

Rapid signaling and structural adaptations to the actin cytoskeleton enable leukocytes to stabilize α4 integrin–mediated adhesion and resist detachment from inflamed endothelium.
Abstract
Leukocytes arrested on inflamed endothelium via integrins are subjected to force imparted by flowing blood. How leukocytes respond to this force and resist detachment is poorly understood. Live-cell imaging with Lifeact-transfected U937 cells revealed that force triggers actin polymerization at upstream α4β1 integrin adhesion sites and the adjacent cortical cytoskeleton. Scanning electron microscopy revealed that this culminates in the formation of structures that anchor monocyte adhesion. Inhibition of actin polymerization resulted in cell deformation, displacement, and detachment. Transfection of dominant-negative constructs and inhibition of function or expression revealed key signaling steps required for upstream actin polymerization and adhesion stabilization. These included activation of Rap1, phosphoinositide 3-kinase γ isoform, and Rac but not Cdc42. Thus, rapid signaling and structural adaptations enable leukocytes to stabilize adhesion and resist detachment forces.
Introduction
The actin cytoskeleton is dynamic and supports a multitude of essential functions in adherent and migrating cells from participating in the formation of protrusions to the generation of tensile forces and cell motility (Vicente-Manzanares and Sánchez-Madrid, 2004). In contrast, the role of the actin cytoskeleton remains poorly understood during recruitment of blood leukocytes into tissues.
Leukocyte recruitment is fundamental to inflammatory and homeostatic processes: host response to infection, pathogenesis of inflammatory disorders, and immune surveillance. Blood leukocytes are recruited through a multistep cascade of highly organized adhesive and signaling steps, including tethering and rolling along vascular endothelium, activation of integrins by chemokine signaling, arrest, stabilization of adhesion, intravascular crawling, and diapedesis (Butcher, 1991; Ley et al., 2007).
Integrins are adhesion and signaling molecules expressed by most nucleated cells (Hynes, 2002). Signaling via chemokine G protein–coupled receptors (GPCRs) triggers rapid up-regulation of leukocyte α4β1 (very late antigen-4 [VLA-4]) and αLβ2 (LFA-1) integrin affinity, which stabilizes bonds with VCAM-1 and ICAM-1, respectively (Constantin et al., 2000; Chan et al., 2001). Arrest is mediated by a relatively small number of integrins and must be rapidly stabilized. One mechanism for stabilization is integrin clustering upon ligand binding and intracellular signaling (outside-in signaling) through adapter proteins (Alon and Dustin, 2007). Linkage of VLA-4 to the actin cytoskeleton through the adapter protein paxillin is critical for stabilization of leukocyte adhesion when exposed to fluid flow and transmission of mechanical force (Alon et al., 2005). Despite this, the actin cytoskeleton in leukocytes has generally been viewed as a relatively static structure that tethers integrins in the cell membrane and restricts their lateral diffusion (Kucik et al., 1996; van Kooyk et al., 1999). Pharmacological inhibition of actin polymerization has been shown to increase LFA-1–mediated adhesion and also resulted in increased accumulation of integrins at the adhesion surface (van Kooyk et al., 1999; Kim et al., 2004). These experiments modeled the prolonged adhesion of contact-dependent cells. On the other hand, leukocytes undergoing recruitment adhere rapidly to the endothelium and resist detachment. There is a paucity of information on whether the actin cytoskeleton participates in adhesion stabilization and whether it adapts to external forces.
Mechanotransduction of fluid forces dramatically influences endothelial cell gene expression in vascular endothelium (Tzima et al., 2005) and neutrophil morphology (Coughlin and Schmid-Schönbein, 2004; Makino et al., 2006; Coughlin et al., 2008). All of these responses involve an intact actin cytoskeleton. Integrins are both force-bearing and force-sensing receptors and, thus, are critical to a variety of cell functions (Brakebusch and Fässler, 2003; Critchley, 2004). Integrin mechanotransduction is a key feature in strengthening integrin-mediated adhesion. For example, when force is applied to magnetic beads attached to cell surface integrins, cells are capable of producing a “stiffening” response thought to be a “strengthening” of the linkage to the actin cytoskeleton (Wang et al., 1993; Choquet et al., 1997).
Several signaling molecules have been implicated in mechanotransduction, including Rap1 and Rho family GTPases and phosphoinositide 3-kinase (PI3K). Rap1 is a small GTPase critical for integrin adhesive function (Bos et al., 2001; Shimonaka et al., 2003) and can be activated by tensile forces (Sawada et al., 2001). Rho small GTPases are critical players in actin polymerization. Rac, a member of the Rho family of small GTPases, has been shown to be critical for F-actin rearrangement in endothelial cells exposed to shear stress (Tzima et al., 2002). PI3K plays a key role in chemokine-induced cell adhesion under fluid flow and cell polarization during chemotactic migration (Sasaki et al., 2000; Smith et al., 2006; Heit et al., 2008). In addition, PI3K has been shown to be important for force-mediated activation of integrins (Katsumi et al., 2005).
Collectively, our data reveal that leukocytes adhering via VLA-4 adapt to external hydrodynamic force by forming upstream structures—“anchors” that stabilize adhesion. This process is dependent on actin polymerization and activation of Rap1 and Rac as well as PI3K-γ. We propose that leukocytes sense and respond to external forces through signaling and structural adaptations, which are indispensible for stabilization of rapid adhesion in the blood stream.
Results
Actin polymerization is critical for stabilization of integrin-mediated leukocyte adhesion and resistance to deformation upon exposure to fluid flow
We set out to investigate the structural and functional role of the actin cytoskeleton in VLA-4–mediated leukocyte adhesion under conditions of fluid flow. Cytochalasin D (CytoD) and Latrunculin B (LatB), two different inhibitors of actin polymerization, were used at concentrations (2 µM) that dramatically reduced F-actin polymerization in lamellipodia of monocyte-like U937 cells but minimally affected total F-actin content and increased clustering of cortical F-actin (Fig. S1, a–d). VLA-4 on the cell surface remained colocalized with F-actin, as measured by immunofluorescence microscopy and flow cytometry of cytoskeleton ghosts, suggesting that the CytoD or LatB did not disrupt the constitutive linkage between integrins and the cortical actin cytoskeleton (Fig. S1, d–f).
In the context of leukocyte recruitment from the blood into tissues, integrins with intermediate affinity can mediate slow rolling, and upon activation of leukocytes by chemokines, the rapid conversion of integrins to high affinity mediates arrest (Campbell et al., 1998; Chan et al., 2001). Therefore, we investigated the role of actin polymerization in VLA-4 affinity modulation by GPCR inside-out signaling. We found that neither CytoD nor LatB pretreatment blocked rapid fMLF (f-Met Leu Phe)-induced VLA-4 affinity up-regulation (Fig. S2 a), consistent with previous studies for VLA-4 and LFA-1 (Chan et al., 2001; Yu et al., 2010).
CytoD or LatB pretreatment inhibited arrest and accumulation of U937 cells induced by fMLF (Fig. S2 b) or chemokine stromal-derived factor 1 α(SDF-1α; Fig. 1 a). In assays in which maximal VLA-4 affinity was induced by manganese (Mn2+), CytoD or LatB also inhibited leukocyte accumulation on VCAM-1–coated flow chambers (Fig. 1 b). Integrin affinity up-regulation by Mn2+ does not require intracellular signaling through chemokine receptors, which can modulate leukocyte actin dynamics (Howard and Meyer, 1984). In detachment assays, both CytoD and LatB dramatically reduced the adhesion of Mn2+-treated U937 cells (Fig. 1 c), human peripheral blood monocytes (Fig. 1 c), and murine lymphocytes (not depicted) to VCAM-1–coated surfaces. Collectively, these data suggest that actin polymerization is required for blood leukocyte resistance to detachment under conditions of flow and implicate a role for actin polymerization in the stabilization of VLA-4–mediated leukocyte arrest but not integrin affinity modulation.
Figure 1.

Actin polymerization is required for leukocyte resistance to fluid flow–induced detachment and deformation. (a and b) Accumulation assays performed with U937 cells infused at 2 dynes/cm2 into a parallel plate flow chamber. After 2 min, the number of adherent cells per high power field was determined (n = 8 per treatment group; two independent experiments (IEs); *, P < 0.001 between indicated groups or relative to the vehicle-treated group). (c) Flow chamber detachment assays with U937 cells or human peripheral blood monocytes pretreated with vehicle, LatB, or CytoD. Leukocytes suspended in an Mn2+-containing buffer were adhered to a VCAM-1–coated surface, and the number of cells remaining adherent after progressively increasing fluid flow was determined (n = 10 per treatment group; three IEs; *, P < 0.001 relative to the vehicle-treated group). (d) Phase-contrast video microscopy images of representative vehicle or CytoD-pretreated U937 cells adhered to VCAM-1 under static conditions and then exposed to fluid flow (2 dynes/cm2). The time relative to the initial image is indicated. Arrowheads point to elongated structures formed upstream of the CytoD-pretreated cell body. (e and f) Representative SEM images of leukocytes adhered and exposed to fluid flow (2 and then 4 dynes/cm2, each for 60 s) and perfusion fixed in situ (three IEs). Arrowheads point to elongated structures upstream of the cell body and thin long structures without associated cell bodies. (d–f) Arrows indicate the direction of fluid flow. Error bars show means ± SD.
Adherent CytoD- or LatB-pretreated leukocytes were deformed after exposure to flow, in contrast to buffer-treated cells that maintained a spherical shape. Leukocytes were displaced from the initial adhesion site, and elongated structures were pulled from the cell body (Fig. 1 d). These structures remained attached to the VCAM-1–coated surface and sporadically would fracture and release tethered leukocytes. Scanning EM (SEM) of leukocytes fixed under flow revealed deformation of CytoD- or LatB-treated cells and abundant elongated structures without associated cell bodies (Fig. 1, e and f).
