

Abstract
The synthesis of several novel chiral phosphoramidite ligands (L1–L8) with C2 symmetric, pseudo C2 symmetric secondary amines and chiral Brønsted acids 1a,b has been achieved. These chiral auxiliaries were obtained from commercially available d-mannitol, and secondary amines in moderate to excellent yields. Excellent diastereoselectivites of ten chiral auxiliaries were obtained. The chiral phosphoramidite ligands and chiral Brønsted acids were fully characterized by spectroscopic methods.
Keywords: phosphoramidite, Brønsted acid, d-mannitol
1. Introduction
Asymmetric catalysis is one of the most cost-effective and environmentally friendly methods for the production of a large variety of enantiomerically enriched molecules [1,2]. An important area of research in asymmetric catalysis involves designing enantiopure ligands and transition metal catalysts which can lead to an efficient and selective transformation. Phosphoramidites (Figure 1) have recently attracted considerable interest as ligands in transition-metal catalyzed organic transformations [3–13]. Phosphoramidites are a versatile ligand class, which can serve as two-, four-, six- or eight-electron donors [14]. Privileged monodentate ligands are often based on chiral BINOL or TADDOL backbones (Figure 1), which are combined with phosphorus (III) reagent and a carbon or heteroatom substituent in a modular way [15–24].
Figure 1.

Chiral phosphoramidite ligands and Brønsted acid derived from BINOL or TADDOL backbone.
The modular assembly makes these ligands suitable for systematic screenings, and that makes general protocols for their rapid synthesis highly desirable. Originally described by Feringa [18], they are increasingly applied as ligands in transition-metal catalyzed organic transformations, such as enantioselective conjugate enone addition reactions [11,25,26], hydrogenations [3,5,6,8], allylic alkylations [9], hydrosilylations [27], vinylations [28], cycloadditions [29–31], Diels-Alder [32] and Heck reactions [33].
We have been developing a new class of chiral monodentate phosphoramidite ligands and chiral Brønsted acid derived from readily accessible enantiopure axially chiral DIOL units (Figure 1). One of the salient features of these novel monodentate phosphorus ligands is their fine-tuning capability through modifications of the R, and Ar groups. This feature is of critical importance because it allows a combinatorial approach to discover the most efficient ligand for a specific reaction or process.
2. Results and Discussion
2.1. Synthesis of Phosphoramidite Ligands
Our aim was to design and synthesize a library of chiral monophosphoramidite ligands decorated with electron-donating as well as electron-withdrawing groups in addition to sterically-demanding substituents. The general procedure is shown in Table 1. The starting optically-active DIOLs I used in these syntheses were prepared according to the literature [34]. The amines used were commercially available or were synthesized from (R)-α-methyl benzyl amine according to the literature [35].
Table 1.
Results of synthesis of chiral phosphoramidite ligands.
| # | Compound | Ligand | Ar | δ P a | Yield [%] b |
|---|---|---|---|---|---|
| 1 | L1 | 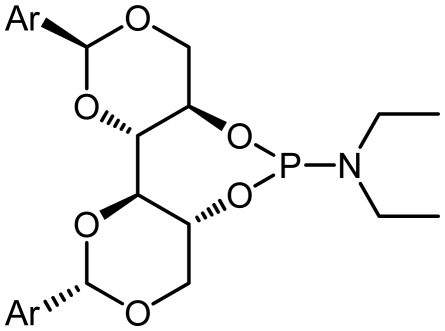 |
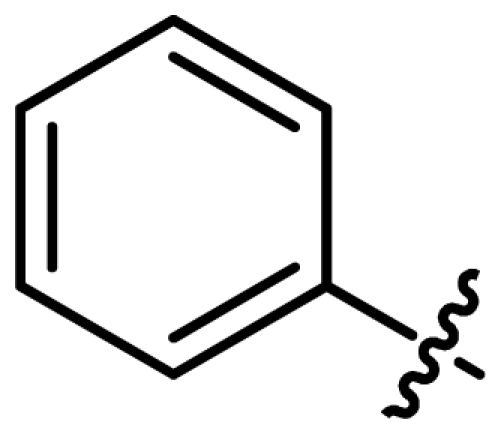 |
127.2 | 55 |
| 2 | L2 | 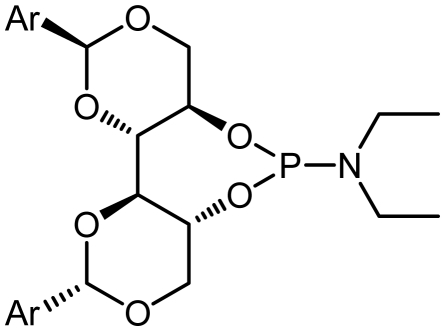 |
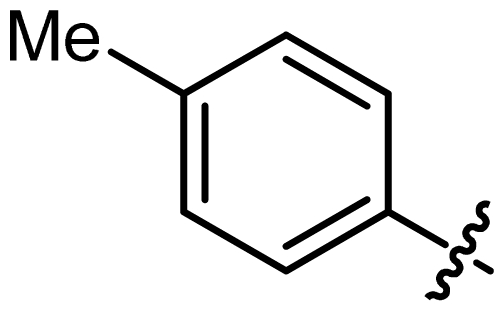 |
127.12 | 35 |
| 3 | L3 | 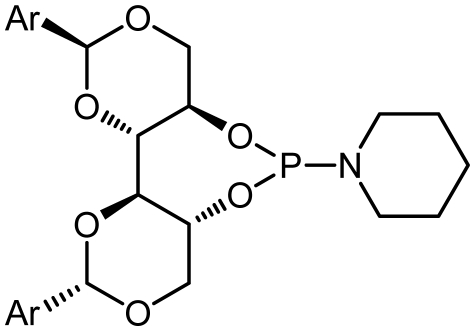 |
 |
122.86 | 45 |
| 4 | L4 | 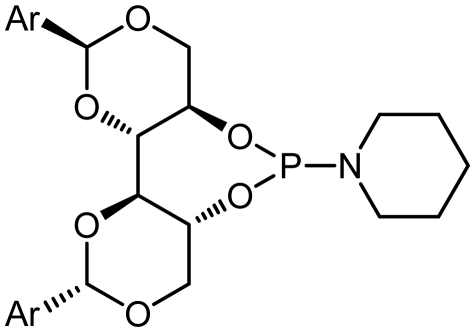 |
 |
122.60 | 40 |
| 5 | L5 | 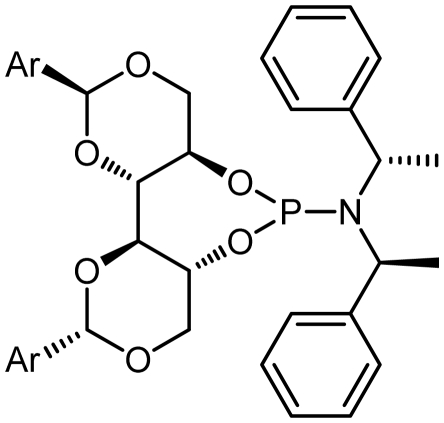 |
 |
134.65 | 31 |
| 6 | L6 | 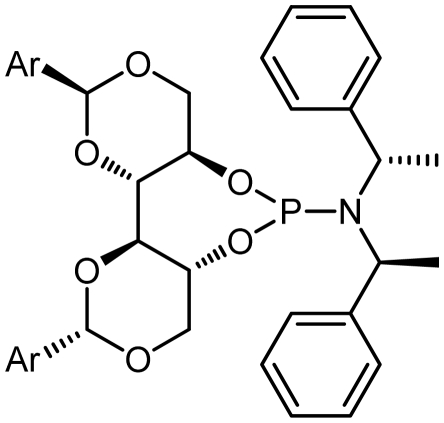 |
 |
132.50 | 45 |
| 7 | L7 | 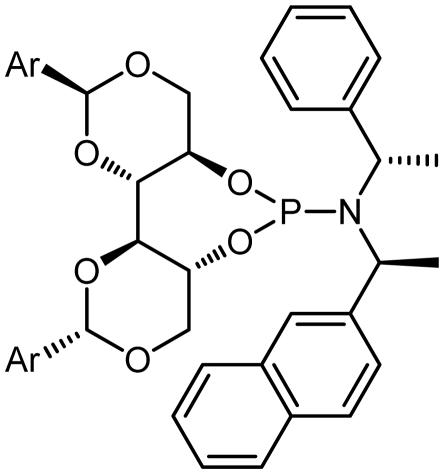 |
 |
135.01 | 50 |
| 8 | L8 | 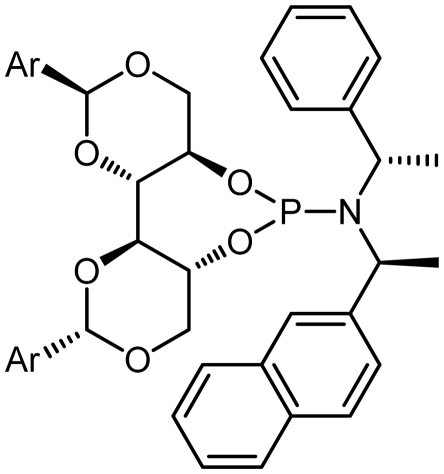 |
 |
134.69 | 38 |
Determined by 31P NMR;
Isolated yield after column chromatography.
The synthetic procedure started with the reaction of amine derivatives with purified PCl3 and Et3N as base in DCM at 0 °C. The resulting intermediate II was treated with one equivalent of DIOLs I. The ligands were obtained as white or pale yellow solids or oily products in moderate to good yields (Scheme 1).
