

Abstract
In our previous publication, a framework for information flow in interaction networks based on random walks with damping was formulated with two fundamental modes: emitting and absorbing. While many other network analysis methods based on random walks or equivalent notions have been developed before and after our earlier work, one can show that they can all be mapped to one of the two modes. In addition to these two fundamental modes, a major strength of our earlier formalism was its accommodation of context-specific directed information flow that yielded plausible and meaningful biological interpretation of protein functions and pathways. However, the directed flow from origins to destinations was induced via a potential function that was heuristic. Here, with a theoretically sound approach called the channel mode, we extend our earlier work for directed information flow. This is achieved by constructing a potential function facilitating a purely probabilistic interpretation of the channel mode. For each network node, the channel mode combines the solutions of emitting and absorbing modes in the same context, producing what we call a channel tensor. The entries of the channel tensor at each node can be interpreted as the amount of flow passing through that node from an origin to a destination. Similarly to our earlier model, the channel mode encompasses damping as a free parameter that controls the locality of information flow. Through examples involving the yeast pheromone response pathway, we illustrate the versatility and stability of our new framework.
Key words: Markov chains, random walks, matrices, proteins, networks, information theory, pathways, channels
1. Introduction
Biological pathways in protein interaction networks have been modeled (Tu et al., 2006; Stojmirović and Yu, 2007; Suthram et al., 2008) as information flows or equivalently random walks between pathway origins and destinations. Ideally, the nodes visited by the flow should suggest a mechanism for the pathway being investigated. For biological specificity of the results, it is important that the flow is directed and localized, that is, the random walks should follow more direct paths from origins to destinations, as opposed to wandering around the whole network. Otherwise, if pathway origins and destinations are distant, many proteins (particularly large network hubs) unrelated to the pathway's biological function may appear as significant. It is therefore necessary to construct a model that is able to controllably pull the information flow towards the pathway destinations.
In an earlier article (Stojmirović and Yu, 2007), we developed a mathematical framework that is capable of directing information flow in interaction networks based on random walks. Via information damping/aging, this framework naturally accommodates information loss/leakage that always occurs in all networks. It requires no prior restriction to the sub-network of interest nor it uses additional (and possibly noisy) information. The framework consisted of two modes absorbing and emitting. Given a set of information sinks, the absorbing mode returns for any network node the likelihood of a random walk starting at that node to terminate at sinks. The emitting mode returns for each network node the expected number of visits to that node by a random walk starting at information sources. The emitting mode can also be used to model biological pathways: given sources and selected destinations (pseudosinks), we introduced heuristic potential functions that adjust the weights of network links to guide the information flow towards pseudosinks (Stojmirović and Yu, 2007).
Although the introduction of potential to direct information flow is novel, the concepts of diffusion and random walks have been extensively used for analysis of protein interaction networks. Nabieva et al. (2005) introduced an algorithm that used truncated diffusion from nodes in interactomes to predict protein function. Tu et al. (2006) used simulations of random walks to infer gene regulatory pathways, while Suthram et al. (2008) modeled the interactome as an electrical network to interpret expression quantitative loci (eQTLs). The latter two approaches are conceptually similar due to the correspondence between random walks on (undirected) graphs and electrical networks (Doyle and Snell, 1984). Missiuro et al. (2009) used the electrical network approach to measure network centrality of each node in several interactomes. Voevodski et al. (2009) proposed a spectral measure of closeness between two proteins based on PageRank to discover functionally related proteins. Most efforts in this direction—for example, the methods proposed by Suthram et al. (2008), Missiuro et al. (2009), and Voevodski et al. (2009)—can be mapped to our absorbing and emitting modes, without potentials (see Section 2.3 for details).
While our earlier model provides very reasonable results on many examples from yeast protein-protein interaction networks (Stojmirović and Yu, 2007), it also has room for improvement. Absent a theory, the potential functions were empirically chosen and the optimal potentials became example-dependent. That is, different potentials might be needed for different networks, sources and pseudosinks. Consequently, the model values (visits) for each node can not be directly interpreted but only in relation to each other. Furthermore, since each choice of the origins and destinations results in a different network graph, rapid computation at large-scale is hindered.
In this sequel, we present a major extension of our previous framework. By appropriately combining the emitting and absorbing modes, we have devised a new, channel, mode that permits directed information flow with probabilistic interpretation. The manuscript is structured as follows. Section 2 presents a succinct review of our previous work and shows how other proposed methods can be mapped to its absorbing or emitting mode. Section 3 details our extension. Section 4 discusses applications of the channel mode to protein interaction networks using the yeast pheromone response pathway as an example. A discussion appears in Section 5, with more technical details provided in the Appendices.
2. Technical Background
2.1. Preliminaries
We will closely follow the notation of Stojmirović and Yu (2007). We represent an interaction network as a weighted directed graph Γ = (V, E, w), where V is a finite set of vertices of size n, E ⊆ V × V is a set of edges, and w is a non-negative real-valued function on V × V that is positive on E, giving the weight of each edge (the weight of non-existing edge is defined to be 0). Assuming an ordering of vertices in V, we represent a real-valued function on V as a state (column) vector 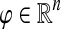 and the connectivity of Γ by the weight matrix W where Wij = w(i, j) (the weight of an edge from i to j). We do not make distinction between a vertex
and the connectivity of Γ by the weight matrix W where Wij = w(i, j) (the weight of an edge from i to j). We do not make distinction between a vertex 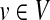 and its corresponding state given by a particular ordering of vertices. Denote by P the n × n matrix such that for all
and its corresponding state given by a particular ordering of vertices. Denote by P the n × n matrix such that for all 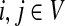 ,
,
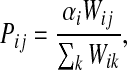 |
(1) |
when 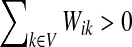 and Pij = 0 otherwise. Here,
and Pij = 0 otherwise. Here, 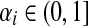 for all i.
for all i.
When αi = 1 for all i, the matrix P is a transition matrix for a random walk or a Markov chain on Γ: for any pair of vertices i and j, Pij gives the transition probability from vertex i to vertex j in one time step. In the general case, the node-specific damping factors αi model dissipation of information: at each step of the random walk there is some probability that the walk leaves the graph. The value αi measures the likelihood for the random walk leaving the vertex i to remain in the graph, or equivalently, the likelihood of dissipation at i is 1 − αi.
For this paper, it will be convenient to express dissipation in terms of the uniform damping coefficient μ, where
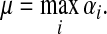 |
(2) |
Let ai = αi/μ and define the matrix Q by P = μQ, that is,
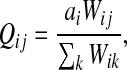 |
(3) |
for 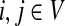 by and 0 < ai ≤ 1. We will consider μ as a free parameter in (0, 1] and the matrix P as dependent on μ.
by and 0 < ai ≤ 1. We will consider μ as a free parameter in (0, 1] and the matrix P as dependent on μ.
2.2. Emitting and absorbing modes
We extract the properties of information flow through a given network by examining the paths of discrete random walks. A random walker starts at an originating node, chosen according to the application domain, and traverses the network, visiting a node at each step. Each walk terminates at an explicit boundary vertex or due to dissipation, which is modeled as reaching an implicit (out-of-network) boundary node.
We distinguish two types of boundary nodes: sources and sinks. Sources emit information, that is, serve as the origins of random walks. All information entering a source from inside the network is dissipated, so a walker is not allowed to visit the source more than once. Sinks absorb information, serving as destinations of walks; information leaving each sink is completely dissipated. The network graph together with a set of boundary nodes and a vector of damping factors α provides the context for the information flow investigated.
The main variable of interest is the (averaged) number of times a vertex is visited by a random walk given the context. Let D denote the set of selected boundary nodes, let T = V\D and let m = |T|. Assuming that the first n − m states correspond to vertices in D, we write the matrix P in the canonical block form:
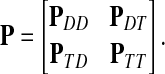 |
(4) |
Here PAB denotes a matrix giving probabilities of moving from the nodes in A to the nodes in B, where A, B stand for either D or T. The states (vertices) belonging to the set T are called transient.
2.2.1. Absorbing mode
Suppose that the boundary set D consists only of sinks. Let F denote an m × (n − m) matrix such that Fij is the total probability that the information originating at 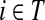 is absorbed at
is absorbed at 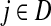 . The matrix F is found by solving the discrete Laplace equation
. The matrix F is found by solving the discrete Laplace equation
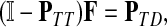 |
(5) |
where  denotes the identity matrix. The matrix
denotes the identity matrix. The matrix 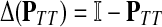 is known as the discrete Laplace operator of the matrix PTT. If
is known as the discrete Laplace operator of the matrix PTT. If 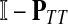 is invertible, Equation (5) has a unique solution
is invertible, Equation (5) has a unique solution
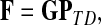 |
(6) |
where 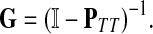
2.2.2. Emitting mode
Now consider the dual problem where D is a set of sources. Let H denote an (n − m) × m matrix such that Hij is the total expected number of times the transient vertex j is visited by a random walk emitted from source i (for all times). Again, H is found by solving the discrete Laplace equation
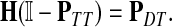 |
(7) |
which, if 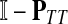 is invertible, has a unique solution
is invertible, has a unique solution
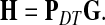 |
(8) |
It is easy to show (Stojmirović and Yu, 2007) that the matrix 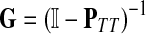 , also known as the Green's function or the fundamental matrix of an absorbing Markov chain (Kemeny and Snell, 1976), exists if every node can be connected to a boundary node or if αi < 1 for all i. The entry Gij represents the mean number of times the random walk reaches vertex
, also known as the Green's function or the fundamental matrix of an absorbing Markov chain (Kemeny and Snell, 1976), exists if every node can be connected to a boundary node or if αi < 1 for all i. The entry Gij represents the mean number of times the random walk reaches vertex 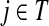 having started in state
having started in state 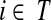 (Kemeny and Snell, 1976). For any transient state i, the value
(Kemeny and Snell, 1976). For any transient state i, the value
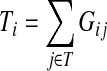 |
(9) |
gives the average length of a path traversed by a random walker starting at i before terminating (Kemeny and Snell, 1976). In this case, the walker is allowed to revisit i after leaving i. In the Markov chain theory, Ti is also known as the average absorption time from i. For the emitting mode, where the walker starts at 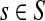 and cannot revisit it, it can be shown that the average path length is
and cannot revisit it, it can be shown that the average path length is
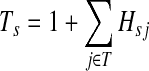 |
(10) |
2.3. Interpretations
If we assume that a random walk deposits a fixed amount of information content each time it visits a node, we can interpret Hij is the overall amount of information content originating from the source s deposited at the transient vertex j. Furthermore, we can interpret Fij as the sum of probabilities (weights) of the paths originating at the vertex 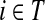 and terminating at the vertex
and terminating at the vertex 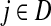 while avoiding all other boundary nodes in the set D, and Hij as the sum of probabilities (weights) of the paths originating at the vertex
while avoiding all other boundary nodes in the set D, and Hij as the sum of probabilities (weights) of the paths originating at the vertex 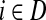 and terminating at the vertex
and terminating at the vertex 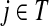 , also avoiding all other nodes in the set D. Each such path has a finite but unbounded length. However, unlike Fij, Hij does not represent a probability because the events of the information being located at j at the times t and t′ are not mutually exclusive (a random walk can be at j at time t and revisit it at time t′). For Fij, the absorbing events at different times are mutually exclusive.
