

Abstract
Infrared irradiation promoted the Diels-Alder cycloadditions of exo-2-oxazolidinone dienes 1–3 with the Knoevenagel adducts 4–6, as dienophiles, leading to the synthesis of new 3,5-diphenyltetrahydrobenzo[d]oxazol-2-one derivatives (7, 9, 11 and 13–17), under solvent-free conditions. These cycloadditions were performed with good regio- and stereoselectivity, favoring the para-endo cycloadducts. We also evaluated the one-pot three-component reaction of active methylene compounds 20, benzaldehydes 21 and exo-2-oxazolidinone diene 2 under the same reaction conditions. A cascade Knoevenagel condensation/Diels-Alder cycloaddition reaction was observed, resulting in the final adducts 13–16 in similar yields. These procedures are environmentally benign, because no solvent and no catalyst were employed in these processes. The regioselectivity of these reactions was rationalized by Frontier Molecular Orbital (FMO) calculations.
Keywords: Diels-Alder cycloadditions, regioselectivity, Knoevenagel, infrared irradiation
1. Introduction
The Diels-Alder cycloaddition is one of the most powerful synthetic methodologies for the construction of cyclic six-membered rings, and tremendous efforts have been focused on expanding the scope of this cycloaddition with various combinations of dienes, dienophiles, catalysts and reaction conditions [1–6]. In this sense, alkenes containing two electron-withdrawing groups have been the target of a large number of recent studies, because many of them can act as Michael acceptors [7–8], as well as hetero-dienes [9–11] or dienophiles [12,13] in Diels-Alder reactions. Similarly, exocyclic dienes have received significant attention in recent years due to their high reactivity in cycloaddition reactions and their synthetic potential [14,15]. We described an efficient cascade methodology, which combines α-diketones and isocyanates in the presence of a dehydrating agent, to afford functionalized N-substituted exo-2-oxazolidinone dienes 1–3 (Tables 1–3). The latter have proved to be stable, and they undergo Diels-Alder cycloadditions with high selectivity [16–20]. In addition, they have shown to be useful synthons in the preparation of carbazoles [21–24], and in the synthesis of new polycyclic compounds by a cascade [4 + 2] cycloaddition/cyclopentannulation/1,5-sigmatropic rearrangement process with Fischer (arylalkynyl)(alkoxy)carbenes [25]. Moreover, dienes 1–3 have been employed to synthesize new η4-diene-Fe(CO)3 complexes, which undergo the addition of alkyllithium reagents to produce stable and unprecedented conjugated enamine-enol ester- and enamido-enol-Fe(CO)3 complexes [26].
Table 1.
Diels-Alder reactions of diene 1 with dienophiles 4–6 a.
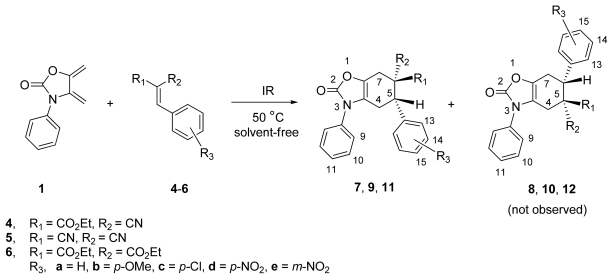 | ||||||
|---|---|---|---|---|---|---|
| Entry | Dienophile | R1 | R2 | R3 | Reaction Time (h) | Product e (%) |
| 1 | 4a | CO2Et | CN | H | 3.5 | 7a (73) |
| 2 b | 4a | CO2Et | CN | H | 20 | 7a (30) |
| 3 c | 4a | CO2Et | CN | H | 24 | 7a (20) |
| 4 d | 4a | CO2Et | CN | H | 24 | 7a (20) |
| 5 | 4b | CO2Et | CN | p-OMe | 4.0 | 7b (50) |
| 6 | 4c | CO2Et | CN | p-Cl | 3.5 | 7c (60) |
| 7 | 4d | CO2Et | CN | p-NO2 | 3.0 | 7d (80) |
| 8 | 4e | CO2Et | CN | m-NO2 | 3.5 | 7e (55) |
| 9 | 5a | CN | CN | H | 4.0 | 9a (80) |
| 10 | 5b | CN | CN | p-OMe | 4.5 | 9b (55) |
| 11 | 5c | CN | CN | p-Cl | 3.0 | 9c (75) |
| 12 | 5d | CN | CN | p-NO2 | 3.0 | 9d (85) |
| 13 | 6a | CO2Et | CO2Et | H | 5.0 | 11a (35) |
| 14 | 6b | CO2Et | CO2Et | p-OMe | 6.0 | 11b (25) |
| 15 | 6c | CO2Et | CO2Et | p-Cl | 5.0 | 11c (30) |
All entries were carried out under IR irradiation at 50 °C and solvent-free conditions, except entries 2–4;
Under thermal (50 °C) and solvent-free conditions;
Under thermal conditions (50 °C) in benzene as the solvent;
Under thermal conditions (50 °C) in THF as the solvent;
After column chromatography.
Table 3.
Domino Knoevenagel condensation/Diels-Alder cycloaddition between diene 2, methylene active compounds 20a–c and benzaldehydes 21a–d a.
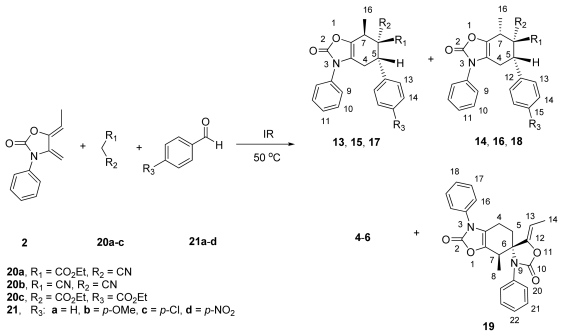 | ||||||
|---|---|---|---|---|---|---|
| Entry | Methylene Active | Benzaldehyde | Reaction Time (min) | By-Products (%) b | Adducts (endo/exo) c | Yield d (%) |
| 1 | 20a | 21a | 35 | 4a/19 (40:5) | 13a/14a (65:35) | 43/12 |
| 2 | 20a | 21b | 50 | 4b/19 (52:8) | 13b/14b (75:25) | 40 e |
| 3 | 20a | 21c | 30 | 4c/19 (35:5) | 13c/14c (68:32) | 60 e |
| 4 | 20a | 21d | 40 | 4d/19 (32:4) | 13d/14d (75:25) | 64 e |
| 5 | 20b | 21a | 30 | 5a/19 (40:5) | 15a/16a (70:30) | 55 e |
| 6 | 20b | 21b | 40 | 5b/19 (40:10) | 15b/16b (85:15) | 50 e |
| 7 | 20b | 21c | 30 | 5c/19 (20:5) | 15c/16c (80:20) | 65/10 |
| 8 | 20b | 21d | 35 | 5d/19 (25:5) | 15d/16d (70:30) | 55/15 |
| 9 | 20c | 21a | 150 | 6a/19 (64:36) | -- | -- |
| 10 | 20c | 21b | 210 | 6b/19 (70:30) | -- | -- |
| 11 | 20c | 21c | 240 | 6c/19 (60:40) | -- | -- |
| 12 | 20c | 21d | 240 | 6d/19 (65:35) | -- | -- |
An equimolar mixture of 2, 20 and 21 was irradiated with IR at 50 °C, under solvent-free conditions;
After column chromatography;
Determined by 1H NMR of the crude reaction;
Yields of the adducts after column chromatography;
Yield of the major adduct.
We have appropriately employed infrared irradiation as an alternative energy source, working under solvent-free conditions and with various types of reactions, including Knoevenagel condensation [27–29], the Fischer indole reaction [30], the Biginelli reaction [31] and, more recently, the molecular rearrangement of perezone into isoperezone [32].
In this context, and as part of our ongoing research into the use of infrared irradiation as the energy source to promote organic reactions, we herein describe a convenient and versatile synthesis of the new substituted tetrahydrobenzo[d]oxazol-2-one derivatives 7, 9, 11 and 13–17, starting from the exo-2-oxazolidinone dienes 1–3 and the Knoevenagel adducts 4–6 (Tables 1 and 2), as the dienophiles, in the Diels-Alder cycloadditions promoted by infrared irradiation, under solvent-free conditions. Moreover, we also carried out an evaluation of how the reactivity and stereoselectivity of these cycloadditions are affected by the structural modifications in the diene, as well as in the Knoevenagel adducts, such as the replacement of the cyano group by the ethoxycarbonyl group (Tables 1 and 2). In addition, we studied the one-pot three-component reactions to obtain the same cycloadducts starting from methylene active compounds 20a–c, benzaldehydes 21a–d and exo-2-oxazolidinone diene 2 via a cascade Knoevenagel/Diels-Alder process under similar reaction conditions.
Table 2.
Diels-Alder reactions of dienes 2 and 3 with dienophiles 4–6 a.
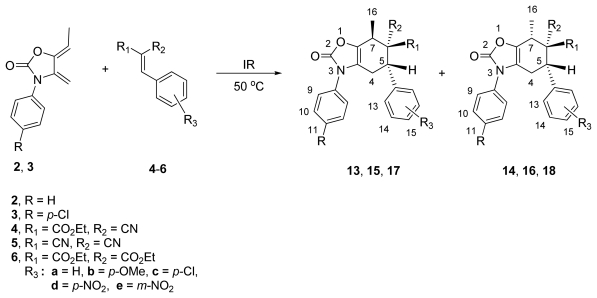 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Diene | Dienophile | R | R1 | R2 | R3 | Reaction Time (h) | Products (endo/exo) b | Yield c (%) |
| 1 | 2 | 4a | H | CO2Et | CN | H | 4.0 | 13a/14a (80:20) | 56/32 |
| 2 | 2 | 4b | H | CO2Et | CN | p-OMe | 5.0 | 13b/14b (75:25) | 65 d |
| 3 | 2 | 4c | H | CO2Et | CN | p-Cl | 4.5 | 13c/14c (68:32) | 60 d |
| 4 | 2 | 4d | H | CO2Et | CN | p-NO2 | 4.0 | 13d/14d (75:25) | 64 d |
| 5 | 3 | 4e | p-Cl | CO2Et | CN | m-NO2 | 4.5 | 13e/14e (75:25) | 70 d |
| 6 | 2 | 5a | H | CN | CN | H | 3.0 | 15a/16a (80:20) | 70 d |
| 7 | 2 | 5b | H | CN | CN | p-OMe | 4.0 | 15b/16b (82:18) | 55 d |
| 8 | 2 | 5c | H | CN | CN | p-Cl | 5.0 | 15c/16c (90:10) | 75 d |
| 9 | 2 | 5d | H | CN | CN | p-NO2 | 2.0 | 15d/16d (80:20) | 75/15 |
| 10 | 3 | 5b | p-Cl | CN | CN | p-OMe | 3.0 | 15e/16e (75:25) | 70/15 |
| 11 | 2 | 6a | H | CO2Et | CO2Et | H | 6.0 | 17a/18a (100:0) | 23 d |
| 12 | 2 | 6b | H | CO2Et | CO2Et | p-OMe | 5.0 | 17b/18b (100:0) | 32 d |
| 13 | 2 | 6c | H | CO2Et | CO2Et | p-Cl | 4.0 | 17c/18c (100:0) | 25 d |
All entries under IR irradiation at 50 °C and solvent-free conditions;
Determined by 1H NMR of the crude reaction mixtures, corresponding to the mixture of stereoisomers;
Yields of the products after column chromatography;
Yield of the major product.
2. Results and Discussion
2.1. Diels-Alder Cycloaddition with Diene 1
As we have previously demonstrated, Knoevenagel adducts can be easily prepared using an infrared irradiation protocol that employs the condensation reaction of benzaldehydes and active methylene compounds, under solvent-free conditions [27–29]. Therefore, we have used this methodology to prepare compounds 4–6. The required dienes 1–3 were synthesized according to the already published procedure [16–20].
We explored synthetic access to the tetrahydrobenzo[d]oxazol-2-one derivatives 7–11 and 13–18, in search of infrared irradiation as a viable promoter of the Diels-Alder cycloadditions, in a two-step synthesis, starting from the exo-heterocyclic dienes 1–3 and the Knoevenagel adducts 4–6.
Initially, the unsubstituted exo-heterocyclic diene 1 was evaluated in terms of reactivity and regioselectivity in the Diels-Alder additions toward derivatives 4a–e, which bear activating substituents such as ethoxycarbonyl (R1) and cyano (R2) groups. Thus, a mixture of diene 1 and olefin 4a (1:1.2 mol-equiv., respectively) was irradiated with an infrared lamp [33] at 50 °C for ca. 3.5 h, under solvent-free conditions, leading to the total conversion of 1 to afford 7a, judging by the 1H NMR analysis of the crude reaction mixture, as a single regioisomeric product in 73% yield. This high regioselectivity contrasts with that observed for the thermal Diels-Alder reaction of 1 with monosubstituted dienophiles, such as methyl vinyl ketone and methyl propiolate, in which the para/meta regioisomeric ratios were lower (from 1:1 up to 8:2) [17].