Scheme 1.

Synthesis of chiral monodentate phosphorus ligands L1–L8.
The ligands synthesized by this method are shown in Table 1. Ligands L1 and L2 were substituted with a diethyl amine group at phosphorus (Table 1, entries 1 and 2). The steric hindrance is even more pronounced in ligand L2, with tolyl instead of phenyl moieties in the DIOL I backbone. This might also account for the rather low chemical yield (35% as compared to 55%). The 1H, 13C and 31P NMR spectra were as expected for these ligands.
Encouraged by these preliminary results, Ligands L3–L8 were efficiently synthesized in one step using the same methodology related Ligands L1 and L2.
The 31P NMR spectroscopic data for ligands L1–L8 are summarized in Table 1. It was found that all phosphoramidite ligands were obtained in excellent isomer purity based on 31P NMR. In some cases, it was observed that minor product isomers of phosphoramidites are evidenced by 31P NMR. Unfortunately, the resulting product oxidized either from aerobic oxidation of the desired phosphoramidite ligands during isolation, or from oxidation of the intermediate dialkylaminophosphorous dichloride (Figure 2). The major and minor isomers of phosphoramidite ligands were not separable by column chromatography. Subsequently, for structure confirmation, the mass spectrum of the new product was recorded. X-ray crystal structure analysis is one possibility to determine the structure unambiguously. Several attempts were made to obtain suitable crystal for X-Ray measurements, but were unsuccessful due to the microcrystalline nature of the products.
Figure 2.

31P NMR data of the mixture isomers of L2.
Ligand L1 was obtained by a similar procedure with diethyl amine, using the DCM as the reaction solvent. Similarly, there are four isomers in the mixture, with one isomer dominating the others. The 31P NMR analysis identified the major isomer at δ = 127.2, while the minor isomers showed chemical shift of δ 134.6, 135.12 respectively.
Given that other L3, L4-phosphoramidites were synthesized, a similar strategy was used with piperidine as secondary amine in 45 and 40% yields respectively. Introduction of C2 symmetric and pseudo C2 symmetric secondary amines of the DIOLs I scaffold would accomplish the same aims as set out. The phosphoramidites ligands L1–L8 are colorless liquids or white solids, which are readily soluble in common organic solvents (Scheme 1). They were fully characterized by 1H, 13C and 31P NMR spectroscopy, mass spectrometry as well as by elemental analysis. Compounds L1–L8 and their solutions must be kept under anhydrous conditions due to their sensitivity to moisture.
2.2. Synthesis of Chiral Brønsted Acids
Chiral Brønsted acids have emerged as efficient enantioselective catalysts for a variety of organic transformations [35–39]. A critical factor in achieving high stereoselectivities in these transformations is the hydrogen bond formed between the donor site of the acid catalyst and the acceptor (basic) site of the electrophilic component, X-H…Y (X and Y are heteroatoms) [40–45]. In this regard, C-H…X (X = O or N) hydrogen bonding interactions have recently been identified as an important factor in some stereoselective transformations [46–49]. Thus, we decided to synthesize 1a–e and evaluate their utility as a recyclable asymmetric organocatalyst (Scheme 2). Thus, the synthesis of chiral Brønsted acids 1a–e was achieved from DIOL I according to procedures set out in the literature [50]. Subsequent reaction of 1a with POCl3 in pyridine at 90 °C, followed by treatment with water and acidification, afforded phosphoric acid 1a in an excellent overall yield (87%). It should be noted that this reaction is very sensitive to both the concentration of acid, and the time as well. Subsequently, for structure confirmation, a melting point 255 °C for phosphoric acid derivatives 1a was observed: the temperature for DIOL I (entry 1, Table 2, Ph) being 192 °C. The resulting chiral phosphoric acid 1a was fully characterized by 1H, 13C, and 31P NMR spectroscopy, mass spectrometry as well as by elemental analysis. The 31P NMR analysis revealed that only one major product at δ = −1.78 was obtained as depicted in Figure 3.