, also avoiding all other nodes in the set D. Each such path has a finite but unbounded length. However, unlike Fij, Hij does not represent a probability because the events of the information being located at j at the times t and t′ are not mutually exclusive (a random walk can be at j at time t and revisit it at time t′). For Fij, the absorbing events at different times are mutually exclusive.
The entry Hij can alternatively be interpreted as equilibrium information content at j for information flow originating from i. In this case we imagine the flow entering the network at node i and leaving the network at i and any other node due to dissipation. The amount of inflow at i is set to 1 and Hij denotes the steady state content at j. Hence, the equilibrium flow rate through an edge (i, j) by the flow entering at 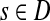 , denoted ψs(i, j), is
, denoted ψs(i, j), is
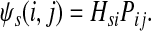 |
(11) |
2.3.1. Electrical networks and heat conduction
A weighted undirected graph Γ = (V, E, w) can be considered as an electrical network with each edge weight (i, j) being associated with resistance Rij =1/Wij. Doyle and Snell (1984) have shown that voltages and currents through nodes and edges can be interpreted in terms of random walks with transition matrix P (where αi = 1 for all 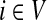 ) and absorbing boundary. Let f denote the voltage vector over all nodes and suppose that a unit voltage is applied between two nodes a and b, so that fa = 1 and fb = 0. Then, the solution for f over T = v\{a, b} according to Kirchhoff's laws is equivalent to the a-th column of the absorbing mode matrix F, that is, fi = Fia. The current flowing through an edge (i, j), which we denote Iij, is then given by
) and absorbing boundary. Let f denote the voltage vector over all nodes and suppose that a unit voltage is applied between two nodes a and b, so that fa = 1 and fb = 0. Then, the solution for f over T = v\{a, b} according to Kirchhoff's laws is equivalent to the a-th column of the absorbing mode matrix F, that is, fi = Fia. The current flowing through an edge (i, j), which we denote Iij, is then given by
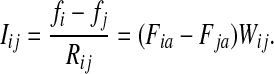 |
(12) |
Therefore, modeling protein interaction networks as resistor networks is equivalent to applying our absorbing mode without dissipation.
However, electrical network paradigm is only applicable to interaction networks where all links can be modeled as undirected edges. This is the case in Missiuro et al., (2009), where the authors only take physical interactions between proteins as links in their networks. On the other hand, the network constructed by Suthram et al. (2008) contained, in addition to physical interactions, the transcription factor-to-gene interactions. These interactions were modeled as directed edges, and Suthram et al. (2008) applied a heuristic approach to model the current flowing through them. In contrast, our absorbing mode can be directly applied to directed networks, although the columns of the matrix F cannot be interpreted as voltages (Fig. 1). We will show in Section 3.5 that, even when edges are directed, F gives rise to potentials.
FIG. 1.

Absorbing mode formalism can be extended beyond resistor networks. Consider, for example, the directed graph shown in (a), where all edges, directed and undirected have weight 1. This graph can be modeled as a resistor network by treating all edges as undirected (b). Applying a unit voltage at node A and grounding at node B leads to the current flowing from A to B. The voltage at each node is indicated by shading (dark means high voltage) while the current at each edge is indicated by the thickness and the direction of the arrow corresponding to that edge. The equivalents of voltage and current can be obtained for the original graph using the absorbing mode with the same boundary (c). Note the qualitative difference between the results in (b) and (c): the node shaped as square conducts significant current in (b) but is totally isolated in (c).
Zhang et al. (2007) applied the same formalism without damping to social networks as a recommendation model. They consider a graph Γ corresponding to a social network, where items under consideration are mapped to nodes, as a heat conduction medium and interpret f as temperature. For each recomendee, by setting his/her favorite items to “high-temperature” and disliked items to “low-temperature” and solving for f over the remaining nodes, they obtain the heat distribution over the entire Γ. The values of f can be used to recommend potential interesting items (other high temperature nodes) to individuals.
2.3.2. Topic-sensitive PageRank
Topic-sensitive PageRank was introduced by Haveliwala (2003) as a context-sensitive algorithm for web search and has been recently applied to protein interaction networks by Voevodski et al. (2009). The PageRank vector P is defined as the unique solution of the equation
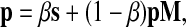 |
(13) |
where M is the transition matrix for a graph (i.e. 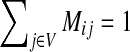 ), 0 < β < 1 and s is a probability vector
), 0 < β < 1 and s is a probability vector 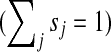 . The vector P is interpreted as the steady state for the random walk governed by M, which at each step has probability β of restarting at a different node. The probability of restarting at the node j is sj. Clearly, P can be written as
. The vector P is interpreted as the steady state for the random walk governed by M, which at each step has probability β of restarting at a different node. The probability of restarting at the node j is sj. Clearly, P can be written as
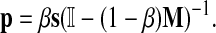 |
(14) |
PageRank can be considered a special case of the emitting mode in the following way. Add an additional vertex v to the graph with no incoming edges and with the weight of each outgoing edge v → i proportional to si. Construct a matrix P using αi = 1 − β for all i in the original graph and αv = β. Let D = {v} be the boundary set. Clearly, (1 − β)M = PTT and βs = PDT, and hence Equation (14) reduces to Equation (7).
2.3.3. Other methods based on random walks
Beyond the analysis of protein interaction networks, approaches based on diffusion and random walks have received attention for a number of applications. We will only mention here a few examples from machine learning to illustrate the point.
A kernel on a space X is a symmetric positive (semi)definite map 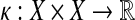 , which can be used to measure similarity between two points in X. A kernel can naturally be treated as an inner product on some feature space. Among other approaches, kernels are the foundation of Support Vector Machines (SVMs), machine learning methods widely used for classification and pattern recognition of data (Schoelkopf and Smola, 2002; Schölkopf et al., 2004).
, which can be used to measure similarity between two points in X. A kernel can naturally be treated as an inner product on some feature space. Among other approaches, kernels are the foundation of Support Vector Machines (SVMs), machine learning methods widely used for classification and pattern recognition of data (Schoelkopf and Smola, 2002; Schölkopf et al., 2004).
A variety of kernels were proposed to compare nodes in undirected graphs (Fouss et al., 2006), mostly derived from discrete Laplacians. Recall that we called the matrix 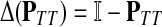 the discrete Laplace operator of the matrix PTT. One can similarly define the matrices
the discrete Laplace operator of the matrix PTT. One can similarly define the matrices 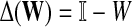 and
and 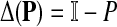 , where W is the adjacency matrix and P is the transition matrix for a weighted undirected graph Γ. Both Δ(W) and Δ(P) were sometimes called the graph Laplacians for Γ.
, where W is the adjacency matrix and P is the transition matrix for a weighted undirected graph Γ. Both Δ(W) and Δ(P) were sometimes called the graph Laplacians for Γ.
Generally, the matrix Δ(W) need not be invertible (in particular, Δ(P) is not invertible [Zhang et al., 2007]). Fouss et al. (2007) proposed using the Moore-Penrose pseudoinverse, which generalizes a matrix inverse to matrices of less than full rank, of Δ(W) as a kernel, with applications to collaborative recommendation. The approach and the application domain of Fouss et al. (2007) are similar to that of Zhang et al. (2007).
The von Neumann diffusion kernel (Schoelkopf and Smola, 2002), proposed by Katz (1953), has the form
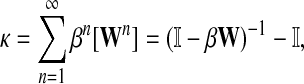 |
(15) |
where β is a damping factor chosen so that 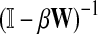 exists. This approach is roughly similar to ours where we compute
exists. This approach is roughly similar to ours where we compute 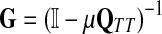 in that both κij and Gij include the sums of the weights for all paths from i to j. The main difference between the two approaches is that the weight of each path of length n included in κ is the product of weights of each link followed, while in our case it is the product of probabilities and therefore has a probabilistic interpretation.
in that both κij and Gij include the sums of the weights for all paths from i to j. The main difference between the two approaches is that the weight of each path of length n included in κ is the product of weights of each link followed, while in our case it is the product of probabilities and therefore has a probabilistic interpretation.
Exponential diffusion kernels, introduced by Kondor and Lafferty (2002), are defined by
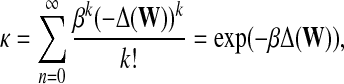 |
(16) |
with a real parameter β. Diffusion kernels can be interpreted to model continuous diffusion through graph, with infinitesimal time steps in contrast to discrete-time diffusion implied by von Neumann diffusion kernel and other similar random-walk based methods. Note that, since every kernel is required to be symmetric, the above formalizations do not extend directly to directed graphs.
3. Theory
Assume V = S ⊔ T ⊔ K, where the set S denotes the sources, K denotes the sinks and T the transient nodes and write the matrix P in the block form as
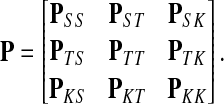 |
(17) |
Let us modify (add context to) the underlying graph Γ so that the random walk can only leave the sources and only enter the sinks. Furthermore, no communication is allowed among sources or among sinks without going through transient nodes. The modified transition matrix, denoted  has the form
has the form
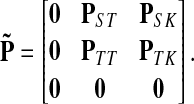 |
(18) |
Treating the vertices in S and T as transient for the absorbing mode in Section 2.2.1, we first derive the matrix F (of size |S ∪ T| × |K|):
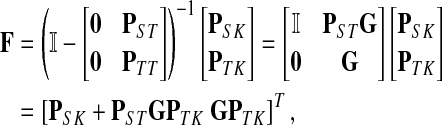 |
where, as before, 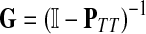 . Similarly, treating the vertices in T and K as transient for the emitting mode in Section 2.2.2, we derive the matrix H (of size |S| × |T ∪ K|):
. Similarly, treating the vertices in T and K as transient for the emitting mode in Section 2.2.2, we derive the matrix H (of size |S| × |T ∪ K|):
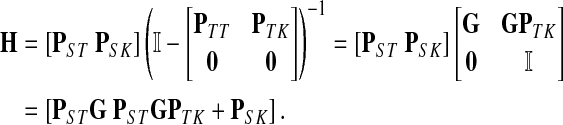 |
The entries of F and H are, as before, interpreted as probabilities of absorption at sinks and average numbers of visits of walks emitted from sources, respectively. Note that the same Green's function, 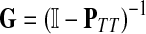 , needs to be computed for both solutions. Also note that the “S” rows of F form the transpose of the “K” columns of H, that is, for all
, needs to be computed for both solutions. Also note that the “S” rows of F form the transpose of the “K” columns of H, that is, for all 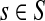 and
and 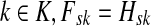 .
.