The structure of compound 7a was established by spectroscopic analysis. The spectrum of High Resolution Mass Spectrometry (HRMS) showed exactly the expected mass (m/z 388.1423); while the IR spectrum showed two carbonyl absorption bands (C=O) at 1757 and 1713 cm−1 and a cyano group absorption at 2362 cm−1. The 1H and 13C NMR spectral data are consistent with the tetrahydrobenzo[d]oxazol-2-one skeleton. It is interesting to note the large difference in the chemical shifts (δ) of the diastereotopic CH2 protons at the C-4 position of the cyclohexene ring, since H-4β appeared at 2.65 ppm as a ddd (J = 17.1, 4.8, 1.5 Hz) due to the geminal, vicinal and homoallylic couplings, respectively; while the signal due to H-4α appeared at 3.13 ppm as a dddd (J = 17.1, 11.4, 4.2, 2.1 Hz). The large difference in the δ value for these protons could be ascribed to the anisotropic effect of the phenyl groups at N-3 and C-5. Decoupling and Nuclear Overhauser Effect (NOE) experiments provided additional support for the structure: H-4α showed a three-bond coupling with H-5 (3J4–5 = 11.4 Hz); while the signal of protons H-5 (3.46 ppm) and H-4α (3.16 ppm) were enhanced when H-4β (2.65 ppm) was irradiated (Figure 1).
Figure 1.

NOE effects observed upon irradiation of proton H-4β for the adduct 7a.
Interestingly, when the reaction was carried out under thermal (50 °C) and solvent-free conditions, the reaction time was longer and the yield lower (Table 1, entry 2). In an attempt to further improve the yield, under thermal conditions (50 °C), benzene and tetrahydrofuran were used as solvents, without yielding better results (Table 1, entries 3 and 4).
Comparing the reaction times (Table 1, entry 1 vs. entries 2–4), it appears that under infrared irradiation the reaction was substantially faster (~3.5 h) and the yield was higher (73%). As for the regioselectivity, it was comparable in both cases, only affording regioisomer 7. Analysis of the crude reaction mixture by 1H NMR did not show evidence of regioisomer 8.
To assess the effect of the substituent R3 in the aromatic ring of the dienophiles on the reactivity and the regioselectivity, several analogues using both electron-poor and electron-rich substituents in 4b–e, were used. When 4b, bearing an electron-releasing group, was irradiated in the presence of 1, the conversion rate slightly decreased (Table 1, entry 5), giving 7b in a lower yield (50%), together with recovered dienophile 4b (50%), However, with the use of dienophile 4d, containing an electron-withdrawing group, a higher yield of the corresponding adduct 7d was obtained. The reactivity trend of the Diels-Alder cycloaddition of dienophiles 4a–e with 1 (Table 1, entries 4–8) met the expectations of a normal electron-demand process [34].
Cycloadduct 7e was isolated as yellow crystals (EtOAc/hexane, 8:2) and its para regiochemistry (as considered for the relative orientation in the cyclohexene ring between the nitrogen atom and the electron-withdrawing groups of the dienophile) was confirmed by X-ray crystallography (Figure 2). The X-ray structure shows that the aryl groups in N-3 and C-5 are almost perpendicular to the heterocycle and to the cyclohexene ring, respectively, presenting the following consistent torsion angles: −59.7(2)° for C(3a)-N(3)-C(8)-C(9) and −129.20(13)° for C(4)-C(5)-C(12)-C(13).
Figure 2.

Molecular structure of 7e with thermal ellipsoids at the 30% probability level.
Complementarily, with the aim of exploring the scope and limitations of the process, as well as of detecting the effect on the cycloadducts induced by the change of the substituents R1 and R2 in the dienophiles, the ethoxycarbonyl group in 4 (R1 = CO2Et) was replaced by a CN group and the cyano group (R2 = CN) by an ethoxycarbonyl group, to produce a series of benzylidenemalononitriles 5a–d (R1 = R2 = CN) and diethyl 2-benzylidenemalonates 6a–c (R1 = R2 = CO2Et), respectively. The reactions were performed under identical conditions to those used for 1 and 4a. The reaction of diene 1 with these two series of analogous dienophiles 5a–d and 6a–c yielded cycloadducts 9a–d and 11a–c, respectively. The fact that in both cases the product was a single para regioisomer indicates a similar behavior in the reactions. It is noteworthy that 1H NMR analysis (300 MHz) of the crude mixtures did not give evidence of the presence of the corresponding regioisomers 10 and 12.
The best yields of the tetrahydrobenzo[d]oxazol-2-one derivatives 9 and 11 corresponded to the reactions between exo-heterocyclic diene 1 with benzylidenemalononitriles 5a–d (Table 1, entries 9–12). In contrast, with the reactions between 1 and the ethyl (E)-2-cyano-3-phenylacrylates 4a–e, the corresponding yields of derivatives 7a–e were lower (Table 1, entries 1–8). When the sterically more demanding diethyl 2-benzylidenemalonates 6a–c were used, the yields of adducts 11a–c were the lowest of all (Table 1, entries 13–15). The reactivity trend found for the Knoevenagel dienophiles can also be explained by the higher electron-withdrawing effect of the cyano group in comparison with the ethoxycarbonyl group [35]. In accordance with previous reports, the regiochemistry of these cycloadditions mainly depends on the electron-donating effect of the nitrogen atom of the heterocycle ring of the diene [16]. However, the exclusive formation of the para regioisomer in our case contrasts with the tendency of the exo-heterocyclic diene 1 to produce a mixture of para/meta regioisomers [17]. This is probably due to the fact that the dienophiles used in the present work are geminally substituted by two electron-withdrawing groups, which enhance the reactivity and, consequently, the regioselectivity [36].
2.2. Diels-Alder Cycloaddition with Dienes 2 and 3
In order to evaluate the effect of the substituent in the exo-heterocyclic diene on the reactivity and selectivity in the course of the Diels-Alder reaction, dienes 2 and 3, bearing a methyl group in the double bond, were added to dienophiles 4–6.
The reactions of dienes 2 and 3 with acrylates 4a–e (R1 = CO2Et, R2 = CN) gave, after IR irradiation and heating at 50 °C for 4–5 h, mixtures of endo/exo cycloadducts 13a–e/14a–e in 55–88% yields (Table 2, entries 1–5). The Diels-Alder reactions were highly regio- and stereoselective, since the para (N-Ar/CO2Et and CN groups) derivatives 13 and 14 were the lone regioisomers, and the para-endo cycloadducts (endo = syn relative configuration between Me/CO2Et groups) 13a–e were obtained in higher yields than the para-exo cycloadducts 14a–e.
The endo/exo ratios of adducts 13a–e/14a–e were determined by integration of the double signals of the methyl groups C-16 in the 1H NMR spectra of the crude mixtures (Table 2, entries 1–5). The separation of these mixtures was achieved by column chromatography on silica gel using hexane as eluent. The structural elucidation of the main products 13a–e was made on the basis of their spectroscopic data (NMR, HRMS and IR). All the data are consistent with the substituted tetrahydrobenzo[d]oxazol-2-one skeleton of 13a–e. The 1H NMR spectrum of 13a shows the presence of ten aromatic protons at 7.26–7.51 ppm, a quartet integrating for two protons (OCH2CH3) at 4.09 ppm, and two overlapped signals attributed to H-7 and H-5 protons at 3.49–3.54 ppm. The proton H-4α appears as a doublet of doublets of doublets (J = 17.1, 11.1, 1.8 Hz) at 2.93 ppm; while the proton H-4β appears as a doublet of doublets (J = 17.1, 5.4 Hz) at 2.62 ppm. There is a signal at 1.35 ppm as a doublet integrating for three protons (H-16) and at 1.11 ppm (OCH2CH3) as a triplet.
The 13C NMR spectrum of 13a displays signals for two carbonyl groups at 164.7 ppm (CO2Et) and 154.0 ppm (C-2), ten signals for vinyl and aromatic carbons at 138.0–120.0 ppm, one signal corresponding to the cyano group at 118.2 ppm, and seven signals at 62.9, 52.2, 40.4, 37.0, 27.0, 15.9 and 13.7 ppm for sp3 carbon atoms. The attributions of the signal were supported by 2D experiments such as Heteronuclear Multiple-Quantum Coherence (HMQC) and Heteronuclear Multiple-Bond Coherence (HMBC).
The relative configuration at C-5, C-6 and C-7 of 13a was determined by NOE experiments (Figure 3), where an enhancement of the signals of protons H-5 and H-4α was observed when the signal of H-4β was irradiated. Likewise, when H-5 was irradiated, an NOE effect was observed for H-4β and H-13. The irradiation of H-4α induced an NOE effect on the signals of H-4β, H-9 and H-13. An enhancement of the signals of protons H-5 and H-7 was observed when the signal of H-16 was irradiated. These data support a syn relationship between H-4β, H-5 and H-16 protons, and justify assigning the structure of the compound 13a as the endo cycloadduct.
Figure 3.

NOE observed upon irradiation of protons H-4β, H-5 and H-16 for the adduct 13a.
The assignment of the stereochemistry of compound 13a was confirmed by X-ray crystallography (Figure 4). The phenyl and ethoxycarbonyl groups at the stereogenic C-5 and C-6 centers have a trans diequatorial orientation. The torsion angle C(12)-C(5)-C(6)-C(6a) of 48.42(18)° supports the gauche conformation for the phenyl and ethoxycarbonyl groups. Meanwhile, the methyl group at the stereogenic C-7 center has a pseudoaxial orientation. Therefore, in the solid state, the carbocyclic six-membered ring adopts a half-chair conformation.
Figure 4.

Molecular structure of 13a with thermal ellipsoids at the 30% probability level.
It is likely that the presence of an electron-donating methyl group in 2 greatly polarizes the π-system of the diene, giving rise to the major para regioisomers 13a–e/14a–e. The endo preference might be due to both steric and electronic factors which favor the endo transition state (vide infra).
Similarly, benzylidenemalonitriles 5a–d (R1 = R2 = CN) reacted with dienes 2 and 3 affording a mixture of anti/syn diastereoisomers 15–16, respectively, in 55–90% yields, favoring the anti (relative configuration between the C-5 phenyl ring with respect to Me-16) cycloadducts 15a–e as determined by 1H NMR (300 MHz) analysis of the crude reaction mixture (Table 2, entries 6–10).
In contrast, the reactions of diene 2 with dienophiles 6a–c (R1 = R1 = CO2Et), under the same experimental condition, provided single diastereoisomers 17a–c in low yields (Table 2, entries 11–13), due in part to the self-dimerization of diene 2 to adduct 19, isolated as a by-product [17]. These results indicate that dienophiles 6a–c are less reactive and more stereoselective that dienophiles 4 and 5.
It appears that these reactions are sterically sensitive, since the use of the more hindered dienophiles 6a–c afforded the corresponding products 17a–c in the poorest yields, although with a better stereoselectivity.
As shown in Tables 1 and 2, the reaction times for diene 2 were similar to those employed for diene 1. This is rather unexpected as previously mentioned [17], since the electron-releasing effect of the methyl substituent of diene 2 should increase the reactivity in Diels-Alder additions according to Alder’s rule. This behavior is also presumably due to the steric effect.
2.3. Multicomponent Reactions
In recent years, the development of multicomponent reactions in order to produce biologically active compounds has been accelerated and thus has become a very important area of research in organic and medicinal chemistry.
As an attempt to obtain compounds 13–18 more efficiently, we turned our attention to a one-pot procedure. Our synthetic strategy was based on the knowledge that the dienophiles 4 and 5 are accessible through a simple Knoevenagel condensation between compounds 20a–b and benzaldehydes 21a–d [27–29], followed by a subsequent Diels-Alder cycloaddition with diene 2, to generate cycloadducts 13–16.
Initially, in this multicomponent approach, a mixture of ethyl 2-cyanoacetate (20a), benzaldehyde (21a) and diene 2 was reacted in a 1:1:1 (mol-equiv.) ratio under infrared irradiation and solvent-free conditions. After 35 min, this reaction led to the desired mixture of tetrahydrobenzo[d]oxazol-2-ones 13a/14a (65:35), albeit in moderate yield (55%), along with some amount of 4a and 19 (40% and 5%, respectively). It is worth noting that the regio- and stereoselectivity was similar (Table 3, entry 1) to those found in the previous methodology (Table 2, entry 1). The Knoevenagel adduct 4a was detected from the crude mixture by H1 NMR analysis, which supports the idea that its initial formation was accomplished before the intermolecular Diels-Alder reaction with diene 2 took place, to give the corresponding adducts.
A similar behavior was observed for the analogous substrates 21b–d with 20a and diene 2, since the cycloadduct mixtures 13b/14b, 13c/14c and 13d/14d were obtained in comparable yields (40–64%) to those obtained via the two-step procedure, confirming the efficiency of the multicomponent approach (Table 3, entries 2–4).
On the other hand, the multicomponent reaction between malononitrile 20b, benzaldehydes 21a–d and diene 2 produced fairly good yields of cycloadducts 15a–d/16a–d. However, in the presence of diethyl malonate (20c), no domino Knoevenagel condensation/Diels-Alder cycloaddition was observed at all. When the reaction temperature was increased to 80 °C, compound 19 was obtained instead of the expected adducts 17/18. These results revealed that the dimerization of 2 is also promoted by IR irradiation to yield 19. The structure of the latter was established by spectroscopic data and corroborated by the study of X-ray diffraction (Figure 5). Previously, we observed the dimerization of diene 2 under thermal conditions (xylene, 120 °C, 10 h) [17]. Comparing the NMR data of these compounds, we found that there were notable differences in chemical shifts, as well as in the difference in their melting points (196–198 °C and 243–244 °C), which suggests that this dimer corresponds to different diastereoisomer. This result can be attributed to the probable influence of infrared radiation as a source of energy.
Figure 5.

Molecular structure of 19 with thermal ellipsoids at the 30% probability level.