Scheme 2.

Synthesis of chiral brønsted acids 1a,e.
Table 2.
Results of synthesis of chiral Brønsted acids having aromatic groups in the auxiliary.
| # | Compound 1 | Ar | δ P a | Yield [%] b |
|---|---|---|---|---|
| 1 | a | C6H5 | −1.78 | 87 |
| 2 | b | p-CH3C6H4 | −1.83 | 81 |
| 3 | c | p-CH3OC6H4 | - | - |
| 4 | e | 2,4-diClC6H3 | - | - |
Determined by 31P NMR;
Isolated yield after column chromatography;
-: no product isolated.
Figure 3.

31P NMR data of the 1a,b.
Having identified the optimal reaction conditions, we next examined the scope and limitations of this reaction using various protecting benzylidine moieties with different substituents on the benzene rings; the results are summarized in Table 2. As is shown in Table 2, in the case of the electron-withdrawing group at the 4-position of the benzene ring of DIOL I, the reactions proceeded smoothly to give an excellent yield of 1b (up to 87%) along with excellent diastereoselectivites (Table 2, entry 2). In the case of electron donating group at 4- or at 2,4-positions of the benzene ring of DIOL I, no products were obtained (Table 2, entries 3 and 4).
We are interested in exploring derivatives with alternative acidic and basic sites to further expand the utility of this fascinating type of organocatalyst [51]. Interestingly, when chiral of Brønsted acid 1a was used to prepare N-morpholino phosphoramidate 2, the reaction failed (Scheme 3).
Scheme 3.

N-Morpholino phosphoramidate as a new motif for asymmetric Brønsted acid catalysis.
2.3. Applications
Chiral dihydropyrimidinethiones (DHPMs) have found increasing applications in the synthesis of pharmaceutically-relevant substances exhibiting a wide range of important pharmacological properties. The Biginelli reaction, one of the most useful multicomponent reactions, offers an efficient way to access multi functionalized 3,4-dihydropyrimidin-2-(1H)-ones (DHPMs). Initial screening experiments were performed by applying a Biginelli reaction initiated with the condensation of an aldehyde with urea or thiourea in the presence of a Brønsted acid (Scheme 4). Utilizing 1 equiv. of 4-chlorobenzaldehyde, 1.2 equiv. of thiourea, 3.0 equiv. of ethyl 3-oxobutanoate, and 10 mol% of 1a in DCM and stirred at RT for 4 days. Formation of dihydropyrimidinethiones (DHPMs) was not observed. Although the reaction was carried out at elevated temperature at 70 °C for 6 days, no catalytic activity was observed. From these initial attempts, it is clear that there is no sign of catalytic activity of 1a towards Biginelli reaction.
Scheme 4.

Biginelli reaction.
3. Experimental Section
General: All the moisture and air sensitive reactions were carried out under an inert atmosphere of an argon-filled glove box and standard Schlenk-line techniques. All the chemicals were purchased from Aldrich, Sigma-Aldrich, Fluka etc., and were used without further purification, unless otherwise stated. Toluene was distilled using Na/benzophenone. CH2Cl2 was dried from CaH2. Silica gel (SiO2; 100–200 mesh) was used for Flash column chromatography. All melting points were measured on a Gallenkamp melting point apparatus in open glass capillaries and are uncorrected. IR Spectra were measured as KBr pellets on a Nicolet 6700 FT-IR spectrophotometer. The NMR spectra were recorded on a Jeol-400 NMR spectrometer. 1H NMR (400 MHz), 13C NMR (100 MHz) and 31P NMR were run in deuterated dimethylsulphoxide (DMSO-d6 or CDCl3). Chemical shifts (δ) are referred in terms of ppm and J-coupling constants are given in Hz. Mass spectra were recorded on a Jeol of JMS-600 H. Elemental analysis was carried out on a Perkin Elmer 2400 Elemental Analyzer; CHN mode. Optical rotations were measured on a Polarimeter, polax-2L.
3.1. General Procedure for the Synthesis of C2 Symmetric and Pseudo C2 Symmetric Secondary Amines (Procedure A) [35]
A mixture of the appropriately substituted ketone (10 mmol, 1.0 eq.) and amine derivatives (10 mmol, 1.0 eq.) in Ti(Oi-Pr)4 (30 mmol, 3.0 eq.) was stirred for 45 min. Pd/C (10%, 200 mg) was added and the mixture stirred under an atmosphere of hydrogen for 48 h. An aqueous solution of NaOH (1 M, 20 mL) was added and the mixture stirred for 45 min. Water (50 mL) was added and the mixture extracted with ethyl acetate (5 × 50 mL). The organic extracts were dried over MgSO4, filtered and concentrated to give the desired amine. If necessary, flash chromatography on silica gel (diethyl ether in petroleum ether) could be used to separate diastereomers, though little, if any separation was observed by thin-layer chromatography so, GC analysis is necessary.
3.2. (R)-Bis((R)-1-Phenylethyl) Amine
Following Procedure A, (R)-bis((R)-1-phenylethyl) amine was obtained from acetophenone (1.20 gm, 10 mmol, 1.0 eq.) and (R)-α-methyl benzyl amine (1.21 gm, 10 mmol, 1.0 eq.) in Ti(Oi-Pr)4 (9.0 mL, 30 mmol, 3.0 eq.) which was obtained as yellowish oil in quantitative yield.
1H NMR (400 MHz, CDCl3, 21 °C): δ = 7.35–7.21 (m, 5 H, C6H5), 3.51 (q, J = 6.6 Hz, 1H, CHCH3), 2.2 (br, 1H, NH), 1.29 (d, J = 6.6 Hz, 3H, CHCH3).
The other analytical data are in accordance with the literature [35].
3.3. (R)-1-(Naphthalen-2-yl)-N-((R)-1-Phenylethyl) Ethanamine
Following Procedure A, (R)-1-(Naphthalen-2-yl)-N-((R)-1-phenylethyl)ethanamine was obtained from 2-acetonaphthone (1.70 gm, 10 mmol, 1.0 eq.) and (R)-α-methyl benzyl amine (1.21 gm, 10 mmol, 1.0 eq.) in Ti(Oi-Pr)4 (9.0 mL, 30 mmol, 3.0 eq.) which was obtained as yellowish oil in quantitative yield.