The matrices F and H can be extended over the whole graph into the matrices 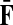 and
and 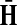 , of sizes n × |K| and |S| × n respectively, by setting
, of sizes n × |K| and |S| × n respectively, by setting 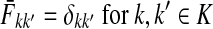 and
and 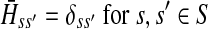 . This is equivalent to setting the K portion of
. This is equivalent to setting the K portion of 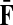 and S portion of
and S portion of 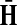 to appropriately sized identity matrices:
to appropriately sized identity matrices:
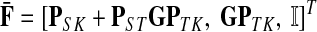 |
(19) |
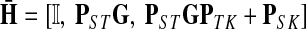 |
(20) |
The matrices 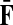 and
and 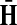 contain explicit boundary conditions with interpretations straightforwardly extended from F and H. Specifically,
contain explicit boundary conditions with interpretations straightforwardly extended from F and H. Specifically, 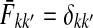 means that a random walk originating from a sink cannot move anywhere else, while
means that a random walk originating from a sink cannot move anywhere else, while 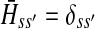 implies that a random walk starting at a source will visit it exactly once and cannot return to it nor to any other source.
implies that a random walk starting at a source will visit it exactly once and cannot return to it nor to any other source.
Remark 3.1. We explicitly assumed that a boundary node can either be a source or a sink. Sometimes, it is desirable to examine flows that both start and terminate at the same point. This case can be reduced to our assumption by introducing for each source that is also a sink an additional node with all the edges of the original node. The new enlarged graph will contain two “logical” nodes for each “physical” source/sink node that plays a dual role and hence it will be possible to have disjoint sets of sources and sinks on the boundary.
3.1. Channel tensor
Define the channel tensor
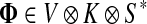 by
by
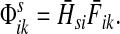 |
(21) |
The entry 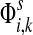 gives the expected number of times a random walk emerging from the source s and terminating at the sink k visits the vertex i (Lemma A.1). In particular,
gives the expected number of times a random walk emerging from the source s and terminating at the sink k visits the vertex i (Lemma A.1). In particular, 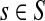 and
and 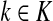 ,
,
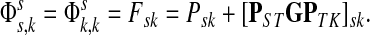 |
(22) |
Hence, the entries of Φ can be interpreted similarly to the entries of 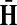 : as expected numbers of visits to nodes in network by random walkers starting at a source node. While
: as expected numbers of visits to nodes in network by random walkers starting at a source node. While 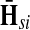 gives the total number of visits to i by a random walker starting at s,
gives the total number of visits to i by a random walker starting at s, 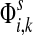 measures only those walkers that ultimately reach the sink k. All other walkers, which either terminate due to dissipation before reaching k, reach other sinks or reach any of the sources, are not considered. Alternatively,
measures only those walkers that ultimately reach the sink k. All other walkers, which either terminate due to dissipation before reaching k, reach other sinks or reach any of the sources, are not considered. Alternatively, 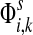 measures the amount of equilibrium flow through the node i by a stream of particles entering through s and leaving from k. The corresponding equilibrium flow through an edge (i, j), denoted ψs,k(i, j) is given by
measures the amount of equilibrium flow through the node i by a stream of particles entering through s and leaving from k. The corresponding equilibrium flow through an edge (i, j), denoted ψs,k(i, j) is given by 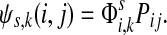 .
.
Suppose s and k are connected through a directed path (equivalently Fsk > 0) and let Tsk denote the expected length of the path traversed by a walker starting at s and terminating at k. Then, it can be shown (Lemma C.1) that,
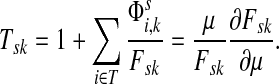 |
(23) |
Other moments and cumulants of the distribution of lengths of paths traversed by walkers starting at s and terminating at k can similarly be expressed in terms of the Green's function G or the derivatives of Fsk with respect to μ (see Appendix C).
3.2. Normalized channel tensor
For brevity we will use a convention that when a set symbol replaces an ordinary index, it means to sum over that entity index of the set in question. For example, for any 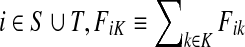 and for all
and for all 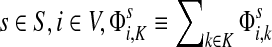 .
.
For 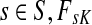 , FsK gives the probability (or expectation) of a random walk emerging from the source s reaching any of the sinks in K. Assuming FsK > 0 for all
, FsK gives the probability (or expectation) of a random walk emerging from the source s reaching any of the sinks in K. Assuming FsK > 0 for all 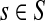 , define the normalized
channel tensor,
, define the normalized
channel tensor, 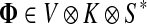 by
by
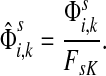 |
(24) |
The normalized channel tensor 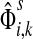 gives the normalized expectation of the number of visits of i by a random walk from s to k. Even though
gives the normalized expectation of the number of visits of i by a random walk from s to k. Even though 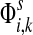 in (21) does not consider any of the random walk paths that return to sources or terminate due to dissipation at transient nodes, the number of visits to any specific node it records is reduced as the dissipation strength increases. The normalization by FsK in (24) takes out the global effect of damping and makes it possible to compare the channel tensors obtained at different dissipation strengths.
in (21) does not consider any of the random walk paths that return to sources or terminate due to dissipation at transient nodes, the number of visits to any specific node it records is reduced as the dissipation strength increases. The normalization by FsK in (24) takes out the global effect of damping and makes it possible to compare the channel tensors obtained at different dissipation strengths.
3.3. Interpretations
Generally, the entries of Φ and 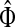 can be interpreted in the same way as the entries of H from the emitting mode. For practical applications, it is sometimes desirable to reduce the amount of available information to a single vector over V, which can be tabulated and graphically visualized using color maps.
can be interpreted in the same way as the entries of H from the emitting mode. For practical applications, it is sometimes desirable to reduce the amount of available information to a single vector over V, which can be tabulated and graphically visualized using color maps.
For a source 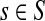 , the source specific content of a node
, the source specific content of a node 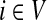 is
is 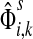 , the (normalized) total number of visits to i by a random walker starting from s and terminating at any of the sinks in K. Equations (22–24) imply that for all
, the (normalized) total number of visits to i by a random walker starting from s and terminating at any of the sinks in K. Equations (22–24) imply that for all 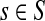 ,
,
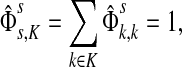 |
(25) |
that is, the entire flow starting at s and reaching one of the sinks is normalized to unity. The total
content vector of 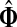 , denoted by
, denoted by 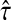 , sums all (normalized) visits for each node:
, sums all (normalized) visits for each node:
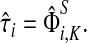 |
(26) |
The concept of destructive interference measures the overlap between visits from different sources for each node. We define the interference vector 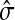 over V by
over V by
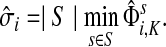 |
(27) |
Hence, 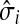 gives the (normalized) total number of times the random walks from all sources co-occur at each node (scaled by the number of sources). The above formulas assume that each source emits the same amount of information. If needed,
gives the (normalized) total number of times the random walks from all sources co-occur at each node (scaled by the number of sources). The above formulas assume that each source emits the same amount of information. If needed, 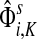 can be weighted by source strength before evaluating total content or interference.
can be weighted by source strength before evaluating total content or interference.
With damping factors less than unity, the random walks from sources to sinks effectively visit a small portion of the entire network. Information Transduction Module (ITM) is a notion that we coined to describe the set of nodes most influenced by the flow. The nodes are ranked using their values for the total content or interference and the most significant nodes are selected. The number of selected nodes depends on the application-specific considerations but we found that the participation ratio
π (Stojmirović and Yu, 2007) of the total content vector 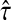 gives a good estimate of the number of nodes whose relative amount of content is significant. It is given by the formula
gives a good estimate of the number of nodes whose relative amount of content is significant. It is given by the formula
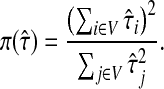 |
(28) |
For undirected graphs, with a context consisting of a single source and a single sink, the values of 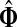 are invariant under interchange of sources and sinks (see Appendix B). In general, however, reversing sources and sinks gives a different result, both due to asymmetry of the weight matrix in directed graphs and because sources and sinks have different roles if more than one of each are present: random walkers originating from different sources can simultaneously visit a transient node while a walk can terminate only at a single sink. Thus, the sinks split the total information flow, that is, compete for it, while sources interfere, either constructively or destructively.
are invariant under interchange of sources and sinks (see Appendix B). In general, however, reversing sources and sinks gives a different result, both due to asymmetry of the weight matrix in directed graphs and because sources and sinks have different roles if more than one of each are present: random walkers originating from different sources can simultaneously visit a transient node while a walk can terminate only at a single sink. Thus, the sinks split the total information flow, that is, compete for it, while sources interfere, either constructively or destructively.
3.4. Path lengths
Damping influences the normalized channel tensor differently from the non-normalized one or the absorbing and emitting solutions. For the non-normalized versions, damping factors control the amount of information reaching the boundary and any intermediate points. In the normalized case, all “normalized” information emitted from the sources reaches sinks (Equation (25)) and damping controls a random walker's average path length, which is always bounded below by the length of the shortest path. Provided each source is connected to at least one sink through a directed path, we have (Corollary C.3)
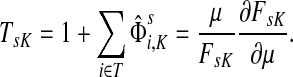 |
(29) |
Small values of μ strongly favor the nodes on the shortest paths, while large values allow random walks to take longer excursions and hence favor the vertices with many connections. As μ ↓ 0, only the nodes at the shortest path receive any flow and TsK → ρ(s, K), the smallest distance between s and any sinks in K. Appendix C contains a more detailed analysis of the role of damping with full statements and proofs.
Note that the μ dependence of TsK allows one to determine the appropriate damping factor for a specified average path length. From the results in Appendix C, it follows that TsK is a smooth function of μ, which is strictly increasing on [0, 1] (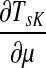 is positive). Therefore, the equation TsK(μ) = x has a unique simple root for ρ(s, K) ≤ x ≤ TsK(1) and any root-finding method can be used to find μ from TsK. When a context contains multiple sources, a desired weighted average of TsK over all
is positive). Therefore, the equation TsK(μ) = x has a unique simple root for ρ(s, K) ≤ x ≤ TsK(1) and any root-finding method can be used to find μ from TsK. When a context contains multiple sources, a desired weighted average of TsK over all 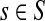 can be set to obtain a global uniform damping factor μ.
can be set to obtain a global uniform damping factor μ.