Blockade of actin polymerization leads to the extrusion of F-actin from the cell body
Visualization of F-actin revealed the effects of CytoD and LatB on U937 cells exposed to fluid flow. Immunostaining showed that elongated upstream structures contained F-actin and VLA-4, which were colocalized (Fig. 2 a). Because actin polymerization was blocked, these data suggest that F-actin was extruded from the cell body, as it was displaced from the attachment site. This possibility was investigated in real time using Lifeact-GFP–transfected U937 cells (Riedl et al., 2008). Upon introduction of flow, F-actin was extruded from the upstream cortical actin cytoskeleton of CytoD- or LatB-treated cells (Fig. 2, b and c; and Videos 1 and 2). In some experiments, CytoD or LatB was added to the perfusion buffer, which recapitulated cell deformation and the extrusion of F-actin and suggested that continuous actin polymerization is required for stable leukocyte adhesion under flow (Fig. 2 d and Video 3).
Figure 2.

Blockade of actin polymerization leads to extrusion of preexisting F-actin from the cell body. (a) U937 cells pretreated with CytoD or LatB were adhered to VCAM-1 under static conditions, exposed to fluid flow (2 dynes/cm2 for 2 min), and perfusion fixed with PFA. F-actin was visualized with 488 phalloidin (green), and VLA-4 was visualized with Cy3-44H6 (red). Asterisks indicate cell bodies. Arrowheads indicate elongated upstream structures with or without an associated cell body. (b and c) U937 cells transfected with Lifeact-GFP were pretreated with CytoD (b) or LatB (c), suspended in a Mn2+-containing buffer, and adhered to a VCAM-1–coated surface in a flow chamber. Fluid flow was introduced (2 dynes/cm2), and sequential images were acquired. The relative time of each image is indicated. The dashed lines provide a spatial reference to the most upstream portion of the cell. See Videos 1 and 2. (d) Addition of LatB into the perfusion buffer. Lifeact-GFP–transfected U937 cells were adhered under static conditions and exposed to fluid flow (2 dynes/cm2). After 2 min, LatB was added, and sequential images were acquired. Differential interference contrast (DIC) and GFP images are shown (the relative time is indicated). See Video 3. Asterisks indicate cell bodies whose position is altered relative to the previous image. Note that occasional cells detach (star). Representative data are from three IEs. Arrows indicate the direction of fluid flow. Bars, 10 µm.
Fluid flow triggers the formation of upstream F-actin–rich anchors
The aforementioned experiments suggest that mechanical force exerted on adherent leukocytes as a consequence of fluid flow induces actin polymerization to withstand detachment and deformation. To directly evaluate this possibility, live-cell imaging was performed using Lifeact-GFP–transfected U937 cells. Each cell was imaged under static conditions and subsequently after flow. Under static conditions, constant random movement of GFP was observed in submembrane regions and microvillous-like projections over the entire cell surface (Videos 4 and 5). These features were maintained in cells exposed to flow, but the most significant difference was the accumulation of F-actin in structures that contacted the adhesion surface upstream of the cell body and in the cortical actin cytoskeleton adjacent to the insertion site of these structures (Fig. 3 a, Fig. S3 d, and Video 6). Upstream accumulation of F-actin did not occur when cells adhered to poly-l-lysine were exposed to flow (unpublished data). We refer to the upstream F-actin–rich structures as anchors because they anchor leukocytes to VCAM-1. Upstream anchors were also observed in human peripheral blood monocytes (Fig. 3 b). Anchors extended from the adhesion surface at ∼1/5 of the cell height, and their mean length was 1.3 µm for monocytes and 1.9 µm for U937 cells (Fig. 3 c and Fig; S3 a). Anchors also formed when monocytes were infused and contacted the VCAM-1–coated surface under constant fluid flow (Fig. 3 d).
Figure 3.

Fluid flow triggers the formation of upstream F-actin–rich anchors. (a and e–h) Live-cell imaging of Lifeact-GFP–transfected U937 cells. Cells suspended in an Mn2+-containing buffer were adhered under static conditions to a VCAM-1–coated surface of a flow chamber and then exposed to fluid flow (2 dynes/cm2 for 2 min). (a) Images of a representative cell at the indicated levels from the adhesion surface before and after exposure to fluid flow. Upstream anchors (solid arrowheads) and anchor insertion sites (open arrowheads) are located within several micrometers of the adhesion surface. The circles represent the cell periphery at the midpoint of the cell as determined by superimposing DIC images. (b) Representative SEM images of perfusion-fixed untransfected U937 cells and monocytes (Mono) exposed to fluid flow. Upstream anchors (solid arrowheads) are visualized clearly when chucks were rotated to obtain a side view. (c) Quantification of anchor dimensions in SEM images (n = 32 and 36; three IEs). Data were normalized to the mean cell height or width (8.03 ± 0.82 and 8.55 ± 0.46 µm for U937 cells, n = 10; and 4.94 ± 0.54 and 5.33 ± 0.57 µm for monocytes, n = 15). (d) SEM showing anchor formation in human monocytes infused into VCAM-1–coated chambers (0.5 dynes/cm2 for 30 s and then 2 dynes/cm2 for 60 s). Upstream anchors are indicated by solid arrowheads. (e and f) Sequential images reveal the kinetics of F-actin accumulation and anchor formation. The time relative to the introduction of a fluid shear force (2 dynes/cm2) is indicated. (e) Top and side views of collapsed z stacks show F-actin–rich anchors (arrowheads) at 15 s. The GFP signal is enhanced in the inset. (f) Images obtained just above the adhesion surface reveal that anchors develop from projections that contacted the adhesion surface under static conditions (solid arrowheads). The accumulation of F-actin in the cortical actin cytoskeleton (open arrowheads) is evident within 1 s after exposure to fluid flow. See Video 6. (g and h) Lifeact-GFP fluorescence intensity analysis across cells. (g) Representative images of a Lifeact-GFP–transfected U937 cell before and after exposure to fluid flow and corresponding GFP fluorescence profiles within yellow lines. Images were obtained at a level at which anchors insert into the cell body. The horizontal dashed red lines correspond to the GFP intensity in the downstream cortical actin cytoskeleton. The graph shows upstream to downstream GFP mean fluorescence intensity (MFI) ratios of 10–15 randomly selected cells (four IEs; *, P < 0.001). (h) Analysis of GFP intensity at different levels from the adhesion surface as shown in the schematic. In each optical slice from a z stack, fluorescence profiles within the yellow lines are shown. Dashed lines at peaks correspond to the downstream (yellow) and upstream (red) cortical actin cytoskeleton and anchors upstream of the cell body (white). The numbers at the top of images refer to the height of each image above the adhesion surface. The upstream to downstream cortical actin mean fluorescence intensity ratios are plotted. The maximal difference in upstream versus downstream cortical actin fluorescence ratio is observed at a level at which anchors insert into the cortical actin cytoskeleton. This representative experiment demonstrates how optical slices from a z stack were selected for analysis in g. Also refer to Fig. S3. Arrows indicate the direction of fluid flow. Error bars show means ± SD.
Increased F-actin appeared in upstream anchors and at sites of anchor insertion within the cortical actin cytoskeleton within seconds of introducing flow and gradually increased over time (Fig. 3, e and f; and Video 6). Exposure to fluid flow induced a twofold increase in F-actin accumulation at anchor insertion into the cortical F-actin relative to the downstream cortical cytoskeleton (Fig. 3, g and h; and Fig. S3 e). Collectively, our data suggest that leukocytes rapidly undergo a structural adaptation involving F-actin reorganization, which enables them to resist detachment forces imparted by fluid flow.
Significance of F-actin–rich upstream anchors
A biomechanical study has established that adhesion contact sites between microvilli at the rear of a spherical leukocyte and a flat surface experience the highest tension upon exposure to laminar flow (Chen and Springer, 1999); thus, it is possible that tension in leukocyte anchors provides a stimulus for actin polymerization. To evaluate this possibility, detachment assays were performed in which Mn2+-treated U937 cells were exposed to a defined hydrodynamic force either directly or in stepwise increments (Fig. 4 a). In the latter group, a significantly higher percentage of leukocytes remained adherent. This effect was not observed in leukocytes pretreated with CytoD or LatB (Fig. 4 a). Live-cell imaging of Lifeact-GFP–transfected cells demonstrated that exposure to stepwise fluid flow correlated with increased F-actin accumulation at upstream anchor insertion points (Fig. 4 b). Collectively, these data suggest that upstream F-actin–rich anchor formation in response to lower hydrodynamic forces preconditions cells to resist a higher force.
Figure 4.

Fluid flow primes leukocytes to resist detachment by inducing and maintaining F-actin in upstream anchors. (a) Detachment assays were performed with vehicle-, CytoD-, or LatB-pretreated U937 cells. Cells were adhered to a VCAM-1–coated surface. Fluid flow was introduced, either directly with 4 dynes/cm2 (open symbols) or in a stepwise fashion (1, 2, and 4 dynes/cm2; solid symbols), and cells remaining adherent were quantified (means ± SD, n = 8; two IEs; *, P < 0.05; **, P < 0.005; ***, P < 0.0001 direct versus stepwise shear). (b) Increase in fluid flow induces F-actin accumulation in anchors. Representative confocal images of a Lifeact-GFP–transfected U937 cell exposed to fluid flow (shear stress: 2 and then 4 dynes/cm2). GFP accumulation is shown within anchors (solid arrowheads) and at their insertions (open arrowheads). The numbers at the bottom of the images refer to the height of each image above the adhesion surface. (c) Removal of fluid flow reduces F-actin accumulation in anchors. U937 cells adhered to VCAM-1 were exposed to fluid flow (2 dynes/cm2 for 2 min), and then flow was stopped. Sequential z stacks of the same cell were obtained before (0 s) and after (300 s) flow was stopped. (left) Top and side view projections of a representative cell demonstrate that upstream anchors (solid arrowheads) diminished after cessation of flow. (right) Sequential images just above the adhesion surface illustrate progressively decreasing F-actin at anchor insertions (open arrowheads). See Videos 7 and 8. Arrows indicate the direction of fluid flow. Bars, 10 µm.