1H NMR (400 MHz, CDCl3, 21 °C): δ = 7.88 (t, J = 9.1 Hz, 2H), 7.76 (d, J = 8.1 Hz, 1H), 7.69 (d, J = 6.9 Hz, 1H), 7.54–7.23 (m, 6H), 7.18–7.14 (m, 2H), 4.39 (q, J = 6.6 Hz, 1H), 3.59 (q, J = 6.6 Hz, 1H), 1.37 (d, J = 6.6 Hz, 3H), 1.34 (d, J = 6.9 Hz, 3H).
The other analytical data are in accordance with the literature [50].
3.4. General Procedure for the Preparation of Phosphoramidites (Procedure B)
Triethylamine (7 mmol, 5.0 eq.) was added dropwise to a solution of phosphorus trichloride (1.4 mmol, 1.0 eq.) in dichloromethane (5 mL) at 0 °C. The solution was warmed to room temperature and the amine (1.4 mmol, 1.0 eq.) was added neat as either the free base or HCl salt. The mixture was stirred for 5 h, at which time DIOL I (1.4 mmol, 1.0 eq.) was added neat and the mixture stirred overnight. The suspension was concentrated and the ligand purified by flash chromatography on silica gel (dichloromethane in petroleum ether with 1% triethylamine) to give the ligand as an oily substance which solidifies on standing or as a foaming solid.
3.5. (4aR,7aR,11aS,11bS)-N,N-Diethyl-2,10-Diphenylhexahydrobis([1,3]Dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepin-6-amine (L1)
Following Procedure B, L1 was obtained from Triethylamine (971 μL, 7 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), diethyl amine (102 mg, 143 μL, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-diphenyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (500 mg, 1.4 mmol, 1.0 eq.) which was obtained as an oily product (355 mg, 0.77 mol, 55%); IR (KBr, cm−1): νmax = 3436, 1612, 1369; 1H NMR (400 MHz, CDCl3): δ = 7.49–7.34 (m, 5H, Ph), 5.54 (s, 1H, PhCH), 4.36 (q, 1H, OCH2), 4.24 (m, 1H, CHO), 3.94 (d, 1H, J = 8.8 Hz, OCH2), 3.81 (m, 1H, CHOP), 3.18 (m, 2H, CH2CH3), 1.10 (t, 3H, J = 7.3 Hz, CH3); 13C NMR (100 MHz, CDCl3): δ = 137.3, 128.3, 126.2, 126.1, 100.7, 100.4, 82.8, 81.6, 38.6, 38.4; 31P NMR (130 MHz, CDCl3): δ = 127.2; MS (m/z): 460.47 [M + 1]+, 47%; Anal. for C24H30NO6P; calcd: C, 62.74; H, 6.58; N, 3.05. Found: C, 62.50; H, 6.49; N, 3.00.
3.6. (4aR,7aR,11aS,11bS)-N,N-Diethyl-2,10-di-p-Tolylhexahydrobis([1,3]Dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepin-6-amine (L2)
Following Procedure B, L2 was obtained from Triethylamine (971 μL, 7 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), diethyl amine (102 mg, 143 μL, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-di-p-tolyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (541 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (265 mg, 0.49 mol, 35%); m.p.: 65 °C; IR (KBr, cm−1): νmax = 3435, 1610, 1345; 1H NMR (400 MHz, CDCl3): δ = 7.37–7.34 (m, 2H, Ph), 7.17–7.14 (m, 2H, Ph), 5.46 (s, 1H, PhCH), 4.33(q, 1H, OCH2), 4.22 (m, 1H, CHO), 3.89 (d, 1H, J = 8.8 Hz, OCH2), 3.76 (m, 1H, CHOP), 3.21–3.16 (m, 2H, CH2CH3), 2.36 (s, 3H, C6H4CH3), 1.09 (t, 3H, J = 6.6 Hz, CH3); 13C NMR (100 MHz, CDCl3): δ = 138.7, 134.6, 128.9, 126.1, 100.7, 100.5, 82.8, 81.5, 38.6, 21.3, 14.8; 31P NMR (130 MHz, CDCl3): δ = 127.1; MS (m/z): 488.55 [M + 1]+, 40%; Anal. for C26H34NO6P; calcd: C, 64.05; H, 7.03; N, 2.87. Found: C, 64.00; H, 7.00; N, 2.95.
3.7. 1-((4aR,7aR,11aS,11bS)-2,10-Diphenylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]dioxaphosphepin-6-yl)piperidine (L3)
Following Procedure B, L3 was obtained from Triethylamine (971 μL, 7 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), piperidine (121 mg, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-diphenyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (500 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (265 mg, 0.49 mol, 35%); m.p.: 110 °C; IR (KBr, cm−1): νmax = 3444, 1607, 1350; 1H NMR (400 MHz, CDCl3): δ = 7.53–7.31 (m, 5H, Ph), 5.50 (s, 1H, PhCH), 4.37 (q, 1H, OCH2), 4.24 (m, 1H, CHO), 3.91 (d, 1H, J = 8.8 Hz, OCH2), 3.79 (m, 1H, CHOP), 3.19 (m, 2H, CH2CH2), 1.63 (m, 2H, CH2CH2CH2), 1.49 (m, 2H, CH2CH2CH2); 13C NMR (100 MHz, CDCl3): δ = 137.7, 129.0, 128.3, 126.2, 100.9, 82.8, 82.1, 76.7, 45.6, 27.2, 25.2; 31P NMR (130 MHz, CDCl3): δ = 122.86; MS (m/z): 472.18 [M + 1]+, 30%; Anal. for C25H30NO6P; calcd: C, 63.69; H, 6.41; N, 2.97. Found: C, 63.55; H, 6.35; N, 2.90.
3.8. 1-((4aR,7aR,11aS,11bS)-2,10-Di-p-Tolylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]dioxaphosphepin-6-yl)piperidine (L4)
Following Procedure B, L4 was obtained from Triethylamine (971 μL, 7 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), piperidine (121 mg, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-di-p-tolyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (541 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (150 mg, 0.30 mol, 40%); m.p.: 100 °C; IR (KBr, cm−1): νmax = 3443, 1600, 1339; 1H NMR (400 MHz, CDCl3): δ = 7.38–7.33 (dd, 2H, Ph), 7.18–7.16 (dd, 2H, Ph), 5.45 (s, 1H, PhCH), 4.37(q, 1H, OCH2), 4.24 (m, 1H, CHO), 3.80 (d, 1H, J = 8.8 Hz, OCH2), 3.73 (m, 1H, CHOP), 3.19 (m, 2H, CH2CH2), 2.32 (s, 3H, CH3), 1.63 (m, 2H, CH2CH2CH2), 1.49 (m, 2H, CH2CH2CH2); 13C NMR (100 MHz, CDCl3): δ = 138.9, 133.3, 128.9, 126.1, 100.9, 82.8, 81.5, 77.4, 28.6, 21.3; 31P NMR (130 MHz, CDCl3): δ = 122.86; MS (m/z): 500.21 [M + 1]+, 75%; Anal. for C27H34NO6P; calcd: C, 64.92; H, 6.86; N, 2.80. Found: C, 65.02; H, 6.75; N, 2.65.