The higher reactivity of ethyl 2-cyanoacetate (20a) and malononitrile (20b) in comparison with diethyl malonate (20c), which successively leads to the Knoevenagel condensation and Diels-Alder reaction under infrared irradiation conditions, may be explained in terms of the difference of acidity constants of the activated methylene: 20c (pKa = 13) [37], 20b (pKa = 11) [38] and 20a (pKa = 9) [39]. This acidity can affect the formation of the Knoevenagel products and, consequently, the final adduct. In addition, these results also suggest that the steric hindrance generated by the ethoxycarbonyl group seems to play a role in controlling the domino reactions and therefore in providing acceptable yields.
2.4. Diels-Alder Regioselectivity and FMO Theory
The regioselectivity of the Diels-Alder additions of dienes 1–3 to dienophiles 4–6 was rationalized in terms of the FMO theory [34]. The geometries of dienes 1 and 2 were previously calculated [17], while the geometries of diene 3 and dienophiles 4–6 were calculated using the B3LYP/6-31G** method [40–42] without any symmetry constraints calculation, and employed as the starting point for the ab initio molecular orbital calculations, using the RHF/6-31G** basis set [43]. It is noteworthy that for derivatives 6, the cis ethoxycarbonyl group to the aryl ring adopts a preferential non-coplanar conformation, leaving the trans acrylate moiety in conjugation with the aromatic substituent. This conjugation is also observed for derivatives 4. This is probably due to the fact that in this conformation the aryl ring is maintained coplanar to the acrylate conjugated π-system, giving rise to a higher stability.
By using the same basis set, the energies of the FMO were calculated for both dienes and dienophiles (Table 4). Since, in the entire series lower, energy gaps were calculated for the interaction between HOMOdiene-LUMOdienophile (Normal Electronic Demand) than between the opposite interaction LUMOdiene-HOMOdienophile (Inverse Electronic Demand), as illustrated by some examples in Table 5, it is then expected that the reaction is conducted under the former interaction.
Table 4.
Ab initio 6-31G** calculations of energies (eV) and coefficients (Ci) of the frontier molecular orbitals for dienes 1–3 and dienophiles 4–6 a.
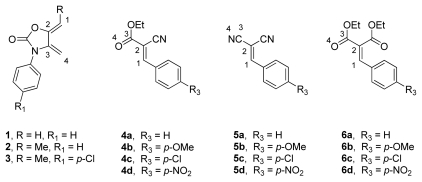 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | |||||||||||
| Compd b | E (eV) | C1 | C2 | C3 | C4 | ΔCic | E (eV) | C1 | C2 | C3 | C4 | ΔCic) |
| 1d | −8.8051 | 0.246 | 0.164 | −0.209 | −0.326 | 0.080 | 2.9065 | 0.263 | −0.245 | −0.245 | 0.258 | −0.005 |
| 2d | −8.5610 | −0.257 | −0.199 | 0.198 | 0.320 | 0.063 | 3.1035 | 0.274 | −0.222 | −0.245 | 0.248 | −0.026 |
| 3 | −8.6408 | −0.277 | −0.220 | 0.199 | 0.324 | 0.047 | 2.7244 | 0.288 | −0.232 | −0.247 | 0.258 | −0.030 |
| 4a | −8.9382 | 0.122 | 0.276 | 0.015 | −0.114 | −0.154 | 1.0104 | 0.296 | −0.210 | −0.131 | 0.113 | 0.086 |
| 4b | −8.4299 | −0.084 | −0.260 | −0.020 | 0.105 | −0.176 | 1.2204 | 0.306 | −0.203 | −0.134 | 0.112 | 0.103 |
| 4c | −9.0541 | 0.116 | 0.265 | 0.014 | −0.109 | −0.149 | 0.7532 | 0.288 | −0.211 | −0.126 | 0.110 | 0.077 |
| 4d | −9.7679 | 0.160 | 0.284 | 0.008 | −0.121 | −0.124 | −0.1056 | 0.219 | −0.196 | −0.096 | 0.092 | 0.023 |
| 5a | −9.2234 | 0.126 | 0.275 | −0.058 | −0.149 | 0.5331 | 0.305 | −0.227 | −0.071 | 0.078 | ||
| 5b | −8.6859 | −0.087 | −0.261 | 0.044 | −0.174 | 0.7611 | 0.315 | −0.219 | −0.073 | 0.096 | ||
| 5c | −9.3227 | 0.118 | 0.263 | −0.055 | −0.145 | 0.2759 | 0.298 | −0.227 | −0.068 | 0.071 | ||
| 5d | −10.0495 | 0.163 | 0.280 | −0.073 | −0.117 | −0.5323 | 0.238 | −0.214 | −0.050 | 0.024 | ||
| 5c | −9.3227 | 0.118 | 0.263 | −0.055 | −0.145 | 0.2759 | 0.298 | −0.227 | −0.068 | 0.071 | ||
| 6a | −8.6751 | 0.112 | 0.257 | 0.013 | −0.100 | −0.145 | 1.7804 | 0.256 | −0.215 | −0.153 | 0.126 | 0.041 |
| 6b | −8.1608 | 0.043 | 0.242 | 0.017 | −0.091 | −0.199 | 1.9179 | 0.272 | −0.210 | −0.151 | 0.122 | 0.062 |
| 6c | −8.8008 | 0.106 | 0.246 | 0.012 | −0.094 | −0.140 | 1.5032 | 0.248 | −0.215 | −0.142 | 0.119 | 0.033 |
| 6d | −9.5564 | 0.148 | 0.271 | 0.005 | −0.108 | −0.123 | 0.5236 | −0.164 | 0.183 | 0.095 | −0.088 | −0.019 |
These are the values of the pz coefficients, the relative pz' contributions and their ΔCi are analogous;
The most stable planar (aryl ring-double bond-numbered trans carbonyl group) s-cis (acrylate moiety) conformation for olefins 4 and 6, and planar (aryl ring-double bond-cyano group) conformation for olefins 5, as shown in the structures at the head of the table;
Carbon 4-carbon 1 for the dienes; carbon 1-carbon 2 for the dienophile;
Reference 17.
Table 5.
Energy gaps (eV) of the frontier molecular orbitals for dienes 1–3 and dienophiles 4a–6a.
| 4a a | 5a a | 6a a | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diene | HOMO-LUMO | LUMO-HOMO | Diff. | HOMO-LUMO | LUMO-HOMO | Diff. | HOMO-LUMO | LUMO-HOMO | Diff. |
| 1 | 9.8155 | 11.8447 | 2.0292 | 9.3382 | 12.1299 | 2.7917 | 10.5855 | 11.5816 | 0.9961 |
| 2 | 9.5714 | 12.0417 | 2.4703 | 9.0941 | 12.3269 | 3.2328 | 10.3414 | 11.7786 | 1.4372 |
| 3 | 9.6512 | 11.6626 | 2.0114 | 9.1739 | 11.9478 | 2.7739 | 10.4212 | 11.3995 | 0.9783 |
HOMOdiene-LUMOdienophile and LUMOdiene-HOMOdienophile.
As expected, the methyl group attached to the diene moiety in dienes 2 and 3 induced an increase of the energy of the HOMO, with respect to the energy of the unsubstituted diene 1. Hence, the reactivity of dienes 2 and 3 should be higher than that of diene 1, as observed for the cycloadditions with mono substituted dienophiles [17]. Nevertheless, in the case of dienophiles 4–6, the reaction times are very similar for all the dienes (Tables 1 and 2), which indicates a similar reactivity as well. It is likely that other factors are involved, such as the steric hindrance generated between the dienes and the substituents in dienophiles 4–6. Although these factors are not sufficiently important to modify the regioselectivity, which is para in the whole series, the preference for the anti relative configuration between the methyl group and the aryl ring in adducts 13–17 seems to support their existence. Moreover, in spite of the presence of electron-withdrawing groups in the aryl ring of the dienophiles, such as the nitro group, which may induce a higher reactivity and higher selectivity [34,36], there is no correlation between the stereoselectivity and the structure of the dienophiles bearing other substituents. Once again, this suggests the significant effect of the steric repulsions at the transition state, and also seems to be the reason for the formation of the single endo stereoisomer (17) in the case of the more hindered dienophiles 6 (Table 2, entries 11–13). The stabilizing secondary orbital interactions eventually present at the endo transition state may reinforce this preference.
The exclusive para regioselectivity (N-Ar/CO2Me or CN groups) observed in all the cycloadditions can be explained on the basis of the coefficient differences for the HOMOdiene-LUMOdienophile interactions (Table 4). These latter should generate the greatest perturbation, since the energy gap is smaller than the inverse interactions (LUMOdiene-HOMOdienophile). Indeed, if the largest FMO coefficients become bonded preferentially at the transition state [44–47], and considering that the relative magnitude of the coefficient of the terminus C-4 is bigger than that of C-1 in the HOMO of dienes 1–3, and that the beta C-1 coefficient is bigger than that of the alpha C-2 in the LUMO of olefins 4–6, a “para” orientation is expected, in agreement with the experimental results. This para regioselectivity supports the idea that the electronic effects also control the course of the reaction, despite the presence of steric interactions generated between the methyl group and the geminal disubstituted carbon at the vicinal carbons in the adducts 13–17.
Therefore, for these cycloadditions, the regio and stereoselectivities can be ascribed to the electronic effects, which are due to the polarization of the π-systems, and to the steric interactions, the latter mainly caused by the polysubstitution of the dienophiles.
3. Experimental Section
3.1. General Procedures and Instrumentation
All reactions were carried out under nitrogen in anhydrous solvents. All glassware was dried in an oven prior to use. All commercially available compounds were used without further purification. Tetrahydrofuran and benzene were distilled from sodium benzophenone ketyl under an N2 atmosphere prior to use. n-Hexane and ethyl acetate were distilled before use. Melting points (uncorrected) were determined with a Fisher-Johns melting point apparatus. 1H NMR and 13C NMR spectra were recorded on a Varian Mercury (300 MHz) and Varian VNMR System (500 MHz) instruments, in CDCl3 as solvent and with TMS as internal reference. High-resolution mass spectra (HRMS) were obtained with a JSM-GCMate II mass spectrometer, and electron impact techniques (70 eV) were employed. X-ray data were collected on Siemens P4 and Oxford Diffraction Xcalibur S single-crystal X-ray difractometers. Thin-layer Chromatography (TLC) analyses were performed using silica plates and were visualized using UV (254 nm) or iodine. The Knoevenagel adducts 4a–e, 5a–d and 6a–c [27–29] and the exo-2-oxazolidinone dienes 1–3 [16–20] were prepared by the methods described in the literature.
3.2. General Procedures for the Synthesis of Adducts 7a–e, 9a–d, 11a–c, 13a–e/14a–e, 15a–e/16a–e and 17a–c via a Two-Step Reaction. Method A
A mixture of the Knoevenagel adducts 4a–e, 5a–d, or 6a–c (1.2 equiv.) and the corresponding dienes, 1, 2, or 3 (1 mol-equiv.) was placed in a 25 mL two-necked, round-bottomed flask (equipped with a reflux condenser, a rubber septum and under nitrogen atmosphere), and the mixture was stirred and was irradiated with an infrared lamp [33] at 50 °C for ~30 min–6 h under solvent-free conditions until the consumption of the diene (tlc). The reaction mixture was allowed to cool to room temperature, and then purified by column chromatography over silica gel (230–400 mesh) using n-hexane/EtOAc (98:2) as eluent, to afford the corresponding cycloadducts 7a–e, 9a–d, 11a–c, 13a–e/14a–e, 15a–e/16a–e and 17a–c.
3.3. General Procedure for the Synthesis of Adducts 13a–d/14a–d and 15a–d/16a–d via a One-Step Reaction. Method B
A mixture of active methylene compounds 20a–c (1 mol-equiv.), benzaldehydes 21a–d (mol-equiv.) and the corresponding diene 2 (1 mol-equiv.), was placed in a 25 mL two-necked, round-bottomed flask (equipped with a reflux condenser, a rubber septum and under nitrogen atmosphere), and the mixture was stirred and was irradiated with an infrared lamp [33] at 50 °C for ~30 min–6 h, under solvent-free conditions, until the consumption of the diene (tlc). The reaction mixture was allowed to cool to room temperature, and then was purified by column chromatography on silica gel (230–400 mesh) using n-hexane/EtOAc (98:2) as eluent, to afford the corresponding cycloadducts 13a–d/14a–d and 15a–d/16a–d.
(5S*,6R*)-6-Ethoxycarbonyl-6-cyano-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (7a). According to Method A, the reaction between 4a (0.330 g, 0.0016 mol) and diene 1 (0.250 g, 0.0013 mol), followed by flash column chromatography, afforded 7a (0.380 g, 73%) as a white solid: mp 145–146 °C; FT-IR (KBr) νmax 2928, 2362, 1757, 1713, 1598 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.87 (t, J = 6.9 Hz, 3H, OCH2CH3), 2.65 (ddd, J = 17.1, 4.8, 1.5 Hz, 1H, H-4β), 3.13 (dddd, J = 17.1, 11.4, 4.2, 2.1 Hz, 1H, H-4α), 3.16 (dd, J = 16.8, 1.5 Hz, 1H, H-7α), 3.43–3.48 (m, 2H, H-5, H-7β), 3.92 (q, J = 6.9, 2H, OCH2CH3), 7.32–7.46 (m, 10H, H-Ar); 13C NMR (125 MHz, CDCl3) δ 13.3 (OCH2CH3), 25.6 (C-4), 31.4 (C-7), 46.5 (C-5), 49.6 (C-6), 63.2 (OCH2CH3), 117.2 (CN), 120.4 (C-3a), 125.0 (C-9), 125.1 (C-10), 128.0 (C-13), 128.1 (C-11), 128.8 (C-14), 129.5 (15), 130.1 (C-7a), 133.3 (C-8), 136.4 (C-12), 154.0 (C-2), 166.5 (CO2CH2CH3); HRMS (EI+) calcd for C22H16N4O4 388.1423, found (M+) 388.1436.
