lung cancer) [87].
2.2.3. Neoangiogenesis and Proliferation
A key component of both primary and secondary (metastatic) tumor growth at any site is angiogenesis [8]. Experimental systems, using breast or melanoma cell lines to model BrM, have revealed that growth may occur by utilizing preexisting vasculature, or co-opting these vessels rather than inducing new vessel formation (neoangiogenesis) [46, 88, 89]. Kusters et al. [90], using a melanoma cell line in a murine metastatic brain tumor model, showed that growth of the metastatic tumor up to 3 mm could occur without inducing the angiogenic switch [91]. Carbonell et al. have also shown that β 1 integrin, expressed by the metastatic tumor cell line, is the key molecule to co-opt adjacent blood vessels to the growing tumor.
Various angiogenic factors have been scrutinized as viable targets for treatment. Vascular endothelia growth factor (VEGF) is the most commonly recognized angiogenic factor. VEGF expression in breast cancer plays a role in metastasis and inhibition with a tyrosine kinase receptor inhibitor-reduced growth and angiogenesis [92]. SSecks (Src-suppressed C kinase substrate) has been observed to decrease VEGF expression. This protein also stimulates proangiogenic angiopoietin 1 and regulates bran angiogenesis and tight junction creation, thus helping to regulate BBB differentiation [93].
MMP-9/gelatinase B complex, a member of the MMP family, and PAI-1, a uPA cell surface receptor, may play roles in angiogenesis [94]. The role in angiogenesis and uniqueness of Plexin D1 expression was explored in tumor cells and vasculogenesis. Neoplastic cells expressed Plexin D1 as well as tumor vasculature, while its expression in nonneoplastic tissue was restricted to a small subset of activated macrophages, which suggests that Plexin D1 may play a significant role in tumor angiogenesis [95]. Overexpression of hexokinase 2 (HK2), which plays a key role in glucose metabolism and apoptosis, may also influence BrM in breast and other cancers. Researchers at the National Cancer Institute found that both mRNA and protein levels of HK2 are elevated in brain metastatic derivative cell lines compared to the parental cell line in vitro. Knockdown of expression reduced cell proliferation, which implies that HK2 contributes to the proliferation and growth of breast cancer metastasis. Finally, increased expression of HK2 is associated with poor survival after craniotomy [96, 97].
At least two tumor suppressor genes that function at the proliferation level of the metastatic cascade have been described. The first, NM23, regulates cell growth by encoding for a nucleotide diphosphate protein kinase that interacts with menin, a TSG encoded by MEN1 [98]. NM23 is thought to reduce signal transduction and thereby decrease anchorage independent colonization, invasion, and motility [99]. In melanoma, decreased expression is correlated with increased brain metastasis [100]. Another tumor suppressor gene, BrMS1, located at 11q13 is altered in many melanomas and breast cancers. BrMS1 prevents disseminated tumor cell growth by restoring the normal gap junction phenotype and maintaining cell-to-cell communication in the primary tumor [101]. Seraj et al. [102] found an inverse correlation between the expression of BrMS1 and the metastatic potential in melanoma.
2.2.4. Cascade-Nonspecific Contributors to Metastasis
There are certain molecular contributions that cannot be attributed to a specific step in the cascade, either because they are active at every level or, as in most cases, their true function is yet to be discovered (see Tables 1 and 2). These molecular entities are on the forefront of cancer research and are worth addressing. Zeb-1, the zinc finger E-box homeobox transcription factor, is overexpressed in metastatic cancers. This overexpression leads to epithelial-mesenchymal transition and increased metastasis. Mutation of Zeb-1 leads to decreases in the proliferation of progenitor cells in mutant mice. This mutation may be a target for metastatic prevention at the progenitor level [103].
Several other genetic markers have been located that pertain to metastasis in particular. A deletion of the 4q arm in lung (both small and nonsmall cell) metastasis to the brain and bone has been documented [104]. Additionally, in NSCLC, overexpression of CDH2 (N-cadherin), KIFC1, and FALZ is highly predictive of metastasis to the brain in early and advanced lung cancer. Therefore, these genes may be used to predict a high risk of metastasis early in the diagnosis [105]. In prostate cancer, increased expression of KLF6-SV1, the Kruppel-like factor tumor suppressor gene, predicts poorer survival and is correlated with increased metastasis to the lymphatic system, the brain, and bone [106]. Finally, overexpression of homeoprotein Six-1, a transcriptional regulator, increased TGF-β signaling and metastasis in breast cancer with significantly shortened relapse times [107]. Gaining a better understanding of the role(s) of these genes and others will be important to deeper knowledge of the metastatic cascade.
