

Abstract
Microarrays can measure the expression of thousands of genes to identify changes in expression between different biological states. Methods are needed to determine the significance of these changes while accounting for the enormous number of genes. We describe a method, Significance Analysis of Microarrays (SAM), that assigns a score to each gene on the basis of change in gene expression relative to the standard deviation of repeated measurements. For genes with scores greater than an adjustable threshold, SAM uses permutations of the repeated measurements to estimate the percentage of genes identified by chance, the false discovery rate (FDR). When the transcriptional response of human cells to ionizing radiation was measured by microarrays, SAM identified 34 genes that changed at least 1.5-fold with an estimated FDR of 12%, compared with FDRs of 60 and 84% by using conventional methods of analysis. Of the 34 genes, 19 were involved in cell cycle regulation and 3 in apoptosis. Surprisingly, four nucleotide excision repair genes were induced, suggesting that this repair pathway for UV-damaged DNA might play a previously unrecognized role in repairing DNA damaged by ionizing radiation.
DNA microarrays contain oligonucleotide or cDNA probes for measuring the expression of thousands of genes in a single hybridization experiment. Although massive amounts of data are generated, methods are needed to determine whether changes in gene expression are experimentally significant. Cluster analysis of microarray data can find coherent patterns of gene expression (1) but provides little information about statistical significance. Methods based on conventional t tests provide the probability (P) that a difference in gene expression occurred by chance (2, 3). Although P = 0.01 is significant in the context of experiments designed to evaluate small numbers of genes, a microarray experiment for 10,000 genes would identify 100 genes by chance. This problem led us to develop a statistical method adapted specifically for microarrays, Significance Analysis of Microarrays (SAM).
SAM identifies genes with statistically significant changes in expression by assimilating a set of gene-specific t tests. Each gene is assigned a score on the basis of its change in gene expression relative to the standard deviation of repeated measurements for that gene. Genes with scores greater than a threshold are deemed potentially significant. The percentage of such genes identified by chance is the false discovery rate (FDR). To estimate the FDR, nonsense genes are identified by analyzing permutations of the measurements. The threshold can be adjusted to identify smaller or larger sets of genes, and FDRs are calculated for each set. To demonstrate its utility, SAM was used to analyze a biologically important problem: the transcriptional response of lymphoblastoid cells to ionizing radiation (IR).
Materials and Methods
Preparation of RNA.
Human lymphoblastoid cell lines GM14660 and GM08925 (Coriell Cell Repositories, Camden, NJ) were seeded at 2.5 × 105 cells/ml and exposed to IR 24 h later. RNA was isolated, labeled, and hybridized to the HuGeneFL GeneChip microarray according to manufacturer's protocols (Affymetrix, Santa Clara, CA).
Microarray Hybridization.
Each gene in the microarray was represented by 20 oligonucleotide pairs, each pair consisting of an oligonucleotide perfectly matched to the cDNA sequence, and a second oligonucleotide containing a single base mismatch. Because gene expression was computed from differences in hybridization to the matched and mismatched probes, expression levels were sometimes reported by the GeneChip analysis suite software as negative numbers.
Northern Blot Hybridization.
Total RNA (15 μg) was resolved by agarose gel electrophoresis, transferred to a nylon membrane, and hybridized to specific radiolabeled DNA probes, which were prepared by PCR amplification.
Results
RNA was harvested from wild-type human lymphoblastoid cell lines, designated 1 and 2, growing in an unirradiated state (U) or in an irradiated state (I) 4 h after exposure to a modest dose of 5 Gy of IR. RNA samples were labeled and divided into two identical aliquots for independent hybridizations, A and B. Thus, data for 6,800 genes on the microarray were generated from eight hybridizations (U1A, U1B, U2A, U2B, I1A, I1B, I2A, and I2B).