Rap1 accumulates at anchor insertions and prevents cell displacement
Rap1 is a known regulator of cell adhesion and cell spreading (Bos et al., 2001). Rap1 can be activated in response to cell stretching (Sawada et al., 2001, 2006). Therefore, we investigated a role for Rap1 in anchor formation. U937 cells were transfected with Rap1-GFP and Lifeact-RFP, adhered to VCAM-1, and exposed to fluid flow. A significant increase in Rap1 accumulation at the anchor insertion into the cortical cytoskeleton was observed (Fig. 5, a and b; and Fig. S4 a). Experiments with constitutively active Rap1-V12–GFP confirmed that accumulated Rap1 was GTP bound (unpublished data). Leukocytes transfected with a dominant negative (dn) Rap1-N17–GFP failed to form F-actin–rich anchors. Furthermore, these cells were displaced from the initial adhesion site (Fig. 5, c–e). F-actin was extruded in a subset of dnRap1-transfected cells, analogous to cells pretreated with CytoD or LatB (unpublished data). These data confirmed a role for Rap1 in the adaptation to fluid flow by leukocytes.
Figure 5.

Rap1 accumulates at anchor insertions and prevents cell displacement. (a and c) Representative images of U937 cells transfected with wild-type Rap1-GFP (a) or Rap1-N17–GFP (dnRap1-GFP; c) and Lifeact-RFP. Cells were adhered to VCAM-1 under static conditions (2 min) and then exposed to a fluid flow (2 dynes/cm2). Flow induced accumulation of Rap1 and F-actin in the region of anchor insertion (open arrowheads). (b) Quantification of Rap1-GFP accumulation at the level of anchor insertion. The ratio of upstream versus downstream Rap1-GFP in the vicinity of the cortical actin cytoskeleton was determined for each cell before and during fluid flow (n = 15 cells; two IEs; *, P < 0.0001). MFI, mean fluorescence intensity. (c–e) In cells transfected with dnRap1-GFP, fluid flow did not induce anchor formation and upstream accumulation of F-actin or dnRap1-GFP. Cells gradually pulled away from their initial adhesion site. The dashed yellow lines in c provide a spatial reference between images. (d) Displacement of Rap1-GFP– versus dnRap1-GFP–transfected U937 cell 45 s after exposure to fluid flow (2 dynes/cm2; n = 10 per group; two IEs; *, P < 0.0001). (e) Displacement over time of representative U937 cells transfected with Rap1-GFP or dnRap1-GFP. Arrows indicate the direction of fluid flow. Error bars show means ± SD. Bars, 10 µm.
Rac-dependent actin polymerization within anchors prevents cell displacement
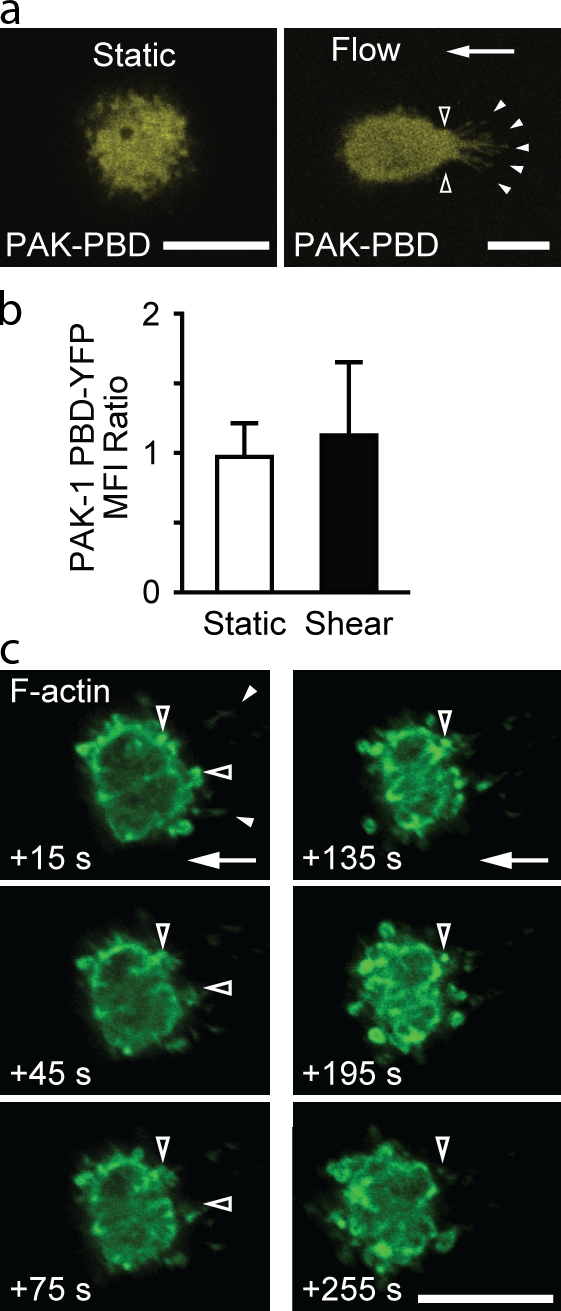
Small GTPases of the Rac and Cdc42 families are well known inducers of actin polymerization (Ridley and Hall, 1992; Ridley et al., 1992; Hall, 1994). Rap1 effectors include guanine exchange factors (GEFs) that activate Rac (Arthur et al., 2004). Therefore, to determine whether fluid flow activated Rac and/or Cdc42, we transfected leukocytes with the p21-binding domain (PBD) of p21-activated kinase (PAK) fused to YFP (PAK-PBD-YFP), which can bind either GTP-bound Rac or Cdc42 (Manser et al., 1994). Leukocytes exposed to fluid flow had a significant increase in the accumulation of PAK-PBD-YFP (Fig. 6, a and b; and Fig. S4 b) at sites of anchor insertion. To determine the role of Rac versus Cdc42, U937 cells were transfected with dnRac-N17–GFP or dnCdc42-N17–GFP. Upon exposure to a fluid flow, cells transfected with dnRac-GFP were deformed and displaced from the initial adhesion site (Fig. 6, c–f; and Fig. S4 c). Stable anchors did not form, and F-actin was extruded in many cells (Fig. S4 c). Cells transfected with dnCdc42-GFP exhibited normal F-actin dynamics and anchor formation in response to flow (Fig. S4 c). These data support a role for Rac-dependent actin polymerization in the formation of anchors and stabilization of leukocyte adhesion.
Figure 6.

Rac, but not Cdc42, is critical for preventing cell displacement. Transfected U937 cells were adhered to VCAM-1 under static conditions (2 min) and exposed to fluid flow (2 dynes/cm2 for 2 min). Images at the level of anchor insertion were selected from confocal z stacks obtained before and after initiation of flow. (a) Representative cells transfected with PAK-PBD-YFP and their fluorescence intensity profiles within the indicated red lines. Each red dashed line provides a reference for PAK-PBD-YFP fluorescence intensity determined at the downstream edge of a cell and facilitates comparison to the fluorescence intensity at the upstream edge. (b) Quantification of PAK-PBD-YFP accumulation in upstream versus downstream regions (n = 10; three IEs; *, P < 0.0001). MFI, mean fluorescence intensity. (c) dnRac-GFP–transfected cells deformed upon exposure to flow. Lifeact-RFP and dnRac-GFP were colocalized in upstream adhesion contacts (arrowheads). (d) Displacement of U937 cells transfected with PAK-PBD-YFP, dnRac-GFP, and dnCdc42-GFP after exposure to fluid flow (2 dynes/cm2 for 60 s; means ± SD; n = 10 cells per group; two IEs; *, P < 0.001). (e) Displacement over time of representative transfected U937 cells. Arrows indicate the direction of fluid flow. Bars, 10 µm.
PI3K activity is critical for leukocyte adhesion stabilization, resistance to deformation, and flow-induced actin polymerization
The PI3K family of lipid kinases can be activated by mechanical stretch (Katsumi et al., 2005) and can regulate Rac-mediated actin polymerization and polarization of cells during chemotaxis and phagocytosis (Sadhu et al., 2003; Srinivasan et al., 2003; Ferrandi et al., 2007). Previously, we observed that blockade of PI3K function did not affect VLA-4 affinity up-regulation by GPCR signaling (Hyduk et al., 2007), but integrin-mediated leukocyte adhesion was dramatically inhibited (Hyduk and Cybulsky, 2002). These observations are analogous to those made after inhibition of actin polymerization.
Several approaches were adopted to elucidate the function of fluid flow-induced PI3K activation. These included utilization of broad spectrum inhibitors, such as Wortmannin (Wort) and LY294002, a γ isoform inhibitor (AS605240), siRNA inhibition of PI3K-γ expression, and isolation of leukocytes from PI3K-γ knockout mice. All of these experiments yielded consistent data showing that PI3K is required for chemokine-induced leukocyte accumulation (Fig. 7 a), adhesion stabilization, and resistance to fluid flow, independent of chemokine signaling (Fig. 7, a and b). Inhibition of PI3K also recapitulated the deformation defects (Fig. 7 c), which we observed in leukocytes treated with inhibitors of actin polymerization (Fig. 2) and dnRac (Fig. 6). Pretreatment or perfusion of Wort onto adherent U937 cells led to a gradual displacement of cells from their initial adhesion site and distortion (Fig. 7, d–f). Wort perfusion experiments confirm that ongoing activation of PI3K by fluid flow is required for maintenance of stable adhesion.
Figure 7.