2.2.5. Overview of microRNAs (miRNAs) and Their Emerging Role in Oncogenesis
Recent evidence has established an important role of microRNAs in cell and tissue development, proliferation and motility via their ability to repress mRNA translation or induce mRNA degradation [108]. The dysregulated expression of a single miRNA can cause a cascade of silencing events capable of eliciting disease development in humans, which includes cancer [109]. Breast cancer is found to possess aberrant regulation of several miRNAs [110]. They also play a prominent role in expression of EMT-related genes. Finally, pseudogenes, which encode RNAs that do not have to produce proteins but can compete for microRNA binding, may play a role in tumorigenesis and metastasis. Poliseno et al. [111]. Recently described the functional relationship between the mRNAs produced by the PTEN tumor suppressor gene and its pseudogene PTENP1. PTENP1 regulates cellular levels of PTEN and can exert a growth-suppressive role and the PTENP1 locus is lost in several human tumors, including prostate and colon cancer. They also showed that other cancer-related genes possess pseudogenes, including oncogenic KRAS. While the role of miRNAs and psuedogenes in metastasis is beyond the scope of this summary, several recent, excellent reports detail this emerging field [21, 111, 112].
3. Conclusion
The metastatic cascade, from its initiation to its completion in the brain, is an extremely complex, multistep process. For patients, the progression in the metastatic cascade to brain colonization is becoming both an increasingly treatable and yet simultaneously and increasingly prevalent feature of their disease, with consequent morbidity. As more evidence regarding the molecular and genetic factors that contribute to the cascade appears, targeting this ominous disease with multiple therapeutic strategies comes closer.
Knowledge of the metastatic process may lead to better detection and treatment of brain metastases. The goal however will be to utilize all the information gained at the genetic and molecular level to stop cancer, at the primary proliferative stage, preventing the initiation of the metastatic cascade and subsequent development of brain metastasis.
Conflict of Interests
Each author declares that he or she has no conflict of interests.
Acknowledgments
This work was supported in part by Grant no. W81XWH-062-0033 from the U.S. Department of Defense Breast Cancer Research Program, to R. J. Weil. The authors wish to thank the Melvin Burkhardt chair in neurosurgical oncology and the Karen Colina Wilson research endowment within the Brain Tumor and Neuro-oncology Center at the Cleveland Clinic Foundation for additional support and funding.
Abbreviations
- AKAP12:
A-kinase anchor protein 12
- ARHGDIB(RhoGD12):
Rho GDP dissociation inhibitor beta
- BBB:
Blood brain barrier
- BKCa:
Big conductance type potassium channel
- BrMS1:
Breast cancer metastasis suppressor 1
- CASP8:
Caspase8
- CDH1:
Cadherin 1
- CD11b:
Cluster of differentiation molecule 11b
- CD45:
Cluster of differentiation 45
- CD44:
cluster of differentiation 44
- CD6:
Cluster of differentiation 68
- CDH2:
Neutral cadherin (N-cadherin), when overexpressed with FALZ and KlFC1 genes) predicts metastasis to brain
- COX2:
Cyclooxygenase 2 (also known as PTGS2)
- CRSP3:
Cofactor required for Sp1 transcriptional activation subunit 3
- CXCR:
Chemokine receptor
- CD87:
uPA binds to the receptor uPA-R
- CNS:
Central nervous system
- CXCL12/CXCR4:
Chemokine/receptor system
- CXCR7:
Alternate receptor to CXCL12
- CSF-1:
Colony stimulating factor-1
- Drg-1:
Differentiation-related, putative metastatic suppressor gene
- ECM:
Extracellular matrix
- EGFR:
Epidermal growth factor receptor
- ERBB2(HER2):
Avian erythroblastic leukemia homolog 2
- F4/80:
Transmembrane protein present on cell surface of mouse
- FALZ:
Overexpression with CDH2 and KlFC1 genes predicts metastasis to the brain
- FAK:
Focal adhesion kinase
- HBEGF:
Heparin binding epidermal growth factor
- HSP27:
Heat shock protein
- HK2:
Hexokinase 2
- HOXB9:
homeobox B9