3.5. Potentials and normalized evolution operators
In Stojmirović and Yu (2007), we used a concept of a potential to redirect the flow towards desired destinations in the emitting mode. To each node 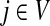 , we associated the value of the total potential Θ(j) such that
, we associated the value of the total potential Θ(j) such that
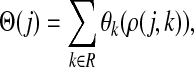 |
(30) |
where R ⊂ T is the set of potential centers, ρ(j, k) is the length of the shortest path from j to k, and θk is an increasing function with a minimum at k. The exponential of the total potential was then used to re-weight the edges incoming to j and form a new matrix 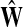 :
:
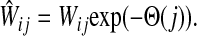 |
(31) |
The matrix 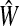 was then normalized to construct the transition matrix to be used (after applying damping) for the emitting mode. It is possible to express the application of the potential Θ as a direct transformation of the transition matrix P (without dissipation included). Let
was then normalized to construct the transition matrix to be used (after applying damping) for the emitting mode. It is possible to express the application of the potential Θ as a direct transformation of the transition matrix P (without dissipation included). Let 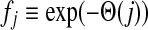 and let
and let 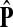 denote the new transition matrix derived from
denote the new transition matrix derived from 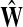 . Then,
. Then, 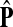 can be written as
can be written as
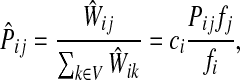 |
(32) |
where
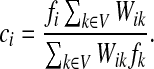 |
(33) |
If ci = 1 for all i, we can express 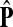 as a similarity transformation of P, where
as a similarity transformation of P, where 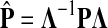 , where Λij = δijfi. In general, this is not the case with the heuristic potentials proposed in Stojmirović and Yu (2007). However, we will now show (with proofs in Appendix D) that there exist a potential derived from the matrix F that transforms the context specific matrix
, where Λij = δijfi. In general, this is not the case with the heuristic potentials proposed in Stojmirović and Yu (2007). However, we will now show (with proofs in Appendix D) that there exist a potential derived from the matrix F that transforms the context specific matrix 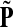 into a stochastic transition matrix over the source and transient nodes. The solution of the emitting mode using the new matrix recovers the normalized channel tensor
into a stochastic transition matrix over the source and transient nodes. The solution of the emitting mode using the new matrix recovers the normalized channel tensor 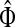 and allows for additional generalizations.
and allows for additional generalizations.
Let 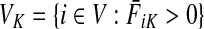 be the set of all nodes in V that are connected with any sink in K by a directed path and denote by SK and TK the sets S ∩ VK and T ∩ VK, respectively. Suppose 0 ≤ μ ≤ 1. For
be the set of all nodes in V that are connected with any sink in K by a directed path and denote by SK and TK the sets S ∩ VK and T ∩ VK, respectively. Suppose 0 ≤ μ ≤ 1. For 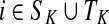 , let
, let
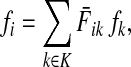 |
(34) |
where fk > 0 are arbitrary for 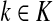 . For
. For 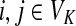 , define
, define
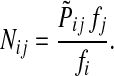 |
(35) |
Since all transient nodes are assumed to be connected to a sink, the matrix N is well defined for 0 < μ ≤ 1. It can be shown using parts of Appendix C.2 that it is also well defined in the limit as μ ↓ 0. Clearly, Nkj = 0 for all 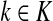 and
and  . Over SK ∪ TK, the matrix N is stochastic (Proposition D.1), that is
. Over SK ∪ TK, the matrix N is stochastic (Proposition D.1), that is  . Hence, N is an evolution operator for flow entering at sources and terminating exclusively at a point in K. The dependence on μ is built in the transition probabilities Nij. Furthermore, Equation (34) is the only way to construct a function over VK so that (35) gives a stochastic transition matrix (Proposition D.1).
. Hence, N is an evolution operator for flow entering at sources and terminating exclusively at a point in K. The dependence on μ is built in the transition probabilities Nij. Furthermore, Equation (34) is the only way to construct a function over VK so that (35) gives a stochastic transition matrix (Proposition D.1).
Denote by G(N), 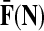 ,
, 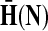 , Φ(N) the quantities corresponding to G, F, H and Φ respectively, when the matrix
, Φ(N) the quantities corresponding to G, F, H and Φ respectively, when the matrix 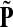 is replaced by the transition matrix N. Since transformation (35) is a similarity transformation from
is replaced by the transition matrix N. Since transformation (35) is a similarity transformation from 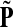 to N, it is easy to establish the following:
to N, it is easy to establish the following:
Proposition 3.2
The following identities hold:
(i) For all i,
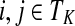 , [G(N)]ij = Gijfj/fi,
, [G(N)]ij = Gijfj/fi,(ii) For all
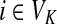 and
and
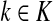 ,
, 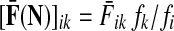 ,
,(iii) For all
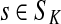 and
and
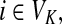
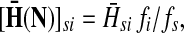
(iv) For all
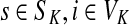 and
and
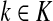 ,
, 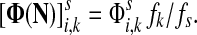
The special case where fk's are equal for all 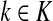 results in
results in 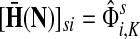 and
and 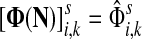 . Hence, N in this case can be considered a “natural” transition operator for random walks or Markov chains that start at sources S and terminate at a point in K. The time evolution of such processes can be followed by raising N to appropriate powers. As demonstrated in the previous sections, the parameter μ, which is implicit in N, controls the how fast the random walkers move towards their destinations. Figure 2 shows a graphical example of the transformation of the operator
. Hence, N in this case can be considered a “natural” transition operator for random walks or Markov chains that start at sources S and terminate at a point in K. The time evolution of such processes can be followed by raising N to appropriate powers. As demonstrated in the previous sections, the parameter μ, which is implicit in N, controls the how fast the random walkers move towards their destinations. Figure 2 shows a graphical example of the transformation of the operator  into N, which directs the flow towards the sink.
into N, which directs the flow towards the sink.
FIG. 2.

Transformation of the evolution operator using potentials. (a) Directed graph from Figure 1 with transition probabilities indicated by edge arrows. Nodes are shaded according to the potential associated with the sink (octagon). (b) Normalized transition operator N resulting from the application of the sink potential to the context specific transition matrix (the single source is indicated as hexagon). (c) Values of the normalized channel tensor as shades and the directional flow through each edge as arrows. Comparison between (b) and (c) shows that edges with large transition probabilities need not carry significant flows.
In general, each value fk represents the sink strength of the sink 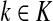 . Equal sink strengths imply no prior preference for any sink while in the case of unequal sink strengths the flow from sources towards sinks is preferentially pulled towards sinks with larger strength. It is also possible to exclude some sinks from consideration by setting their strength to 0. Since the scaling of fk's does not affect the transition matrix, they can be considered as probabilities over K and, in the Bayesian framework, as priors. Indeed, the equation
. Equal sink strengths imply no prior preference for any sink while in the case of unequal sink strengths the flow from sources towards sinks is preferentially pulled towards sinks with larger strength. It is also possible to exclude some sinks from consideration by setting their strength to 0. Since the scaling of fk's does not affect the transition matrix, they can be considered as probabilities over K and, in the Bayesian framework, as priors. Indeed, the equation
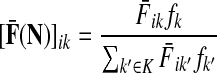 |
(36) |
can be easily recognized as Bayes' formula for posterior likelihood. Here 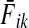 can be interpreted as the likelihood of a random walk from i being absorbed at sink k, given that k is absorbing; fk is the prior probability that k is absorbing; while
can be interpreted as the likelihood of a random walk from i being absorbed at sink k, given that k is absorbing; fk is the prior probability that k is absorbing; while  is the likelihood that a walker starting at i is absorbed at k, given that it is absorbed at any of the “active” sinks (i.e., sinks with fk > 0). This suggests a use of the absorbing and channel modes as Bayesian inference frameworks for forming and testing hypotheses. For example, sinks can be associated with mutually exclusive hypotheses. The likelihood of each source being associated with a hypothesis can then be evaluated using (36).
is the likelihood that a walker starting at i is absorbed at k, given that it is absorbed at any of the “active” sinks (i.e., sinks with fk > 0). This suggests a use of the absorbing and channel modes as Bayesian inference frameworks for forming and testing hypotheses. For example, sinks can be associated with mutually exclusive hypotheses. The likelihood of each source being associated with a hypothesis can then be evaluated using (36).
The matrix N can also be expressed in terms of potentials. Suppose fk > 0 for each 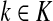 and set the potential of each node
and set the potential of each node 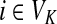 by
by
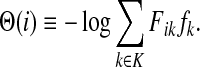 |
(37) |
Then, N can be written as
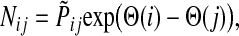 |
(38) |
with the straightforward interpretation of the information flow moving from high- to low- potential nodes. Unlike our earlier potential (31), which was totally heuristic, this new potential is theoretically founded.
4. Applications to Cellular Networks
In the recent years, development of high-throughput genomic and proteomic techniques resulted in proteome-wide interaction networks (interactomes) in a number of model organisms (Ito et al., 2001; Uetz et al., 2000; Giot et al., 2003; Li et al., 2004; Stelzl et al., 2005; Rual et al., 2005; Ptacek et al., 2005). Databases such as the BioGRID (Breitkreutz et al., 2008), IntAct (Kerrien et al., 2007), DIP (Salwinski et al., 2004), and MINT (Chatr-Aryamontri et al., 2007) have been established to collect and curate sets of interactions from different experiments and make them publicly available. Most databases contain physical binding interactions, while the BioGRID additionally includes genetic interactions (such as synthetic lethality) and biochemical interactions, which describe a biochemical effect of one protein upon another.
A protein (or a protein state) is mapped to a node in a cellular protein network. Hence, the solution of a channel mode context (as tensors Φ and 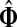 ) will highlight an ITM consisting of the proteins most visited by a directed flow from sources to sinks, that is, the proteins lying on the most likely paths connecting sources and sinks. Clearly, biological interpretations of the model results will depend on the nature of interactions ascribed for links within the network graphs: the interpretation for an ITM from a genetic or functional network and that for an ITM from a physical network should be different. Here, we will mainly focus on the physical networks where interactions correspond to binding between two proteins (undirected) or a post-translational modification of one protein by another (directed). Each step of a random walk in such a network is equivalent to a physical event and dissipation naturally corresponds to protein degradation by a protease and negative feedback mechanisms that limit transmission of information. It is thus plausible that the information channels obtained by solving the channel mode with suitable sources and sinks may correspond to (portions of) actual signaling or gene regulation pathways. However, it is important to note that the biological validity of a network path is contingent upon the transitivity of biochemical effect along that path as not all protein-protein interactions have the same downstream effect. Also, even in the best case, the information obtained from a random walk models would be primarily qualitative since cellular processes in general do not correspond to linear models.
) will highlight an ITM consisting of the proteins most visited by a directed flow from sources to sinks, that is, the proteins lying on the most likely paths connecting sources and sinks. Clearly, biological interpretations of the model results will depend on the nature of interactions ascribed for links within the network graphs: the interpretation for an ITM from a genetic or functional network and that for an ITM from a physical network should be different. Here, we will mainly focus on the physical networks where interactions correspond to binding between two proteins (undirected) or a post-translational modification of one protein by another (directed). Each step of a random walk in such a network is equivalent to a physical event and dissipation naturally corresponds to protein degradation by a protease and negative feedback mechanisms that limit transmission of information. It is thus plausible that the information channels obtained by solving the channel mode with suitable sources and sinks may correspond to (portions of) actual signaling or gene regulation pathways. However, it is important to note that the biological validity of a network path is contingent upon the transitivity of biochemical effect along that path as not all protein-protein interactions have the same downstream effect. Also, even in the best case, the information obtained from a random walk models would be primarily qualitative since cellular processes in general do not correspond to linear models.