3.9. (4aR,7aR,11aS,11bS)-2,10-Diphenyl-N,N-bis((S)-1-phenylethyl)hexahydrobis([1,3]dioxino) [5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L5)
Following Procedure B, L5 was obtained from Triethylamine (971 μL, 7.0 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), (R)-bis((R)-1-phenylethyl) amine (315 mg, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-diphenyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (500 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (200 mg, 0.44 mmol, 31%); m.p.: 103 °C; IR (KBr, cm−1): νmax = 3423, 1625, 1310; 1H NMR (400 MHz, CDCl3): δ = 7.53–7.34 (m, 10H, Ph), 5.50 (s, 1H, PhCH), 4.65 (m, 1H, CHCH3), 4.25(q, 1H, OCH2), 3.97 (m, 1H, CHO), 3.91 (d, 1H, J = 8.8 Hz, OCH2), 3.80 (m, 1H, CHOP), 1.21 (d, 3H, J = 8.8 Hz, CH3); 13C NMR (100 MHz, CDCl3): δ = 137.0, 128.3, 126.2, 100.8, 82.5, 80.6, 69.5, 31.0, 29.7; 31P NMR (130 MHz, CDCl3): δ = 134.65; MS (m/z): 612.24 [M + 1]+, 64%; Anal. for C36H38NO6P; calcd: C, 70.69; H, 6.26; N, 2.29. Found: C, 70.69; H, 6.45; N, 2.33.
3.10. (4aR,7aR,11aS,11bS)-N,N-Bis((S)-1-Phenylethyl)-2,10-di-ptolylhexahydrobis([ 1,3]dioxino)[5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L6)
Following Procedure B, L6 was obtained from Triethylamine (971 μL, 7.0 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), (R)-bis((R)-1-phenylethyl) amine (315 mg, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-di-p-tolyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (541 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (400 mg, 0.62 mmol, 45%); m.p.: 80–82 °C; IR (KBr, cm−1): νmax = 3441, 1618, 1343; 1H NMR (400 MHz, CDCl3): δ = 7.43–7.04 (m, 9H, Ph), 5.52 (s, 1H, PhCH), 4.61 (m, 1H, CHO), 4.42(q, 1H, OCH2), 4.25(q, 1H, OCH2), 4.04 (m, 1H, CHCH3), 3.80 (m, 1H, CHOP), 2.33(s, 3H, CH3), 1.21 (d, 3H, J = 8.8 Hz, CH3); 13C NMR (100 MHz, CDCl3): δ = 143.0, 139.5, 134.5, 128.9, 127.9, 127.8, 126.7, 100.7, 82.9, 81.7, 29.7, 21.3; 31P NMR (130 MHz, CDCl3): δ = 132.5; MS (m/z): 640.22 [M + 1]+, 55%; Anal. for C38H42NO6P; calcd: C, 71.35; H, 6.62; N, 2.19. Found: C, 71.29; H, 6.50; N, 2.13.
3.11. (4aR,7aR,11aS,11bS)-N-((S)-1-(Naphthalen-2-yl)ethyl)-2,10-diphenyl-N-((S)-1- Phenylethyl)hexahydrobis([1,3]dioxino)[5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L7)
Following Procedure B, L7 was obtained from triethylamine (971 μL, 7.0 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), (R)-1-(naphthalen-2-yl)-N-((R)-1-phenylethyl) ethanamine (315 mg, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-diphenyl-[4,4′-bi(1,3-dioxane)]-5,5′-diol (500 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (463 mg, 0.7 mmol, 50%); m.p.: 98 °C; IR (KBr, cm−1): νmax = 3435, 1632, 1299; 1H NMR (400 MHz, CDCl3): δ = 7.88–7.35 (m, 12H, Ph), 5.53 (s, 1H, PhCH), 4.57 (m, 1H, CHCH3), 4.25(q, 1H, OCH2), 4.11 (m, 1H, CHO), 4.00 (d, 1H, J = 8.8 Hz, OCH2), 3.79 (m, 1H, CHOP), 1.31 (d, 3H, J = 8.8 Hz, CH3), 1.21 (d, 3H, J = 8.8 Hz, CH3) ; 13C NMR (100 MHz, CDCl3): δ = 137.5, 129.1, 128.3, 126.2, 126.1, 100.8, 82.5, 80.7, 69.7, 61.8,53.2, 21.3; 31P NMR (130 MHz, CDCl3): δ = 135.01; MS (m/z): 662.26 [M + 1]+, 35%; Anal. for C40H40NO6P; calcd: C, 72.60; H, 6.09; N, 2.12. Found: C, 72.48; H, 6.00; N, 2.08.
3.12. (4aR,7aR,11aS,11bS)-N-((S)-1-(Naphthalen-2-yl)ethyl)-N-((S)-1-phenylethyl)-2,10-di-ptolylhexahydrobis([ 1,3]dioxino)[5,4-d:4′,5′-f][1,3,2]dioxaphosphepin-6-amine (L8)
Following Procedure B, L8 was obtained from Triethylamine (971 μL, 7.0 mmol, 5.0 eq.), phosphorus trichloride (123 μL, 1.4 mmol, 1.0 eq.), (R)-1-(naphthalen-2-yl)-N-((R)-1-phenylethyl) ethanamine (315 mg, 1.4 mmol, 1.0 eq.), and (2S,2′S,4R,4′R,5R,5′R)-2,2′-di-p-tolyl-[4,4′-bi(1,3- dioxane)]-5,5′-diol (541 mg, 1.4 mmol, 1.0 eq.) which was obtained as a foaming white solid (366 mg, 0.53 mmol, 38%); m.p.: 85 °C; IR (KBr, cm−1): νmax = 3436, 1615, 1378; 1H NMR (400 MHz, CDCl3): δ = 7.88–7.16 (m, 12H, Ph), 5.47 (s, 1H, PhCH), 4.50 (m, 1H, CHCH3), 4.39(q, 1H, OCH2), 4.24 (m, 1H, CHO), 4.10 (d, 1H, J = 8.8 Hz, OCH2), 3.80 (m, 1H, CHOP), 2.36 (s, 3H, CH3), 1.31 (d, 3H, J = 8.8 Hz, CH3), 1.25 (d, 3H, J = 8.8 Hz, CH3); 13C NMR (100 MHz, CDCl3): δ = 137.5, 129.1, 128.3, 126.2, 126.1, 100.8, 82.5, 80.7, 69.7, 61.8,53.2, 21.5, 21.3; 31P NMR (130 MHz, CDCl3): δ = 134.69; MS (m/z): 690.29 [M + 1]+, 70%; Anal. for C42H44NO6P; calcd: C, 73.13; H, 6.43; N, 2.03. Found: C, 73.40; H, 6.27; N, 2.05.