- HRAS:
Harvey rat sarcoma viral oncogene homolog
- Iba1:
Ionized calcium binding adaptor molecule 1
- KAI1 (CD82) gene:
Tumor suppressor gene
- KCNMA1:
tumor suppressor gene
- KIFC1:
Overexpression with CDH2 and FALZ genes predicts metastasis to brain
- KISS-1:
Gene that encodes metastin
- KLF6-SV1:
Kruppel-like factor tumor suppressor gene
- KRAS:
Kirsten rat sarcoma viral oncogene homolog
- LEF1:
Lymphoid enhancing factor 1
- MIB-1:
Mindbomb homolog 1 labeling index
- MMAC1:
Multiple advanced cancers
- MMPs:
Matrix metalloprotease
- MEN1:
Multiple endocrine neoplasia type 1
- MTA1:
Metastasis-associated protein 1
- NM23(NME1):
Metastasis suppressor gene also known as NDP kinase
- NSCLC:
Nonsmall cell lung cancer
- NTs:
Neurotrophins
- PAI-1/2:
plasminogen activator inhibitor type 1 and 2
- PTEN:
Chromosome 10
- PIGF:
Phosphatidylinositol glycan, class F
- PTPN11:
Protein-tyrosine phosphatase, nonreceptor type 11
- PTEN:
Phosphatase and tensin homologue deleted on chromosome 10
- Ras:
Oncogene (rat sarcoma) gene
- RRM1:
Ribonucleotide reductase, M12 subunit
- sHSP:
Small heat shock protein
- SSecks:
Src-suppressed C kinase substrate
- Six-1:
Homeoprotein transcriptional regulator
- ST6GALNAC5:
α2,6-sialyltransferase
- S100A4:
S100 calcium-binding protein A4
- SMAD4:
Sma- and Ma-related protein 4
- SRC:
Rous sarcoma virus protein
- TIAM1:
T-cell lymphoma invasion and metastasis 1
- TIMP2:
Tissue inhibitor of metalloproteinase 2
- TCF:
T-cell factor pathway
- TGF:
Transforming growth factor
- TIMP-1:
Tissue inhibitor of metalloprotease 1
- tPA:
Tissue plasminogen activator
- uPA:
Urokinase-type plasminogen activator
- uPA-R (CD87):
Urokinase-type plasminogen activator receptor
- VBM:
Vascular basement membrane
- VEGF:
Vascular endothelial growth factor
- VDUP:
Vitamin D3-upregulated protein
- VHL:
Von Hippel-Lindau tumor suppressor
- WNT:
Wingless integration gene
- Zeb-1:
Zinc finger E-box-binding homeobox 1
- OMIM no.:
Online Mendelian Inheritance in Man Identification Number (http://www.ncbi.nlm.nih.gov/), which provides detailed information and references for these genes, their protein products, and potential functions.
References
- 1.Gavrilovic IT, Posner JB. Brain metastases: epidemiology and pathophysiology. Journal of Neuro-Oncology. 2005;75(1):5–14. doi: 10.1007/s11060-004-8093-6. [DOI] [PubMed] [Google Scholar]
- 2.Patchell RA. The management of brain metastases. Cancer Treatment Reviews. 2003;29(6):533–540. doi: 10.1016/s0305-7372(03)00105-1. [DOI] [PubMed] [Google Scholar]
- 3.Nathoo M, Chahlavi A, Barnett GH, Toms SA. Pathobiology of brain metastases. Journal of Clinical Pathology. 2005;58(3):237–242. doi: 10.1136/jcp.2003.013623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landis SH, Murray T, Bolden S, Wingo PA. Cancer statistics, 1998. Ca-A Cancer Journal for Clinicians. 1998;48(1):6–29. doi: 10.3322/canjclin.48.1.6. [DOI] [PubMed] [Google Scholar]
- 5.Wen PY, Loeffler JS. Brain metastases. Current Treatment Options in Oncology. 2000;1(5):447–458. doi: 10.1007/s11864-000-0072-3. [DOI] [PubMed] [Google Scholar]
- 6.Kim SH, Chao ST, Toms SA, et al. Stereotactic radiosurgical treatment of parenchymal brain metastases from prostate adenocarcinoma. Surgical Neurology. 2008;69(6):641–646. doi: 10.1016/j.surneu.2007.05.035. [DOI] [PubMed] [Google Scholar]
- 7.Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer and Metastasis Reviews. 1989;8(2):98–101. [PubMed] [Google Scholar]
- 8.Fidler IJ, Yano S, Zhang RD, Fujimaki T, Bucana CD. The seed and soil hypothesis: vascularisation and brain metastases. Lancet Oncology. 2002;3(1):53–57. doi: 10.1016/s1470-2045(01)00622-2. [DOI] [PubMed] [Google Scholar]
- 9.Ewing J. Neoplastic Diseases. WB Saunders; 1922. [Google Scholar]
- 10.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 11.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 12.Delattre JY, Krol G, Thaler HT, Posner JB. Distribution of brain metastases. Archives of Neurology. 1988;45(7):741–744. doi: 10.1001/archneur.1988.00520310047016. [DOI] [PubMed] [Google Scholar]