The simplest way to use the channel mode is for knowledge retrieval by exploring large networks. In many model organisms, it is possible to construct physical protein interaction networks that integrate proteome-wide data collected from results of multiple experiments from different sources using a variety of techniques. All three modes discussed in this paper, emitting, absorbing and channel, can be used to explore network neighborhoods of proteins of interest and learn more about their function(s). In particular, given two proteins, one set as a source and the other as a sink, one may use the channel mode to extract a sub-network containing only the proteins most relevant to the possible functional relation between them. By using graphical tools to visualize the sub-network and by examining the annotations for the individual proteins within it, one can learn about their role within the cell and hence understand the cellular context of the query proteins.
More complex settings of the channel mode can be used for hypothesis forming and confirmation. For example, using destructive interference between flows from multiple sources may reveal the points of crosstalk between different biological pathways that can be selected for further experimental investigation. Given one or more proteins of interest one can explore the hypothesis about their function by using the property that sinks split the flow. Set these proteins of interest as sources and set several sinks, each associated with an a different biological role. After running a channel mode, the sinks attracting most of the flow would point to the most likely cellular role of the proteins, given all alternatives. Of course, if all alternatives are biologically invalid, no valid functional inference can be made.
Since it is possible to arbitrarily specify sources and sinks and obtain model results that may not correspond to any cellular role, it is desirable to be able to check whether retrieved ITMs can be associated with any existing annotation. A common way to do so is through enrichment analysis (Huang et al., 2009), which assigns terms from a controlled vocabulary such as Gene Ontology (Ashburner et al., 2000) or KEGG (Kanehisa et al., 2010) to a set of genes or proteins with weights. Each term from a controlled vocabulary annotates one or more proteins and enrichment analysis aims to retrieve, by statistical inference, those terms that best describe the set of submitted proteins with weights. While many enrichment tools were developed for analysis of microarrays (Huang et al., 2009), we found that none of them are entirely suitable for analyzing the results of our model. We have therefore developed a novel tool, called SaddleSum (Stojmirović and Yu, 2010), which is based on asymptotic approximation of tail probabilities (Lugannani and Rice, 1980). For each term, it computes the probability that a score greater than or equal to the sum of weights, for all the proteins associated with that term, can arise by chance. In the context of the channel mode, the quantities that can serve as input to SaddleSum are source specific content, total content, and destructive interference.
4.1. Example: yeast pheromone pathway
As an illustration, we will consider the mating pheromone response pathway in Saccharomyces cerevisiae, the one of the best understood signalling pathways in eukaryotes (Bardwell, 2005). The mating signal is transferred from a membrane receptor to a transcription factor in nucleus, leading to transcription of mating genes. We will only provide a very brief overview of the pathway necessary for discussing our examples; more details are available in the review by Bardwell (2005).
A mating pheromone binds the transmembrane G-protein coupled pheromone receptors Ste2p/Ste3p. This leads to dissociation of Ste4p and Ste18p, the membrane bound subunits of the G-protein complex, which also contains the subunit Gpa1p. Ste4p then binds to the protein kinase Ste20p, which is recruited to the membrane through Cdc42p, and the scaffold protein Ste5p. On the scaffold, a MAPK (mitogen activated protein kinase) cascade occurs, where each protein kinase in the cascade is activated by being phosphorylated by the previous kinase and in turn activates the next protein. In this case, the cascade goes Ste20p → Ste11p → Ste7p → Fus3p or Kss1p. The final activated kinase Fus3p or Kss1p then migrates to the nucleus where it phosphorylates the proteins Dig1p and Dig2p, the repressors of the Ste12p transcription factor activity. The Ste12p transcription factor can then, in coordination with other transcription factors such as Tec1p, promote the transcription of the mating genes.
As a basis for the underlying network, we used all physical yeast protein-protein interactions from the BioGRID-3.0.65 (Breitkreutz et al., 2008). To improve the fidelity of the network, we removed every interaction reported by a single publication unless that publication described a low-throughput experiment, which we assumed to be more targeted. We considered an experiment low-throughput if it reported fewer than 300 interactions in total. We also removed all interactions labelled with the “Affinity Capture-RNA” experimental system since they were not protein-to-protein. The physical binding interactions were given a weight 1 in both directions while the interactions labeled as “Biochemical Activity” were interpreted as directional (bait → prey) and given a weight of 2. In cases where both physical and biochemical interactions were reported, only biochemical were considered. Since it is known (Steffen et al., 2002) that proteins with a large number of non-specific interaction partners might overtake the true signaling proteins in the information flow modeling, we excluded a set of 165 nodes corresponding to cytoskeleton proteins, histones and chaperones. We found that the excluded nodes do not strongly affect the results of the particular examples presented here. For each example, we computed the normalized channel tensor summed over all sinks, that is 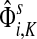 in our notation.
in our notation.
Figure 3 focuses solely on the MAPK cascade portion of the pheromone pathway, with Ste20p as a single source and Ste12p as a single sink. Selection of top proteins by participation ratio captures all important participants of the cascade but emphasizes a “shortcut” through Slt2p, which is a MAP kinase involved in a different signalling pathway. Upon examination of the reference (Zarzov et al., 1996) used by the BioGRID to support the Ste20p → Slt2p link, we discovered that it does not anywhere claim existence of such interaction. Hence, we removed Slt2p from our network for all subsequent queries and reran the query. In addition to the true pathway, the second ITM emphasized a path through Nup53p (a nuclear core protein). We examined the publication (Lusk et al., 2007) indicated by the BioGRID to support the Ste20p → Nup53p link and found that while it is true that Ste20p phosphorylates Nup53p in vitro, another kinase was mainly responsible for its phosphorylation in vivo. We therefore felt justified to exclude Nup53p as well. The final ITM resulting from the same query with Slt2p and Nup53p excluded in addition to the 165 proteins mentioned before is shown in Figure 3. Enrichment analysis using the GO database (Fig. 5, column D) gives “receptor signaling protein serine/threonine kinase activity” as a top term under “Molecular Function” and “filamentous growth” as a top term under “Biological Process.” Hence, the final ITM agrees well with the canonical understanding of the MAPK cascade.
FIG. 3.

ITMs for the MAPK cascade part of the yeast pheromone response obtained by running the normalized channel mode with Ste20p as the source and Ste12p as the sink (μ = 0.85). In addition to the “standard” excluded nodes (histones, chaperones, cytoskeleton), we also removed the nodes for Slt2p and Nup53p as discussed in the main text. Gray shading of each node indicates its total content (darker nodes represent more visits). The number of nodes shown is determined by the participation ratio.
FIG. 5.

Gene Ontology term enrichment analysis of examples from Figures 3 and 4 using SaddleSum. The most significant GO terms from the Biological Process and Molecular Function categories are shown on the left (number of annotated proteins is in brackets), with their E-values indicated by shading of the squares on the right. Each column corresponds to a single example: (A) Fig. 4c (μ = 1). (B) Fig. 4b (μ = 0.85); (C) Fig. 4d (μ = 0.55). (D) Fig. 3 (μ = 0.85). The input weights for columns A–C were obtained from the interference values at all non-excluded nodes except sources and sinks, while total content was used for column D. E-values larger than the cutoff of 0.01 are shown as white squares.
To obtain an ITM best describing the entire pheromone response pathway, it is necessary to include two sources, the receptor Ste2p and the membrane-bound protein Cdc42p (Fig. 4). Including only Ste2p is not sufficient because of the shortcut through the link Gpa1p → Fus3p, which avoids the MAPK cascade. Furthermore, inclusion of Cdc42p is biologically sensible because Cdc42p activates Ste20p (Bardwell, 2005) and is hence necessary for the MAPK cascade. Since the information flows from Ste2p and Cdc42p to Ste12p share some but definitely not all paths in common (Fig. 4a), interference between them (Fig. 4b), rather than total visits, is most appropriate to highlight the most important proteins in the signalling pathway.
FIG. 4.

Yeast pheromone response ITMs obtained by running the normalized channel mode with Ste2p and Cdc42p as the sources and Ste12p as the sink with damping factors μ = 0.85 (a, b), μ = 1 (c), and μ = 0.55 (d). Part (a) shows flow intensity from each source using a separate base color, while (b–c) show interference (darker nodes indicate stronger interference). All images show the top 30 nodes in terms of the total content for the case of μ = 0.85.
Figure 4b–d illustrates the effect of changing the damping factor μ. With μ = 1 (Fig. 4c), the flows from sources visit a much larger portion of the network (the average path length 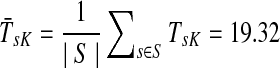 ) than with μ = 0.85 (Fig. 4b,
) than with μ = 0.85 (Fig. 4b, 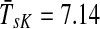 ) or μ = 0.55 (Fig. 4d,
) or μ = 0.55 (Fig. 4d, 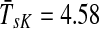 ). The lower bound on path length is 3, the shortest distance from both sources to Ste12p. In terms of enrichment analysis with GO (Fig. 5, columns A–C), all three cases pick as significant the terms related to cell growth but with different statistical significance. In addition, both the μ = 0.85 and μ = 1 cases can be associated with terms related to MAP kinase and signal transduction. Hence, results for large μ tend to give lower GO term E-values but with lower specificity while results for small μ give very specific results but with less significant E-values. The μ-dependence of E-values for any given term is not surprising since different μs correspond to different null models. Based on the images in Figure 4, the enrichment results as well as our experience in other model contexts, the values of μ around 0.85, corresponding to a random walk visiting about four more nodes than the minimum necessary to reach the sink, appear to give the best results in emphasizing biologically relevant channels.
). The lower bound on path length is 3, the shortest distance from both sources to Ste12p. In terms of enrichment analysis with GO (Fig. 5, columns A–C), all three cases pick as significant the terms related to cell growth but with different statistical significance. In addition, both the μ = 0.85 and μ = 1 cases can be associated with terms related to MAP kinase and signal transduction. Hence, results for large μ tend to give lower GO term E-values but with lower specificity while results for small μ give very specific results but with less significant E-values. The μ-dependence of E-values for any given term is not surprising since different μs correspond to different null models. Based on the images in Figure 4, the enrichment results as well as our experience in other model contexts, the values of μ around 0.85, corresponding to a random walk visiting about four more nodes than the minimum necessary to reach the sink, appear to give the best results in emphasizing biologically relevant channels.
5. Discussion
We have described the channel mode for modeling context-specific information flow in interaction networks. It supports discovery of the most likely channels through networks between user-specified origins (sources) and destinations (sinks) of information. The transition operator N, constructed by applying potentials centered on sinks to the original transition operator, fully describes the dynamics of the flow within the channels. The mathematical formulation of the channel mode is flexible and can be easily modified for related cases. For example, it is possible to model the flow through a sequence of “checkpoints” by using destination from one context as the origin for another.