3.13. General Procedure for the Preparation of Chiral Brønsted Acid (Procedure C) [20]
To a solution of DIOL I (0.5 g, 1.29 mmol) in dry pyridine (10 mL) was slowly added phosphoryl chloride (178 μL, 1.94 mmol, 1.5 equiv.) at room temperature and the mixture was heated to reflux for 2 h. The reaction mixture was then allowed to cool to room temperature. Distilled water (0.83 mL) was added, and then the mixture was heated to 95 °C for 30 min and cooled again to room temperature. Pyridine was removed in vacuo, and 6 M HCl was added to the mixture. The mixture was extracted with CH2Cl2, and the combined organic extracts were again washed with 6 M HCl solution 3 times, and dried over anhydrous Na2SO4, and concentrated in vacuo. The crude residue was purified by column chromatography on SiO2 (hexane:AcOEt = 3:1→CH2Cl2:MeOH = 4:1, v:v) to give the desired compound.
3.14. (4aR,7aR,11aS,11bS)-6-Hydroxy-2,10-diphenylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepine 6-Oxide (1a)
Following Procedure C, 1a was obtained from (2S,2′S,4R,4′R,5R,5′R)-2,2′-diphenyl-[4,4′-bi(1,3- dioxane)]-5,5′-diol as a white solid (471 mg, 1.12 mmol, 87%); m.p.: 270 °C; IR (KBr, cm−1): νmax = 3450, 1610, 1355, 1200; [α] D24 = +77° (c = 1.0 g/dL, DMSO); 1H NMR (400 MHz, CDCl3): δ = 7.41–7.35 (m, 5H, Ph), 5.65 (s, 1H, PhCH), 4.60(brs, 1H, OH), 4.28(q, 1H, J = 11.0 Hz, OCH2), 4.17 (m, 1H, OCH), 4.04 (d, 1H, J = 8.8 Hz, OCH2), 3.79 (t, 1H, J = 10.2 Hz, CHOP); 13C NMR (100 MHz, CDCl3): δ = 137.7, 129.5, 128.7, 126.8, 100.4, 80.6, 68.4, 68.3, 65.7; 31P NMR (130 MHz, CDCl3): δ = −1.78; MS (m/z): 421.10 [M + 1]+, 85%; Anal. for C20H21O8P; calcd: C, 57.15; H, 5.04. Found: C, 57.20; H, 5.00.
3.15. (4aR,7aR,11aS,11bS)-6-Hydroxy-2,10-di-p-tolylhexahydrobis([1,3]dioxino)[5,4-d:4′,5′- f][1,3,2]Dioxaphosphepine 6-Oxide (1b)
Following Procedure C, 1b was obtained from (2S,2′S,4R,4′R,5R,5′R)-2,2′-di-p-tolyl-[4,4′-bi(1,3- dioxane)]-5,5′-diol as a white solid (470 mg, 1.04 mmol, 81%); m.p.: 255 °C; IR (KBr, cm−1): νmax = 3451, 1612, 1369, 1210; [α]D24 = +58° (c = 1.0 g/dL, DMSO); 1H NMR (400 MHz, CDCl3): δ = 7.27 (d, 2H, J = 8.0 Hz, Ph), 7.16 (d, 2H, J = 8.0 Hz, Ph), 5.58 (s, 1H, PhCH), 4.60 (brs, 1H, OH), 4.25 (q, 1H, J = 11.0 Hz, OCH2), 4.13 (m, 1H, OCH), 4.04 (dd, 1H, J = 8.8 Hz, OCH2), 3.79 (t, 1H, J = 10.2 Hz, CHOP), 2.27 (s, 3H, CH3); 13C NMR (100 MHz, CDCl3): δ = 138.8, 134.9, 129.1, 126.7, 100.5, 80.6, 68.4, 65.7, 21.3; 31P NMR (130 MHz, CDCl3): δ = −1.83; MS (m/z): 449.13 [M + 1]+, 76%; Anal. for C22H25O8P; calcd: C, 58.93; H, 5.62. Found: C, 58.73; H, 5.55.
4. Conclusions
We have designed chiral phosphoramidites L1–L8 and Brønsted acid 1a,b as a new motif for asymmetric catalysis. The potentially broad utility of this motif will be further explored in our laboratory.
Acknowledgments
The authors extend their appreciation to the Deanship of Scientific Research, at King Saud University for funding the work through the research group project No. RGP-VPP-044.