- 13.Chang EL, Wefel JS, Maor MH, et al. A pilot study of neurocognitive function in patients with one to three new brain metastases initially treated with stereotactic radiosurgery alone. Neurosurgery. 2007;60(2):277–283. doi: 10.1227/01.NEU.0000249272.64439.B1. [DOI] [PubMed] [Google Scholar]
- 14.Lockman PR, Mittapalli RK, Taskar KS, et al. Heterogeneous blood-tumor barrier permeability determines drug efficacy in experimental brain metastases of breast cancer. Clinical Cancer Research. 2010;16(23):5664–5678. doi: 10.1158/1078-0432.CCR-10-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beasley KD, Toms SA. The molecular pathobiology of metastasis to the brain: a review. Neurosurgery Clinics of North America. 2011;22(1):7–14. doi: 10.1016/j.nec.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 16.Eichler AF, Chung E, Kodack DP, Loeffler JS, Fukumura D, Jain RK. The biology of brain metastases-translation to new therapies. Nature Reviews Clinical Oncology. 2011;8(6):344–356. doi: 10.1038/nrclinonc.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mehta MP, Rodrigus P, Terhaard CHJ, et al. Survival and neurologic outcomes in a randomized trial of motexafin gadolinium and whole-brain radiation therapy in brain metastases. Journal of Clinical Oncology. 2003;21(13):2529–2536. doi: 10.1200/JCO.2003.12.122. [DOI] [PubMed] [Google Scholar]
- 18.Chiang AC, Massagué J. Molecular basis of metastasis. The New England Journal of Medicine. 2008;359(26):2752–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Junttila MR, Evan GI. P53 a Jack of all trades but master of none. Nature Reviews Cancer. 2009;9(11):821–829. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 20.Jones PA, Baylin SB. The Epigenomics of Cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the rosetta stone of a hidden RNA language? Cell. 2011;146(3):353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Current Opinion in Biotechnology. 2007;18(5):460–466. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 23.Marcato P, Dean CA, Da P, et al. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29(1):32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 24.Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature. 2008;456(7222):593–598. doi: 10.1038/nature07567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pang R, Law WL, Chu ACY, et al. A subpopulation of CD26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell. 2010;6(6):603–615. doi: 10.1016/j.stem.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer and Metastasis Reviews. 2009;28(1-2):15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 27.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Zavadil J, Böttinger EP. TGF-β and epithelial-to-mesenchymal transitions. Oncogene. 2005;24(37):5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 29.Savagner P, Arnoux V. Epithelio-mesenchymal transition and cutaneous wound healing. Bulletin de l’Academie Nationale de Medecine. 2009;193(9):1981–1992. [PubMed] [Google Scholar]
- 30.Graham TR, Zhau HE, Odero-Marah VA, et al. Insulin-like growth factor-I—dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Research. 2008;68(7):2479–2488. doi: 10.1158/0008-5472.CAN-07-2559. [DOI] [PubMed] [Google Scholar]
- 31.Leong KG, Niessen K, Kulic I, et al. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. Journal of Experimental Medicine. 2007;204(12):2935–2948. doi: 10.1084/jem.20071082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mani SA, Guo W, Liao MJ, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133(4):704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pietras K, Östman A. Hallmarks of cancer: interactions with the tumor stroma. Experimental Cell Research. 2010;316(8):1324–1331. doi: 10.1016/j.yexcr.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 34.Östman A, Augsten M. Cancer-associated fibroblasts and tumor growth—bystanders turning into key players. Current Opinion in Genetics and Development. 2009;19(1):67–73. doi: 10.1016/j.gde.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432(7015):332–337. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orimo A, Gupta PB, Sgroi DC, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 39.Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Medicine. 2008;5(1, article 019):0123–0138. doi: 10.1371/journal.pmed.0050019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bristow RG, Hill RP. Hypoxia and metabolism: hypoxia, DNA repair and genetic instability. Nature Reviews Cancer. 2008;8(3):180–192. doi: 10.1038/nrc2344. [DOI] [PubMed] [Google Scholar]