Unlike other models based on random walks and/or electrical networks proposed in the literature (Tu et al., 2006; Suthram et al., 2008; Missiuro et al., 2009; Voevodski et al., 2009) that can be reduced to either emitting or absorbing modes, our channel mode allows for “directed” information flow. Furthermore, it can readily accommodate networks containing directed links and multiple sources and sinks. Most importantly, like our original framework (absorbing and emitting modes), the channel mode uses damping to retain the information flow in the portion of the network most relevant to the specified context and prevent visits to distant nodes. Damping is controlled by a free parameter μ (or more generally, node specific parameters αi), which in the case of the channel mode controls the amount of path deviation from the shortest one. In statistical physics terms, this is equivalent to using fugacity to control the number of particles of the system. Computation of the model solution requires only a solution to a (sparse) system of linear equations, without needing to simulate random walks as was done in Tu et al. (2006). If computation of multiple contexts with the same damping coefficients is required, it is possible, using well known results from linear algebra (Press et al., 2007), to re-use the Green's function for one context to efficiently compute the Green's function for another.
Applied to physical protein interaction networks, the channel mode can be used as a framework for knowledge retrieval through network exploration and hypothesis formation and confirmation. The node weights obtained can be interpreted directly as well as submitted to an enrichment tool for further analysis. Note however that, in many cases, the annotation of a protein by a term is directly tied to publications reporting its physical interactions.
As illustrated by our pheromone pathway example, the results of our model are sensitive to “shortcuts” between biologically unrelated protein nodes. Therefore, to obtain valid conclusions from the ITMs retrieved, the underlying interaction network must be constructed from high-quality data relevant to the biological context under investigation. The nodes with many non-specific interactions, as well as links that may not represent actual in vivo interactions under the query context, should be removed from the network. The damping factor μ also needs to be selected appropriately for the biological context investigated, depending on whether the coverage (high μ) or the selectivity (low μ) of the channel are desired more. The results of enrichment analysis for the example shown in Figure 4 indicate that at least some interpretations are robust to the change of μ.
We have already deployed a software implementation of our framework, called ITM Probe, to the web for the use of biomedical researchers (Stojmirović and Yu, 2009). In future, we plan to extend our information flow framework to a platform for network-based context-specific qualitative analysis of cellular process. The improved models will take into account additional biological information, such as protein complex memberships, post-translational modification states and abundances, possibly leading to non-linear transition operators. Generally, while wishing to improve accuracy by incorporating more specific information, we aim to preserve the simplicity of the present framework.
6. Appendix
A. Channel tensor as expectation
Lemma A.1
Let
 be a random variable denoting the total number of times a random walk
starting at
be a random variable denoting the total number of times a random walk
starting at
 and absorbed at
and absorbed at
 visits
visits
 . Then,
. Then,
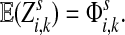 |
(39) |
Proof
Consider a path 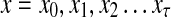 from
from 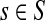 to
to 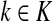 of total length τ where
of total length τ where 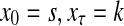 and
and 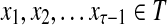 . The total weight or probability associated with x is
. The total weight or probability associated with x is 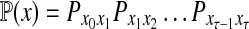 . For any
. For any 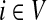 , let Xi(x, t) = 1 if xt = i and 0 otherwise. Then, the total number of times x visits i is
, let Xi(x, t) = 1 if xt = i and 0 otherwise. Then, the total number of times x visits i is 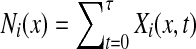 and
and
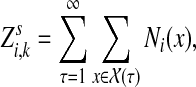 |
where  denotes the set of all paths from s to k of length τ. Therefore,
denotes the set of all paths from s to k of length τ. Therefore,
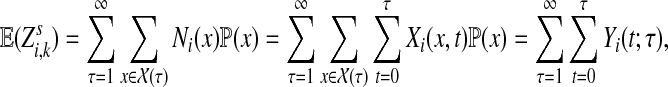 |
(40) |
where  . There are three cases to consider depending on whether i is a source, a sink or a transient node.
. There are three cases to consider depending on whether i is a source, a sink or a transient node.
If i is a source, a path from s can visit i only if i = s and t = 0. Therefore, Xi(x, t) = δsiδt0 and hence
 |
(41) |
Here  is exactly the total weight of paths of length τ − 2 that start at
is exactly the total weight of paths of length τ − 2 that start at  , visit nodes in T and terminate at
, visit nodes in T and terminate at 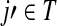 . Hence,
. Hence,
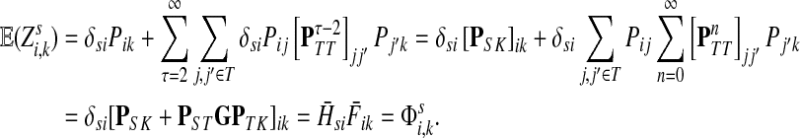 |
(42) |
Similarly, if i is a sink, a walker from s can visit i and terminate at k only if i = k and 0 < t = τ. Thus, Xi(x, t) = δikδtτ and
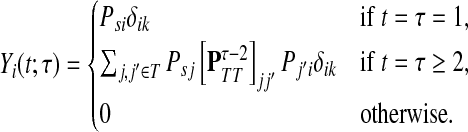 |
(43) |
Therefore,
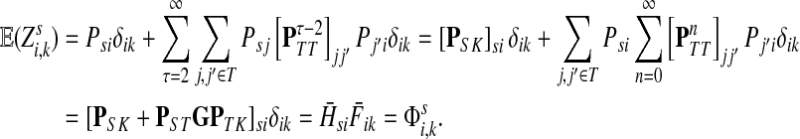 |
(44) |
Finally, suppose  . In order to visit i at time t and terminate at k at time τ, a path in
. In order to visit i at time t and terminate at k at time τ, a path in  must take one step to reach T, spend there t − 1 steps before arriving at i, then take another τ − t − 1 steps in T and an additional step to terminate at k. Considering all possible paths that visit i at time t, we have
must take one step to reach T, spend there t − 1 steps before arriving at i, then take another τ − t − 1 steps in T and an additional step to terminate at k. Considering all possible paths that visit i at time t, we have
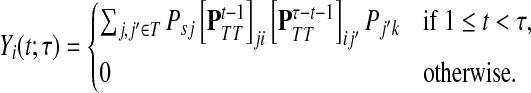 |
(45) |
It follows that
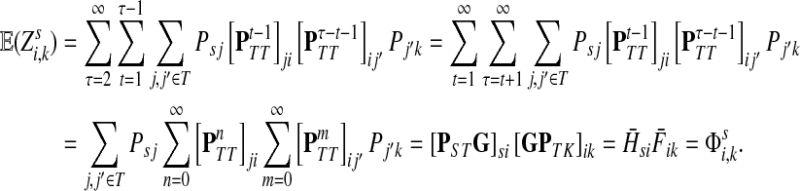 |
▪
B. Reversibility of sources and sinks
It is easy to see that in general, reversing sources and sinks produces different values for the normalized channel tensor. One important exception, however, is the case when the underlying graph is undirected and there is a single source and a single sink.
Lemma B.1
Let Γ = (V, E, w) be an undirected weighted graph with a weight matrix
W
and
transition matrix
P
as defined in (1), with
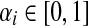 for all
for all
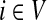 . Suppose Γ is connected and let
. Suppose Γ is connected and let
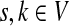 . Denote by
. Denote by
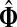 the normalized channel tensor over Γ with s as a single source and k as a
single sink, and denote by
the normalized channel tensor over Γ with s as a single source and k as a
single sink, and denote by
 the normalized channel tensor over Γ with k as a single source and s
as a single sink. Then, for all
the normalized channel tensor over Γ with k as a single source and s
as a single sink. Then, for all
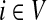 ,
,
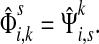 |
(46) |
Proof
Since Γ is an undirected graph, it satisfies the detailed balance equation πyPxy = Pyxπx for all 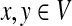 , where
, where 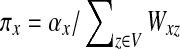 . It directly follows that
. It directly follows that
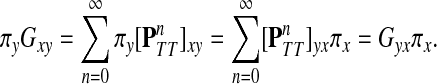 |
(47) |
For i = s or i = k, one can immediately see that  . Observing that the transient state is the same for both
. Observing that the transient state is the same for both  and
and 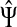 , we have for each
, we have for each  ,
,
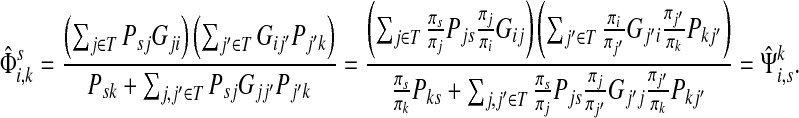 |
▪
C. The role of the damping factor in the channel mode
Recall that P = μQ, where 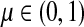 is the uniform damping factor and Q is given in (3). For this range of μ, the Green's function
is the uniform damping factor and Q is given in (3). For this range of μ, the Green's function 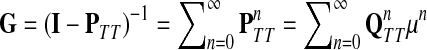 is well-defined (see (Stojmirović and Yu, 2007), Proposition 2.2) and hence the solution matrices
is well-defined (see (Stojmirović and Yu, 2007), Proposition 2.2) and hence the solution matrices 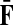 and
and  from Equations (19–20) are well defined and continuous as functions of μ. As μ ↓ 0, all the damping factors in α uniformly tend to 0 and P → 0. However, we will show in C.2 that the normalized channel tensor is well-defined in the limit as μ ↓ 0 (provided it is well defined for other values of μ).
from Equations (19–20) are well defined and continuous as functions of μ. As μ ↓ 0, all the damping factors in α uniformly tend to 0 and P → 0. However, we will show in C.2 that the normalized channel tensor is well-defined in the limit as μ ↓ 0 (provided it is well defined for other values of μ).
At the other extreme, as μ ↑ 1 and P → Q, the Green's function may not exist for every choice of boundary nodes, since the spectral radius of QTT may be equal to 1. If the vertex set is restricted to V (K), the set of all nodes connected through a directed path to at least one sink, then by Proposition 2.1 of (Stojmirović and Yu, 2007), the Green's function is well-defined for μ = 1 as well. Also note that for a channel tensor Φ to be non-trivial (i.e. non-zero everywhere), it is also necessary that each source is connected to at least one sink through a directed path, or equivalently, that FsK > 0 for all  .
.
C.1. Path lengths
The damping parameter μ controls the distribution of lengths of the paths (or the times) a random walk emitted from a source takes before being absorbed at a sink.
