

Abstract
Recently, the genomes of two Mycoplasma fermentans strains, namely M64 and JER, have been completely sequenced. Gross comparison indicated that the genome of M64 is significantly bigger than the other strain and the difference is mainly contributed by the repetitive sequences including seven families of simple and complex transposable elements ranging from 973 to 23,778 bps. Analysis of these repeats resulted in the identification of a new distinct family of Integrative Conjugal Elements of M. fermentans, designated as ICEF-III. Using the concept of “reaction connectivity”, the metabolic capabilities in M. fermentans manifested by the complete and partial connected biomodules were revealed. A comparison of the reported M. pulmonis, M. arthritidis, M. genitalium, B. subtilis, and E. coli essential genes and the genes predicted from the M64 genome indicated that more than 73% of the Mycoplasmas essential genes are preserved in M. fermentans. Further examination of the highly and partly connected reactions by a novel combinatorial phylogenetic tree, metabolic network, and essential gene analysis indicated that some of the pathways (e.g. purine and pyrimidine metabolisms) with partial connected reactions may be important for the conversions of intermediate metabolites. Taken together, in light of systems and network analyses, the diversity among the Mycoplasma species was manifested on the variations of their limited metabolic abilities during evolution.
Introduction
Mycoplasma, a member of the class Mollicutes, is a genus lacking a rigid bacterial cell wall. A number of species of this genus are medically and agriculturally important. Extensive genetic and genomic investigations had been carried out to shed light on their biology and pathogenicity [1], [2], [3], [4], [5], [6], [7], [8], [9], [10]. In addition, Mycoplasmas are genetically simple bacteria with drastically reduced genomes and thus are interesting to study because of the presumably limited but crucial metabolic capabilities and biological activities. To date the genomes of at least twenty species, with sizes ranging from 0.58 (M. genitalium) to 1.36 Mbp (M. penetrans), have been completely determined [4], [5], [6], [7], [10], [11], [12], [13], [14], [15], [16], [17], [18], [19], [20], [21], [22], [23], [24] (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi). These sequences contained the basic and crucial information for better understanding of this interesting group of microorganisms.
In spite of intensive basic and clinical research on M. fermentans in recent years, its role and involved molecular mechanism in HIV pathogenesis, sexually-transmitted genital tract infection, systemic infection, rheumatic disorders, chronic fatigue syndrome and other diseases has remained elusive [25], [26], [27], [28], [29], [30], [31], [32], [33], [34], [35]. M. fermentans is a fastidious microorganism isolated or detected commonly from human genitourinary and respiratory tracts [36], [37] and correlated with diseases in both healthy individuals and AIDS patients. It was first described several decades before its detection in patients with AIDS in the late 1980s and was considered an opportunistic pathogen or a sexually transmissible cofactor contributing to the pathology and pathogenesis of HIV-associated diseases [38]. The detection of this species in the peripheral blood lymphocytes and urine of AIDS patients in a previous study suggested that it might have the ability to act as polyclonal activators of both T and B lymphocytes to stimulate the replication of HIV. It is thought to behave as a cofactor or immunomodulator in HIV-related diseases [39]. M. fermentans might be a systemic pathogen, causing fatal disease owing to the infection of the bone marrow in non-HIV-infected patients [40]. It may actively invade cultured cells such as HeLa cells and survive as an intracellular pathogen [41]. They had also been found to be present intracellularly or adhered to cell surface [42]. On the other hand, M. fermentans may play a critical role in genital tract infection and rheumatic disorders such as rheumatoid arthritis [43], [44]. In addition, several studies have been conducted to elucidate the relationship of Mycoplasma and chronic fatigue syndrome (CFS) [45]. These studies showed Mycoplasma species including M. fermentans, M. hominis, and M. penetrans could be detected in the patients with CFS, however no solid evidence that these organisms act as a cause of CFS has been reported. In conclusion, M. fermentans is a clinically interesting Mycoplasma species and recent studies had brought the attention towards its possible involvement in several critical human diseases.