- 41.Hirohashi S, Kanai Y. Cell adhesion system and human cancer morphogenesis. Cancer Science. 2003;94(7):575–581. doi: 10.1111/j.1349-7006.2003.tb01485.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bremnes RM, Veve R, Hirsch FR, Franklin WA. The E-cadherin cell-cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer. 2002;36(2):115–124. doi: 10.1016/s0169-5002(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 43.Hulit J, Suyama K, Chung S, et al. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Research. 2007;67(7):3106–3116. doi: 10.1158/0008-5472.CAN-06-3401. [DOI] [PubMed] [Google Scholar]
- 44.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110(6):673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 45.Yoshimasu T, Sakurai T, Oura S, et al. Increased expression of integrin α3β1 in highly brain metastatic subclone of a human non-small cell lung cancer cell line. Cancer Science. 2004;95(2):142–148. doi: 10.1111/j.1349-7006.2004.tb03195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carbonell WS, Ansorga O, Sibson N, Muschel R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE. 2009;4(6) doi: 10.1371/journal.pone.0005857. Article ID e5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao X, Guan J-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Advanced Drug Delivery Reviews. 2011;63(8):610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng Y, Lu Z. Paradoxical roles of FAK in tumor cell migration and metastasis. Cell Cycle. 2009;8(21):3474–3479. doi: 10.4161/cc.8.21.9846. [DOI] [PubMed] [Google Scholar]
- 49.Marchetti D, Nicolson GL. Human heparanase: a molecular determinant of brain metastasis. Advances in Enzyme Regulation. 2001;41:343–359. doi: 10.1016/s0065-2571(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 50.Marchetti D, Aucoin R, Blust J, Murry B, Greiter-Wilke A. p75 neurotrophin receptor functions as a survival receptor in brain-metastatic melanoma cells. Journal of Cellular Biochemistry. 2004;91(1):206–215. doi: 10.1002/jcb.10649. [DOI] [PubMed] [Google Scholar]
- 51.Zucker S, Cao J, Chen WT. Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene. 2000;19(56):6642–6650. doi: 10.1038/sj.onc.1204097. [DOI] [PubMed] [Google Scholar]
- 52.Finger EC, Giaccia AJ. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer and Metastasis Reviews. 2010;29(2):285–293. doi: 10.1007/s10555-010-9224-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes and Development. 2006;20(19):2673–2686. doi: 10.1101/gad.1451806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stamenkovic I. Extracellular matrix remodelling: the role of matrix metalloproteinases. Journal of Pathology. 2003;200(4):448–464. doi: 10.1002/path.1400. [DOI] [PubMed] [Google Scholar]
- 55.Jäälinojä J, Herva R, Korpela M, Höyhtyä M, Turpeenniemi-Hujanen T. Matrix metalloproteinase 2 (MMP-2) immunoreactive protein is associated with poor grade and survival in brain neoplasms. Journal of Neuro-Oncology. 2000;46(1):81–90. doi: 10.1023/a:1006421112839. [DOI] [PubMed] [Google Scholar]
- 56.Krüger A, Sanchez-Sweatman OH, Martin DC, et al. Host TIMP-1 overexpression confers resistance to experimental brain metastasis of a fibrosarcoma cell line. Oncogene. 1998;16(18):2419–2423. doi: 10.1038/sj.onc.1201774. [DOI] [PubMed] [Google Scholar]
- 57.Danø K, Behrendt N, Høyer-Hansen G, et al. Plasminogen activation and cancer. Thrombosis and Haemostasis. 2005;93(4):676–681. doi: 10.1160/TH05-01-0054. [DOI] [PubMed] [Google Scholar]
- 58.Bindal AK, Hammoud M, Wet Ming Shi, Wu SZ, Sawaya R, Rao JS. Prognostic significance of proteolytic enzymes in human brain tumors. Journal of Neuro-Oncology. 1994;22(2):101–110. doi: 10.1007/BF01052886. [DOI] [PubMed] [Google Scholar]