For nodes 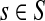 and
and 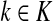 , let Lsk (more precisely, Lsk(μ)) denote the random variable giving the length of the path (or a number of steps) taken by a random walk originating at s and terminating at k. At least one such path from s to k exists if and only if Fsk > 0. The underlying probability density
, let Lsk (more precisely, Lsk(μ)) denote the random variable giving the length of the path (or a number of steps) taken by a random walk originating at s and terminating at k. At least one such path from s to k exists if and only if Fsk > 0. The underlying probability density 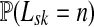 is given by
is given by
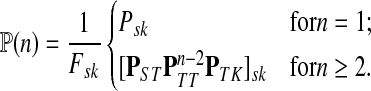 |
(48) |
Let 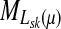 denote the moment generating function for Lsk and let
denote the moment generating function for Lsk and let 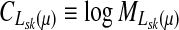 denote its cumulant generating function. Let us write Fsk as a function of μ:
denote its cumulant generating function. Let us write Fsk as a function of μ:
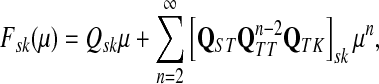 |
(49) |
and observe that
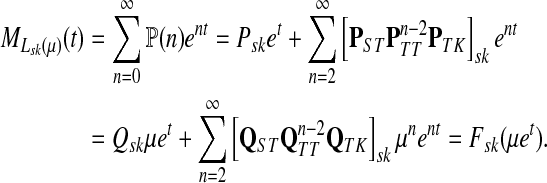 |
(50) |
Thus, all moments and cumulants of Lsk can be expressed in terms of the Green's function G and its related quantities F, H and Φ, both directly and in terms of derivatives of their entires with respect to μ. In particular,
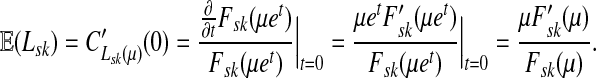 |
(51) |
Using the easily provable identity 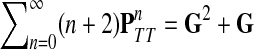 , we have
, we have
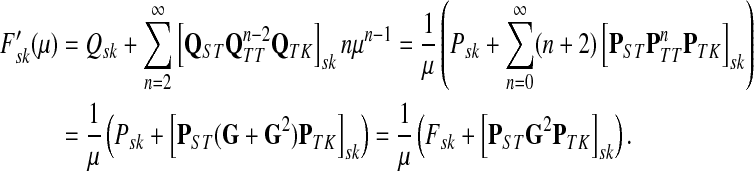 |
(52) |
Therefore, by (51),
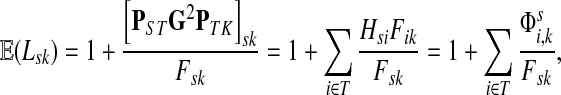 |
(53) |
and we obtain the following
Lemma C.1
Let
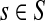 , let
, let
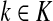 and let
and let
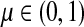 . Suppose Fsk > 0. Then,
. Suppose Fsk > 0. Then,
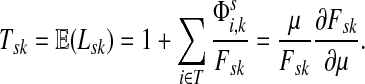 |
(54) |
Similarly,
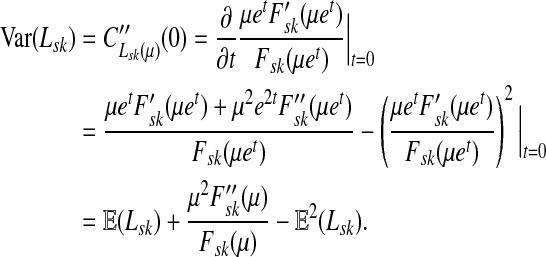 |
(55) |
Using another easily provable identity 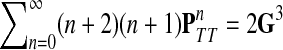 , and Equation (52), we have
, and Equation (52), we have
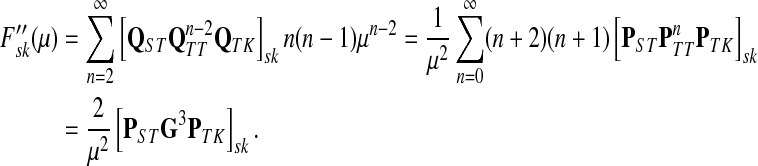 |
(56) |
Hence, we obtain
Lemma C.2
Let
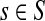 , let
, let
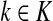 and let
and let
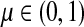 . Suppose Fsk > 0. Then,
. Suppose Fsk > 0. Then,
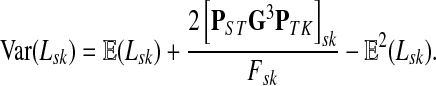 |
(57) |
Denote by LsK the random variable giving the length of the path (or the number of steps) taken by a random walk originating at s and terminating at any sink in K. This random variable is well-defined provided s is connected with at least one 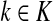 through a directed path, or equivalently, if
through a directed path, or equivalently, if 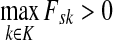 . Let
. Let 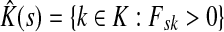 Then, LsK can be expressed as a weighted sum of Lsk over
Then, LsK can be expressed as a weighted sum of Lsk over 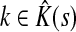 :
:
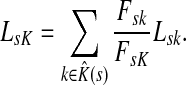 |
(58) |
Here Fsk/FsK gives the conditional probability of a random walker from s reaching sink k, given that it reaches any of the sinks in 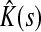 . Through properties of mean, this leads to the following corollary.
. Through properties of mean, this leads to the following corollary.
Corollary C.3
Let
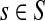 and let
and let
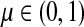 . Suppose
. Suppose
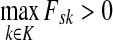 . Then,
. Then,
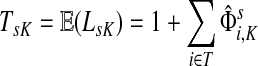 |
(59) |
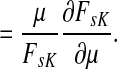 |
(60) |
Since  and
and  can be expressed in terms of sums and products of entries of G, they are continuous and increasing functions of
can be expressed in terms of sums and products of entries of G, they are continuous and increasing functions of 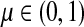 . The value of
. The value of 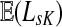 is bounded from below by the length of the shortest path from the source to any of the sinks. If the graph nodes are restricted to V (K), G is well-defined for μ = 1 and
is bounded from below by the length of the shortest path from the source to any of the sinks. If the graph nodes are restricted to V (K), G is well-defined for μ = 1 and 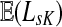 is bounded and attains its maximum there. The value of the maximum varies depending on the underlying network graph and the particular context.
is bounded and attains its maximum there. The value of the maximum varies depending on the underlying network graph and the particular context.
C.2. Large dissipation asymptotics
For all 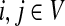 , let ρ(i, j) denote the (unweighted) length of the shortest directed path between i and j. We allow ρ(i, j) = ∞ if there exists no directed path between i and j. It is well-known that ρ is a (not necessarily symmetric) distance that satisfies the triangle inequality, that is, for all i, j,
, let ρ(i, j) denote the (unweighted) length of the shortest directed path between i and j. We allow ρ(i, j) = ∞ if there exists no directed path between i and j. It is well-known that ρ is a (not necessarily symmetric) distance that satisfies the triangle inequality, that is, for all i, j, 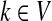 , ρ(i, j) + ρ(j, k) ≥ ρ(i, k). For any source
, ρ(i, j) + ρ(j, k) ≥ ρ(i, k). For any source 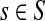 , recall that
, recall that 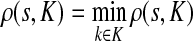 and let
and let 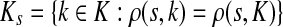 , the set of all the sinks closest to s.
, the set of all the sinks closest to s.
Theorem C.4
Let
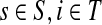 and
and
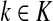 such that ρ(s, i) and ρ(i, k) are both finite. Then, if
such that ρ(s, i) and ρ(i, k) are both finite. Then, if
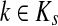 and i lies on the shortest path from s to k,
and i lies on the shortest path from s to k,
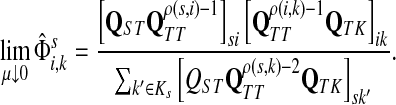 |
(61) |
Otherwise,
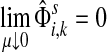
Proof
Let 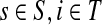 and
and 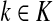 . Since, ρ(s, i) and ρ(i, k) are finite, it follows that ρ(s, k) is also finite, that is, k is reachable from s through i and the normalized channel tensor
. Since, ρ(s, i) and ρ(i, k) are finite, it follows that ρ(s, k) is also finite, that is, k is reachable from s through i and the normalized channel tensor 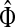 is well defined for all
is well defined for all 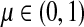 . Recall that
. Recall that
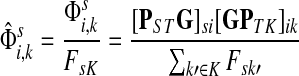 |
(62) |
where Fsk′ = [PSK + PSTGPTK]sk′.
Let u,  and let d = ρ(u, v). It can be easily shown (see Lemma A.3 from (Stojmirović and Yu, 2007) for a partial proof) that
and let d = ρ(u, v). It can be easily shown (see Lemma A.3 from (Stojmirović and Yu, 2007) for a partial proof) that 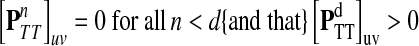 . Therefore,
. Therefore,
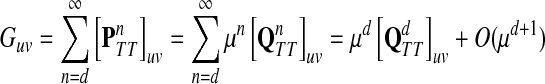 |
as μ ↓ 0. Hence,
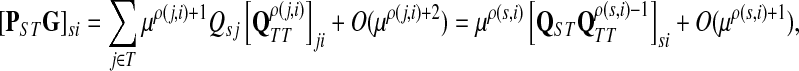 |
(63) |
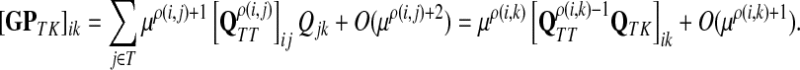 |
(64) |
Let ξ = ρ(s, k″), where 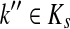 . We will consider the denominator of Equation (62) under two separate cases, ξ = 1 and ξ > 1.
. We will consider the denominator of Equation (62) under two separate cases, ξ = 1 and ξ > 1.
If ξ > 1, for all 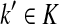 , the vertices s and k′ are not adjacent and thus Psk′ = 0. Hence, since s and k′ are connected, there exist
, the vertices s and k′ are not adjacent and thus Psk′ = 0. Hence, since s and k′ are connected, there exist 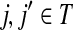 such that ρ(s, k′) = ρ(s, j) + ρ(j, j′) + ρ(j′, k′) = ρ(j, j′) + 2, implying
such that ρ(s, k′) = ρ(s, j) + ρ(j, j′) + ρ(j′, k′) = ρ(j, j′) + 2, implying
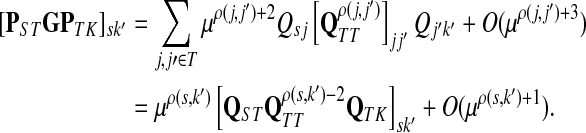 |
(65) |
Similarly,
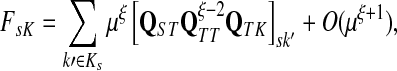 |
(66) |
and, as μ ↓ 0,
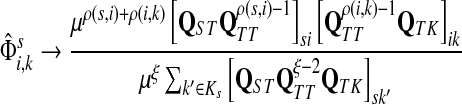 |
(67) |
By the triangle inequality and our assumptions on s, i and k,
 |
(68) |
The first inequality becomes an equality if and only if i lies on the shortest path between s and k while the second is an equality if and only if  . Therefore, if the assumption of the theorem is satisfied, the value of
. Therefore, if the assumption of the theorem is satisfied, the value of 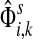 converges to the value of the right hand side of Equation (61), while otherwise
converges to the value of the right hand side of Equation (61), while otherwise 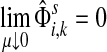 . ▪
. ▪
On the other hand, if 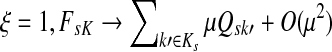 and therefore, since
and therefore, since 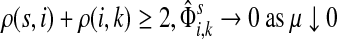 . ▪
. ▪
We have therefore shown that, as μ ↓ 0, only the nodes associated with the shortest path from each source to the sink(s) closest to it will have positive values of the normalized channel tensor – all other entries will be exactly 0.
Corollary C.5
Let
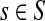 and suppose the normalized channel tensor
and suppose the normalized channel tensor
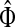 is well defined for all
is well defined for all
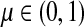 . Then,
. Then,
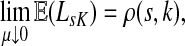 |
(69) |
where
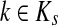 .