We scaled the data from different hybridizations as follows. A reference data set was generated by averaging the expression of each gene over all eight hybridizations. The data for each hybridization were compared with the reference data set in a cube root scatter plot. We chose the cube root scatter plot because it resolved the vast majority of genes that are expressed at low levels and permitted the inclusion of negative levels of expression that are sometimes generated by the GeneChip software. A linear least-squares fit to the cube root scatter plot was then used to calibrate each hybridization.
After scaling, a linear scatter plot was generated for average gene expression in the four A aliquots (U1A, I1A, U2A, and U2A) vs. the average in the four B aliquots (U1B, I1B, U2B, and U2B), a partitioning of the data that eliminates biological changes in gene expression (Fig. 1A). The linear scatter plot confirmed that the data were generally reproducible but failed to resolve genes expressed at low levels. Better resolution of these genes was achieved by the cube root scatter plot (Fig. 1B), which revealed three salient features: the large percentage of genes (24%) assigned negative levels of expression, the large percentage of genes with low levels of expression, and the low signal-to-noise ratio at low levels of expression.
Figure 1.

Gene expression measured by microarrays. (A) Linear scatter plot of gene expression. Each gene (i) in the microarray is represented by a point with coordinates consisting of average gene expression measured from the four A hybridizations (avg xA) and the average gene expression in the four B hybridizations (avg xB). (B) Cube root scatter plot of gene expression. The average gene expression from the A and B hybridizations have been plotted on a cube root scale to resolve genes expressed at low levels. (C) Cube root scatter plot of average gene expression from the four hybridizations with uninduced cells (avg xU) and induced cells 4 h after exposure to 5 Gy of IR (avg xI). Some of the genes that responded to IR are indicated by arrows.
To assess the biological effect of IR, a scatter plot was generated for average gene expression in the four irradiated states vs. the four unirradiated states (compare Fig. 1 B and C). A few of the potentially significant changes in gene expression are indicated by arrows in Fig. 1C, but the effect was not easily quantified, and a method was needed to identify changes with statistical confidence.
Our approach was based on analysis of random fluctuations in the data. In general, the signal-to-noise ratio decreased with decreasing gene expression (Fig. 1). However, even for a given level of expression, we found that fluctuations were gene specific. To account for gene-specific fluctuations, we defined a statistic based on the ratio of change in gene expression to standard deviation in the data for that gene. The “relative difference” d(i) in gene expression is:
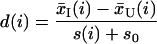 |
1 |
where x̄I(i) and x̄U(i) are defined as the average levels of expression for gene (i) in states I and U, respectively. The “gene-specific scatter” s(i) is the standard deviation of repeated expression measurements:
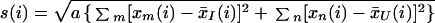 |
2 |
where Σm and Σn are summations of the expression measurements in states I and U, respectively, a = (1/n1 + 1/n2)/(n1 + n2 − 2), and n1 and n2 are the numbers of measurements in states I and U (four in this experiment).
To compare values of d(i) across all genes, the distribution of d(i) should be independent of the level of gene expression. At low expression levels, variance in d(i) can be high because of small values of s(i). To ensure that the variance of d(i) is independent of gene expression, we added a small positive constant s0 to the denominator of Eq. 1. The coefficient of variation of d(i) was computed as a function of s(i) in moving windows across the data. The value for s0 was chosen to minimize the coefficient of variation. For the data in this paper, this computation yielded s0 = 3.3.
Scatter plots of d(i) vs. s(i) are shown in Fig. 2. The scatter plot for relative difference between states I and U is shown in Fig. 2A. By contrast, the scatter plot for relative difference between cell lines 1 and 2 shows more marked changes in Fig. 2B. These relative differences exceeded random fluctuations in the data, as measured by the relative difference between hybridizations A and B in Fig. 2C.
Figure 2.