PI3K activity is required for stable adhesion and resistance to flow-induced detachment and deformation. (a and b) Accumulation assays (2 dynes/cm2 for 2 min) with U937 cells pretreated with vehicle or LY294002 (LY) or transfected with control or PI3K-γ siRNA. The extent of PI3K-γ knockdown by siRNA as determined by Western blotting was 85% (n = 3; see Fig. S5). The number of U937 cells that underwent arrest and remained adherent was quantified (n = 12 per group; three IEs; *, P < 0.001 relative to the control group). (b) Detachment assays with leukocytes (indicated) treated with PI3K inhibitors, 1 µM Wortmannin (Wort), 50 µM LY294002, and 100 µM AS605240 and transfected with control or PI3K-γ siRNA and CD8+ T cells isolated from wild-type and PI3K-γ knockout mice. The buffer contained Mn2+ (n = 10; three IEs; *, P < 0.05 relative to the control group). (c) Representative SEM images of 1 µM Wort-pretreated U937 cells and human monocytes adhered to VCAM-1 and then exposed to flow (2 dynes/cm2 for 2 min). Arrowheads indicate the extrusion of F-actin upstream of the cell body. (d) Wort perfusion induces cell displacement. U937 cells adhered to VCAM-1 were exposed to fluid flow (2 dynes/cm2 for 2 min). Vehicle or Wort (1 µM final concentration) was then added to the perfusion buffer (at time = 0 s). Representative sequential DIC images show Wort-induced cell displacement and distortion. Arrowheads indicate initial adhesion contact sites. (e) Displacement of U937 cells after exposure to fluid flow (2 dynes/cm2) for 40 s. Treatments are indicated (n = 3–10 cells per group; two IEs; *, P < 0.005; **, P < 0.0001). (f) Displacement over time of representative U937 cells. Error bars are means ± SD. Arrows indicate the direction of fluid flow.
Blockade of PI3K prevents the accumulation of active Rac and F-actin in structural anchors
Because PI3K is a regulator of Rac activity (Han et al., 1998), we investigated whether it is required for Rac and F-actin accumulation within anchors. U937 cells transfected with PAK-PBD-YFP were pretreated with Wort and exposed to fluid flow. Blockade of PI3K reduced the accumulation of PAK-PBD-YFP at sites of anchor insertion (Fig. 8, a and b) in contrast to untreated cells (Fig. 6, a and b). Perfusion of Wort onto adherent Lifeact-GFP–transfected U937 cells led to a gradual degradation of F-actin within anchors (Fig. 8 c and Video 9). These cells were displaced from the VLA-4–mediated attachment site. Collectively, these data support a role for force-induced PI3K in regulating F-actin polymerization and adhesion stabilization.
Figure 8.
Inhibition of PI3K blocks the accumulation of PAK-PBD and F-actin in anchors and their insertion sites. (a and b) U937 cells transfected with PAK-PBD-YFP were pretreated with 1 µM Wort (30 min), adhered under static conditions to VCAM-1 (2 min), and then exposed to fluid flow (2 dynes/cm2 for 2 min). Confocal z stacks were obtained just before and after exposure to flow. (a) Optical slices at the level of anchor insertion (open arrowheads) showed that Wort inhibited PAK-PBD-YFP accumulation. Solid arrowheads indicate anchors. (b) Quantification of upstream versus downstream PAK-PBD-YFP accumulation (means ± SD; n = 5 cells per group; three IEs). MFI, mean fluorescence intensity. (c) Lifeact-GFP–transfected U937 cells were adhered to VCAM-1 and exposed to fluid flow (2 dynes/cm2). Wort was then added to the perfusion buffer (at time = 0 s). Sequential images show a gradual diminution of F-actin in upstream anchor insertions (open arrowheads). Closed arrowheads indicate anchors. See Video 9. Arrows indicate the direction of fluid flow. Bars, 10 µm.
Fluid flow activates PI3K at the upstream cell membrane and in anchors
We investigated spatial and temporal evidence for PI3K activation during formation of anchors. U937 cells were transfected with a GFP probe consisting of the pleckstrin homology (PH) domain of protein kinase B (AKT). The AKT-PH probe specifically binds phosphatidylinositol 3,4,5-triphosphate (PIP3), the main product catalyzed by PI3Ks (Yu et al., 2003). The basal level of PIP3 in cells is low, and thus, AKT-PH is evenly distributed. Local activation of PI3Ks converts PIP2 to PIP3 in the cell membrane and leads to an increased local concentration of AKT-PH (Vanhaesebroeck and Waterfield, 1999; Botelho et al., 2000). U937 cells adhered under static conditions to VCAM-1 showed no preferential distribution of AKT-PH or a PLC-δ PH domain GFP fusion probe that preferentially binds PIP2 (Fig. 9, a and b). In contrast, upon exposure of cells to flow, the AKT-PH, but not the PLC-δ–PH, preferentially accumulated within upstream anchors and at their insertion sites (Fig. 9, c–g). These findings are consistent with the rapid activation of PI3K in upstream anchors by fluid flow.
Figure 9.

Fluid flow activates PI3K at the upstream cell membrane and in anchors. (a–d) U937 cells transfected with probes that bind to PIP3 (AKT-PH-GFP) or PIP2 (PLC-δ–PH-GFP). Cells were adhered to a VCAM-1–coated surface and imaged before and after exposure to fluid flow (2 dynes/cm2 for 2 min). Images of representative cells at levels above the adhesion surface to approximately the midpoint, with the corresponding GFP fluorescence profile at the white dashed lines. A difference between upstream and downstream GFP intensity was only observed in cells transfected with AKT-PH-GFP after exposure to fluid flow (dashed red lines in c vs. d). (e) Quantification of GFP fluorescence (means ± SD; n = 9 for flow and 4 for static; three IEs; *, P < 0.001) MFI, mean fluorescence intensity. (f and g) Accumulation of the AKT-PH-GFP (PIP3), but not PLC-δ–PH-GFP (PIP2), probe in anchors. Images of collapsed z stacks (top and side views) of representative transfected U937 cells that were exposed to flow as in a–d. Each red dashed line is a reference line that was set at the downstream peak of AKT-PH or PLC-δ–PH-GFP probe fluorescence intensity. This line facilitates comparison to the peak fluorescence intensity at the upstream edge of the cell. Increased upstream fluorescence is seen only with the AKT-PH-GFP probe. The corresponding GFP fluorescence profiles within the yellow boxes are shown below. Solid arrowheads indicate anchors, and open arrowheads show regions of anchor insertion. Arrows indicate the direction of fluid flow. Bars, 10 µm.
Discussion
Contact-dependent cells, such as fibroblasts and endothelial cells, continually generate internal traction forces via integrin-mediated adhesion to extracellular matrix (Ridley et al., 2003). In contrast, leukocytes in the vascular compartment must arrest on inflamed endothelium. This suddenly exposes them to high external forces from flowing blood, which requires leukocytes to resist this detaching force. We investigated the cellular and molecular responses of adherent leukocytes subjected to hydrodynamic forces. Our data reveal a mechanism that allows leukocytes to sense and resist these forces (Fig. 10). Exposure of blood leukocytes adherent via high affinity VLA-4 to an external force stretches microvilli and triggers localized accumulation of active Rap1 and PI3K, which regulate Rac-dependent actin polymerization. As a result, F-actin–enriched anchors form and provide the tensile strength necessary to reinforce and resist a detaching force.
Figure 10.

Schematic illustration of leukocyte anchor formation and stabilization mechanism in response to hydrodynamic force. Leukocytes attached to VCAM-1 via high affinity VLA-4 stretch microvilli in response to fluid flow. Tension generated through integrins along F-actin activates PI3K and Rap1. Rap1 and/or PI3K localizes Rac GEFs to sites of force generation. Rac-dependent F-actin polymerization stabilizes the actin cytoskeleton. Leukocyte structural anchors form stabilizing VLA-4–mediated arrest and adhesion.
PSGL-1–mediated rolling results in a rapid elongation of microvilli and microridges and the formation of tethers (Schmidtke and Diamond, 2000; Ley et al., 2007). Tethers form as a result of separation of the lipid bilayer from the cortical cytoskeleton (Ramachandran et al., 2004), and tether formation has been associated with lower rolling velocities. In part, this may be because each tether acts as a fulcrum that promotes cell rotation, which changes the moment of force, and decreases the strain experienced by PSGL-1–selectin bonds (Schmidtke and Diamond, 2000). Recently, total internal reflectance fluorescence microscopy was used to visualize the formation of membrane tethers that extended as much as 16 µm upstream of rolling neutrophils and counteracted hydrodynamic forces (Sundd et al., 2010). However, unlike anchors, tethers are transient structures consistent with the transient nature of PSGL-1–selectin bonds. In contrast, high affinity integrin bonds are more stable and maintain prolonged contact with activated endothelium. This leads to anchor formation. When we inhibited Rap1, PI3K, Rac, or actin polymerization, the cell body was displaced from the initial site of adhesion. Moreover, anchors were destabilized, and F-actin was extruded from the cortical cytoskeleton, yet integrin-mediated adhesive contacts persisted (Figs. 2, 5, 6, and 7).
Rapid accumulation of F-actin and anchor formation was observed in real time when fluid flow was applied to adherent cells (Fig. 3). Anchors were formed upon stretching of an upstream microvillus or microridge. Anchors were maintained for as long as cells were exposed to a hydrodynamic force (Fig. 4). The length and height of anchors can act as lever arms dissipating forces transmitted by integrin bonds, as previously described for selectin-mediated rolling (Alon et al., 1995). Anchor dimensions were proportionally similar in human monocytes and larger U937 cells, suggesting a potentially universal structural response in leukocytes to defined hydrodynamic forces (Fig. 3 c and Fig. S3, b and c).
The contribution of tensile forces to outside-in integrin signaling initiated by ligand binding and integrin clustering is poorly understood. We did not observe significant changes in cell morphology or in the distribution of F-actin when leukocytes with high affinity integrins were adhered to VCAM-1–coated surfaces under static conditions (Fig. 3 and Videos 4 and 5). Only after the application of fluid flow did F-actin accumulation and the formation of anchors occur. VLA-4 located at the tips of anchors bound to VCAM-1 on the adhesion surface, yet we observed F-actin accumulation throughout anchors and at their insertion in to the cortical cytoskeleton. This suggests that tension transmitted throughout anchors initiates signaling ultimately leading to actin polymerization. Potential mechanosensory molecules include adaptor proteins, such as p130cas (Sawada et al., 2006) stretch-activated receptors and/or channels (Matthews et al., 2006).