(5S*,6R*)-6-Ethoxycarbonyl-6-cyano-5-(4-methoxyphenyl)-3-(phenyl)-4,5,6,7-tetrahydrobenzo[d] oxazol-2-one (7b). According to Method A, the reaction between 4b (0.44 g, 0.0019 mol) and diene 1 (0.300 g, 0.0016 mol), followed by flash column chromatography, afforded 7b (0.330 g, 50%) as a pale yellow solid: mp 168–169 °C; FT-IR (KBr) νmax 2934, 2244, 1769, 1716, 1598 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.94 (t, J = 7.2 Hz, 3H, OCH2CH3), 2.61 (ddd, J = 16.2, 4.8, 1.2 Hz, 1H, H-4β), 3.06 (dddd, J = 16.2, 11.4, 4.2, 1.2 Hz, 1H, H-4α), 3.14 (dd, J = 16.2, 2.1 Hz, 1H, 7α), 3.39–3.46 (m, 2H, H-5, H-7β), 3.78 (s, 3H, OCH3), 3.96 (q, J = 7.2 Hz, 2H, OCH2CH3), 6.85 (d, J = 8.7 Hz, 2H, H-14), 7.30 (d, J = 8.7 Hz, 2H, H-13), 7.36–7.48 (m, 5H, H-Ar); 13C NMR (125 MHz, CDCl3) δ 13.4 (OCH2CH3), 25.6 (C-4), 30.1 (C-7), 45.6 (C-5), 49.8 (C-6), 55.2 (OCH3), 63.1 (OCH2CH3), 114.3 (C-14), 117.5 (CN), 120.8 (C-3a), 125.4 (C-9), 128.3 (C-11), 128.6 (C-12), 129.6 (C-13), 129.9 (C-10), 130.4 (C-7a), 133.5 (C-8), 154.0 (C-2), 159.7 (C-15), 166.5 (CO2CH2CH3); HRMS (EI+) calcd for C16H16N2O3 418.1529, found (M+) 418.1526.
(5S*,6R*)-6-Ethoxycarbonyl-5-(4-chlorophenyl)-6-cyano-3-(phenyl)-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (7c). According to Method A, the reaction between 4c (0.452 g, 0.0019 mol) and diene 1 (0.300 g, 0.0016 mol), followed by flash column chromatography, afforded 7c (0.400 g, 60%) as a pale yellow solid: mp 175–177 °C; FT-IR (KBr) νmax 2910, 2256, 1762, 1710, 1566 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.96 (t, J = 7.0 Hz, 3H, OCH2CH3), 2.63 (ddd, J = 16.8, 4.8, 1.8 Hz, 1H, H-4β), 3.05 (dddd, J = 16.8, 11.4, 4.8, 2.1 Hz, 1H, H-4α), 3.16 (dd, J = 16.5, 1.8 Hz, 1H, H-7α), 3.41 (ddd, J = 16.5, 4.2, 1.8 Hz, 1H, H-7β), 3.45 (dd, J = 11.4, 4.8 Hz, 1H, H-5), 3.98 (qd, J = 7.2, 2.0 Hz, 2H, OCH2CH3), 7.30–7.47 (m, 9H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.5 (OCH2CH3), 25.5 (C-4), 31.3 (C-7), 45.7 (C-5), 49.4 (C-6), 63.3 (OCH2CH3), 116.9 (CN), 120.2 (C-3a), 125.1 (C-9), 128.2 (C-11), 129.0 (C-10), 129.6 (C-13), 129.7 (C-14), 130.0 (C-7a), 133.2 (C-8), 134.8 (C-15), 134.9 (C-12), 154.0 (C-2), 166.4 (CO2CH2CH3); HRMS (EI+) calcd for C23H19N2O4Cl 422.1033, found (M+) 422.1032.
(5S*,6R*)-6-Ethoxycarbonyl-6-cyano-5-(4-nitrophenyl)-3-(phenyl)-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (7d). According to Method A, the reaction between 4d (0.47 g, 0.0019 mol) and diene 1 (0.300 g, 0.0016 mol), followed by flash column chromatography, afforded 7d (0.550 g, 80%) as a pale yellow solid: mp 161–163 °C; FT-IR (KBr) νmax 2982, 2246, 1770, 1743, 1600 cm−1; 1H NMR (300 MHz, CDCl3) δ 0.97 (t, J = 6.5 Hz, 3H, OCH2CH3), 2.68 (ddd, J = 18.5, 4.5, 1.5 Hz, 1H, H-4β), 3.09 (dddd, J = 18.5, 11.5, 4.5, 2.0 Hz, 1H, H-4α), 3.22 (dd, J = 16.5 Hz, 0.5, 1H, H-7α), 3.43 (ddd, J = 16.5, 4.5, 2.0 Hz, 1H, H-7β), 3.61 (dd, J = 12.0, 4.5 Hz, 1H, H-5), 4.01 (qd, J = 6.5, 1.6 Hz, 2H, OCH2CH3), 7.36–7.48 (m, 5H, H-Ar), 7.27 (d, J = 9.0 Hz, 2H, H-13), 8.21 (d, J = 9.0 Hz, 2H, H-14); 13C NMR (75.4 MHz, CDCl3) δ 13.5 (OCH2CH3), 25.4 (C-4), 30.5 (C-7), 45.7 (C-5), 49.0 (C-6), 63.6 (OCH2CH3), 116.5 (CN), 119.9 (C-3a), 123.9 (C-9), 125.0 (C-10), 128.3 (C-11), 129.4 (C-13), 129.7 (C-14), 129.7 (C-8), 133.0 (C-7a), 143.6 (C-12), 148.0 (C-15), 153.8 (C-2), 166.0 (CO2CH2CH3); HRMS (EI+) calcd for C23H19N3O6 433.1274, found (M+) 433.1272.
(5S*,6R*)-6-Ethoxycarbonyl-6-cyano-5-(3-nitrophenyl)-3-(phenyl)-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (7e). According to Method A, the reaction between 4e (0.413 g, 0.0019 mol) and diene 1 (0.300 g, 0.0016 mol), followed by flash column chromatography, afforded 7e (0.380 g, 55%) as a pale yellow solid: mp 180–181 °C; FT-IR (KBr) νmax 2925, 2244, 1771, 1741, 1531 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.96 (t, J = 7.2 Hz, 3H, OCH2CH3), 2.69 (ddd, J = 16.8, 4.8, 1.5 Hz, 1H, H-4β), 3.12 (dddd, J = 16.8, 11.4, 5.7, 2.1 Hz, 1H, H-4α), 3.22 (dd, J = 16.8, 1.5 Hz, 1H, H-7α), 3.45 (ddd, J = 16.8, 3.9, 2.1 Hz, 1H, H-7β), 3.65 (dd, J = 11.4, 4.8 Hz, 1H, H-5), 4.01 (qd, J = 7.2, 1.8 Hz, 2H, OCH2CH3), 7.35–7.50 (m, 5H, H-Ar), 7.57 (t, J = 7.5 Hz, 1H, H-16), 7.83 (d, J = 7.5 Hz, 1H, H-17), 8.21 (dd, J = 7.5, 1.8 Hz, 1H, H-15), 8.26 (dd, J = 7.5, 1.8 Hz, 1H, H-13); 13C NMR (125 MHz, CDCl3) δ 13.9 (OCH2CH3), 24.7 (C-4), 31.9 (C-7), 45.8 (C-5), 49.5 (C-6), 64.0 (OCH2CH3), 116.3 (CN), 120.0 (C-3a), 123.7(C-13), 123.8 (C-15), 125.0 (C-9), 128.2 (C-11), 129.4 (C-10), 130.2 (C-7a), 130.3 (C-17), 133.3 (C-8), 134.2 (C-16), 139.0 (C-12), 148.2 (C-14), 153.8 (C-2), 166.2 (CO2CH2CH3). HRMS (EI+) calcd for C23H19N3O6 433.1273, found (M+) 433.1273.
6,6-Dicyano-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (9a). According to Method A, the reaction between 5a (0.345 g, 0.0022 mol) and diene 1 (0.350 g, 0.0018 mol), followed by flash column chromatography, afforded 9a (0.510 g, 80%) as a pale yellow solid: mp 150–151 °C; FT-IR (KBr) νmax 2933, 2251, 1769, 1720, 1501 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.76 (dd, J = 17.1, 5.4 Hz, 1H, H-4β), 3.02–3.13 (m, 2H, H-4α, H-7α), 3.41–3.49 (m, 1H, H-7β), 3.46 (dd, J = 10.8, 5.4 Hz, 1H, H-5), 7.35–7.50 (m, 10H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 24.5 (C-4), 32.5 (C-6), 37.3 (C-7), 46.6 (C-5), 113.3 (CN), 113.6 (CN), 121.1 (C-3a), 125.3 (C-9), 127.9 (C-15), 128.0 (C-14), 128.5 (C-11), 129.7 (C-13), 129.8 (C-10), 129.8 (C-7a), 132.8 (C-8), 134.9 (C-12), 153.6 (C-2); HRMS (EI+) calcd for C21H15N3O2 341.1164, found (M+) 341.1165.
6,6-Dicyano-5-(4-methoxyphenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (9b). According to Method A, the reaction between 5b (0.436 g, 0.0023 mol), and diene 1 (0.370 g, 0.0019 mol) followed by flash column chromatography, afforded 9b (0.400 g, 55%) as a pale yellow solid: mp 157–159 °C; FT-IR (KBr) νmax 2935, 2252, 2217, 1768, 1598 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.73 (dd, J = 16.8, 4.8 Hz, 1H, H-4β), 2.97–3.03 (m, 1H, H-4α), 3.39–4.01 (m, 2H, H-7α, H-7β), 3.44 (dd, J = 10.5, 5.1 Hz, 1H, H-5), 3.78 (OCH3), 6.92 (d, J = 8.7 Hz, 2H, H-14), 7.33–7.39 (m, 5H, H-Ar), 7.45 (d, J = 8.5 Hz, 2H, H-13); 13C NMR (75.4 MHz, CDCl3) δ 24.4 (C-4), 32.2 (C-7), 37.6 (C-6), 45.7 (C-5), 55.2 (OCH3), 113.4 (CN), 113.7 (CN), 114.5 (C-14), 121.0 (C-3a), 125.1 (C-9), 126.8 (C-11), 127.9 (C-13), 129.3 (C-10), 129.4 (C-12), 130.8 (C-7a), 132.8 (C-8), 153.6 (C-2), 160.3 (C-15); HRMS (EI+) calcd for C22H17N3O3 371.1270, found (M+) 371.1270.
5-(4-Chlorophenyl)-6,6-dicyano-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (9c). According to Method A, the reaction between 5c (0.482 g, 0.0025 mol) and diene 1 (0.400 g, 0.0021 mol), followed by flash column chromatography, afforded 9c (0.60 g, 75%) as a pale yellow solid: mp 179–181 °C; FT-IR (KBr) νmax 2923, 2215, 1778, 1549 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.75 (dd, J = 17.5, 5.0 Hz, 1H, H-4β), 3.01–3.07 (m, 1H, H-4α), 3.42–3.47 (m, 3H, H-5, H-7α, H-7β), 7.35–7.50 (m, 9H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 24.4 (C-4), 29.6 (C-7), 37.2 (C-6), 46.1 (C-5), 113.1 (CN), 113.4 (CN), 120.8 (C-3a), 125.3 (C-9), 127.9 (C-7a), 128.6 (C-11), 129.7, 129.7, 129.8, 129.9 (C-10, C-13, C-14), 132.8 (C-8), 133.3 (C-15), 136.0 (C-12), 153.6 (C-2); HRMS (EI+) calcd for C21H14ClN3O2 375.0774, found (M+) 375.0774.
6,6-Dicyano-5-(4-nitrophenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (9d). According to Method A, the reaction between 5d (0.383 g, 0.0019 mol) and diene 1 (0.300 g, 0.0016 mol) followed by flash column chromatography, afforded 9d (0.525 g, 85%) as a pale yellow solid: mp 166–167 °C; FT-IR (KBr) νmax 2918, 2220, 1772, 1599 cm−1; 1H NMR (300 MHz, CDCl3) δ 2.80 (dd, J = 17.7, 5.1 Hz, 1H, H-4β), 3.01–3.16 (m, 1H, H-4α), 3.45–3.50 (m, 2H, H-7α, H-7β), 3.60 (dd, J = 10.5, 5.1 Hz, 1H, H-5), 7.30–7.55 (m, 5H, H-Ar), 7.62 (d, J = 8.5 Hz, 2H, H-13), 8.30 (d, J = 8.5 Hz, 2H, H-14); 13C NMR (75.4 MHz, CDCl3) δ 24.3 (C-4), 32.6 (C-7), 36.8 (C-6), 46.3 (C-5), 113.0 (CN), 113.3 (CN), 120.5 (C-3a), 124.5 (C-14), 125.3 (C-9), 127.8 (C-7a), 128.8 (C-11), 129.4 (C-13), 129.9 (C-10), 133.0 (C-8), 141.5 (C-12), 149.0 (C-15), 153.0 (C-2); HRMS (EI+) calcd for C21H14N4O4 386.1015, found (M+) 386.1015.