Scatter plots of relative difference in gene expression d(i) vs. gene-specific scatter s(i). The data were partitioned to calculate d(i), as indicated by the bar codes. The shaded and unshaded entries were used for the first and second terms in the numerator of d(i) in Eq. 1. (A) Relative difference between irradiated and unirradiated states. The statistic d(i) was computed from expression measurements partitioned between irradiated and unirradiated cells. (B) Relative difference between cell lines 1 and 2. The statistic d(i) was computed from expression measurements partitioned between cell lines 1 and 2. (C) Relative difference between hybridizations A and B. The statistic d(i) was computed from the permutation in which the expression measurements were partitioned between the equivalent hybridizations A and B. (D) Relative difference for a permutation of the data that was balanced between cell lines 1 and 2.
Although the relative difference computed from hybridizations A and B provided a control for random fluctuations, additional controls were needed to assign statistical significance to the biological effect of IR. Instead of performing more experiments, which are expensive and labor intensive, we generated a large number of controls by computing relative differences from permutations of the hybridizations for the four irradiated and four unirradiated states. To minimize potentially confounding effects from differences between the two cell lines, we analyzed the data by using the 36 permutations that were balanced for cell lines 1 and 2. Permutations were defined as balanced when each group of four experiments contained two experiments from cell line 1 and two experiments from cell line 2. Fig. 2 C and D are examples of balanced permutations.
To find significant changes in gene expression, genes were ranked by magnitude of their d(i) values, so that d(1) was the largest relative difference, d(2) was the second largest relative difference, and d(i) was the ith largest relative difference. For each of the 36 balanced permutations, relative differences dp(i) were also calculated, and the genes were again ranked such that dp(i) was the ith largest relative difference for permutation p. The expected relative difference, dE(i), was defined as the average over the 36 balanced permutations, dE(i) = Σpdp(i)/36.
To identify potentially significant changes in expression, we used a scatter plot of the observed relative difference d(i) vs. the expected relative difference dE(i) (Fig. 3A). For the vast majority of genes, d(i) ≅ dE(i), but some genes are represented by points displaced from the d(i) = dE(i) line by a distance greater than a threshold Δ. For example, the threshold Δ = 1.2 illustrated by the broken lines in Fig. 3A yielded 46 genes that were “called significant.” These 46 genes are shown in the context of the scatter plot for d(i) vs. s(i) (Fig. 3B) and in the scatter plot for the cube root of gene expression x̄I(i) vs. x̄U(i) (Fig. 3C). Genes identified by d(i) do not necessarily have the largest changes in gene expression.
Figure 3.

Identification of genes with significant changes in expression. (A) Scatter plot of the observed relative difference d(i) versus the expected relative difference dE(i). The solid line indicates the line for d(i) = dE(i), where the observed relative difference is identical to the expected relative difference. The dotted lines are drawn at a distance Δ = 1.2 from the solid line. (B) Scatter plot of d(i) vs. s(i). (C) Cube root scatter plot of average gene expression in induced and uninduced cells. The cutoffs for 2-fold induction and repression are indicated by the dashed lines. In A–C, the 46 potentially significant genes for Δ = 1.2 are indicated by the squares.
To determine the number of falsely significant genes generated by SAM, horizontal cutoffs were defined as the smallest d(i) among the genes called significantly induced and the least negative d(i) among the genes called significantly repressed. The number of falsely significant genes corresponding to each permutation was computed by counting the number of genes that exceeded the horizontal cutoffs for induced and repressed genes. The estimated number of falsely significant genes was the average of the number of genes called significant from all 36 permutations. For Δ = 1.2, the permuted data sets generated an average of 8.4 falsely significant genes, compared with 46 genes called significant, yielding an estimated FDR of 18% (Table 1). As Δ decreased, the number of genes called significant by SAM increased but at the cost of an increasing FDR. (Omitting s0 from Eq. 1 produced higher FDRs of 45, 35, and 28% for Δ = 0.6, 0.9, and 1.2.)
Table 1.