Three types of transposable elements including insertion sequences (IS), ICEFs (integrative conjugal element of M. fermentans), and prophage had been found in some M. fermentans strains [46], [47], . These elements had a wide range of size distribution ranging from 973 to 23,778 bp and some were present as multiple copies. For instance, nine copies of IS1630, which encoded an IS30 family like transposase and had diverse target site specificity, have been detected in M. fermentans [47]. IS1550, also known as ISMi 1, had been demonstrated to be a possibly active element in E. coli. In addition, IS1550 had been found to be integrated into IS1630 and be inserted by the large transposable element ICEF [46], [47], [48], [49]. ICEF is similar to conjugative self-transmissible integrating elements (constins), but due to the absence of the homologs of integrases, transposases or recombinases, it is considered to be distinct from typical constins in the mechanism of integration-excision. Moreover, the approximately 16-kb M. fermentans prophage ΦMFV1 might be integrated as single or multiple copies in the genome [49]. Taken together, the presence of these elements might be important for the plasticity of the genome and evolution of M. fermentans [49], [50], [51].
To increase our understandings of the biology of M. fermentans, which might be helpful for revealing its pathogenic roles in the suspected diseases, a comprehensive comparative genomic analysis was performed on M. fermentans strains M64 and JER and the other Mycoplasmas. We examined the DNA sequences which contributed to the dramatic difference in size between the M64 and JER genomes. The metabolic abilities of M. fermentans M64 were analyzed by a systems analysis method which was based on the evaluation reaction connectivity. Finally, an integrated analysis of a phylogenetic tree, metabolic network, and essential genes was carried out to uncover the essentiality and diversity of metabolic reactions in M. fermentans M64 during evolution.
Results and Discussion
M. fermentans M64 Harbors a Large Number of Transposable Elements
Similar to Mycoplasma mycoides subsp. mycoides [16], sequence analyses indicated that the M. fermentans M64 genome also possessed a high density of transposable elements (Figures S1 and S2) [52]. Nine copies of two types of large repetitive sequences and many copies of relative small insertion sequence (IS) elements accounted for 21.6% of the genome (Table 1 and Figure 1) [46], [47], [48], [49]. Among the large repeats were three families of ICEF (22.3 to 23.8 kb) which included four copies of the previously sequenced ICEF-1A (23.8 kb) and the partly characterized ICEF-IIA, B, and C (22.3 kb), and two complete (20.2 and 20.8 kb) and one partial (20.7 kb; 52 bp truncation at the 3′-end) copies of ICEF-III (ICEF-IIIA and B) belonging to a new family discovered in our recent study [52]. Two copies (20.2 and 20.8 kb) of ICEF-III were complete, whereas another one was truncated by IS1550A. In addition, there were two copies of ΦMFV1 prophage DNAs (15.8 and 15.6 kb) inserted in the chromosome. A total of 15 complete and 4 incomplete copies of three families of IS elements including eight IS1550 (973 to 1,416 bp; 6 complete copies, one copy interrupted by ICEF-IIA, and another copy might have been truncated by the transposition of ICEF-IA), seven IS1630 (1,178 to 1,384 bp; one interrupted by ICEF-IIIA), and four ISMf1 (1,395 to 1,570 bp; three complete and one incomplete elements) were identified.
Table 1. General features of M. fermentans M64 genome.
| Repetitive sequences included* | Repetitive sequences excluded* | |
| Genome size (bp) | 1,118,751 | 887,179 |
| GC content (%) | 26.9 | 27 |
| Gene density (rRNA and, tRNA genes included) (%) | 89.7 | 90.6 |
| Total number of CDSs | 1050 | 820 |
| Number of CDSs with annotated function | 576 (54.9%) | 507 (61.8%) |
| Number of hypothetical proteins | 332 (31.6%) | 210 (25.6%) |
| Number of conserved hypothetical proteins | 142 (13.5%) | 103 (12.6%) |
| Average protein length (a.a.) | 314 | 321 |
| Start codon usage | ||
| AUG | 963 (91.7%) | 752 (91.7%) |
| UUG | 55 (5.2%) | 43 (5.2%) |
| GUG | 32 (3.1%) | 25 (3.1%) |
| Total number of rRNA genes | 5 | 3* |
| Total number of tRNA genes | 35 | 35 |
| Repetitive transposable elements | ||
| IS elements | 19 (4 incomplete) | - |
| ICEFs | 7 | - |
| Prophage, ΦMFV1 | 2 | - |
| Total length of repetitive sequences (bp) | 241,287 | - |
| Repetitive sequences (%) | 21.6 | - |
Repetitive sequences included the transposable elements and the duplicated 16 and 23S rRNA.