6,6-Diethoxycarbonyl-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (11a). According to Method A, the reaction between 6a (0.445 g, 0.0018 mol) and diene 1 (0.280 g, 0.0015 mol), followed by flash column chromatography, afforded 11a (0.243 g, 35%) as a white solid: mp 124–125 °C; FT-IR (KBr) νmax 2926, 1770, 1732, 1502 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.16 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.20 (t, J = 6.9 Hz, 3H, OCH2CH3), 2.58 (d, J = 16.8 Hz, 1H, H-4α), 3.11 (ddd, J = 16.8, 3.9, 2.4 Hz, 1H, H-7β), 3.22–3.32 (m, 2H, H-4α, H-7α), 3.94 (dd, J = 7.2, 2.4 Hz, 1H, H-5), 4.09 (qd, J = 7.2, 4.8 Hz, 2H, OCH2CH3), 4.19 (qd, J = 6.9, 4.8 Hz, 2H, OCH2CH3), 7.13–7.45 (m, 10H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (OCH2CH3), 13.8 (OCH2CH3), 24.5 (C-4), 25.4 (C-7), 42.5 (C-5), 58.3 (C-6), 61.8 (OCH2CH3), 62.1 (OCH2CH3), 120.2 (C-3a), 125.1 (C-13), 127.8 (C-11), 127.8 (C-15), 128.0 (C-10), 128.7 (C-9), 129.4 (C-14), 132.1 (C-7a), 133.6 (C-8), 139.8 (C-12), 154.5 (C-2), 168.3 (CO2CH2CH3), 169.5 (CO2CH2CH3); HRMS (EI+) calcd for C25H25NO6 435.1681, found (M+) 435.1681.
6,6-Diethoxycarbonyl-5-(4-methoxyphenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (11b). According to Method A, the reaction between 6b (0.624 g, 0.0022 mol) and diene 1 (0.350 g, 0.0018 mol), followed by flash column chromatography, afforded 11b (0.215 g, 25%) as a white solid: mp 140–141 °C; FT-IR (KBr) νmax 2929 1769, 1729, 1504 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.20–1.23 (m, 6H, OCH2CH3), 2.85 (d, J = 16.8, 1H, H-4α), 3.11 (ddd, J = 16.8, 3.6, 2.2 Hz, 1H, H-7β), 3.23–3.32 (m, 2H, H-4β, H-7α), 3.48–3.56 (m, 1H, H-5), 3.76 (s, 3H, OCH3), 4.08–4.18 (m, 4H, OCH2CH3), 6.78 (d, J = 9.0 Hz, 2H, H-13), 7.07 (d, J = 9.0 Hz, 2H, H-14), 7.31–7.42 (m, 5H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (OCH2CH3), 13.9 (OCH2CH3), 24.8 (C-7), 25.4 (C-4), 42.5 (C-5), 55.1 (C-6), 61.8 (OCH2CH3), 62.1 (OCH2CH3), 113.8 (C-14), 120.2 (C-3a), 125.1 (C-9), 127.8 (C-11), 129.0 (C-13), 129.4 (C-12), 131.8 (C-10), 132.1 (C-7a), 154.4 (C-2), 159.0 (C-15), 168.5 (CO2CH2CH3), 169.5 (CO2CH2CH3); HRMS (EI+) calcd for C26H27NO7 465.1787, found (M+) 465.1787.
5-(4-Chlorophenyl)-6,6-diethoxycarbonyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (11c). According to Method A, the reaction between 6c (0.542 g, 0.0019 mol) and diene 1 (0.300 g, 0.0016 mol), followed by flash column chromatography, afforded 11c (0.225 g, 30%) as a white solid: mp 167–169 °C; FT-IR (KBr) νmax 2981, 1770, 1732, 1503 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.18 (t, J = 7.5 Hz, 3H, OCH2CH3), 1.19 (t, J = 7.5 Hz, 3H, OCH2CH3), 2.57 (dd, J = 17.1, 2.4 Hz, 1H, H-4α), 3.08 (ddd, J = 17.1, 3.3, 2.4 Hz, 1H, H-7β), 3.18–3.30 (m, 2H, H-4β, H-7α), 3.90 (dd, J = 6.9, 2.4 Hz, 1H, H-5), 4.05–4.21 (m, 2H, OCH2CH3), 7.10 (d, J = 8.7 Hz, 2H, H-13), 7.24 (d, J = 8.7 Hz, 2H, H-14), 7.30–7.46 (m, 5H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (OCH2CH3), 13.9 (OCH2CH3), 24.8 (C-7), 25.4 (C-4), 42.2 (C-5), 58.2 (C-6), 61.9 (OCH2CH3), 62.2 (OCH2CH3), 120.0 (C-3a), 125.1 (C-9), 127.9 (C-11), 128.8 (C-14), 129.4 (C-13), 129.5 (C-10), 132.0 (C-15), 133.7 (C-7a), 138.3 (C-12), 154.4 (C-2), 168.2 (CO2CH2CH3), 169.3 (CO2CH2CH3); HRMS (EI+) calcd for C25H24NO6Cl 469.1292, found (M+) 469.1292.
(5R*,6S*,7R*)-6-Cyano-6-ethoxycarbonyl-7-methyl-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (13a). (5R*,6S*,7S*)-6-Cyano-6-ethoxycarbonyl-7-methyl-3,5-diphenyl-4,5,6,7-tetrahydrobenzo [d]oxazol-2-one (14a). According to Method A, the reaction between 4a (0.480 g, 0.0023 mol) and diene 2 (0.400 g, 0.0020 mol) gave a mixture of isomers 13a/14a (80:20) as a white solid. The isomers were separated by flash column chromatography, giving 0.450 g (56%) of 13a as white solid, mp 165–166 °C and 0.250 g (32%) of 14a as a pale yellow solid, mp 165–167 °C. Data of 13a: FT-IR (KBr) νmax 2984, 2362, 1769, 1750, 1500 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.11 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.35 (d, J = 6.9 Hz, 3H, H-16), 2.62 (dd, J = 17.1, 5.4 Hz, 1H, H-4β), 2.93 (ddd, J = 17.1, 11.1, 1.8 Hz, 1H, H-4α), 3.49–3.54 (m, 2H, H-5, H-7), 4.09 (q, J = 7.2 Hz, 2H, OCH2CH3), 7.26–7.51 (m, 10H, H-Ar); 13C NMR (125 MHz, CDCl3) δ 13.7 (OCH2CH3), 15.9 (C-16), 27.0 (C-4), 37.0 (C-7), 40.4 (C-5), 52.2 (C-6), 62.9 (OCH2CH3), 118.2 (CN), 120.0 (C-3a), 125.2 (C-9), 128.1 (C-11), 128.2 (C-15), 128.4 (C-14), 128.7 (C-13), 129.6 (C-10), 133.3 (C-7a), 134.6 (C-8), 138.0 (C-12), 154.0 (C-2), 164.7 (CO2CH2CH3); Data of 14a. FT-IR (KBr) νmax 2984, 2262, 1769, 1750, cm−1; 1H NMR (500 MHz, CDCl3) δ 0.86 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.42 (d, J = 6.9 Hz, 3H, H-16), 3.13 (ddd, J = 17.1, 11.7, 4.5 Hz, 1H, H-4α), 3.48–3.53 (m, 2H, H-5, H-7), 3.95 (q, J = 7.2 Hz, 2H, OCH2CH3), 7.24–7.58 (m, 10H, H-Ar); 13C NMR (125 MHz, CDCl3) δ 13.4 (OCH2CH3), 15.7 (C-16), 26.8 (C-4), 36.4 (C-7), 154.1 (C-2); HRMS (EI+) calcd for C24H22N2O4 402.1579, found (M+) 402.1580.
Method B. Reaction of ethyl 2-cyanoacetate 20a (0.200 g, 0.0017 mol), benzaldehyde 21a (0.184 g, 0.0017 mol) with exo-2-oxazolidinone diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 13a/14a (65:35). The isomers were separated by flash column chromatography, giving 0.340 g (43%) of 13a and 0.095 g (12%) of 14a.
(5R*,6S*,7R*)-6-Cyano-6-ethoxycarbonyl-5-(4-methoxyphenyl)-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (13b). (5R*,6S*,7S*)-6-Cyano-6-ethoxycarbonyl-5-(4-methoxyphenyl)-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (14b). According to Method A, the reaction between 4b (0.480 g, 0.0020 mol) and diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 13b/14b (75:25) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.520 g (65%) of major isomer 13b as pale yellow solid: mp 172–174 °C. Data of 13b: FT-IR (KBr) νmax 2933, 2200, 1767, 1754, 1501 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.14 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.37 (d, J = 6.9 Hz, 3H, H-16), 2.59 (dd, J = 17.1, 4.8 Hz, 1H, H-4β), 2.86–2.95 (m, 1H, H-4α), 3.46–3.51 (m, 2H, H-5, H-7), 3.83 (s, 3H, OCH3), 4.09 (q, J = 7.2 Hz, 2H, OCH2CH3), 6.84 (d, J = 8.7 Hz, 2H, H-14), 7.27–7.47 (m, 7H, H-Ar). Signals attributed to minor isomer 14b: 0.92 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.42 (d, J = 6.9 Hz, 3H, H-16), 2.38 (dd, J = 17.1, 4.8 Hz, 1H, H-4β), 3.08 (dd, J = 16.8, 10.9 Hz, 1H, H-4α), 3.43–3.60 (m, 2H, H-5, H-7), 3.96 (q, J = 7.2 Hz, 2H, OCH2CH3); 13C NMR (75.4 MHz, CDCl3) δ 14.0 (OCH2CH3), 16.2 (C-16), 27.2 (C-7), 31.2 (C-4), 37.1 (C-5), 52.8 (C-6), 55.4 (OCH3), 63.1 (OCH2CH3), 114.2 (C-14), 118.0 (CN), 120.3 (C-3a), 125.4 (C-9), 127.5 (C-11), 129.5 (C-13), 129.8 (C-10), 130.0 (C-8), 133.4 (C-7a), 134.8 (C-12), 152 (C-2), 159.5 (CO2CH2CH3), 165.0 (C-15); HRMS (EI+) calcd for C25H24N2O5 432.1685, found (M+). 432.1681.
Method B. Reaction of ethyl 2-cyanoacetate 20a (0.190 g, 0.0017 mol) and benzaldehyde 21b (0.230 g, 0.0017 mol) with exo-2-oxazolidinone diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 13b/14b (75:25). The isomers were separated by flash column chromatography, giving 0.300 g (40%) of major isomer 13b.
(5R*,6S*,7R*)-5-(4-Chlorophenyl)-6-cyano-6-ethoxycarbonyl-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (13c). (5R*,6S*,7S*)-5-(4-Chlorophenyl)-6-cyano-6-ethoxycarbonyl-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (14c). According to Method A, the reaction between 4c (0.500 g, 0.0020 mol) and diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 13c/14c (90:10) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.570 g (75%) of major isomer 13c as pale yellow solid: mp 182–184 °C. Data of 13c: FT-IR (KBr) νmax 2931, 2230, 1773, 1719, 1544 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.16 (t, J = 6.9 Hz, 3H, OCH2CH3), 1.33 (d, J = 6.9 Hz, 3H, H-16), 2.58 (dd, J = 16.8, 5.1 Hz, 1H, H-4β), 2.89 (ddd, J = 16.8, 11.1, 1.6 Hz, 1H, H-4α), 3.46–3.54 (m, 2H, H-5, H-7), 4.11 (qd, J = 6.9, 1.5 Hz, 2H, OCH2CH3), 7.28–7.47 (m, 9H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.7 (OCH2CH3), 15.8 (C-16), 27.0 (C-4), 37.0 (C-7), 40.1 (C-5), 52.2 (C-6), 62.9 (OCH2CH3), 118.2 (CN), 120.0 (C-3a), 125.2 (C-9), 128.1 (C-10), 128.4 (C-11), 129.4 (C-13), 129.6 (C-14), 133.2 (C-8), 134.5 (C-7a), 146.0 (C-12), 148.0 (C-15), 154.0 (C-2), 164.2 (CO2CH2CH3); HRMS (EI+) calcd for C24H21N2O4Cl 436.1190, found (M+) 436.1186.
Method B. Reaction of ethyl 2-cyanoacetate 20a (0.17 g, 0.0015 mol) and benzaldehyde 21c (0.200 g, 0.0015 mol) with exo-2-oxazolidinone diene 2 (0.300 g, 0.0015 mol) gave a mixture of isomers 13c/14c (68:32). The isomers were separated by flash column chromatography, giving 0.456 g (64%) of major isomer 13c.