Comparison of methods for identifying changes in gene expression
| Parameter | Number falsely significant | Number called significant | FDR |
|---|---|---|---|
| SAM | |||
| Δ = 0.4 | 134.9 | 288 | 47% |
| Δ = 0.5 | 78.1 | 192 | 41% |
| Δ = 0.6 | 56.1 | 162 | 35% |
| Δ = 0.9 | 19.1 | 80 | 24% |
| Δ = 1.2 | 8.4 | 46 | 18% |
| Δ = 1.2; R = 1.5 | 4.5 | 34 | 12% |
| Fold change | |||
| R = 2.0 | 283.1 | 348 | 81% |
| R = 2.5 | 137.8 | 169 | 82% |
| R = 3.0 | 76.8 | 99 | 78% |
| R = 3.5 | 46.7 | 64 | 73% |
| R = 4.0 | 29.3 | 35 | 84% |
| Pairwise fold change | |||
| R = 1.2 | 245.6 | 355 | 69% |
| R = 1.3 | 155.4 | 220 | 71% |
| R = 1.5 | 76.2 | 118 | 65% |
| R = 1.7 | 44.8 | 70 | 64% |
| R = 2.0 | 22.8 | 38 | 60% |
To increase the stringency for calling significant changes in gene expression, parameters for each method (Δ and R) were increased, as described in the text. The false discovery rate (FDR) was defined as the percentage of falsely significant genes compared to the genes called significant.
Our method for setting thresholds provides asymmetric cutoffs for induced and repressed genes. The alternative is the standard t test, which imposes a symmetric horizontal cutoff, with d(i) > c for induced genes and d(i) <− c for repressed genes. However, the asymmetric cutoff is preferred because it allows for the possibility that d(i) for induced and repressed genes may behave differently in some biological experiments.
SAM proved to be superior to conventional methods for analyzing microarrays (Table 1 and Fig. 4A). First, SAM was compared with the approach of identifying genes as significantly changed if an R-fold change was observed. In this “fold change” method, r(i) = x̄I(i)/x̄U(i), and gene (i) was called significantly changed if r(i) > R or r(i) < 1/R. To permit computation of r(i) from negative values for gene expression, x̄I(i) and x̄U(i) were converted to 10 when their values were negative or less than 10. The results of this procedure yielded unacceptably high FDRs of 73–84%.
Figure 4.

Comparison of SAM to conventional methods for analyzing microarrays. (A) Falsely significant genes plotted against number of genes called significant. Of the 57 genes most highly ranked by the fold change method, 5 were included among the 46 genes most highly ranked by SAM. Of the 38 genes most highly ranked by the pairwise fold change method, 11 were included among the 46 genes most highly ranked by SAM. These results were consistent with the FDR of SAM compared to the FDRs of the fold change and pairwise fold change methods. (B) Northern blot validation for genes identified by the fold change method. Values of r(i) are plotted for genes chosen at random from the 57 genes most highly ranked by the fold change method. (C) Validation for genes identified by SAM. Results are plotted for genes chosen at random from the 46 genes most highly ranked by SAM. Genes analyzed by Northern blot are represented by circles. TNF-α was validated by using a PreDeveloped TaqMan assay (PE Biosystems) and is represented by a square. The straight lines in B and C indicate the position of exact agreement between Northern blot and microarray results.
Another approach attempts to account for uncertainty in the data by identifying genes as significantly changed if an R-fold change is observed consistently between paired samples (4). To apply this “pairwise fold change” method to our four data sets before IR and four data sets after IR, changes in gene expression were declared significant if 12 of 16 pairings satisfied the criteria r(i) > R or r(i) < 1/R. Despite the demand for consistent changes between paired samples, this method yielded FDRs of 60–71%.
To understand why fold-change methods fail, note that the vast majority of genes are expressed at low levels where the signal-to-noise ratio is very low (Fig. 3C). Thus, 2-fold changes in gene expression occur at random for a large number of genes. Conversely, for higher levels of expression, smaller changes in gene expression may be real, but these changes are rejected by fold-change methods. The pairwise fold-change method provides modest improvement but remains inferior to SAM.