Figure 1. Terminal and junction sequences of the IS elements, ICEFs, and ΦMFV1 prophage DNAs in the M. fermentans M64 genome.

The terminal inverted repeats and flanking sequences of 15 complete and 4 partial copies of three families of IS elements including the IS1550, IS1630, and ISMf1, 7 copies of three families of ICEFs, and 2 prophage genomes are shown. Bold uppercase letters with arrows on top indicate the terminal inverted repeats. The target site duplications are shown in underlined bold lowercase. The elements interrupted by other transposable elements are indicated.
M. fermentans strains M64 and JER had Significant Difference in Large Transposable Elements
Sequence comparison indicated that the genome of M. fermentans M64 (1,118,751 bp) [52] is 141 and 115 kb larger than that of the JER strain (977,524 bp) [21] and the nearly complete PG18 strain (1,004,014 bp; Accession no. AP009608) [53]. The differences are mainly contributed by the differences between the copy numbers of ICEF and ΦMFV1 prophage DNAs in these genomes (Figure 2). Notably, there are still gaps of unknown size and potential assembly problems in the PG18 genome, thus the actual difference between it and M64 genome remains to be accurately demonstrated. The M. fermentans JER genome essentially lacked all of the previously identified ICEF elements and ΦMFV1 prophage DNA but contained two partial copies of the newly identified ICEF-III elements [48], [49]. Regardless of these differences, the organization of the M64 and JER genome sequence were largely similar. On the other hand, at least two apparently complete ICEFs and one ΦMFV1 prophage DNA were found within the almost complete PG18 genome. Nonetheless, expectably, the organization of the PG18 genome is considerably different from that of the M64 possibly due to the gaps and sequence arrangement problems in an incomplete genome.
Figure 2. Dotplot of the genomes of M. fermentans strains M64 versus JER (A) and PG18 (B).

The graphs were generated with the Gepard 1.21 with word length of 20 and window size of 0 [78]. The long repetitive sequences are indicated by horizontal arrows: ICEF-I and -II (unfilled arrows), ICEF-III (black arrows), and ΦMFV1 (gray arrows). The locations of the IS elements of the ISMf1, IS1550 and IS1630 families are indicated by small black and gray (elements interrupted by ICEFs) triangles. The two types of non-IS small duplications are indicated by small filled (16–23S rRNA operon) and open triangles (uncharacterized repetitive sequence containing a hypothetical protein-coding gene).
Analyses of the integrity and flanking sequences of the IS elements and ICEFs suggested that the current M. fermentans M64 genome architecture at least in part might result from the transposition and/or recombination events of the elements in different periods of time. Almost all of the elements, except for members of IS1550A-1, -2 and -3 subfamilies, are polymorphic in DNA sequences. As shown in Figure 1, the four ICEFs (ICEF-I, -IIA, -IIB, and -IIC) are terminated with 24-bp imperfect inverted repeats and flanked by distinct 8-bp target site duplications. The new ICEF-IIIs, except for the incomplete ICEF-IIIB which is disrupted by IS1550A, are all terminated with 23-bp imperfect inverted repeats and flanked by 8-bp target site duplications. Sequence comparison of ICEF-IA, IIA, and IIIA and phylogenetic analysis of 11 intact ICEs (Integrative Conjugal Elements) in six Mycoplasma species indicated that ICEF-III is distinct from the other two ICEF families in M. fermentans and unique in all reported ICEs (Figure S3). Both copies of ΦMFV1 prophage genomes are terminated with 4-bp inverted repeats and flanked by identical 6-bp (TTTTTA) target site duplications, suggesting that this could be the preferred transposition target sequence, as reported previously [49]. The seven complete IS1550 are terminated with 29-bp imperfect inverted repeats and four of them are flanked by 3-bp direct repeats. The seven complete IS1630 are terminated with 27-bp heterogeneous inverted repeats and four of these elements are flanked by 16-, 23-, 25-, and 26-bp duplicated junction sequences. The three complete copies of ISMf1 contain 47-bp imperfect terminal inverted repeat but none of them have noticeable target site duplication. The absence of target site duplication in some of these elements suggested that post-transposition DNA rearrangements or additional transposition events might have occurred prior to strain evolution since those families of transposable elements, except for ΦMFV1 prophage, are present in M. fermentans strains M64, JER, and PG18. The high frequency of DNA rearrangements in M. fermentans has been shown by Hu et al. in a previous study [51].