- 59.Yamaguchi H, Wyckoff J, Condeelis J. Cell migration in tumors. Current Opinion in Cell Biology. 2005;17(5):559–564. doi: 10.1016/j.ceb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 60.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer and Metastasis Reviews. 2009;28(1-2):113–127. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buccione R, Caldieri G, Ayala I. Invadopodia: specialized tumor cell structures for the focal degradation of the extracellular matrix. Cancer and Metastasis Reviews. 2009;28(1-2):137–149. doi: 10.1007/s10555-008-9176-1. [DOI] [PubMed] [Google Scholar]
- 62.Artym VV, Kindzelskii AL, Chen WT, Petty HR. Molecular proximity of seprase and the urokinase-type plasminogen activator receptor on malignant melanoma cell membranes: dependence on β1 integrins and the cytoskeleton. Carcinogenesis. 2002;23(10):1593–1602. doi: 10.1093/carcin/23.10.1593. [DOI] [PubMed] [Google Scholar]
- 63.Oxmann D, Held-Feindt J, Stark AM, Hattermann K, Yoneda T, Mentlein R. Endoglin expression in metastatic breast cancer cells enhances their invasive phenotype. Oncogene. 2008;27(25):3567–3575. doi: 10.1038/sj.onc.1211025. [DOI] [PubMed] [Google Scholar]
- 64.Wyckoff J, Wang W, Lin EY, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Research. 2004;64(19):7022–7029. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 65.Verkman AS. Aquaporins: translating bench research to human disease. Journal of Experimental Biology. 2009;212(11):1707–1715. doi: 10.1242/jeb.024125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khaitan D, Sankpal UT, Weksler B, et al. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer. 2009;9, article no. 258 doi: 10.1186/1471-2407-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee JH, Miele ME, Hicks DJ, et al. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. Journal of the National Cancer Institute. 1996;88(23):1731–1737. doi: 10.1093/jnci/88.23.1731. [DOI] [PubMed] [Google Scholar]
- 68.Shirasaki F, Takata M, Hatta N, Takehara K. Loss of expression of the metastasis suppressor gene KiSS1 during melanoma progression and its association with LOH of chromosome 6q16.3-q23. Cancer Research. 2001;61(20):7422–7425. [PubMed] [Google Scholar]
- 69.Dong JT, Suzuki H, Pin SS, et al. Down-regulation of the KAI1 metastasis suppressor gene during the progression of human prostatic cancer infrequently involves gene mutation or allelic loss. Cancer Research. 1996;56(19):4387–4390. [PubMed] [Google Scholar]
- 70.Guan RJ, Ford HL, Fu Y, Li Y, Shaw LM, Pardee AB. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Research. 2000;60(3):749–755. [PubMed] [Google Scholar]
- 71.Shah MA, Kemeny N, Hummer A, et al. Drg1 expression in 131 colorectal liver metastases: correlation with clinical variables and patient outcomes. Clinical Cancer Research. 2005;11(9):3296–3302. doi: 10.1158/1078-0432.CCR-04-2417. [DOI] [PubMed] [Google Scholar]
- 72.Nam DH, Jeon HM, Kim S, et al. Activation of Notch signaling in a xenograft model of brain metastasis. Clinical Cancer Research. 2008;14(13):4059–4066. doi: 10.1158/1078-0432.CCR-07-4039. [DOI] [PubMed] [Google Scholar]
- 73.Mareel M, Oliveira MJ, Madani I. Cancer invasion and metastasis: interacting ecosystems. Virchows Archiv. 2009;454(6):599–622. doi: 10.1007/s00428-009-0784-0. [DOI] [PubMed] [Google Scholar]
- 74.Stafford LJ, Vaidya KS, Welch DR. Metastasis suppressors genes in cancer. International Journal of Biochemistry and Cell Biology. 2008;40(5):874–891. doi: 10.1016/j.biocel.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 75.Dollé L, Depypere HT, Bracke ME. Anti-invasive and anti-metastasis strategies: new roads, new tools and new hopes. Current Cancer Drug Targets. 2006;6(8):729–751. doi: 10.2174/156800906779010263. [DOI] [PubMed] [Google Scholar]
- 76.Davies PF, Spaan JA, Krams R. Shear stress biology of the endothelium. Annals of Biomedical Engineering. 2005;33(12):1714–1718. doi: 10.1007/s10439-005-8774-0. [DOI] [PubMed] [Google Scholar]