Of the 46 genes most highly ranked by SAM (Δ = 1.2), 36 increased or decreased at least 1.5-fold (R = 1.5). The number of falsely significant genes that met these two criteria was 4.5, corresponding to a FDR of 12% (Table 1). Fas was identified three times as alternately spliced forms, leaving 34 independent genes (Table 2). As an indication of biological validity, 10 of the 34 genes have been reported in the literature as part of the transcriptional response to IR. TNF-α was reported to be induced by other investigators (5) but was repressed here. Quantitative reverse transcription–PCR confirmed this result.
Table 2.
Genes with changes in expression called significant by SAM
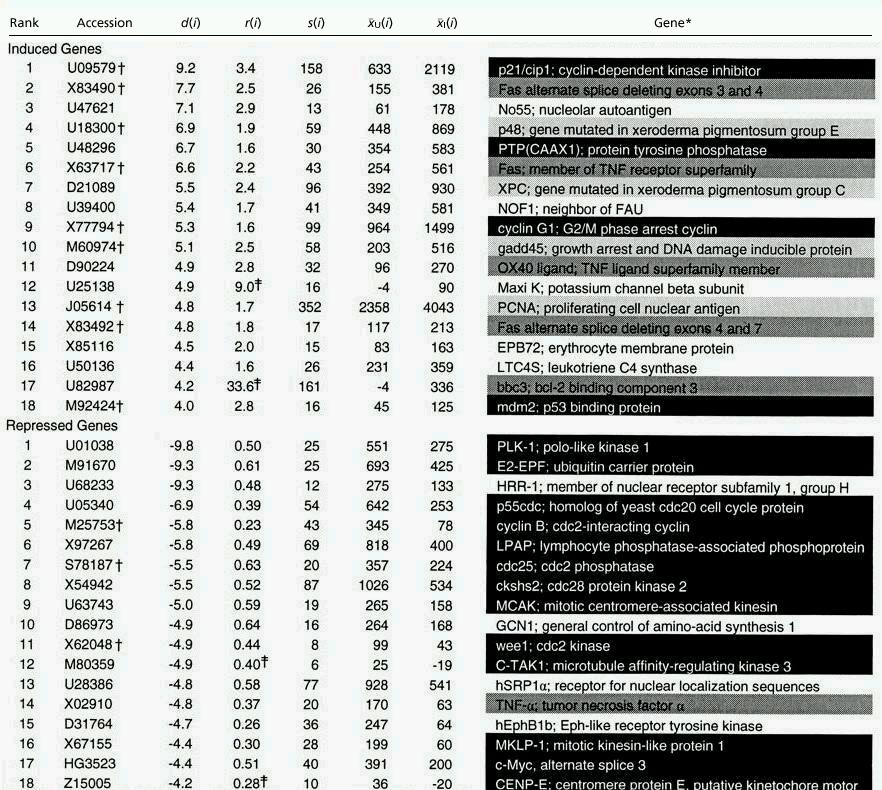 |
Gene functions: Black = cell cycle; Dark gray = apoptosis; Light gray = DNA repair.
Genes previously reported to respond transcriptionally to IR.
‡To compute r(i) = x̄I(i)/x̄U(i), negative levels of expression were reset to a value of 10.
To test the validity of SAM directly, we performed Northern blots for genes that were randomly selected from the 46 and 57 genes most highly ranked by SAM (Δ = 1.2) and the fold-change method (at least 3.6-fold change), respectively. Northern blots showed little correlation with the genes identified by the fold change method (Fig. 4B), but strong correlation with the genes identified by SAM (Fig. 4C). Indeed, Northern blots contradicted only 1 (maxiK) of 11 genes identified by SAM, consistent with our estimated FDR.