(5R*,6S*,7R*)-6-Cyano-6-ethoxycarbonyl-7-methyl-5-(4-nitrophenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (13d). (5R*,6S*,7S*)-6-Cyano-6-ethoxycarbonyl-7-methyl-5-(4-nitrophenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (14d). According to Method A, the reaction between 4d (0.513 g, 0.0020 mol) and diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 13d/14d (85:15) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.660 g (80%) of major isomer 13d as white solid: mp 178–180 °C. Data of 13d: FT-IR (KBr) νmax 3078, 2983, 2240, 1764, 1710, 1520 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.19 (t, J = 6.9 Hz, 3H, OCH2CH3), 1.34 (d, J = 7.0 Hz, 3H, H-16), 2.64 (dd, J = 16.5, 4.8 Hz, 1H, H-4β), 2.94 (ddd, J = 16.5, 11.4, 1.5 Hz, 1H, H-4α), 3.56–3.67 (m, 2H, H-5, H-7), 4.14 (qdd, J = 6.9, 4.5, 2.7 Hz, 2H, OCH2CH3), 7.37–7.45 (m, 5H, H-Ar), 7.71 (d, J = 8.7 Hz, 2H, H-13), 8.19 (d, J = 8.7 Hz, 2H, H-14). Signals attributed to minor isomer 14d: 0.95 (t, J = 6.9 Hz, 3H, OCH2CH3), 1.47 (d, J = 7.0 Hz, 3H, H-16), 3.12 (ddd, J = 16.5, 11.4, 1.5 Hz, 1H, H-4α), 3.99 (qdd, J = 6.9, 4.5, 2.7 Hz, 2H, OCH2CH3); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (OCH2CH3), 15.7 (C-16), 26.7 (C-4), 37.0 (C-7), 40.0 (C-5), 51.7 (C-6), 63.4 (OCH2CH3), 117.6 (CN), 119.4 (C-3a), 123.7 (C-9), 125.2 (C-10), 128.3 (C-11), 129.4 (C-13), 129.6 (C-14), 133.0 (C-8), 133.1 (C-7a), 134.3 (C-12), 147.6 (C-15), 153.8 (C-2), 164.4 (CO2CH2CH3); HRMS (EI+) calcd for C24H21N3O6 447.1430, found (M+). 447.1450.
Method B. Reaction of ethyl 2-cyanoacetate 20a (0.17 g, 0.0015 mol) and benzaldehyde 21d (0.220 g, 0.0015 mol) with exo-2-oxazolidinone diene 2 (0.300 g, 0.0015 mol) gave a mixture of isomers 13d/14d (75:25). The isomers were separated by flash column chromatography, giving 0.419 g (64%) of major isomer 13d.
(5R*,6S*,7R*)-3-(4-Chlorophenyl)-6-cyano-6-ethoxycarbonyl-7-methyl-5-(3-nitrophenyl)-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (13e). (5R*,6S*,7S*)-3-(4-Chlorophenyl)-6-cyano-6-ethoxycarbonyl-7-methyl-5-(3-nitrophenyl)-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (14e). According to Method A, the reaction between 4e (0.280 g, 0.0015 mol) and diene 3 (0.300 g, 0.0012 mol) gave a mixture of isomers 13e/14e (75:25) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.430 g (70%) 13e as a pale yellow solid: mp 180–182 °C; FT-IR (KBr) νmax 2926, 2230, 1772, 1749, 1529 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.19 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.34 (d, J = 6.9 Hz, 3H, H-16), 2.67 (dd, J = 17.2, 5.4 Hz, 1H, H-4β), 2.95 (ddd, J = 17.2, 11.1, 1.8 Hz, 1H, H-4α), 3.57 (q, J = 6.9 Hz, 1H, H-7), 3.65 (dd, J = 11.1, 5.4 Hz, 1H, H-5), 4.13 (q, J = 7.2 Hz, 2H, OCH2CH3), 7.33 (d, J = 8.7 Hz, 2H, H-9), 7.43 (d, J = 8.7 Hz, 2H, H-10), 7.55 (t, J = 8.1 Hz, 1H, H-17), 7.91 (d, J = 8.1 Hz, 1H, H-18), 8.18 (d, J = 8.4, 3 Hz, 1H, H-15), 8.36 (dd, J = 1.8, 1.2 Hz, 1H, H-13). Signals attributed to minor isomer 14e: 0.92 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.42 (d, J = 6.9 Hz, 3H, H-16), 3.17 (ddd, J = 17.1, 11.1, 1.8 Hz, 1H, H-4α), 4.02 (qd, J = 7.2, 2.1 Hz, 2H, OCH2CH3), 7.82 (d, J = 8.1 Hz, 1H, H-17), 8.21 (d, J = 8.4 Hz, 1H, H-15); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (OCH2CH3), 15.8 (C-16), 26.7 (C-4), 36.9 (C-7), 39.9 (C-5), 51.8 (C-6), 63.4 (OCH2CH3), 117.5 (CN), 119.2 (C-3a), 123.3 (C-11), 124.3 (C-17), 126.4 (C-13), 129.9 (C-10), 134.0 (C-8), 134.1 (C-9), 134.7 (C-15), 139.9 (C-18), 144.0 (C-14), 148.0 (C-2), 164.3 (CO2CH2CH3); HRMS (EI+) calcd for C24H20ClN3O6 481.1040, found (M+) 481.1039.
(5R*,7R*)-6,6-Dicyano-7-methyl-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (15a). (5R*,7S*)-6,6-Dicyano-7-methyl-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (16a). According to Method A, the reaction between 5a (0.386 g, 0.0025 mol) and diene 2 (0.420 g, 0.0020 mol) gave a mixture of isomers 15a/16a (80:20) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.520 g (70%) of major isomer 15a as pale yellow solid: mp 145–147 °C. Data of 15a: FT-IR (KBr) νmax 2979, 2210, 2215, 1777, 1523 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.71 (d, J = 7.2 Hz, 3H, H-16), 2.70 (ddd, J = 17.1, 4.8, 2.1 Hz, 1H, H-4β), 3.14 (ddd, J = 17.1, 11.4, 4.0 Hz, 1H, H-4α), 3.48 (dd, J = 11.4, 4.8 Hz, 1H, H-5), 3.54–3.58 (m, 1H, H-7), 7.35–7.50 (m, 10H, H-Ar). Signals attributed to minor isomer 16a: 1.67 (d, J = 7.2 Hz, 3H, H-16); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (C-16), 24.8 (C-4), 38.6 (C-7), 45.7 (C-6), 47.6 (C-5), 112.0 (CN), 113.5 (CN), 120.7 (C-3a), 125.2 (C-9),128.0 (C-11), 128.5 C (15), 129.4 (C-14), 129.7 (C-13), 129.8 (C-10), 131.9 (C-7a), 132.2 (C-12), 135.3 (C-8), 154.0 (C-2); HRMS (EI+) calcd for C22H17N3O2 355.1320, found (M+) 355.1319.
Method B. Reaction of malononitrile 20b (0.114 g, 0.0017 mol) and benzaldehyde 21a (0.184 g, 0.0017 mol) with exo-2-oxazolidinone diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 15a/16a (70:30). The isomers were separated by flash column chromatography, giving 0.340 g (55%) of major isomer 15a.
(5R*,7R*)-6,6-Dicyano-5-(4-methoxyphenyl)-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (15b). (5R*,7R*)-6,6-Dicyano-5-(4-methoxyphenyl)-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo [d]oxazol-2-one (16b). According to Method A, the reaction between 5b (0.60 g, 0.0030 mol) and diene 2 (0.500 g, 0.0024 mol) gave a mixture of isomers 15b/16b (82:18) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.528 g (55%) of major isomer 15b as pale yellow solid: mp 150–152 °C. Data of 15b: FT-IR(KBr) νmax 2977, 2936, 2235, 2230, 1776, 1597, cm−1; 1H NMR (300 MHz, CDCl3) δ 1.68 (d, J = 7.2 Hz, 3H, H-16), 2.66 (ddd, J = 17.1, 4.8, 2.1 Hz, 1H, H-4β), 3.08 (ddd, J = 17.1, 11.4, 3.9 Hz, 1H, H-4α), 3.47 (dd, J = 11.4, 4.8 Hz, 1H, H-5) 3.52 (qdd, J = 7.2, 3.9, 2.1 Hz, 1H, H-7), 3.80 (s, 3H, OCH3), 6.92 (d, J = 8.7 Hz, 2H, H-14), 7.34–7.48 (m, 7H, H-9, H-10, H-11, H-13). Signals attributed to minor isomer 16b: 1.67 (d, J = 7.2 Hz, 3H, H-16); 13C NMR (75.4 MHz, CDCl3) δ 13.8 (C-16), 24.7 (C-4), 38.3 (C-7), 46.1 (C-6), 46.7 (C-5), 55.2 (OCH3), 111.8 (CN), 113.4 (CN),114.5 (C-14), 120.4 (C-3a), 125.2 (C-9), 127.2 (C-12), 128.4 (C-11), 129.3 (C-13), 129.7 (C-10), 132.0 (C-7a), 132.9 (C-8), 153.7 (C-2), 160.3 (C-15); HRMS (EI+) calcd for C23H19N3O3 385.1426, found (M+) 385.1411.
Method B. Reaction of malononitrile 20b (0.114 g, 0.0017 mol) and benzaldehyde 21b (0.236 g, 0.0017 mol) with exo-2-oxazolidinone diene 2 (0.350 g, 0.0017 mol) gave a mixture of isomers 15b/16b (85:15). The isomers were separated by flash column chromatography, giving 0.478 g (50%) of major isomer 15b.
(5R*,7R*)-5-(4-Chlorophenyl)-6,6-dicyano-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (15c). (5R*,7S*)-5-(4-Chlorophenyl)-6,6-dicyano-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d] oxazol-2-one (16c). According to Method A, the reaction between 5c (0.336 g, 0.0017 mol) and diene 2 (0.300g, 0.0015 mol) gave a mixture of isomers 15c/16c (90:10) as a pale yellow solid, which was purified by flash column chromatography, to yield 0.435 g (75%) of major isomer 15c as pale yellow solid: mp 172–173 °C. Data of 15c: FT-IR (KBr) νmax 2926, 2230, 1754, 1520 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.62 (d, J = 6.9 Hz, 3H, H-16), 2.94 (dd, J = 8.5, 3.0 Hz, 1H, H-4β), 3.31 (ddd, J = 17.1, 11.4, 3.9, Hz, 1H, H-4α), 3.91–3.99 (m, 1H, H-7), 4.03 (dd, J = 11.4, 5.1 Hz, 1H, H-5), 7.40–7.66 (m, 9H, H-Ar). Signals attributed to minor isomer 16c; 13C NMR (125 MHz, CDCl3) δ 14.5 (C-16), 24.3 (C-4), 38.6 (C-7), 43.2 (C-5), 47.3 (C-6), 112.1 (CN), 113.0 (CN), 125.7 (C-9), 128.2 (C-14), 128.4 (C-11), 129.0 (C-13), 130.3, (C-10), 131.0 (C-15), 132.0 (C-7a), 133.4 (C-8), 134.0 (C-12), 154.0 (C-2); HRMS (EI+) calcd for C22H16N3O2Cl 389.0931, found (M+) 389.0901.
Method B. Reaction of malononitrile 20b (0.131 g, 0.0019 mol) and benzaldehyde 21c (0.027 g, 0.0019 mol) with exo-2-oxazolidinone diene 2 (0.400 g, 0.0019 mol) gave a mixture of isomers 15c/16c (70:30). The isomers were separated by flash column chromatography, giving 0.425 g (55%) of 15c and 0.11 g (15%) of 16c.
(5R*,7R*)-6,6-Dicyano-7-methyl-5-(4-nitrophenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (15d). (5R*,7S*)-6,6-Dicyano-7-methyl-5-(4-nitrophenyl)-3-phenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (16d). According to Method A, the reaction between 5d (0.53 g, 0.0026 mol) and diene 2 (0.450 g, 0.0022 mol) gave a mixture of isomers 15d/16d (80:20) as a pale yellow solid. The isomers were separated by flash column chromatography, giving 0.670 g (75%) of 15d as pale yellow solid, mp 169–171 °C and 0.134 g (15%) of 16d as a pale yellow solid, mp 170–171 °C. Data of 16d: FT-IR (KBr) νmax 2925, 2215, 1772, 1599, cm−1; 1H NMR (300 MHz, CDCl3) δ 1.66 (d, J = 6.9 Hz, 3H, H-16), 3.03 (ddd, J = 16.8, 4.8, 2.1 Hz, 1H, H-4β), 3.41 (ddd, J = 16.8, 11.4, 3.9 Hz, 1H, H-4α), 3.92–4.01 (m, 1H, H-7), 4.27 (dd, J = 11.4, 4.8 Hz, 1H, H-5), 7.48–7.57 (m, 5H, H-9, H-10, H-11), 7.95 (d, J = 8.9 Hz, 2H, H-13), 8.35 (d, J = 8.9 Hz, 2H, H-14). Signals attributed to minor isomer 16d: 1.61 (d, J = 6.9 Hz, 3H, H-16), 3.09 (ddd, J = 16.8, 4.8, 2.1 Hz, 1H, H-4β), 3.29 (ddd, J = 16.8, 11.4, 3.9 Hz, 1H, H-4α), 3.81–3.84 (m, 1H, H-7), 4.24 (dd, J = 8.4, 4.8 Hz, 1H, H-5); 13C NMR (75.4 MHz, DMSO-d6) δ 13.9 (C-16), 24.6 (C-4), 38.2 (C-7), 46.0 (C-6), 46.5 (C-5), 112.8 (CN), 114.2 (CN), 121.2 (C-3a), 124.7 (C-14), 126.1 (C-9), 128.7 (C-11), 128.8 (C-10), 130.1 (C-13), 132.5 (C-7a), 131.1 (C-8), 134.4(C-12), 144.5 (C-8), 149.2 (C-15), 154.0 (C-2); HRMS (EI+) calcd for C22H16N4O4 400.1171, found (M+) 400.1165.
Method B. Reaction of malononitrile 20b (0.170 g, 0.0015 mol) and benzaldehyde 21d (0.300 g, 0.0019 mol) with exo-2-oxazolidinone diene 2 (0.300 g, 0.0019 mol) gave a mixture of isomers 15d/16d (70:30). The isomers were separated by flash column chromatography, giving 0.431 g (55%) of 15d and 0.165 g (15%) of 16d.