References
- 1.Noyori R. Centenary Lecture. Chemical multiplication of chirality: Science and applications. Chem. Soc. Rev. 1989;18:187–208. [Google Scholar]
- 2.Jacobsen E.N., Pfaltz A., Yamamoto H. Comprehensive Asymmetric Catalysis I–III. Springer; Berlin, Germany: 1999. [Google Scholar]
- 3.Eberhardt L., Armspach D., Harrowfield J., Matt D. BINOL-derived phosphoramidites in asymmetric hydrogenation: Can the presence of a functionality in the amino group influence the catalytic outcome? Chem. Soc. Rev. 2008;37:839–864. doi: 10.1039/b714519e. [DOI] [PubMed] [Google Scholar]
- 4.Toselli N., Martin D., Achard M., Tenaglia A., Burgi T., Buono G. Enantioselective cobalt-catalyzed [6+2] cycloadditions of cycloheptatriene with alkynes. Adv. Synth. Catal. 2008;350:280–286. [Google Scholar]
- 5.Kostas I.D., Vallianatou K.A., Holz J., Borner A. A new easily accessible chiral phosphate-phosphoramidite ligand based on 2-anilinoethanol and R-BINOL moieties for Rh-catalyzed asymmetric olefin hydrogenation. Tetrahedron Lett. 2008;49:331–334. [Google Scholar]
- 6.Mršić N., Lefort L., Boogers J.A.F., Minnaard A.J., Feringa B.L., de Vries J.G. Asymmetric hydrogenation of quinolines catalyzed by iridium complexes of monodentate BINOL-derived phosphoramidites. Adv. Synth. Catal. 2008;350:1081–1089. [Google Scholar]
- 7.Minnaard A.J., Feringa B.L., Lefort L., de Vries J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res. 2007;40:1267–1277. doi: 10.1021/ar7001107. [DOI] [PubMed] [Google Scholar]
- 8.Zhang W., Zhang X. Highly enantioselective hydrogenation of α-dehydroamino esters and itaconates with triphosphorous bidentate ligands and the unprecedented solvent effect thereof. J. Org. Chem. 2007;72:1020–1023. doi: 10.1021/jo0622429. [DOI] [PubMed] [Google Scholar]
- 9.Shekar S., Trantow B., Leitner A., Hartwig J.F. Sequential catalytic isomerization and allylic substitution. Conversion of racemic branched allylic carbonates to enantioenriched allylic substitution products. J. Am. Chem. Soc. 2006;128:11770–11771. doi: 10.1021/ja0644273. [DOI] [PubMed] [Google Scholar]
- 10.Zhao B., Wang Z., Ding K. Practical by ligand design: A new class of monodentate phosphoramidite ligands for rhodium-catalyzed enantioselective hydrogenations. Adv. Synth. Catal. 2006;348:1049–1057. [Google Scholar]
- 11.Alexakis A., Albrow V., Biswas K., d’Augustin M., Prietob O., Woodward S. Highly enantioselective copper(I)-phosphoramidite-catalysed additions of organoaluminium reagents to enones. Chem. Commun. 2005:2843–2845. doi: 10.1039/b503074a. [DOI] [PubMed] [Google Scholar]
- 12.van den Berg M., Minnaard A.J., Haak R.M., Leeman M., Schudde E.P., Meetsma A., Feringa B.L., de Vries A.H.M., Maljaars C.E.P., Willans C.E., et al. Monodentate phosphoramidites: A breakthrough in rhodium-catalysed asymmetric hydrogenation of olefins. Adv. Synth. Catal. 2003;345:308–323. [Google Scholar]
- 13.DeVries A.H.M., Meetsma A., Feringa B.L. Enantioselective conjugate addition of dialkylzinc reagents to cyclic and acyclic enones catalyzed by chiral copper complexes of new phosphorus amidites. Angew. Chem. Int. Ed. 1996;35:2375–2376. [Google Scholar]
- 14.Mikhel I.S., Ruegger H., Butti P., Camponovo F., Huber D., Mezzetti A. A chiral phosphoramidite beyond monodentate coordination: Secondary π-interactions turn a dangling aryl into a two-, four-, or six-electron donor in d6 and d8 complexes. Organometallics. 2008;27:2937–2948. [Google Scholar]
- 15.Sakaki J.-I., Schweizer W.B., Seebach D. Catalytic enantioselective hydrosilylation of aromatic ketones using rhodium complexes of TADDOL-derived cyclic phosphonites and phosphites. Helv. Chim. Acta. 1993;76:2654–2665. [Google Scholar]
- 16.Alexakis A., Frutos J., Mangeney P. Chiral phosphorus ligands for the asymmetric conjugate addition of organocopper reagents. Tetrahedron Asymmetry. 1993;4:2427–2430. [Google Scholar]
- 17.Alexakis A., Bäckvall J.E., Krause N., Pàmies O., Diéguez M. Enantioselective copper-catalyzed conjugate addition and allylic substitution reactions. Chem. Rev. 2008;108:2796–2823. doi: 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]
- 18.Feringa B.L. Phosphoramidites: Marvellous ligands in catalytic asymmetric conjugate addition. Acc. Chem. Res. 2000;33:346–353. doi: 10.1021/ar990084k. [DOI] [PubMed] [Google Scholar]
- 19.Minnaard A.J., Feringa B.L., Lefort L., de Vries J.G. Asymmetric hydrogenation using monodentate phosphoramidite ligands. Acc. Chem. Res. 2007;40:1267–1277. doi: 10.1021/ar7001107. [DOI] [PubMed] [Google Scholar]
- 20.Teichert J.F., Feringa B.L. Phosphoramidites: Privileged ligands in asymmetric catalysis. Angew. Chem. Int. Ed. 2010;49:2486–2528. doi: 10.1002/anie.200904948. [DOI] [PubMed] [Google Scholar]
- 21.Peña D., Minnaard A.J., de Vries J.G., Feringa B.L. Highly enantioselective rhodium-catalyzed hydrogenation of beta-dehydroamino acid derivatives using monodentate phosphoramidites. J. Am. Chem. Soc. 2002;124:14552–14553. doi: 10.1021/ja028235t. [DOI] [PubMed] [Google Scholar]
- 22.Burks H.E., Liu S., Morken J.P. Development, mechanism, and scope of the palladium-catalyzed enantioselective allene diboration. J. Am. Chem. Soc. 2007;129:8766–8773. doi: 10.1021/ja070572k. [DOI] [PubMed] [Google Scholar]
- 23.Albicker M.R., Cramer N. Enantioselective palladium-catalyzed direct arylations at ambient temperature: Access to indanes with quaternary stereocenters. Angew. Chem. Int. Ed. 2009;48:9139–9142. doi: 10.1002/anie.200905060. [DOI] [PubMed] [Google Scholar]
- 24.Bayer A., Thewalt U., Rieger B. Novel monodentate phosphoramidites by chiral pool synthesis starting from d-mannitol, and their PdII and RhI complexes. Eur. J. Inorg. Chem. 2002;1:199–203. [Google Scholar]
- 25.Maciá B., Fernández-Ibáñez A., Mršić N., Minnaard A.M., Feringa B.L. Copper-catalyzed enantioselective conjugate addition of Grignard reagents to acyclic enones using monodentate phosphoramidite ligands. Tetrahedron Lett. 2008;49:1877–1880. [Google Scholar]