Nineteen of the 34 genes most highly ranked by SAM appear to be involved in the cell cycle. Three are known to be induced in a p53-dependent manner: p21, cyclin G1, and mdm2 (6–8). Six cell cycle genes were repressed: E2-EPF, p55cdc, cyclin B, ckshs2, cdc25, and wee1 (9, 10). Five genes encoding the mitotic machinery were also repressed: PLK-1, MKLP-1, MCAK, C-TAK1, CENP-E (11–13). Three genes involved in cell proliferation were induced or repressed: PTP(CAAX1), LPAP, and c-myc (14–18). Some responses appeared paradoxical. For example, cdc25 phosphatase and wee1 kinase have antagonistic effects on the phosphorylation state of cdc2, but both genes were repressed. Repression of these genes together with the mitotic genes may represent a damage response that dismantles the cell cycle machinery until the cell has repaired the damaged DNA.
Four of the 34 genes play roles in DNA repair, but none are involved in the repair of IR-induced double-strand breaks. Instead, the genes (p48, XPC, gadd45, PCNA) have roles in nucleotide excision repair, a pathway conventionally associated with UV-induced damage (19–22). We confirmed the induction of these genes by Northern blot (23–25). Fornace et al. reported defective removal of base damage induced by IR in xeroderma pigmentosum cells (26). Leadon et al. reported that a novel DNA repair pathway involving long excision repair patches of at least 150 nucleotides is activated by IR but not UV (27). Our results suggest that this novel pathway might include p48, XPC, gadd45, and PCNA.
Four of the 34 genes play roles in apoptosis (Fas, bbc3, TNF-α, OX40 ligand). The remaining genes may have previously unsuspected roles in the DNA damage response or may be among the estimated set of four falsely detected genes.
The 34 genes most highly ranked by SAM are only a subset of all of the genes that change 1.5-fold with IR. Indeed, we calculated the difference between the number of genes called significant and the number of falsely significant genes for decreasing Δ = 0.3, 0.2, and 0.1, and found the differences to be 92, 170, and 184, respectively. Thus, SAM suggests that approximately 180 of the 6,800 genes on the microarray were induced or repressed by 5 Gy IR.
Discussion
SAM is a method for identifying genes on a microarray with statistically significant changes in expression, developed in the context of an actual biological experiment. SAM was successful in analyzing this experiment as well as several other experiments with oligonucleotide and cDNA microarrays (data not shown).
In the statistics of multiple testing (28–30), the family-wise error rate (FWER) is the probability of at least one false positive over the collection of tests. The Bonferroni method, the most basic method for bounding the FWER, assumes independence of the different tests. An acceptable FWER could be achieved for our microarray data only if the corresponding threshold was set so high that no genes were identified. The step-down correction method of Westfall and Young (29), adapted for microarrays by Dudoit et al. (http://www.stat.berkeley.edu/users/terry/zarray/Html/matt.html), allows for dependent tests but still remains too stringent, yielding no genes from our data.
Westfall and Young (29) define “weak control” to be control of the FWER when all of the null hypotheses are true (i.e., when there are no changes in gene expression). “Strong control” is control of the FWER when any subset of the null hypotheses is true. Under certain conditions, weak control implies strong control. In fact, the step-down correction method exerts both weak and strong control.
The method of Benjamini and Hochberg (31) assumes independent tests and guarantees an upper bound for the FDR (with both weak and strong control) by a step-up or step-down procedure applied to the individual P values. For our data, the P value for each gene is calculated from permutations of the eight experiments. Because of the limited number of permutations, the FDR is too “granular”, and we identified either zero or 300 significant genes, depending on how the P value was defined. A similar granular result was obtained for the adaptation to dependent tests by Benjamini et al. [The Control of the False Discovery Rate in Multiple Testing Under Dependency (Department of Statistics and Operations Research, Tel Aviv University, Tel Aviv). http://www.math.tau.ac.il/∼ybenja/].
SAM does not have strong or weak control of the FWER. Instead, SAM provides an estimate of the FDR for each value of the tuning parameter Δ. The estimated FDR is computed from permutations of the data and hence assumes that all null hypotheses are true, allowing for the possibility of dependent tests. It seems plausible that this estimated FDR approximates the strongly controlled FDR when any subset of null hypotheses is true. However, we have not proven this in general. It is possible for SAM to give an estimate of the FDR that is greater than 1. However, this has not occurred in our experience. Indeed, SAM provides a reasonably accurate estimate for the true FDR. To confirm this, we constructed artificial data sets in which a subset of genes was induced over a background of noise. SAM successfully identified the induced genes and estimated the FDR with reasonable accuracy.