Consistent with a previous study (Sawada et al., 2001), we found that force induced active Rap1 accumulation at sites of anchor insertion, which was critical for adhesion stabilization (Fig. 5). The role of Rap1 in leukocyte arrest is controversial. Mn2+-induced T lymphocyte adhesion to ICAM-1 or VCAM-1 has been reported to be both dependent (de Bruyn et al., 2002) and independent (Ghandour et al., 2007) of Rap1. In the context of chemokine-induced arrest mediated by VLA-4, Rap1 was shown to be dispensable (Ghandour et al., 2007). The reasons for this discrepancy remain unclear, but it is possible that differences in expression or activation of Rap1 GEFs and GTPase-activating proteins are responsible.
Previous studies indicate that Rap1 signaling functions to activate PI3K (Fukuyama et al., 2006; Kortholt et al., 2010). It is possible that amplification loops might exist that regulate their activity. In Dictyostelium discoideum, activated Rap1 binds and activates PI3K, which in turn is critical for Rac1 activation, cell adhesion, and subsequent pseudopod formation (Kortholt et al., 2010). In E-cadherin–mediated cell–cell adhesion, PI3K was required for Rap1-dependent activation of Rac via Vav2, a Rac-GEF; in addition, overexpression of constitutively active Rap1 could not overcome the effects of PI3K inhibition, suggesting that PI3K is downstream of Rap1 (Fukuyama et al., 2006). Whether PI3K regulates Rap1-GEFs is not currently well understood.
Previous studies have demonstrated the activation of PI3K in cells exposed to tensile forces (Suzuma et al., 2002; Katsumi et al., 2005). In our study, leukocyte structural adaptations in response to fluid flow are dependent on force-induced activation of PI3K and localization of its product, PIP3, within anchors (Fig. 9). PI3K activation was also required for Rac-dependent F-actin accumulation in anchors (Fig. 8). This finding is consistent with the established role of PI3K as a regulator of actin polymerization (Vanhaesebroeck and Waterfield, 1999; Pollard et al., 2000). The reinforcement of tension-bearing structures by actin polymerization is critical for adaptation of cells to external forces.
Materials and methods
Reagents and antibodies
Anti–human IgG F(ab′)2 (mouse adsorbed) for coating of adhesion surfaces was purchased from Invitrogen. Human SDF-1α was purchased from PeproTech, and human VCAM-1/fragment crystallizable (Fc) and ICAM-1/Fc were purchased from R&D Systems. HBSS (containing Ca2+ and Mg2+), manganese chloride, fMLF (the bacterial chemoattractant f-Met-Leu-Phe), and PKH-26 were purchased from Sigma-Aldrich. CytoD, LatB, LY294002, Wort, and PI3K-γ inhibitor II AS605240 were purchased from EMD. 488 phalloidin was purchased from Invitrogen. Primary antibodies included mouse anti–human CD49d clone HP2/1 (AbD Serotec) and clone 44H6 (gift from M. Letarte, Hospital for Sick Children, Toronto, Ontario, Canada), FITC- or allophycocyanin (APC)-conjugated CD49d clone 9F10 (BioLegend), APC-conjugated anti–human CD14 clone 61D3 (eBioscience), and FITC-conjugated anti–mouse CD8 (BioLegend). Goat anti–mouse-Cy3 secondary antibodies were purchased from Jackson ImmunoResearch Laboratories, Inc.
Leukocytes
Cell culture.
The myelomonocytic U937 cell line was obtained from American Type Culture Collection. A line of U937 cells transfected with human formyl peptide receptor (FPR; U937-FPR) was obtained from G.P. Downey (University of Toronto, Toronto, Ontario, Canada). U937 cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated FBS. 500 g/ml geneticin was added to the culture medium when culturing U937-FPR cells. Cell culture reagents were purchased from Invitrogen.
Human monocytes.
Human peripheral blood mononuclear cells depleted of platelets were isolated by an adapted density gradient centrifugation protocol (OptiPrep; Axis-Shield). Blood sample collection from healthy human volunteers was approved by the Research Ethics Board at University Health Network. In brief, venous blood anticoagulated with 2 mM EDTA was mixed with 1.85 g OptiPrep and 2 ml of solution B for every 5 ml of blood (100 ml of solution B = 3.42 ml NaCl, 1 ml Hepes, 2 ml EDTA, 1 ml plasma, and 92.5 ml distilled H2O) to create a 1.091-g/ml density layer. Layered over the top was a 1.078-g/ml density layer made up of 2 ml of solution B (for every 5 ml of blood) and 3.02 g OptiPrep. Enriched mononuclear leukocytes were separated by centrifugation at 1,600 rpm for 30 min at 4°C. Monocytes were purified from peripheral blood mononuclear cells using the human monocyte enrichment kit (EasySep; STEMCELL Technologies) according to the manufacturer’s protocol and used immediately. The purity of monocytes was determined by flow cytometry with the anti–human CD14 antibody and was routinely >90%.
Mouse lymphocytes.
Axillary, cervical, and inguinal lymph nodes were harvested and gently dispersed, and cells were filtered through a 70-µm mesh. After a PBS wash, a negative selection process was performed using a CD8+ T cell isolation kit (MACS; Miltenyi Biotec) according to the manufacturer’s instructions. The purity of CD8+ T cells was determined by flow cytometry and routinely was >90%.
Cell transfection
U937 cells were suspended in 100 µl Nucleofector solution supplemented with 1 or 2 µg of each construct (Lifeact-GFP, Lifeact-RFP, PH-AKT-GFP, PH-PLC-δ–GFP, dnRac-GFP, dnCdc42-GFP, PAK-PBD-YFP, Rap1-GFP, Rap1-N17–GFP, and Rap1-V12–GFP). For double transfections, 1 µg of each construct was used. Cells were transfected using the Cell line Nucleofector Kit V (Lonza) and the Nucleofector II program V-001. Cells were recovered in 500 µl RPMI 1640 with 10% FBS and transferred into 1-ml preincubated FBS-supplemented RPMI 1640 6-well plates. Cells were allowed to recover in a humidified 5% CO2, 37°C chamber for 24 or 48 h. The GFP- and/or RFP-expressing cells were analyzed by fluorescence microscopy (FluoView A; Olympus) and flow cytometry (Cytomics FC500; Beckman Coulter) to monitor transfection efficiency. At 24 or 48 h, a trypan blue viability assay was performed before infusion into a parallel plate flow chamber confocal microscopy setup. Predesigned siRNA targeting PI3K-γ was purchased from Invitrogen (Silencer; catalog no. 16708; ID no. 143809). siRNA was transfected using Nucleofector as described in this paragraph. After transfection with siRNA, cells were cultured in RPMI 1640 and 10% FBS for 72 h. Efficiency of knockdown was determined by Western blotting with an anti–p110-γ antibody (Cell Signaling Technology).
DNA constructs.
The first 17 amino acids of the actin-binding protein 140 probe, referred to as Lifeact, fused to GFP or RFP were gifts from M. Sixt (Institute of Science and Technology, Klosterneuburg, Austria). Plasmids encoding the PH domain of AKT and PLC-δ fused to GFP (PH-AKT-GFP and PH-PLC-δ–GFP, respectively), PAK-PBD-YFP, dnRac-N17–GFP, and dnCdc42-N17–GFP were gifts from S. Grinstein (Hospital for Sick Children, Toronto, Ontario, Canada). Rap1-GFP, dnRap1-N17–GFP, and constitutively active Rap1-V12–GFP were gifts from M. Philips (New York University School of Medicine, New York, NY).
Parallel plate flow chamber assays
VCAM-1/Fc was immobilized on 35-mm polystyrene tissue-culture dishes as previously described (Hyduk et al., 2007). In brief, goat anti–human IgG F(ab)2 was incubated at a low (10 µg/ml) or high (100 µg/ml) density for 60 min at 22°C. In some experiments with low density coating, 20 µg/ml human SDF-1α was co-coated (for 120 min at 22°C). Nonspecific binding sites were blocked with 5% FBS for 60 min followed by VCAM-1/Fc for 60 min (5 µg/ml for low density or 20 µg/ml for high density experiments). Leukocyte accumulation and detachment assays were performed using parallel plate flow chambers (Glycotech) as previously described (Hyduk et al., 2007). Adhesion molecule–coated dishes served as the bottom surface, and a silicone gasket formed the flow path (0.254-mm height and 2.5-mm width). Flow chambers were maintained at 37°C with an infrared heat lamp. Cells were observed with an inverted phase-contrast microscope (Diaphot 300; Nikon), recorded with a video camera (DXC-151A; Sony) and time-lapse video cassette recorder (SVT-S3100; Sony), and analyzed offline using Image 4.0.2 (Scion). Where indicated, leukocytes were pretreated (for 30 min at 37°C) with either 2 µM CytoD, 2 µM LatB, 50 µM LY294002, 1 µM Wort, 100 µM PI3K-γ inhibitor II AS605240, or DMSO carrier. Alternatively, inhibitors were included within the perfusion assay buffer.
Accumulation assays.
Leukocytes in assay buffer (HBSS with Ca2+ and Mg2+, 0.5% FBS, and 10 mM Hepes) were infused (at a concentration of 1.0 × 106 cells/ml) with a programmable syringe pump (Kent Scientific) to achieve a wall shear stress of 1 or 2 dynes/cm2. After 2 min of infusion, the number of leukocytes stationary for >5 s was determined in three to six nonoverlapping high power fields. In some experiments, leukocytes were infused at 0.5 dynes/cm2 for 30 s followed by an increased flow (2 dynes/cm2). After 60 s, cells were perfusion fixed by infusing 2.5% glutaraldehyde and 2.0% formaldehyde in 0.1 M phosphate buffer, pH 7.4, for 5 min.
Detachment assays.
Leukocytes were infused at a concentration of 1.0 × 106 cells/ml via the outflow port into a flow chamber and allowed to settle under static conditions onto the adhesion surface for 2 min. After static adhesion, flow was introduced by pulling assay buffer through the flow chamber with a programmable syringe pump. Incremental increases in shear stress occurred at 30-s intervals. The number of cells remaining adherent after each interval was determined in three to six nonoverlapping high power fields. Data were expressed as a percentage of input cells remaining adherent.