.
Proof
Let 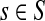 , let
, let 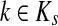 and let d = ρ(s, k). For
and let d = ρ(s, k). For 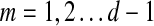 , let
, let 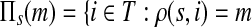 and
and 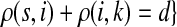 . The set Πs(m) consists of all transient nodes that are at the distance m from s on a shortest path from s to any of the sinks closest to s. By Theorem C.4,
. The set Πs(m) consists of all transient nodes that are at the distance m from s on a shortest path from s to any of the sinks closest to s. By Theorem C.4,
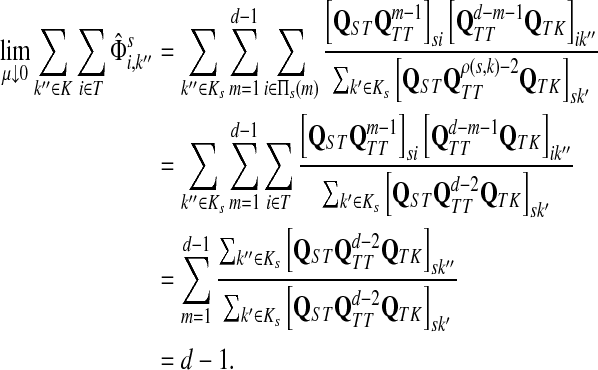 |
Therefore, by Equation (59),
 |
▪
D. Normalized evolution operator
To make our arguments more concise we will here additionally assume, without loss of generality, that every node is connected to a sink via a directed path, that is, that VK = V.
Note that N is indeed well defined in the limit as μ ↓ 0. For example, if i,  , we have from (64)
, we have from (64)
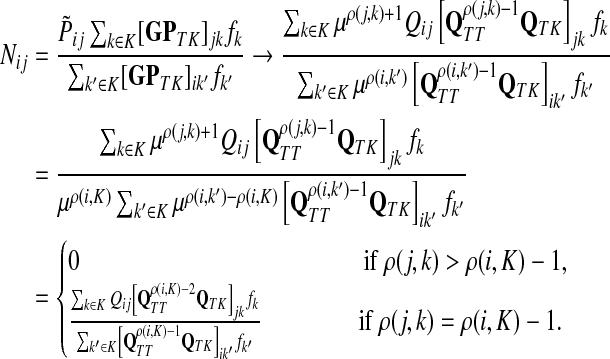 |
(70) |
Similar well-defined limits for Ni,j, with i,j∈V, can also be easily demonstrated using the results from Appendix C.2.
Proposition D.1
Let
f
denote an arbitrary vector over V. Suppose i
 . Then,
. Then,
 |
(71) |
Proof
Write the vector f as f = [fS, fT, fK]T and the matrix 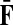 as
as  , where
, where 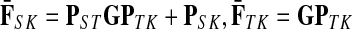 and
and 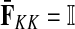 . The right equality from (71) can then be written in the block matrix form as
. The right equality from (71) can then be written in the block matrix form as 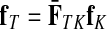 , and
, and 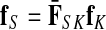 .
.
By definition of N, our premise 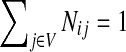 is equivalent to
is equivalent to
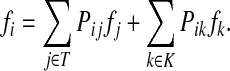 |
(72) |
For 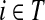 , Equation (72) can be expressed in matrix form as fT = PTTfT + PTKfK, that is,
, Equation (72) can be expressed in matrix form as fT = PTTfT + PTKfK, that is, 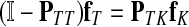 . Since the matrix
. Since the matrix 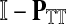 is invertible by our assumption of connectivity, this is further equivalent to
is invertible by our assumption of connectivity, this is further equivalent to
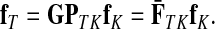 |
(73) |
For i ∈ S, Equation (72) can be written as fS = PSTfT + PSKfK, which using (73) is equivalent to
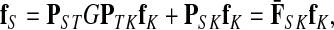 |
(74) |
as required. ▪
Proof of Proposition 3.2
Proof
All properties follow from the fact that the transformation from 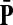 to N is a similarity transformation.
to N is a similarity transformation.
- (i) Let i,
 . We have
. We have
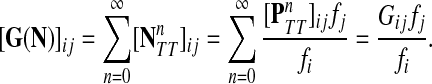
-
(ii) Let
 and suppose
and suppose 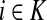 . Then
. Then 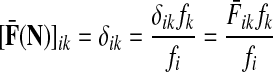 . Now suppose
. Now suppose  . Then,
. Then,
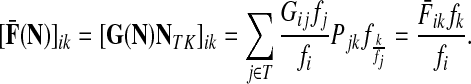 If
If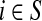 , we have
, we have
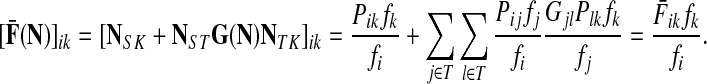
- (iii) Let
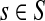 and suppose
and suppose 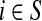 . Then
. Then 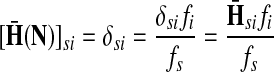 . Now suppose
. Now suppose  . Then
. Then  . If
. If 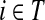 ,
,
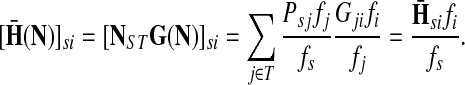
- (iv) Let
 and
and  . Then,
. Then,
▪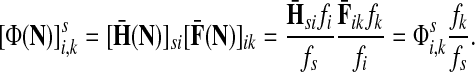
Acknowledgments
This work was supported by the Intramural Research Program of the National Library of Medicine at the National Institutes of Health.
Disclosure Statement
No competing financial interest exist.
References
- Ashburner M. Ball C.A. Blake J.A., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell L. A walk-through of the yeast mating pheromone response pathway. Peptides. 2005;26:339–350. doi: 10.1016/j.peptides.2004.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz B. Stark C. Reguly T., et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Res. 2008;36:D637–D640. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A. Ceol A. Palazzi L., et al. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35:D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle P.G. Snell J.L. Random Walks and Electric Networks. Mathematical Association of America; Washington, D. C: 1984. [Google Scholar]
- Fouss F. Yen L. Pirotte A., et al. An experimental investigation of graph kernels on a collaborative recommendation task. Proc. ICDM 2006. 2006:863–868. [Google Scholar]
- Fouss F. Pirotte A. Renders J.-M., et al. Random-walk computation of similarities between nodes of a graph with application to collaborative recommendation. IEEE Trans. Knowl. Data Eng. 2007;19:355–369. [Google Scholar]
- Giot L. Bader J.S. Brouwer C., et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- Haveliwala T.H. Topic-sensitive PageRank: a context-sensitive ranking algorithm for web search. IEEE Trans. Knowl. Data Eng. 2003;15:784–796. [Google Scholar]
- Huang D.W. Sherman B.T. Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T. Chiba T. Ozawa R., et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M. Goto S. Furumichi M., et al. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L. A new status index derived from sociometric analysis. Psychometrika. 1953;18:39–43. [Google Scholar]
- Kemeny J.G. Snell J.L. Finite Markov Chains. Springer-Verlag; New York: 1976. [1960]. [Google Scholar]
- Kerrien S. Alam-Faruque Y. Aranda B., et al. IntAct—open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–D565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondor R.I. Lafferty J.D. Diffusion kernels on graphs and other discrete input spaces. Proc. ICML 2002. 2002:315–322. [Google Scholar]
- Li S. Armstrong C.M. Bertin N., et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugannani R. Rice S. Saddle point approximation for the distribution of the sum of independent random variables. Adv. Appl. Probab. 1980;12:475–490. [Google Scholar]
- Lusk C.P. Waller D.D. Makhnevych T., et al. Nup53p is a target of two mitotic kinases, Cdk1p and Hrr25p. Traffic. 2007;8:647–660. doi: 10.1111/j.1600-0854.2007.00559.x. [DOI] [PubMed] [Google Scholar]
- Missiuro P. Liu K. Zou L., et al. Information flow analysis of interactome networks. PLoS Comput. Biol. 2009;5:e1000350. doi: 10.1371/journal.pcbi.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabieva E. Jim K. Agarwal A., et al. Whole-proteome prediction of protein function via graph-theoretic analysis of interaction maps. Bioinformatics. 2005;21(Suppl 1):302–310. doi: 10.1093/bioinformatics/bti1054. [DOI] [PubMed] [Google Scholar]
- Press W.H. Teukolsky S.A. Vetterling W. T., et al. Numerical Recipes: The Art of Scientific Computing. 3rd. Cambridge University Press; Cambridge, UK: 2007. [Google Scholar]
- Ptacek J. Devgan G. Michaud G., et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–684. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- Rual J.-F. Venkatesan K. Hao T., et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Salwinski L. Miller C. S. Smith A.J., et al. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:449–451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoelkopf B. Smola A. J. Learning with Kernels. MIT Press; Cambridge, MA: 2002. [Google Scholar]
- Schölkopf B. Tsuda K. Vert J.-P. Kernel Methods in Computational Biology. MIT Press; Cambridge, MA: 2004. [Google Scholar]
- Steffen M. Petti A. Aach J., et al. Automated modelling of signal transduction networks. BMC Bioinform. 2002;3:34. doi: 10.1186/1471-2105-3-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U. Worm U. Lalowski M., et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122:957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Stojmirović A. Yu Y.-K. Information flow in interaction networks. J. Comput. Biol. 2007;14:1115–1143. doi: 10.1089/cmb.2007.0069. [DOI] [PubMed] [Google Scholar]
- Stojmirović A. Yu Y.-K. ITM Probe: analyzing information flow in protein networks. Bioinformatics. 2009;25:2447–2449. doi: 10.1093/bioinformatics/btp398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojmirović A. Yu Y.-K. Robust and accurate data enrichment statistics via distribution function of sum of weights. Bioinformatics. 2010;26:2752–2759. doi: 10.1093/bioinformatics/btq511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthram S. Beyer A. Karp R., et al. eQED: an efficient method for interpreting eQTL associations using protein networks. Mol. Syst. Biol. 2008;4:162. doi: 10.1038/msb.2008.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z. Wang L. Arbeitman M., et al. An integrative approach for causal gene identification and gene regulatory pathway inference. Bioinformatics. 2006;22:e489–496. doi: 10.1093/bioinformatics/btl234. [DOI] [PubMed] [Google Scholar]
- Uetz P. Giot L. Cagney G., et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Voevodski K. Teng S. Xia Y. Spectral affinity in protein networks. BMC Syst. Biol. 2009;3:112. doi: 10.1186/1752-0509-3-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarzov P. Mazzoni C. Mann C. The SLT2(MPK1) MAP kinase is activated during periods of polarized cell growth in yeast. EMBO J. 1996;15:83–91. [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.-C. Blattner M. Yu Y.-K. Heat conduction process on community networks as a recommendation model. Phys. Rev. Lett. 2007;99:154301. doi: 10.1103/PhysRevLett.99.154301. [DOI] [PubMed] [Google Scholar]