Although this paper analyzes a simple two-state experiment, SAM can be generalized to other types of experiments by defining d(i) in a different way. Suppose the data includes gene expression xj(i) and a response parameter yj, in which i = 1, 2, … , m genes, j = 1, 2, … , n states. The generalized statistical parameter still takes the form d(i) = r(i)/[s(i) + s0], except that the definitions of r(i) and s(i) change.
To identify genes with changes in expression in an experiment with three or more states, the parameter d(i) is defined in terms of the Fisher's linear discriminant. One goal might be to identify genes whose expression in one type of tumor is different from its expression in other types of tumors. Suppose that a set of n samples consists of K nonoverlapping subsets, such that the response parameter yj ε {1, … , K}. Define C(k) = {j : yj = k}. Let nk = number of observations in C(k). The average gene expression in each subset is x̄k(i) = Σj∈C(k)xj(i)/nk and the average gene expression for all n samples is x̄(i) = Σjxj(i)/n. Then define:
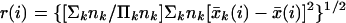 |
3 |
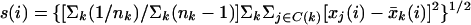 |
4 |
SAM can be adapted for still other types of experimental data. For example, to identify genes whose expression correlates with survival time, d(i) is defined in terms of Cox's proportional hazards function, in which some of the patients remain alive or are lost to follow-up at the time of the study. To identify genes whose expression correlates with a quantitative parameter, such as tumor stage, d(i) can be defined in terms of the Pearson correlation coefficient. Another example includes the definition of d(i) for paired data, such as gene expression in tumors before and after chemotherapy. In each case, the FDR is estimated by random permutation of the data for gene expression among the different experimental arms, i.e., permutations among the n arms of yj. Thus, SAM is a robust and straightforward method that can be adapted to a broad range of experimental situations. SAM and the adaptations discussed above are available for use at http://www-stat-class.stanford.edu/SAM/SAMServlet.
Acknowledgments
We thank Peter Jackson, Ron Davis, James Ferrell, Dean Felsher, Lisa DeFazio, Joe Budman, Jean Tang, Tom Tan, and Kerri Rieger for helpful discussions. This work was supported by the Burroughs Wellcome Clinical Scientist Award and by National Institutes of Health (NIH) Grant CA77302 to G.C., by NIH Small Business Technology Transfer grant CA75675 to G.C. and Affymetrix, and by the Stanford Genome Training Grant to V.T.
Abbreviations
- SAM
significance analysis of microarrays
- FDR
false discovery rate
- IR
ionizing radiation
- FWER
family-wise error rate
References
- 1.Eisen M, Spellman P, Brown P, Botstein D. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts C, Nelson B, Marton M, Stoughton R, Meyer M, Bennett H, He Y, Dai H, Walker W, Hughes T, Tyers M, Boone C, Friend S. Science. 2000;287:873–880. doi: 10.1126/science.287.5454.873. [DOI] [PubMed] [Google Scholar]
- 3.Galitski T, Saldanha A, Styles C, Lander E, Fink G. Science. 1999;285:251–254. doi: 10.1126/science.285.5425.251. [DOI] [PubMed] [Google Scholar]
- 4.Ly D, Lockhart D, Lerner R, Schultz P. Science. 2000;287:2486–2492. doi: 10.1126/science.287.5462.2486. [DOI] [PubMed] [Google Scholar]
- 5.Weill D, Gay F, Tovey M, Chouaib S. J Interferon Cytokine Res. 1996;16:395–402. doi: 10.1089/jir.1996.16.395. [DOI] [PubMed] [Google Scholar]