Pan-genome Analysis Across the M64, JER, and PG18 Strains
To unveil the variations in gene content among M. fermentans M64, JER, and PG18, sequence similarity searches across all predicted proteins were carried out by using BLASTP to dissect the gene repertoire in these strains. The homologs were recognized under the criterion of E-value <10−5. Due to differential activities of the events of gene duplication and/or horizontal gene transfer, the number of genes shared by any two or present in all three strains might be different. For instance, the gene MfeM64YM_0098 of M64 had a total of 8 homologs (MfeM64YM_0098, MfeM64YM_0108, MfeM64YM_0229, MfeM64YM_0238, MfeM64YM_0385, MfeM64YM_0474, MfeM64YM_0483, and MfeM64YM_0992), whereas PG18 and JER, respectively, had only 4 (MBIO_0117, MBIO_0267, MBIO_0276, and MBIO_0750) and 5 (MFE_08000, MFE_02760, MFE_02860, MFE_04903, and MFE_04960) gene homologs. Thus, pan-genome analysis exhibited 762∼837 genes constituting the core genome of M. fermentans strains M64 (837), JER (762), and PG18 (809) (Figure 3B). In agreement with the phylogenetic relationship among these three strains (Figure 3A), more gene homologs are shared by M64 (955) and PG18 (872) than M64 (849) and JER (774), corroborating the closer relationship between M64 and PG18 obtained from the analysis of the concatenated sequence of dnaE, pyk, and rpoA (Figure 3B, Table S1). Expectably, M64 with the largest chromosome rich in mobile elements possesses much more strain specific genes (83 genes) than JER (17) and PG18 (11). Further sequence analysis indicated that most strain specific genes encoded proteins without annotated function. Based on the annotations in the KEGG Orthology database, the gene pool in the core genome (762∼837 genes) contained 378 well-annotated orthologous groups of genes with approximately half belonging to the genetic information processing biomodules (Figure 3C).
Figure 3. Overview of the pan-genome analysis across the M. fermentans strains M64, JER, and PG18.

A. The phylogenetic tree of the M. fermentans strains M64, JER, and PG18. The concatenated sequences of the genes dnaE, pyk, and rpoA shared by the three strains of M. fermentans were used for phylogenetic analysis. The maximum-likelihood method was performed to reconstruct this tree using PHYML 3.0. The scale bar stands for the estimated number of nucleotide substitutions per site. B. The pan-genome of the M. fermentans strains M64, JER, and PG18. Venn diagram exhibits the number (including duplications) of genes unique to each strain or shared by these strains (BLASTP E-value <10−5). Superscripts represent the name initials of the strains which show the indicated numbers of genes with homologies to other strains. The numbers of genes in the nearly complete PG18 genome may be slightly underestimated due to the presence of gaps and potential unresolved duplications in genome assembly. C. Functional distribution of 378 orthologous groups of genes in the core genome. Some of the proteins have more than one KEGG pathway/biomodule annotation.
M. fermentans M64 Metabolic Capabilities Revealed by Reaction Connectivity-Based Analyses
Global analyses of M. fermentans M64 metabolism indicated only 131 of the 1,050 predicted proteins mapped to the metabolic pathways in KEGG database (KEGG Mapper; http://www.genome.jp/kegg/tool/map_pathway1.html). Apparently, it had a relatively large number of proteins in carbohydrate (57 proteins) and nucleotide (31 proteins) metabolic networks, and a scattered distribution of proteins in amino acid (25 proteins), cofactors and vitamin (18 proteins), and lipid (8 proteins) metabolic networks (Figure S4). A gross examination of the reactions with predicted proteins in the global metabolic network immediately perceived that M. fermentans could only synthesize or degrade a limited number of the metabolites as established for other Mycoplasma species.