Fixation under flow.
U937 cells or monocytes (1.0 × 106 cells/ml) adhered under flow to a VCAM-1–coated 33-mm glass coverslip mounted in parallel plate flow chamber were also fixed under flow. For SEM, 2.5% glutaraldehyde and 2.0% formaldehyde in 0.1 M phosphate buffer, pH 7.4, was infused for 5 min. For immunoconfocal experiments, leukocytes were fixed with 2.0% PFA.
SEM
Glass coverslips perfused with 2.5% glutaraldehyde and 2.0% formaldehyde were placed in the same fixative for 1 h (22°C). After washing, coverslips were postfixed with 1% osmium tetroxide in 0.1 M phosphate buffer, pH 7.4, for 1 h. Samples were dehydrated through an ascending graded ethanol series, infiltrated with a graded hexamethyldisilizane (Sigma-Aldrich) in absolute ethanol, and dried overnight in a fume hood. Coverslips were mounted on aluminum stubs, sputter coated with gold-palladium (SCD 050; Bal-Tec), and examined using a scanning electron microscope (S-2500; Hitachi) operated at 15 kV. Digital images were acquired using a frame grabber and PCI (Quartz Imaging Corp.). To visualize the side profile of leukocytes and anchors, glass coverslips from flow chamber experiments processed for EM were scored with a diamond scriber, and half the coverslip was removed to rotate the specimen and image it at a 90° angle. Side-view micrographs were characterized in ImageJ (National Institutes of Health) for anchor thickness, anchor insertion height, and anchor length. Veil-type upstream structures were characterized as an individual anchor.
Membrane extraction EM
U937 cells were plated on a 5-mm coverslip coated with 20 µg/ml VCAM-1/Fc for a period of 30 min in assay buffer. Upon cell spreading, leukocyte cell membranes were extracted as previously described (Hartwig and Shevlin, 1986). In brief, before detergent extraction, cells were washed in PHEM buffer (60 mM Pipes, 25 mM Hepes, 20 mM EGTA, and 2 mM MgCl2, pH 6.9). The PHEM buffer was aspirated, and 200 µl PHEM with 0.75% Triton X-100 and 1 µM phalloidin was added for 2 min. The solution was removed and washed three times with PHEM–1 µM phalloidin. Thereafter, cells were fixed with PHEM–1% glutaraldehyde for 10 min, removed, and replaced with 0.01% sodium azide. Coverslips were processed for EM as previously described (Hartwig and DeSisto, 1991). In brief, coverslips were washed extensively in distilled water, rapidly frozen on a helium-cooled copper block, freeze dried at −80°C, and rotary coated with 1.5 nm tantalum/tungsten at a 25° angle and 5 nm carbon at 90° without rotation. The metal replicas were floated off the coverslip using 25% hydrofluoric acid, washed in water, and collected on carbon-coated Formvar 200-mesh copper grids. Grids were photographed in an electron microscope (1200-EX; JEOL) at 80 kV.
Fluorescence microscopy analysis of VLA-4 colocalization with F-actin
Leukocytes were adhered to coverslips coated with poly-l-lysine or VCAM-1. In static assays, cells were washed with assay buffer and incubated with 5 µg/ml mouse anti–human α4 integrin antibody (HP2/1 or 44H6; for 1 h at 22°C). Coverslips were blocked with 5% FBS followed by a series of washes. Cells were simultaneously fixed in 2% PFA and permeabilized in 0.1% Triton X-100 (5 min at 22°C), washed three times, and incubated with 488 phalloidin (1:1,000 dilution; Invitrogen) and goat anti–mouse secondary antibody (1:500 dilution). In fluid flow experiments, leukocytes were preincubated for 5 min on ice with the 44H6 antibody before the parallel plate flow chamber assay. At the end of the assay, cells were perfusion fixed with 2% PFA, washed, and stained with a secondary antibody and 488 phalloidin. Coverslips were mounted in antifade mounting medium (Vectashield; Vector Laboratories) and imaged with a laser-scanning confocal microscope (FluoView 1000; Olympus) with a 60× oil immersion objective (Plan Apochromat; 1.42 NA).
Live-cell imaging
Real-time imaging was performed on a confocal microscope (FluoView 1000) equipped with a 60× oil immersion objective, FV10-ASW acquisition software (Olympus), and an environmental chamber that maintained parallel plate flow chambers at 37°C throughout the experiment. The frame acquisition rate ranged from 0.4 to 1.0 s per frame (indicated in the video legend). When required, a digital zoom ranging from 5.0 to 9.0 was used (indicated in the video legend; refer to scale bars). Z stacks were acquired by obtaining 0.2–0.5-µM-thick serial sections of an entire cell. 3D reconstructions from z stacks were performed using FV10-ASW acquisition software.
U937 cells were transfected with GFP-, RFP-, or YFP-tagged constructs using the Nucleofector 24 or 48 h before imaging. Transfected cells were infused into a flow chamber and allowed to settle for 1–2 min. At this time, a transfected U937 cell that was brightly fluorescent was identified, and images (including a z stack) were acquired under static conditions for 1 min. Fluid flow was introduced, and continuous real-time frame acquisition was continued for 2–5 min followed by a z stack. If the cell detached upon exposure to fluid flow or shortly thereafter, the experiment was terminated. Therefore, postshear serial z stacks and 3D reconstructions were obtained only when a selected cell did not detach. Occasionally, real-time imaging continued for 2–5 min once fluid flow was stopped. In some experiments, transfected U937 cells were pretreated with a pharmacological inhibitor (30 min at 37°C) before infusion into the flow chamber, whereas in other experiments, inhibitors were introduced into the assay buffer after stabilizing anchors formed (in response to fluid flow), and real-time imaging revealed the effect of the inhibitor on preformed anchors.
Flow cytometry
Integrin and F-actin quantification.
To quantify total leukocyte F-actin and α4β1 expression, U937 cells at a concentration of 1.0 × 106 cells/ml were placed in assay buffer and stained with 5 µg/ml anti–human CD49d (clone 44H6) or APC-conjugated anti–human CD49d (clone 9F10) for 30 min on ice. Cells were washed with PBS and fixed in 2% PFA. Cells were then blocked in 5% FBS for 1 h, permeabilized for 5 min with 0.1% Triton X-100, and incubated with Cy3-conjugated anti–mouse IgG secondary antibody (1:500 dilution) and 488 phalloidin (1:1,000 dilution; for 1 h at 4°C in the dark). Cells were washed, centrifuged, and reconstituted in 500 µl PBS. A flow cytometer (LSR II; BD) was used.
Integrin affinity assay.
Soluble LDV-FITC binding assay FITC-conjugated peptide 4-((N′-2-methylphenyl)ureido)-phenylacetyl-l-leucyl-l-α-aspartyl-l-valyl-l-prolyl-l-alanyl-l-alanyl-l-lysine (LDV-FITC) was synthesized by Commonwealth Biotechnologies, Inc. and used as a ligand for VLA-4 integrins. The LDV-FITC binding assay was performed as previously described (Hyduk et al., 2011). In brief, U937-FPR cells (0.5 × 106 cells in 50 µl) pretreated with vehicle, CytoD, LatB, or LY294002 and siRNA-transfected cells were suspended in HBSS (with 1 mM Ca2+/Mg2+) supplemented with 20 mM Hepes and 0.5% FBS (assay buffer) and were incubated with 3 nM LDV-FITC (30 min at 37°C). Cells were stimulated at 37°C with 100 nM fMLF (2 min), 50 nM PMA (30 min), or 1 mM MnCl2 (5 min), diluted with 1 ml HBSS, and immediately fixed by adding 1 ml of 2% PFA at 22°C. Cells were washed with HBSS, and bound LDV-FITC was detected using biotin-conjugated anti-FITC followed by streptavidin-fluorochrome (Jackson ImmunoResearch Laboratories, Inc.). Cells were analyzed by flow cytometry using a flow cytometer (Cytomics FC500).
Leukocyte cytoskeleton ghosts.
The association of VLA-4 with the actin cytoskeleton was assessed by extracting U937 cell membranes with 1% Triton X-100 (5 min on ice) and preparing cytoskeletal ghosts. This approach was adapted from Nebe et al. (1997). Initially, cells were stained with PKH-26, a dye that labels membrane lipids, because the loss of PKH-26 fluorescence indicated the extent of membrane extraction. Once membrane extraction was optimized, experiments were then performed to assess VLA-4 association with the actin cytoskeleton. Cells were treated with inhibitors (CytoD or LatB for 30 min), stained with mouse anti–human α4 integrin antibody (9F10), and washed, and the membrane was extracted. Cells were washed, stained with a phycoerythrin-conjugated secondary antibody (goat anti–mouse IgG), washed, fixed with PFA (20 min), washed, and analyzed with a flow cytometer (LSR II).
Image analysis
The displacement of cells from the initial adhesion site after exposure to fluid flow in a parallel plate chamber was quantified using ImageJ software. The mean displacement of cells that resisted detachment for >30 s was determined. ImageJ software was also used to quantify fluorescence intensity in images. The appropriate image slice was selected, and a rectangular box or line was drawn along the x axis that intersected a leukocyte. The box encompassed the upstream anchors and the downstream cortical actin cytoskeleton as well as regions of upstream and downstream background fluorescence. A fluorescence intensity profile within the box was generated across the entire cell. Cells transfected with different fluorescent probes were analyzed, and multiple cells were analyzed in each experiment. In some experiments, analyses were performed on sequential image slices using the same line or rectangle to gain insight into the cell’s height as a measure of the anchor fluorescence profile. A plot profile analysis was applied in ImageJ to determine the fluorescence distribution. Pixel intensity values were obtained and imported into an Excel microscopy worksheet (Microsoft). A mean of the most intense pixels coinciding with the upstream location of anchors was compared with the downstream cortical actin cytoskeleton, and a ratio of upstream to downstream mean fluorescence intensity was obtained. This analysis consistently showed higher Lifeact-GFP fluorescence signals within upstream anchors relative to the downstream cortical actin cytoskeleton, a region where anchors do not form. 3D deconvolution was performed using AutoQuant deconvolution software (Media Cybernetics) with the following settings: deconvolution method with adaptive point spread function (PSF; blind); PSF setting with theoretical PSF; and deconvolution setting with 12 iterations, noise level high, and a noise value of 200.