- 6.Harper J W, Adami G R, Wei N, Keyomarsi K, Elledge S J. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 7.Okamoto K, Beach D. EMBO J. 1994;13:4816–4822. doi: 10.1002/j.1460-2075.1994.tb06807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prives C. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- 9.Furnari B, Rhind N, Russell P. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Diaz L, Haas A, Giudice G. J Biol Chem. 1992;267:15829–15835. [PubMed] [Google Scholar]
- 11.Lee K, Yuan Y, Kuriyama R, Erikson R. Mol Cell Biol. 1995;15:7143–7151. doi: 10.1128/mcb.15.12.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maney T, Hunter A, Wagenbach M, Wordeman L. J Cell Biol. 1998;142:787–801. doi: 10.1083/jcb.142.3.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood K, Sakowicz R, Goldstein L, Cleveland D. Cell. 1997;91:357–366. doi: 10.1016/s0092-8674(00)80419-5. [DOI] [PubMed] [Google Scholar]
- 14.Ding I, Bruyns E, Li P, Magada D, Paskind M, Rodman L, Seshadri T, Alexander D, Giese T, Schraven B. Eur J Immunol. 1999;29:3956–3961. doi: 10.1002/(SICI)1521-4141(199912)29:12<3956::AID-IMMU3956>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 15.Cates C, Michael R, Stayrook K, Harvey K, Burke Y, Randall S, Crowell P, Crowell D. Cancer Lett. 1996;110:49–55. doi: 10.1016/s0304-3835(96)04459-x. [DOI] [PubMed] [Google Scholar]
- 16.Godfrey W, Fagnoni R, Harara M, Buck D, Engleman E. J Exp Med. 1994;180:757–762. doi: 10.1084/jem.180.2.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lord J, McIntosh B, Greenberg P, Nelson B. J Immunol. 2000;164:2533–2541. doi: 10.4049/jimmunol.164.5.2533. [DOI] [PubMed] [Google Scholar]
- 18.Prevot D, Voeltzel T, Birot A, Morel A, Rostan M, Magaud J, Corbo L. J Biol Chem. 2000;275:147–153. doi: 10.1074/jbc.275.1.147. [DOI] [PubMed] [Google Scholar]
- 19.Aboussekhra A, Biggerstaff M, Shivji M, Vilpo J, Moncollin V, Podust V, Protic M, Hubscher U, Egly J, Wood R. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 20.Smith M, Ford J, Hollander M, Bortnick R, Amundson S, Seo Y, Deng C, Hanawalt P, Fornace A J. Mol Cell Biol. 2000;20:3705–3714. doi: 10.1128/mcb.20.10.3705-3714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugasawa K, Ng J, Masutani C, Iwai S, van der Spek P, Eker A, Hanaoka F, Bootsma D, Hoeijmakers J. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 22.Tang J, Hwang B, Ford J, Hanawalt P, Chu G. Mol Cell. 2000;5:737–744. doi: 10.1016/s1097-2765(00)80252-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kastan M, Zhan Q, El-Deiry F, Jacks T, Walsh W, Plunkett B, Vogelstein B, Fornace A. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 24.Hwang B J, Ford J, Hanawalt P C, Chu G. Proc Natl Acad Sci USA. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Morris G. Mol Cell Biol. 1999;19:12–20. doi: 10.1128/mcb.19.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fornace A, Dobson P, Kinsella T. Radiat Res. 1986;106:73–77. [PubMed] [Google Scholar]
- 27.Leadon S, Dunn A, Ross C. Radiat Res. 1996;146:123–130. [PubMed] [Google Scholar]
- 28.Hochberg Y, Tamhane A. Multiple Comparisons Procedures. New York: Wiley; 1987. [Google Scholar]
- 29.Westfall P, Young S. Resampling-Based Multiple Testing. New York: Wiley; 1993. [Google Scholar]
- 30.Hsu J. Multiple Comparisons: Theory and Methods. London: Chapman & Hall; 1996. [Google Scholar]
- 31.Benjamini Y, Hochberg Y. J R Stat Soc B. 1995;57:289–300. [Google Scholar]