Evaluation of the functionality of the 58 pathways with M64 proteins via the examination of connectivity of reactions [54] found only nine pathways contained five or more connected reactions (Table 2). Several carbohydrate metabolic pathways seemed to have sufficient connected reactions to drive the synthesis or degradation of the pertinent metabolites. The presence of a complete glycolysis/gluconeogenesis pathway (18 connected reactions) suggested that M. fermentans M64 is capable of catabolizing D-glucose to pyruvate using the Embden-Meyerhoff-Parnas (EMP) pathway. The pyruvate may then be converted to acetyl-CoA, acetate, or D-lactate with the pyruvate metabolism pathway which contained a single cluster of nine connected reactions driven by ten predicted proteins. Due to the lack of citrate cycle enzymes, it may use D-glucose by fermentative degradation. On the other hand, the pentose phosphate pathway, which contained a large cluster of 13 and a small cluster of two connected reactions, may be utilized to synthesize phosphoribosyl diphosphate (PRPP) that may then enter the purine (11 connected reactions) and pyrimidine (10 reactions) metabolism pathways for nucleotide synthesis. Although only 6 genes were mapped to the fructose and mannose metabolism pathway, these encoded proteins form a cluster of six connected reactions to drive the conversion of D-fructose to glyceraldehyde-3-phosphate, which may then enter the glycolysis pathway for further degradation.
Table 2. Summary of the number of M. fermentans M64 predicted genes, reactions, connected reactions in KEGG metabolic pathways.
| Metabolism | KEGG Pathway | # Genes | # Reactions | Connectivity in Reactions |
| Amino Acid | Arginine and proline metabolism | 5 | 5 | 3,1,1 |
| Cysteine and methionine metabolism | 2 | 2 | 2 | |
| Carbohydrate | Glycolysis/Gluconeogenesis | 15 | 18 | 17,1 |
| Pentose phosphate pathway | 12 | 15 | 13,2 | |
| Pyruvate metabolism | 9 | 8 | 8 | |
| Amino sugar and nucleotide sugar metabolism | 10 | 11 | 7,2,2 | |
| Fructose and mannose metabolism | 5 | 6 | 6 | |
| Galactose metabolism | 9 | 7 | 2,1,1,1,1,1 | |
| Pentose and glucuronate interconversions | 6 | 6 | 2,1,1,1,1 | |
| Starch and sucrose metabolism | 4 | 5 | 2,1,1,1 | |
| Propanoate metabolism | 2 | 2 | 2 | |
| Cofactors and Vitamins | Nicotinate and nicotinamide metabolism | 5 | 6 | 5,1 |
| Pantothenate and CoA biosynthesis | 3 | 3 | 3 | |
| One carbon pool by folate | 3 | 4 | 4 | |
| Thiamine metabolism | 2 | 2 | 2 | |
| Riboflavin metabolism | 1 | 2 | 2 | |
| Lipoic acid metabolism | 1 | 2 | 2 | |
| Energy | Oxidative phosphorylation | 9 | 2 | 2 |
| Lipid | Glycerophospholipid metabolism | 6 | 5 | 5 |
| Glycerolipid metabolism | 3 | 2 | 2 | |
| Nucleotide | Pyrimidine metabolism | 22 | 22 | 8,3,2,2,2,1,1(×4) |
| Purine metabolism | 20 | 27 | 6,6,3,3,3,2,2,1,1 | |
| Other Amino Acids | Selenoamino acid metabolism | 3 | 5 | 2,1,1,1 |
The number of reactions within each “group of linked reactions” and “orphan reaction” is separated by commas.
Neither the purine nor the pyrimidine pathways for de novo nucleotides synthesis seemed to be complete and thus M. fermentans M64 must rely on external supply of nucleotides or nucleotide synthesis intermediates to produce the building blocks of DNA and RNA (Table 2 and Figure S5). Apparently, M. fermentans M64 possesses the enzymes to form subnetworks of connected reactions between the RNA synthesis and the metabolism of (i) guanosine, guanine, GMP, GDP, and GTP, (ii) adenosine, adenine, AMP, ADP, and ATP, and (iii) UTP and CTP; and between DNA synthesis and the metabolisms of (iv) guanosine, guanine, deoxyguanosine, dGMP, dGDP, and dGTP, and (v) dAMP, dADP, and dATP.