Statistics
All data reported are expressed as mean values ± SD. Multiple group analysis was performed using one-way analysis of variance with Tukey-Kramer multiple comparisons posthoc test applied. Group comparisons were determined to be significant for P < 0.05.
Online supplemental material
Fig. S1 shows the effects of actin polymerization inhibitors on F-actin content, distribution in spread cells, VLA-4 expression, and VLA-4 colocalization with F-actin. Fig. S2 shows the effects of actin polymerization inhibitors on fMLF-induced VLA-4 affinity up-regulation and accumulation of U937 cells in a flow chamber assay. Fig. S3 shows additional SEM images of leukocyte anchors, quantification of anchor dimensions, and supporting images of leukocytes transfected with Lifeact-GFP demonstrating anchor formation. Fig. S4 shows additional supporting images of U937 cells transfected with Rap1-GFP, PAK-PBD-YFP, dnRac-GFP, and dnCdc42-GFP. Fig. S5 shows the effects of inhibiting PI3K on VLA-4 affinity up-regulation, the extent of PI3K-γ knockdown with siRNA, and the colocalization of VLA-4 and F-actin. Videos 1 and 2 show that pretreatment of two different U937 cells with LatB results in F-actin extrusion from cells after exposure to fluid flow. Video 3 shows that stable U937 cell adhesion is disrupted upon addition of LatB to the flow buffer. Videos 4 and 5 show actin dynamics in representative Lifeact-GFP–transfected U937 cells that are adhered under static conditions to VCAM-1 (Video 4) or poly-l-lysine (Video 5). Video 6 shows the formation of upstream F-actin–enriched anchors upon introduction of fluid flow. Videos 7 and 8 show that anchors retract and that the upstream F-actin content diminishes when fluid flow is turned off in two different cells. Video 9 shows degradation of F-actin–enriched anchors when Wort is perfused onto of a stably adherent U937 cell. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201107140/DC1. Online supplemental material is also provided in the JCB DataViewer.
Acknowledgments
We would like to thank Henry Hong and the imaging facility in the Department of Cell and Systems Biology at the University of Toronto for their expertise and assistance with EM imaging.
This work was supported by the Canadian Institutes of Health Research (grant MOP-89740 to M.I. Cybulsky and Canada Graduate Scholarship Doctoral Award to J. Rullo) and Heart and Stroke Foundation of Ontario (Career Investigator Award to M.I. Cybulsky). M.I. Cybulsky is a member of the Heart and Stroke/Richard Lewar Centre of Excellence at the University of Toronto.
Footnotes
Abbreviations used in this paper:
- APC
- allophycocyanin
- CytoD
- Cytochalasin D
- DIC
- differential interference contrast
- dn
- dominant negative
- Fc
- fragment crystallizable
- FPR
- formyl peptide receptor
- GEF
- guanine exchange factor
- GPCR
- G protein–coupled receptor
- IE
- independent experiment
- LatB
- Latrunculin B
- PAK
- p21-activated kinase
- PBD
- p21-biding domain
- PH
- pleckstrin homology
- PI3K
- phosphoinositide 3-kinase
- PIP3
- phosphatidylinositol (3,4,5)-trisphosphate
- PSF
- point spread function
- SEM
- scanning EM
- VLA-4
- very late antigen-4
- Wort
- Wortmannin
References
- Alon R., Dustin M.L. 2007. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 26:17–27 10.1016/j.immuni.2007.01.002 [DOI] [PubMed] [Google Scholar]
- Alon R., Hammer D.A., Springer T.A. 1995. Lifetime of the P-selectin-carbohydrate bond and its response to tensile force in hydrodynamic flow. Nature. 374:539–542 10.1038/374539a0 [DOI] [PubMed] [Google Scholar]
- Alon R., Feigelson S.W., Manevich E., Rose D.M., Schmitz J., Overby D.R., Winter E., Grabovsky V., Shinder V., Matthews B.D., et al. 2005. α4β1-dependent adhesion strengthening under mechanical strain is regulated by paxillin association with the α4-cytoplasmic domain. J. Cell Biol. 171:1073–1084 10.1083/jcb.200503155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur W.T., Quilliam L.A., Cooper J.A. 2004. Rap1 promotes cell spreading by localizing Rac guanine nucleotide exchange factors. J. Cell Biol. 167:111–122 10.1083/jcb.200404068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos J.L., de Rooij J., Reedquist K.A. 2001. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol. 2:369–377 10.1038/35073073 [DOI] [PubMed] [Google Scholar]
- Botelho R.J., Teruel M., Dierckman R., Anderson R., Wells A., York J.D., Meyer T., Grinstein S. 2000. Localized biphasic changes in phosphatidylinositol-4,5-bisphosphate at sites of phagocytosis. J. Cell Biol. 151:1353–1368 10.1083/jcb.151.7.1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakebusch C., Fässler R. 2003. The integrin-actin connection, an eternal love affair. EMBO J. 22:2324–2333 10.1093/emboj/cdg245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher E.C. 1991. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 67:1033–1036 10.1016/0092-8674(91)90279-8 [DOI] [PubMed] [Google Scholar]
- Campbell J.J., Hedrick J., Zlotnik A., Siani M.A., Thompson D.A., Butcher E.C. 1998. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science. 279:381–384 10.1126/science.279.5349.381 [DOI] [PubMed] [Google Scholar]
- Chan J.R., Hyduk S.J., Cybulsky M.I. 2001. Chemoattractants induce a rapid and transient upregulation of monocyte α4 integrin affinity for vascular cell adhesion molecule 1 which mediates arrest: an early step in the process of emigration. J. Exp. Med. 193:1149–1158 10.1084/jem.193.10.1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Springer T.A. 1999. An automatic braking system that stabilizes leukocyte rolling by an increase in selectin bond number with shear. J. Cell Biol. 144:185–200 10.1083/jcb.144.1.185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choquet D., Felsenfeld D.P., Sheetz M.P. 1997. Extracellular matrix rigidity causes strengthening of integrin-cytoskeleton linkages. Cell. 88:39–48 10.1016/S0092-8674(00)81856-5 [DOI] [PubMed] [Google Scholar]
- Constantin G., Majeed M., Giagulli C., Piccio L., Kim J.Y., Butcher E.C., Laudanna C. 2000. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 13:759–769 10.1016/S1074-7613(00)00074-1 [DOI] [PubMed] [Google Scholar]
- Coughlin M.F., Schmid-Schönbein G.W. 2004. Pseudopod projection and cell spreading of passive leukocytes in response to fluid shear stress. Biophys. J. 87:2035–2042 10.1529/biophysj.104.042192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin M.F., Sohn D.D., Schmid-Schönbein G.W. 2008. Recoil and stiffening by adherent leukocytes in response to fluid shear. Biophys. J. 94:1046–1051 10.1529/biophysj.107.107102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley D.R. 2004. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem. Soc. Trans. 32:831–836 10.1042/BST0320831 [DOI] [PubMed] [Google Scholar]
- de Bruyn K.M., Rangarajan S., Reedquist K.A., Figdor C.G., Bos J.L. 2002. The small GTPase Rap1 is required for Mn(2+)- and antibody-induced LFA-1- and VLA-4-mediated cell adhesion. J. Biol. Chem. 277:29468–29476 10.1074/jbc.M204990200 [DOI] [PubMed] [Google Scholar]
- Ferrandi C., Ardissone V., Ferro P., Rückle T., Zaratin P., Ammannati E., Hauben E., Rommel C., Cirillo R. 2007. Phosphoinositide 3-kinase gamma inhibition plays a crucial role in early steps of inflammation by blocking neutrophil recruitment. J. Pharmacol. Exp. Ther. 322:923–930 10.1124/jpet.107.123026 [DOI] [PubMed] [Google Scholar]
- Fukuyama T., Ogita H., Kawakatsu T., Inagaki M., Takai Y. 2006. Activation of Rac by cadherin through the c-Src-Rap1-phosphatidylinositol 3-kinase-Vav2 pathway. Oncogene. 25:8–19 [DOI] [PubMed] [Google Scholar]
- Ghandour H., Cullere X., Alvarez A., Luscinskas F.W., Mayadas T.N. 2007. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood. 110:3682–3690 10.1182/blood-2007-03-077628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. 1994. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu. Rev. Cell Biol. 10:31–54 10.1146/annurev.cb.10.110194.000335 [DOI] [PubMed] [Google Scholar]
- Han J., Luby-Phelps K., Das B., Shu X., Xia Y., Mosteller R.D., Krishna U.M., Falck J.R., White M.A., Broek D. 1998. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science. 279:558–560 10.1126/science.279.5350.558 [DOI] [PubMed] [Google Scholar]
- Hartwig J.H., DeSisto M. 1991. The cytoskeleton of the resting human blood platelet: structure of the membrane skeleton and its attachment to actin filaments. J. Cell Biol. 112:407–425 10.1083/jcb.112.3.407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig J.H., Shevlin P. 1986. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J. Cell Biol. 103:1007–1020 10.1083/jcb.103.3.1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit B., Liu L., Colarusso P., Puri K.D., Kubes P. 2008. PI3K accelerates, but is not required for, neutrophil chemotaxis to fMLP. J. Cell Sci. 121:205–214 10.1242/jcs.020412 [DOI] [PubMed] [Google Scholar]
- Howard T.H., Meyer W.H. 1984. Chemotactic peptide modulation of actin assembly and locomotion in neutrophils. J. Cell Biol. 98:1265–1271 10.1083/jcb.98.4.1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyduk S.J., Cybulsky M.I. 2002. Alpha 4 integrin signaling activates phosphatidylinositol 3-kinase and stimulates T cell adhesion to intercellular adhesion molecule-1 to a similar extent as CD3, but induces a distinct rearrangement of the actin cytoskeleton. J. Immunol. 168:696–704 [DOI] [PubMed] [Google Scholar]