- 77.Thamilselvan V, Craig DH, Basson MD. FAK association with multiple signal proteins mediates pressure-induced colon cancer cell adhesion via a Src-dependent PI3K/Akt pathway. FASEB Journal. 2007;21(8):1730–1741. doi: 10.1096/fj.06-6545com. [DOI] [PubMed] [Google Scholar]
- 78.Von Sengbusch A, Gassmann P, Fisch KM, Enns A, Nicolson GL, Haier J. Focal adhesion kinase regulates metastatic adhesion of carcinoma cells within liver sinusoids. American Journal of Pathology. 2005;166(2):585–596. doi: 10.1016/S0002-9440(10)62280-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nieswandt B, Hafner M, Echtenacher B, Männel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Research. 1999;59(6):1295–1300. [PubMed] [Google Scholar]
- 80.Sahai E. Illuminating the metastatic process. Nature Reviews Cancer. 2007;7(10):737–749. doi: 10.1038/nrc2229. [DOI] [PubMed] [Google Scholar]
- 81.Lalor PF, Lai WK, Curbishley SM, Shetty S, Adams DH. Human hepatic sinusoidal endothelial cells can be distinguished by expression of phenotypic markers related to their specialised functions in vivo. World Journal of Gastroenterology. 2006;12(34):5429–5439. doi: 10.3748/wjg.v12.i34.5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brown DM, Ruoslahti E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell. 2004;5(4):365–374. doi: 10.1016/s1535-6108(04)00079-0. [DOI] [PubMed] [Google Scholar]
- 83.Saito N, Hatori T, Murata N, et al. Comparison of metastatic brain tumour models using three different methods: the morphological role of the pia mater. International Journal of Experimental Pathology. 2008;89(1):38–44. doi: 10.1111/j.1365-2613.2007.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Huysentruyt LC, Mukherjee P, Banerjee D, Shelton LM, Seyfried TN. Metastatic cancer cells with macrophage properties: evidence from a new murine tumor model. International Journal of Cancer. 2008;123(1):73–84. doi: 10.1002/ijc.23492. [DOI] [PubMed] [Google Scholar]
- 85.Bos PD, Zhang XHF, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. doi: 10.1038/nature08021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khanna C, Wan X, Bose S, et al. The membrane-cytoskeleton linker ezrin is necessary for osteosarcoma metastasis. Nature Medicine. 2004;10(2):182–186. doi: 10.1038/nm982. [DOI] [PubMed] [Google Scholar]
- 87.Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 88.Fitzgerald DP, Palmieri D, Hua E, et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clinical and Experimental Metastasis. 2008;25(7):799–810. doi: 10.1007/s10585-008-9193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leenders WPJ, Küsters B, De Waal RMW. Vessel co-option: how tumors obtain blood supply in the absence of sprouting angiogenesis. Endothelium. 2002;9(2):83–87. doi: 10.1080/10623320212006. [DOI] [PubMed] [Google Scholar]
- 90.Kusters B, Westphal JR, Smits D, et al. The pattern of metastasis of human melanoma to the central nervous system is not influenced by integrin αvβ3 expression. International Journal of Cancer. 2001;92(2):176–180. doi: 10.1002/1097-0215(200102)9999:9999<::aid-ijc1173>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 91.Folkman J, Hanahan D. Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symposia. 1991;22:339–347. [PubMed] [Google Scholar]
- 92.Lee SK, Huang S, Lu W, Lev DC, Price JE. Vascular endothelial growth factor expression promotes the growth of breast cancer brain metastases in nude mice. Clinical and Experimental Metastasis. 2004;21(2):107–118. doi: 10.1023/b:clin.0000024761.00373.55. [DOI] [PubMed] [Google Scholar]
- 93.Xia W, Unger P, Miller L, Nelson J, Gelman IH. The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Research. 2001;61(14):5644–5651. [PubMed] [Google Scholar]
- 94.Bergers G, Brekken R, McMahon G, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nature Cell Biology. 2000;2(10):737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Roodink I, Raats J, Van Der Zwaag B, et al. Plexin D1 expression is induced on tumor vasculature and tumor cells: a novel tarqet for diagnosis and therapy? Cancer Research. 2005;65(18):8317–8323. doi: 10.1158/0008-5472.CAN-04-4366. [DOI] [PubMed] [Google Scholar]