(5R*,7R*)-3-(4-Chlorophenyl)-6,6-dicyano-5-(4-methoxyphenyl)-7-methyl-4,5,6,7-tetrahydrobenzo [d]oxazol-2-one (15e). (5R*,7S*)-3-(4-Chlorophenyl)-6,6-dicyano-5-(4-methoxyphenyl)-7-methyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (16e). According to Method A, the reaction between 5b (0.280 g, 0.0015 mol) and diene 3 (0.300 g, 0.0012 mol) gave a mixture of isomers 15e/16e (75:25) as a pale yellow solid. The isomers were separated by flash column chromatography, giving 0.387 g (70%) of 15e as pale yellow solid, mp 162–163 °C and 0.083 g (15%) of 16e as a pale yellow solid, mp 162–163 °C. Data of 15e: FT-IR (KBr) νmax 2935, 2250, 1760, 1496 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.67 (d, J = 6.6 Hz, 3H, H-16), 2.69 (ddd, J = 17.4, 5.1, 2.1 Hz, 1H, H-4β), 3.09 (ddd, J = 17.4, 11.7, 3.9 Hz, 1H, H-4α), 3.45 (dd, J = 10.8, 5.1 Hz,1H, H-5), 3.47–3.82 (m, 1H, H-7), 3.82 (s, 3H, OCH3), 6.96 (d, J = 9.0 Hz, 2H, H-14), 7.33 (d, J = 9.0 Hz, 4H, H-13, H-9), 7.40 (d, J = 9.0 Hz, 2H, H-9), 7.46 (d, J = 9.0 Hz, 2H, H-10); 13C NMR (75.4 MHz, CDCl3) δ 14.1 (C-16), 25.1 (C-4), 38.7 (C-7), 46.3 (C-6), 47.1 (C-5), 55.6 (OCH3), 112.1 (CN), 113.6 (CN), 114.9 (C-14), 120.4 (C-3a), 126.7 (C-9), 127.3 (C-12), 129.5 (C-13), 130.2 (C-10), 131.7 (C-8), 132.5 (C-7a), 134.6 (C-11), 153.7 (C-2), 160.5 (C-15). Data of 16e. Yield: 57% (pale yellow solid, mp 163–165 °C); FT-IR (KBr) νmax 2935, 2235, 1760, 1609 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.63 (d, J = 6.9 Hz, 3H, H-16), 2.83–2.98 (m, 2H, H-4), 3.37–3.39 (m, 1H, H-7), 3.54–3.57 (m, 1H, H-5), 3.82 (s, 3H, OCH3), 6.94 (d, J = 8.7 Hz, 2H, H-14), 7.31 (d, J = 8.7 Hz, 4H, H-13, H-9), 7.45 (d, J = 9.0 Hz, 2H, H-10); 13C NMR (75.4 MHz, CDCl3) δ 15.1 (C-16), 24.5 (C-4), 35.1 (C-7), 42.3 (C-6), 43.3 (C-5), 55.3 (OCH3), 112.8 (CN), 113.6 (CN), 114.6 (C-14), 119.6 (C-3a), 126.4 (C-12), 126.5 (C-9), 129.4 (C-13), 129.9 (C-10), 131.3 (C-8), 133.0 (C-7a), 134.3 (C-11), 153.4 (C-2), 160.4 (C-15); HRMS (EI+) calcd for C23H18N3O3Cl 419. 1036, found (M+) 419. 1036.
(5R*,7R*)-6,6-Diethoxycarbonyl-7-methyl-3,5-diphenyl-4,5,6,7-tetrahydrobenzo[d]oxazol-2-one (17a). According to Method A, the reaction between 6a (0.44 g, 0.0017 mol) and diene 2 (0.300 g, 0.0015 mol) produced only the isomer 17a (0.154 g, 23%) as pale yellow solid: mp 132–133 °C. FT-IR: νmax 2980, 1770, 1727, 1503 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.18 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.38 (d, J = 6.9 Hz, 1H, C-16), 2.42 (ddd, J = 17.4, 4.8, 2.1 Hz, H-4α), 3.49–3.59 (m, 2H, H-7, H-4β), 3.82 (dd, J = 6.6, 2.1 Hz, 1H, H-5), 4.04–4.27 (m, 4H, OCH2CH3), 7.23–7.45 (m, 10H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.6 (C-16), 13.8 (OCH2CH3), 14.0 (OCH2CH3), 26.3 (C-7), 29.5 (C-4), 43.3 (C-5), 61.1 (C-6), 61.3 (OCH2CH3), 61.4 (OCH2CH3), 119.5 (C-3a), 125.2 (C-9), 127.7 (C-11), 128.0 (C-12), 128.4 (C-14), 128.6 (C-13), 129.3 (C-10), 133.6 (C-8), 135.1 (C-7a), 154.7 (C-2), 168.5 (CO2CH2CH3) 169.3 (CO2CH2CH3). HRMS (EI+) calcd for C26H27NO6 449.1838, found (M+) 449.1838.
(5R*,7R*)-6,6-Diethoxycarbonyl-5-(4-methoxyphenyl)-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo [d]oxazol-2-one (17b). According to Method A, the reaction between 6b (0.58 g, 0.0028mol) and diene 2 (0.350 g, 0.0017 mol) produced only the isomer 17b (0.269 g, 32%) as a pale yellow solid: mp 143–145 °C. FT-IR: νmax 2980, 1768, 1727, 1504 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.20 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.37 (d, J = 6.6 Hz, 3H, H-16), 2.38 (ddd, J = 17.1, 4.8, 2.4 Hz, 1H, H-4α), 3.47–3.56 (m, 2H, H-4β, H-7), 3.75 (s, 3H, OCH3), 3.78 (dd, J = 6.9, 2.4 Hz, 1H, H-5), 4.10–4.26 (m, 4H, OCH2CH3), 6.77 (d, J = 8.7 Hz, 2H, H-14), 7.04 (d, J = 8.7 Hz, 2H, H-15), 7.28–7.45 (m, 5H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.6 (C-16), 13.8 (OCH2CH3), 13.9 (OCH2CH3), 26.5 (C-4), 29.5 (C-7), 42.5 (C-5), 55.1 (OCH3), 61.2 (C-6), 61.2 (OCH2CH3), 61.3 (OCH2CH3), 113.8 (C-14), 119.5 (C-3a), 125.2 (C-9), 127.7 (C-11), 129.1 (C-10), 129.3 (C-13), 132.5 (C-12), 133.7 (C-7a), 135.2 (C-8), 154.7 (C-2), 158.9 (C-15), 168.6 (CO2CH2CH3), 169.3 (CO2CH2CH3). HRMS (EI+) calcd for C26H27NO6 479.1944, found (M+) 479.1898.
(5R*,7R*)-5-(4-Chlorophenyl)-6,6-diethoxycarbonyl-7-methyl-3-phenyl-4,5,6,7-tetrahydrobenzo[d] oxazol-2-one (17c). According to Method A, the reaction between 4a (144 mg, 2.0 mmol) and diene 2 (162 mg, 1.0 mmol) produced only the isomer 17c (0.121 g, 25%) as a pale yellow solid: mp 166–168 °C. FT-IR: νmax 2980, 1770, 1729, 1597 cm−1; 1H NMR (300 MHz, CDCl3) δ 1.20 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.25 (t, J = 7.2 Hz, 3H, OCH2CH3), 1.38 (d, J = 6.6 Hz, 1H, H-16), 2.40 (dd, J = 17.4, 2.4 Hz, 1H, H-4α), 3.44–3.52 (m, 2H, H-4β, H-7), 3.80 (dd, J = 6.9, 2.4 Hz, 1H, H-5), 4.08–4.26 (m, 4H, OCH2CH3), 7.09 (d, J = 8.4 Hz, 2H, H-14), 7.22 (d, J = 8.4 Hz, 1H, H-13), 7.31–7.45 (m, 5H, H-Ar); 13C NMR (75.4 MHz, CDCl3) δ 13.7 (C-16), 13.8 (OCH2CH3), 13.8 (OCH2CH3), 29.6 (C-7), 31.4 (C-4), 42.5 (C-5), 60.8 (C-6), 61.4 (OCH2CH3), 61.5 (OCH2CH3), 119.2 (C-3a), 125.0 (C-9), 127.7 (C-11), 128.6 (C-10), 129.3 (C-13), 129.5 (C-14), 130.4 (C-7a), 133.5 (C-15), 135.0 (C-8), 138.8 (C-12), 154.5 (C-2), 168.3 (CO2CH2CH3), 168.9 (CO2CH2CH3). HRMS (EI+) calcd for C26H26NO6Cl 483.1448, found (M+) 483.1445.
Dimerization of Diene 2. According to Method B, the reaction between 20a (0.250 g, 0.0023 mol), 21c (0.375 g, 0.0023 mol) and diene 2 (0.474 g, 0.0023 mol), produced two products: 4a (0.375 g, 64%) and dimer 19 (0.0.340 g, 36%) as a pale yellow crystal (acetone/hexane). 19: mp 196–198 °C; IR (KBr) νmax 2924, 17850, 1762, 1706, 1501, 1245 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.38 (d, J = 6.9 Hz, 3H, H-8), 1.81 (d, 1H, J = 6.9 Hz, 3H, H-14), 2.00–2.10 (m, 1H, H-4β), 2.14–2.20 (m, 1H, H-5β), 2.34–2.41 (m, 1H, H-4α), 2.62–2.75 (m, 1H, H-5α), 2.95–3.08 (m, 1H, H-7α), 4.70 (q, J = 6.9 Hz, 1H, H-13), 7.22–7.48 (m, 10H, H-Ar); 13C NMR (125 MHz, CDCl3) δ 10.2 (C-14), 10.3 (C-8), 17.3 (C-4), 32.2 (C-5), 34.0 (C-7), 67.5 (C-6), 99.5 (C-13), 118.9 (C-3a), 124.9 (C-16), 127.9 (C-20), 128.0 (C-22), 129.5 (C-18), 129.8 (C-21), 130.0 (C-17), 133.0 (C-15), 133.4 (C-19), 134.9 (C-7a), 145.6 (C-12), 154.2 (C-2), 154.3 (C-10); HRMS (EI+) calcd for C24H22N2O4 402.1579, found (M+) 402.1578.
3.4. X-ray Structure Study of 7e, 13a and 19
Single crystals were obtained by slow evaporation of concentrated solutions of 7e (n-hexane/AcOEt, pale yellow solid), 13a (n-hexane/CH2Cl2, white solid), and 19 (n-hexane/AcOEt, pale yellow). These were mounted on glass fibers. Crystallographic measurements were performed on a Siemens P-4 difractometer using Mo KR radiation (graphite crystal monochromator, λ = 71073 Ǻ) at room temperature. Three standard reflections, which were monitored periodically, showed no change during data collection. Unit cell parameters were obtained from least-squares refinement of 26 reflections in the range 2° < 2θ < 20°. Intensities were corrected for Lorentz and polarization effects. No absorption correction was applied. Anisotropic temperature factors were introduced for all non-hydrogen atoms. Hydrogen atoms were placed in idealized positions and their atomic coordinates refined. Structures were solved using the SHELXTL [48], SHELX97 [49], or SIR92 [50] programs as implemented in the WinGX suite [51] and refined using SHELXTL or SHELX97 within WinGX, on a personal computer. In all cases ORTEP and packing diagrams were made with PLATON and ORTEP-3 [52–53].
3.5. Theoretical Calculations
The ab initio HF/6-31G(d,p) and DFT B3LYP/6-31G(d,p) calculations were carried out using Gaussian 03 [43] (PC-Linux). Geometries were calculated at the B3LYP/6-31G(d,p) level, and these were employed as the starting point for optimizations at the same level. The energies and coefficients of the frontier molecular orbitals were obtained at single point from the HF/6-31G(d,p) level.
4. Conclusions
In summary, we have successfully developed a new, efficient, regio- and stereoselective Diels-Alder reaction between a series of Knoevenagel adducts as dienophiles and exo-2-oxazolidinone dienes. This process was also satisfactorily carried out via the one-pot, three-component reaction between the corresponding benzaldehydes, the active methylene compounds and the exo-2-oxazolidinone dienes. Both methodologies were promoted by infrared irradiation, as an eco-friendly energy source for the first time, under solvent-free conditions. In all the cases, the para-endo cycloadducts were favored, with respect to the meta or para-exo adducts. An additional advantage of these methods is the fact that the use of a solvent and the activation of the reactions by an acid catalyst were unnecessary, finding environmentally friendly protocols.
Supporting Information
Acknowledgments
We thank Alberto V. Jerezano for their help in spectrometric analyses, and Bruce Allan Larsen for revising the English of the manuscript. J.T. acknowledges SIP-IPN (Grants 20100236 and 20110172) and CONACYT (Grant 83446) and F.D. SIP-IPN (Grants 20101131 and 20110175) and CONACYT (Grant 156933) for financial support. M.I.F-C. thanks CONACYT for a graduate scholarship (No. 206928). F.D. and J.T. are fellows of the EDI-IPN and COFAA-IPN programs.