- 26.Pfretzschner T., Kleemann L., Janza B., Harms K., Schrader T. On the role of PIII ligands in the conjugate addition of diorganozinc derivatives to enones. Chem. Eur. J. 2004;10:6048–6057. doi: 10.1002/chem.200400316. [DOI] [PubMed] [Google Scholar]
- 27.Jensen J.F., Svendsen B.Y., la Cour T.V., Pedersen H.L., Johannsen M. Highly enantioselective hydrosilylation of aromatic alkenes. J. Am. Chem. Soc. 2002;124:4558–4559. doi: 10.1021/ja025617q. [DOI] [PubMed] [Google Scholar]
- 28.Duursma A., Boiteau J.-G., Lefort L., Boogers J.A.F., de Vries A.H.M., de Vries J.G., Minnaard A.J., Feringa B.L. Highly enantioselective conjugate additions of potassium organotrifluoroborates to enones by use of monodentate phosphoramidite ligands. J. Org. Chem. 2004;69:8045–8052. doi: 10.1021/jo0487810. [DOI] [PubMed] [Google Scholar]
- 29.Trost B.M., Silverman S.M., Stambuli J.P. Palladium-catalyzed asymmetric [3+2] cycloaddition of trimethylenemethane with imines. J. Am. Chem. Soc. 2007;129:12398–12399. doi: 10.1021/ja0753389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trost B.M., Cramer N., Silverman S.M. Enantioselective construction of spirocyclic oxindolic cyclopentanes by palladium-catalyzed trimethylenemethane-[3+2]-cycloaddition. J. Am. Chem. Soc. 2007;129:12396–12397. doi: 10.1021/ja075335w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trost B.M., Stambuli J.P., Silverman S.M., Schwörer U. Palladium-catalyzed asymmetric [3+2] trimethylenemethane cycloaddition reactions. J. Am. Chem. Soc. 2006;128:13328–13329. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faller J.W., Fontaine P.P. Resolution and diels-alder catalysis with planar chiral arene-tethered ruthenium complexes. Organometallics. 2005;24:4132–4138. [Google Scholar]
- 33.Imbos R., Minnaard A.J., Feringa B.L. Monodentate phosphoramidites: Versatile ligands in catalytic asymmetric intramolecular Heck reactions. Dalton Trans. 2003;10 doi: 10.1039/b300565h. [DOI] [Google Scholar]
- 34.Baggett N., Stribblehill P. Asymmetric reduction of ketones by using complexes of lithium tetrahydridoaluminate(III) with 1,4:3,6-dianhydro-d-mannitol and 1,3:4,6-di-O-benzylidene-d-mannitol. J. Chem. Soc. Perkin Trans. 1997;1:1123–1126. [Google Scholar]
- 35.Trost B.M., Silverman S.M., Stambuli J.P. Development of an asymmetric trimethylenemethane cycloaddition reaction: Application in the enantioselective synthesis of highly substituted carbocycles. J. Am. Chem. Soc. 2011;133:19483–19497. doi: 10.1021/ja207550t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Doyle A.G., Jacobsen E.N. Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev. 2007;107:5713–5743. doi: 10.1021/cr068373r. [DOI] [PubMed] [Google Scholar]
- 37.Akiyama T. Stronger Brønsted acids. Chem. Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]
- 38.Yu X., Wang W. Hydrogen-bond-mediated asymmetric catalysis. Chem. Asian J. 2008;3:516–532. doi: 10.1002/asia.200700415. [DOI] [PubMed] [Google Scholar]
- 39.Terada M. Binaphthol-derived phosphoric acid as a versatile catalyst for enantioselective carbon-carbon bond forming reactions. Chem. Commun. 2008:4097–4112. doi: 10.1039/b807577h. [DOI] [PubMed] [Google Scholar]
- 40.Vachal P., Jacobsen E.N. Structure-based analysis and optimization of a highly enantioselective catalyst for the strecker reaction. J. Am. Chem. Soc. 2002;124:10012–10014. doi: 10.1021/ja027246j. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X., Du H., Wang Z., Wu Y.-D., Ding K. Experimental and theoretical studies on the hydrogen-bond-promoted enantioselective hetero-Diels-Alder reaction of Danishefsky’s diene with benzaldehyde. J. Org. Chem. 2006;71:2862–2869. doi: 10.1021/jo060129c. [DOI] [PubMed] [Google Scholar]
- 42.Gridnev I.D., Kouchi M., Sorimachi K., Terada M. On the mechanism of stereoselection in direct Mannich reaction catalyzed by BINOL-derived phosphoric acids. Tetrahedron Lett. 2007;48:497–500. [Google Scholar]
- 43.Yamanaka M., Itoh J., Fuchibe K., Akiyama T. Chiral Brønsted acid catalyzed enantioselective mannich-type reaction. J. Am. Chem. Soc. 2007;129:6756–6764. doi: 10.1021/ja0684803. [DOI] [PubMed] [Google Scholar]
- 44.Simón L., Goodman J.M. Theoretical study of the mechanism of hantzsch ester hydrogenation of imines catalyzed by chiral BINOL-phosphoric acids. J. Am. Chem. Soc. 2008;130:8741–8747. doi: 10.1021/ja800793t. [DOI] [PubMed] [Google Scholar]
- 45.Anderson C.D., Dudding T., Gordillo R., Houk K.N. Origin of enantioselection in hetero-Diels-Alder reactions catalyzed by naphthyl-TADDOL. Org. Lett. 2008;10:2749–2752. doi: 10.1021/ol800875k. [DOI] [PubMed] [Google Scholar]
- 46.Corey E.J., Lee T. The formyl C–H…O hydrogen bond as a critical factor in enantioselective Lewis-acid catalyzed reactions of aldehydes. Chem. Commun. 2001:1321–1329. [Google Scholar]
- 47.Castellano R.K. Progress toward understanding the nature and function of C–H…O interactions. Curr. Org. Chem. 2004;8:845–865. [Google Scholar]
- 48.Washington I., Houk K.N. CH…O Hydrogen bonding influences π-facial stereoselective epoxidations. Angew. Chem. Int. Ed. 2001;40:4485–4488. doi: 10.1002/1521-3773(20011203)40:23<4485::aid-anie4485>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 49.Terada M., Soga K., Momiyama N. Enantioselective activation of aldehydes by chiral phosphoric acid catalysts in an Aza-ene-type reaction between glyoxylate and enecarbamate. Angew. Chem. Int. Ed. 2008;47:4122–4125. doi: 10.1002/anie.200800232. [DOI] [PubMed] [Google Scholar]
- 50.Kashikura W., Mori K., Akiyama T. Chiral phosphoric acid catalyzed enantioselective synthesis of β-amino-α,α-difluoro carbonyl compounds. Org. Lett. 2011;13:1860–1863. doi: 10.1021/ol200374m. [DOI] [PubMed] [Google Scholar]
- 51.Li J., Subramaniam K., Smith D., Qiao J.X., Li J.J., Q-Cutrone J., Kadow J.F., Vite G.D., Chen B-C. AlMe3-promoted formation of amides from acids and amines. Org. Lett. 2012;14:214–217. doi: 10.1021/ol203007s. [DOI] [PubMed] [Google Scholar]