- 96.Palmieri D, Fitzgerald D, Shreeve SM, et al. Analyses of resected human brain metastases of breast cancer reveal the association between up-regulation of hexokinase 2 and poor prognosis. Molecular Cancer Research. 2009;7(9):1438–1445. doi: 10.1158/1541-7786.MCR-09-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Weil RJ, Palmieri DC, Bronder JL, Stark AM, Steeg PS. Breast cancer metastasis to the central nervous system. American Journal of Pathology. 2005;167(4):913–920. doi: 10.1016/S0002-9440(10)61180-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yaguchi H, Ohkura N, Tsukada T, Yamaguchi K. Menin, the multiple endocrine neoplasia type 1 gene product, exhibits GTP-hydrolyzing activity in the presence of the tumor metastasis suppressor nm23. The Journal of Biological Chemistry. 2002;277(41):38197–38204. doi: 10.1074/jbc.M204132200. [DOI] [PubMed] [Google Scholar]
- 99.Ouatas T, Salerno M, Palmieri D, Steeg PS. Basic and translational advances in cancer metastasis: Nm23. Journal of Bioenergetics and Biomembranes. 2003;35(1):73–79. doi: 10.1023/a:1023497924277. [DOI] [PubMed] [Google Scholar]
- 100.Sarris M, Scolyer RA, Konopka M, Thompson JF, Harper CG, Lee SC. Cytoplasmic expression of nm23 predicts the potential for cerebral metastasis in patients with primary cutaneous melanoma. Melanoma Research. 2004;14(1):23–27. doi: 10.1097/00008390-200402000-00004. [DOI] [PubMed] [Google Scholar]
- 101.Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Research. 2000;60(11):2764–2769. [PubMed] [Google Scholar]
- 102.Seraj MJ, Harding MA, Gildea JJ, Welch DR, Theodorescu D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clinical and Experimental Metastasis. 2000;18(6):519–525. doi: 10.1023/a:1011819621859. [DOI] [PubMed] [Google Scholar]
- 103.Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development. 2008;135(3):579–588. doi: 10.1242/dev.007047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wrage M, Ruosaari S, Eijk PP, et al. Genomic profiles associated with early micrometastasis in lung cancer: relevance of 4q deletion. Clinical Cancer Research. 2009;15(5):1566–1574. doi: 10.1158/1078-0432.CCR-08-2188. [DOI] [PubMed] [Google Scholar]
- 105.Grinberg-Rashi H, Ofek E, Perelman M, et al. The expression of three genes in primary non-small cell lung cancer is associated with metastatic spread to the brain. Clinical Cancer Research. 2009;15(5):1755–1761. doi: 10.1158/1078-0432.CCR-08-2124. [DOI] [PubMed] [Google Scholar]
- 106.Narla G, DiFeo A, Fernandez Y, et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. Journal of Clinical Investigation. 2008;118(8):2711–2721. doi: 10.1172/JCI34780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Micalizzi DS, Christensen KL, Jedlicka P, et al. The Six1 homeoprotein induces human mammary carcinoma cells to undergo epithelial-mesenchymal transition and metastasis in mice through increasing TGF-β signaling. Journal of Clinical Investigation. 2009;119(9):2678–2690. doi: 10.1172/JCI37815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Blenkiron C, Miska EA. miRNAs in cancer: approaches, aetiology, diagnostics and therapy. Human Molecular Genetics. 2007;16(1):R106–R113. doi: 10.1093/hmg/ddm056. [DOI] [PubMed] [Google Scholar]
- 109.Wendt MK, Allington TM, Schiemann WP. Mechanisms of the epithelial-mesenchymal transition by TGF-β . Future Oncology. 2009;5(8):1145–1168. doi: 10.2217/fon.09.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dalmay T, Edwards DR. MicroRNAs and the hallmarks of cancer. Oncogene. 2006;25(46):6170–6175. doi: 10.1038/sj.onc.1209911. [DOI] [PubMed] [Google Scholar]
- 111.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465(7301):1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.White NMA, Fatoohi E, Metias M, Jung K, Stephan C, Yousef GM. Metastamirs: a stepping stone towards improved cancer management. Nature Reviews Clinical Oncology. 2011;8(2):75–84. doi: 10.1038/nrclinonc.2010.173. [DOI] [PubMed] [Google Scholar]