Footnotes
Supporting Information Available
Tables summarizing 1H and 13C NMR data of the adducts 7a–e, 9a–d, 11a–d, 13a–e, 14a–e, 15a–e, 16a–e and 17a–d including images of IR, 1H and 13C NMR (HMQC, HMBC and NOE experiments) and mass spectra for most of the products. Cartesian Coordinates (B3LYP/6-31G**), energies, and lowest vibrational frequencies (RHF/6-31G**) of the optimized geometries of diene 3 and dienophiles 4a–d, 5a–d and 6a–d. Crystallographic information for 7e, 13a and 19 in CIF format, including X-ray diffraction data, atomic coordinates, thermal parameters and complete bond distances and angles. This material is available free of charge via the Internet at the Cambridge Crystallographic Data Centre (e-mail: deposit@ccdc.cam.ac.uk) as supplementary publication: CCDC 756200 (7e), CCDC 756201 (13a), and CCDC 829554 (19).
References
- 1.Sabot C., Oueis E., Brune X., Renard P.-Y. Synthesis of polisubstituted 3-hydroxypyridines via the revisited hetero-Diels-Alder reaction of 5-alkoxyoxazoles whit dienophiles. Chem. Commun. 2012;48:768–770. doi: 10.1039/c1cc16562c. [DOI] [PubMed] [Google Scholar]
- 2.Suárez-Moreno G.V., González-Zamora E., Méndez F. Oxazole as an electron-deficient diene in the Diels-Alder reaction. Org. Lett. 2011;13:6358–6361. doi: 10.1021/ol202648r. [DOI] [PubMed] [Google Scholar]
- 3.Nawrat C.C., Lewis W., Moody C.J. Synthesis of amino-1,4-benzoquinones and their use in Diels-Alder approaches to the aminonaphthoquinone antibiotics. J. Org. Chem. 2011;76:7872–7881. doi: 10.1021/jo201320g. [DOI] [PubMed] [Google Scholar]
- 4.Martin N, Seoane C., Hanack M. Recent advanced in o-quinodimethane chemistry. Org. Prep. Proc. Int. 1991;23:237–272. [Google Scholar]
- 5.Charlton J.L., Alauddin M.M. Orthoquinodimethanes. Tetrahedron. 1987;43:2873–2889. [Google Scholar]
- 6.Fringuelli F., Taticchi A. The Diels-Alder Reaction Selected Practical Methods. John Wiley; New York, NY, USA: 2002. [Google Scholar]
- 7.Sabitha G., Reddy G.S., Kiran K., Rajkumar M., Yadav J.S., Ramakrishna K.V.S., Kunwar A.C. Iodotrimethysilane induced diastereoselective synthesis of tetrahydropyranones by a tandem Knoevenagel-Michael reaction. Tetrahedron Lett. 2003;44:7455–7457. [Google Scholar]
- 8.Ramachary D.B., Barbas C. Towards organo-click chemistry. Development of organocatalytic multicomponent reactions through combinations of Aldol, Wittig, Knoevenagel, Michael, Diels-Alder and Huisgen cycloaddition reactions. Chem. Eur. J. 2004;10:5323–5331. doi: 10.1002/chem.200400597. [DOI] [PubMed] [Google Scholar]
- 9.Palasz A., Palasz T. Knoevenagel condensation of cyclic ketones with benzoylacetonitrile and N,N'-dimethylbarbituric acid. Application of sterically hindered condensation products in the synthesis of spiro and dispiropyrans by hetero-Diels-Alder reactions. Tetrahedron. 2011;67:1422–1431. [Google Scholar]
- 10.Kuttruff C.A., Zipse H., Trauner D. Concise total syntheses of variecolortides A and B through an unusual Hetero-Diels-Alder reaction. Angew. Chem. Chem. Int. Ed. 2011;50:1402–1405. doi: 10.1002/anie.201006154. [DOI] [PubMed] [Google Scholar]
- 11.Kim I., Kim S.G., Choi J., Lee G.H. Facile synthesis of benzo-fused 2,8-dioxabiclyclo [3.3.1]nonane derivatives via a domino Knoevenagel condensation/hetero-Diels-Alder reaction sequence. Tetrahedron. 2008;64:664–671. [Google Scholar]
- 12.Pizzirani D., Roberti M., Recanatini M. Domino Knoevenagel/Diels-Alder sequence coupled to Suzuki reaction: A valuable synthethic platform for chemical biology. Tetrahedron Lett. 2007;48:7120–7124. [Google Scholar]
- 13.Amantini D., Fringuelli F., Piermatti O., Pizzo F., Vaccaro L. Water, a clean, inexpensive, and re-usable reaction medium. One-pot synthesis of (E)-2-aryl-1-cyano-1-nitroethenes. Green Chem. 2001;3:229–232. [Google Scholar]
- 14.Fernandez I., Dyker C.A., DeHope A., Donnadieu B., Frenking G., Bertrand G. Exocyclic delocalization at the expense of aromaticity in 3,5-bis(π-donor) substituted pirazolium ions and corresponding cyclic bent allene. J. Amer. Chem. Soc. 2009;131:11875–11881. doi: 10.1021/ja903396e. [DOI] [PubMed] [Google Scholar]
- 15.Sikervar V., Fuchs P.L. SN2' addition/1,2-elimination of dimethylsulfonium methilide with epoxy vinyl sulfones: Synthesis of exocyclic cross-conjugated dienyl sulfones. Chem. Commun. 2011;47:3472–3474. doi: 10.1039/c0cc05405d. [DOI] [PubMed] [Google Scholar]
- 16.Hernández R., Sánchez J.M., Gómez A., Trujillo G., Aboytes R., Zepeda G., Bates R.W., Tamariz J. Novel heterocyclic outer-ring dienes: N-alkyl- and N-aryl substituted 4,5-dimethylene-2-oxazolidinones. Heterocycles. 1993;36:1951–1956. [Google Scholar]
- 17.Mandal A.B., Gómez A., Trujillo G., Méndez F., Jiménez H.A., Rosales M.J., Martínez R., Delgado F., Tamariz J. One-step synthesis and highly regio- and stereoselective Diels-Alder of novel exo-2-oxazolidinone dienes. J. Org. Chem. 1997;62:4105–4115. [Google Scholar]
- 18.Fuentes A., Martínez-Palou R., Jiménez-Vázquez H.A., Delgado F., Reyes A., Tamariz J. Diels-Alder reactions of 2-oxazolidinone dienes in polar solvents using catalysis or non-conventional energy sources. Monatsh. Chem. 2005;136:177–192. [Google Scholar]
- 19.Martínez R., Jiménez-Vázquez H.A., Reyes A., Tamariz J. Stereoselective synthesis of 4,5-diethylidene-oxazolidinones as new dienes in Diels-Alder reactions. Helv. Chim. Acta. 2002;85:464–482. [Google Scholar]
- 20.Martínez R., Jiménez-Vázquez H.A., Delgado F., Tamariz J. Synthesis and highly Diels-Alder cycloadditions of the new dienes N-susbtituted 2,3,5,6-tetrahydrobenzoxazol-2-ones. Tetrahedron. 2003;59:481–492. [Google Scholar]
- 21.Benavides A., Peralta J., Delgado F., Tamariz J. Total synthesis of the natural carbazoles murrayanine and murrayafoline A, based on the regioselective Diels-Alder addition of exo-2-oxazolidinone dienes. Synthesis. 2004:2499–2504. [Google Scholar]
- 22.Bernal P., Benavides A., Bautista R., Tamariz J. Exo-2-oxazolidinone dienes in the total synthesis of the natural carbazoles, 6-methoxymurrayanine and clausenine. Synthesis. 2007:1943–1948. [Google Scholar]
- 23.Bernal P., Tamariz J. Total synthesis of murrayanine involving 4,5-dimethyleneoxazolidin-2-ones and a palladium(0)-catalyzed diaryl insertion. Helv. Chim. Acta. 2007;90:1449–1454. [Google Scholar]
- 24.Bautista R., Bernal P., Montiel L.E., Delgado F., Tamariz J. Total synthesis of the natural carbazoles glycozolicine, mukoline, and mukolidine, startingfrom 4,5-dimethyleneoxazolidin-2-ones. Synthesis. 2011:929–933. [Google Scholar]
- 25.Reyes L., Mendoza H., Vázquez M.A., Ortega-Jiménez F., Fuentes-Benítes A., Jiménez-Vázquez H., Flores-Conde M.I., Miranda R., Tamariz J., Delgado F. Synthesis of new polycyclic oxazol-2-one derivatives by a tandem [4+2] cycloaddition/cyclopentannulation/ 1,5-sigmatropic rearrangement process of Fischer (arylalkynyl)(alkoxy)carbenes and exo-2-oxazolidinone dienes. Organometallics. 2008;27:4334–4345. [Google Scholar]
- 26.Ortega-Jiménez F., Benavides A., Delgado F., Jiménez-Vázquez H.A., Tamariz J. Synthesis and reactivity of η4-Diene-Fe(CO)3 complexes from exo-2-oxazolidinone dienes. A facile generation of stable conjugated enol-enamido species. Organometallics. 2010;29:149–159. [Google Scholar]
- 27.Delgado F., Tamariz J., Zepeda G., Landa M., Miranda R., García J. Knoevenagel condensation catalyzed by a mexican bentonite using infrared irradiation. Synth. Commun. 1995;25:753–759. [Google Scholar]
- 28.Obrador E., Castro M., Tamariz J., Zepeda G., Miranda R., Delgado F. Knovenagel condensation in heterogeneous phase catalyzed by IR radiation and tonsil actisil FF. Synth. Commun. 1998;28:4649–4663. [Google Scholar]
- 29.Alcerreca G., Sanabria R., Miranda R., Arroyo G., Tamariz J., Delgado F. Preparation of benzylidene barbituric acids promoted by infrared irradiation, in the absence of solvent. Synth. Commun. 2000;30:1295–1301. [Google Scholar]
- 30.Penieres G., Miranda R., García J., Aceves J., Delgado F. Modification of the Fischer indole synthesis. Heterocycl. Commun. 1996;2:401–402. [Google Scholar]
- 31.Osnaya R., Arroyo G., Parada L., Delgado F., Trujillo J., Salmón S., Miranda R. Biginelli vs Hantzsch esters study under infrared radiation and solventless conditions. Arkivoc. 2003;xi:112–117. [Google Scholar]
- 32.Martínez J., Velasco-Bejarano B., Delgado F., Pozas R., Torres Domínguez H.M., Trujillo J., Arroyo G.A., Miranda R. Eco-contribution to the chemistry of perezone, a comparative study, using different modes of activation and solventless conditions. Nat. Prod. Commun. 2008;3:1465–1468. [Google Scholar]
- 33.Pool G.C., Teuben J.H. IR Radiation as a Heat Source in Vacuum Sublimation. In: Wayda A.L., Darensbourg M.Y.W., editors. Practical Organometallic Chemistry; Symposium Series; Washington DC, USA. 1987. pp. 30–33. [Google Scholar]
- 34.Fleming I. Frontier Orbitals and Organic Chemical Reactions. John Wiley & Sons; Chichester, UK: 1976. [Google Scholar]
- 35.Smith M.B., March J. March’s Advanced Organic Chemistry. Reactions, Mechanisms, and Structure. 5th ed. John Wiley & Sons, Inc; New York, NY, USA: 2001. p. 370. [Google Scholar]
- 36.Argile A., Ruasse M.-F. Reactivity and selectivity control by reactants and products. A general relationship between the selectivity and the position of the transition state. Tetrahedron Lett. 1980;21:1327–1330. [Google Scholar]
- 37.Bowden K., Stewart R. Strongly basic systems—V:H-acidity scale based on the ionization of carbon acids. Tetrahedron. 1965;21:261–266. [Google Scholar]
- 38.Pearson R.G., Dillon R.L. Rates of ionization of pseudo acids. Relation between rates and equilibria. J. Am. Chem. Soc. 1953;75:2439–2443. [Google Scholar]
- 39.Bell R.P. The Proton in Chemistry. Cornell University Press; Ithaca, NY, USA: 1959. [Google Scholar]
- 40.Becke A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 41.Becke A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]
- 42.Lee C.T., Yang W.T., Parr R.G. Development of the Colle-Savetti correlational-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 43.Gaussian 03, revision E.01. Gaussian Inc; Wallingford, CT, USA: 2004. [Google Scholar]
- 44.Fukui K. Recognition of stereochemical paths by orbital interaction. Accounts Chem. Res. 1971;4:57–64. [Google Scholar]
- 45.Eisenstein O., Lefour J.M., Anh N.T., Hudson R.F. Simple prediction of cycloaddition orientation. I. Diels-Alder reactions. Tetrahedron. 1977;33:523–531. [Google Scholar]
- 46.Houk K.N. Generalized frontier orbitals of alkenes and dienes. Regioselectivity in Diels-Alder reactions. J. Am. Chem. Soc. 1973;95:4092–4094. [Google Scholar]
- 47.Sustmann R. Orbital energy control of cycloaddition reactivity. Pure Appl. Chem. 1974;40:569–593. [Google Scholar]
- 48.Sheldrick G.M. A short history of SHELX. Acta Crystallogr. 2008;A64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 49.SHELX97, release 97-2; programs for crystal structure analysis. Institüt für Anorganische Chemie der Universität; Göttingen, Germany: 1997. [Google Scholar]
- 50.Altomare A., Cascarano G., Giacovazzo C., Guagliardi A. Completion and refinement of crystal structures with SIR92. J. Appl. Crystallogr. 1993;26:343–350. [Google Scholar]
- 51.Farrugia L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999;32:837–838. [Google Scholar]
- 52.Spek A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003;36:7–13. [Google Scholar]
- 53.Farrugia L.J. ORTEP-3 for Windows-a version of ORTEP-III with a Graphical User Interface (GUI) J. Appl. Crystallogr. 1997;30:565. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.