- Hyduk S.J., Chan J.R., Duffy S.T., Chen M., Peterson M.D., Waddell T.K., Digby G.C., Szaszi K., Kapus A., Cybulsky M.I. 2007. Phospholipase C, calcium, and calmodulin are critical for alpha4beta1 integrin affinity up-regulation and monocyte arrest triggered by chemoattractants. Blood. 109:176–184 10.1182/blood-2006-01-029199 [DOI] [PubMed] [Google Scholar]
- Hyduk S.J., Rullo J., Cano A.P., Xiao H., Chen M., Moser M., Cybulsky M.I. 2011. Talin-1 and kindlin-3 regulate alpha4beta1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J. Immunol. 187:4360–4368 10.4049/jimmunol.1003725 [DOI] [PubMed] [Google Scholar]
- Hynes R.O. 2002. Integrins: bidirectional, allosteric signaling machines. Cell. 110:673–687 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- Katsumi A., Naoe T., Matsushita T., Kaibuchi K., Schwartz M.A. 2005. Integrin activation and matrix binding mediate cellular responses to mechanical stretch. J. Biol. Chem. 280:16546–16549 10.1074/jbc.C400455200 [DOI] [PubMed] [Google Scholar]
- Kim M., Carman C.V., Yang W., Salas A., Springer T.A. 2004. The primacy of affinity over clustering in regulation of adhesiveness of the integrin αLβ2. J. Cell Biol. 167:1241–1253 10.1083/jcb.200404160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortholt A., Bolourani P., Rehmann H., Keizer-Gunnink I., Weeks G., Wittinghofer A., Van Haastert P.J. 2010. A Rap/phosphatidylinositol 3-kinase pathway controls pseudopod formation [corrected]. Mol. Biol. Cell. 21:936–945 (published erratum appears in Mol Biol Cell. 2010. 21:1314) 10.1091/mbc.E09-03-0177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucik D.F., Dustin M.L., Miller J.M., Brown E.J. 1996. Adhesion-activating phorbol ester increases the mobility of leukocyte integrin LFA-1 in cultured lymphocytes. J. Clin. Invest. 97:2139–2144 10.1172/JCI118651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley K., Laudanna C., Cybulsky M.I., Nourshargh S. 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7:678–689 10.1038/nri2156 [DOI] [PubMed] [Google Scholar]
- Makino A., Prossnitz E.R., Bünemann M., Wang J.M., Yao W., Schmid-Schönbein G.W. 2006. G protein-coupled receptors serve as mechanosensors for fluid shear stress in neutrophils. Am. J. Physiol. Cell Physiol. 290:C1633–C1639 10.1152/ajpcell.00576.2005 [DOI] [PubMed] [Google Scholar]
- Manser E., Leung T., Salihuddin H., Zhao Z.S., Lim L. 1994. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature. 367:40–46 10.1038/367040a0 [DOI] [PubMed] [Google Scholar]
- Matthews B.D., Overby D.R., Mannix R., Ingber D.E. 2006. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J. Cell Sci. 119:508–518 10.1242/jcs.02760 [DOI] [PubMed] [Google Scholar]
- Nebe B., Bohn W., Pommerenke H., Rychly J. 1997. Flow cytometric detection of the association between cell surface receptors and the cytoskeleton. Cytometry. 28:66–73 10.1002/(SICI)1097-0320(19970501)28:1<66::AID-CYTO8>3.0.CO;2-F [DOI] [PubMed] [Google Scholar]
- Pollard T.D., Blanchoin L., Mullins R.D. 2000. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu. Rev. Biophys. Biomol. Struct. 29:545–576 10.1146/annurev.biophys.29.1.545 [DOI] [PubMed] [Google Scholar]
- Ramachandran V., Williams M., Yago T., Schmidtke D.W., McEver R.P. 2004. Dynamic alterations of membrane tethers stabilize leukocyte rolling on P-selectin. Proc. Natl. Acad. Sci. USA. 101:13519–13524 10.1073/pnas.0403608101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A.J., Hall A. 1992. Distinct patterns of actin organization regulated by the small GTP-binding proteins Rac and Rho. Cold Spring Harb. Symp. Quant. Biol. 57:661–671 10.1101/SQB.1992.057.01.072 [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Paterson H.F., Johnston C.L., Diekmann D., Hall A. 1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 70:401–410 10.1016/0092-8674(92)90164-8 [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Schwartz M.A., Burridge K., Firtel R.A., Ginsberg M.H., Borisy G., Parsons J.T., Horwitz A.R. 2003. Cell migration: integrating signals from front to back. Science. 302:1704–1709 10.1126/science.1092053 [DOI] [PubMed] [Google Scholar]
- Riedl J., Crevenna A.H., Kessenbrock K., Yu J.H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T.A., Werb Z., et al. 2008. Lifeact: a versatile marker to visualize F-actin. Nat. Methods. 5:605–607 10.1038/nmeth.1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhu C., Masinovsky B., Dick K., Sowell C.G., Staunton D.E. 2003. Essential role of phosphoinositide 3-kinase delta in neutrophil directional movement. J. Immunol. 170:2647–2654 [DOI] [PubMed] [Google Scholar]
- Sasaki T., Irie-Sasaki J., Jones R.G., Oliveira-dos-Santos A.J., Stanford W.L., Bolon B., Wakeham A., Itie A., Bouchard D., Kozieradzki I., et al. 2000. Function of PI3Kgamma in thymocyte development, T cell activation, and neutrophil migration. Science. 287:1040–1046 10.1126/science.287.5455.1040 [DOI] [PubMed] [Google Scholar]
- Sawada Y., Nakamura K., Doi K., Takeda K., Tobiume K., Saitoh M., Morita K., Komuro I., De Vos K., Sheetz M., Ichijo H. 2001. Rap1 is involved in cell stretching modulation of p38 but not ERK or JNK MAP kinase. J. Cell Sci. 114:1221–1227 [DOI] [PubMed] [Google Scholar]
- Sawada Y., Tamada M., Dubin-Thaler B.J., Cherniavskaya O., Sakai R., Tanaka S., Sheetz M.P. 2006. Force sensing by mechanical extension of the Src family kinase substrate p130Cas. Cell. 127:1015–1026 10.1016/j.cell.2006.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke D.W., Diamond S.L. 2000. Direct observation of membrane tethers formed during neutrophil attachment to platelets or P-selectin under physiological flow. J. Cell Biol. 149:719–730 10.1083/jcb.149.3.719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimonaka M., Katagiri K., Nakayama T., Fujita N., Tsuruo T., Yoshie O., Kinashi T. 2003. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J. Cell Biol. 161:417–427 10.1083/jcb.200301133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.F., Deem T.L., Bruce A.C., Reutershan J., Wu D., Ley K. 2006. Leukocyte phosphoinositide-3 kinase gamma is required for chemokine-induced, sustained adhesion under flow in vivo. J. Leukoc. Biol. 80:1491–1499 10.1189/jlb.0306227 [DOI] [PubMed] [Google Scholar]
- Srinivasan S., Wang F., Glavas S., Ott A., Hofmann F., Aktories K., Kalman D., Bourne H.R. 2003. Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160:375–385 10.1083/jcb.200208179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundd P., Gutierrez E., Pospieszalska M.K., Zhang H., Groisman A., Ley K. 2010. Quantitative dynamic footprinting microscopy reveals mechanisms of neutrophil rolling. Nat. Methods. 7:821–824 10.1038/nmeth.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuma I., Suzuma K., Ueki K., Hata Y., Feener E.P., King G.L., Aiello L.P. 2002. Stretch-induced retinal vascular endothelial growth factor expression is mediated by phosphatidylinositol 3-kinase and protein kinase C (PKC)-zeta but not by stretch-induced ERK1/2, Akt, Ras, or classical/novel PKC pathways. J. Biol. Chem. 277:1047–1057 10.1074/jbc.M105336200 [DOI] [PubMed] [Google Scholar]
- Tzima E., Del Pozo M.A., Kiosses W.B., Mohamed S.A., Li S., Chien S., Schwartz M.A. 2002. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 21:6791–6800 10.1093/emboj/cdf688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E., Irani-Tehrani M., Kiosses W.B., Dejana E., Schultz D.A., Engelhardt B., Cao G., DeLisser H., Schwartz M.A. 2005. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 437:426–431 10.1038/nature03952 [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B., Waterfield M.D. 1999. Signaling by distinct classes of phosphoinositide 3-kinases. Exp. Cell Res. 253:239–254 10.1006/excr.1999.4701 [DOI] [PubMed] [Google Scholar]
- van Kooyk Y., van Vliet S.J., Figdor C.G. 1999. The actin cytoskeleton regulates LFA-1 ligand binding through avidity rather than affinity changes. J. Biol. Chem. 274:26869–26877 10.1074/jbc.274.38.26869 [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M., Sánchez-Madrid F. 2004. Role of the cytoskeleton during leukocyte responses. Nat. Rev. Immunol. 4:110–122 10.1038/nri1268 [DOI] [PubMed] [Google Scholar]
- Wang N., Butler J.P., Ingber D.E. 1993. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 260:1124–1127 10.1126/science.7684161 [DOI] [PubMed] [Google Scholar]
- Yu T., Wu X., Gupta K.B., Kucik D.F. 2010. Affinity, lateral mobility, and clustering contribute independently to beta 2-integrin-mediated adhesion. Am. J. Physiol. Cell Physiol. 299:C399–C410 10.1152/ajpcell.00039.2009 [DOI] [PubMed] [Google Scholar]
- Yu W., O’Brien L.E., Wang F., Bourne H., Mostov K.E., Zegers M.M. 2003. Hepatocyte growth factor switches orientation of polarity and mode of movement during morphogenesis of multicellular epithelial structures. Mol. Biol. Cell. 14:748–763 10.1091/mbc.E02-06-0350 [DOI] [PMC free article] [PubMed] [Google Scholar]