M. fermentans M64, which is similar to the other Mycoplasma species, had very few amino acid metabolism proteins predicted from the genome. Only two of the amino acid metabolic pathways had connected reactions – the “arginine and praline” and “cysteine and methionine” pathways had clusters of 3 and 2, respectively, reactions. The presence of arginine deiminase (ArcA), ornithine carbamoyltransferase (ArcB), and carbamate kinase (ArcC) formed a cluster of reactions catalyzing the degradation of arginine to ammonia in the “arginine and proline metabolism” is in agreement with the previous study in which M. fermentans was shown to be capable of utilizing arginine (62). Notwithstanding some of the proteins did not form highly connected reaction clusters, they might still be important for the survival of the microorganism. For instance, the proline iminopeptidase (Pip) may release proline from peptide and the aspartate-ammonia ligase (AsnA) catalyzes the interconversion of aspartate and asparagine. In addition, S-adenosylmethionine synthetase (MetK) is an indispensable enzyme for the conversion of methionine to S-adenosyl-L-methionine, which can then be used as the substrate of cytosine-specific methyltransferase (Dcm) for DNA, and rRNA methylation. Therefore, the overall network and connectivity analysis suggested that M. fermentans M64 must rely on external supply of all amino acids for protein synthesis.
Conservation of Genes and Biomodules among the Mycoplasmas
Clustering analysis of the proteins from 27 sequenced Mycoplasma and Phytoplasma species indicated that most genes responsible for DNA replication, nucleotide excision, homologous recombination, transcription, and translation and a significant fraction of the membrane transporters including some of the ABC transporters and members of protein transport and bacterial secretion system are highly conserved among these species (Figure 4). The genes related to carbohydrate metabolism are conserved in almost all species except for M. haemofelis, M. suis, M. arthritidis, and M. hominis. Several genes accounting for pyruvate metabolism and other carbohydrate metabolisms were lost in them, such as pdhA, pdhB, pdhC, and pdhD. Moreover, distinct patterns of other metabolisms could also be observed for M. haemofelis and M. suis. Intriguingly, M. haemofelis and M. suis are hemotropic Mycoplasmas. They belong to a special group of Mycoplasmas, also known as hemoplasma, with a tropism for red blood cells [55], [56]. This distinctive propensity may be associated with the evolution of the pool of metabolism- and cellular process-related genes. M. arthritidis and M. hominis, whose hosts are rodent and human respectively, also form a unique phylogenetic clade, referred to as the M. hominis cluster, in Mycoplasmas. They both cannot carry out the glucose fermentation but capable of arginine hydrolysis [57], [58]. All of the Mycoplasma species exhibited conserved energy metabolism-related genes including genes encoding NADH dehydrogenase and F-type ATPase aside from cytochrome-containing complex, indicating a truncated electron transport chain. Thus, the ATP generation proceeds via inefficient substrate-level phosphorylation in the flavin-terminated respiratory pathway rather than oxidative phosphorylation [59], [60], [61]. In addition, the pentose phosphate pathway, an alternative to glycolysis, may generate NADPH for reductive biosynthesis reactions in Mycoplasma species. The presence of only a limited number of recognizable genes encoding the proteins in the biomodules for metabolisms of most carbohydrates, amino acids, and cofactors and vitamins are conserved, suggesting that most, if not all, of these metabolic pathways are defective unless there are cryptic genes which can carry out the missed function [62]. This observation is in agreement with the requirements of a wide spectrum of substrates and factors for the growth of Mycoplasma.
Figure 4. Clustering analysis of the metabolic and cellular process proteins of 27 Mycoplasma species/strains and Phytoplasma OY.

The order of the Mycoplasma species is according to their phylogenetic relationship (23S rRNA tree). Each column represents the proteins of one single species/strain and each row (horizontal line) represents the copy number (grey color scale on top) of one gene. The sizes of the genomes in Mbp are indicated below the 3-letter abbreviated names of the organisms. Functional clustering and comparison of the proteins among species were based on KO (KEGG Orthology) assignments. The conservation of each gene among the Mycoplasma species (i.e. the number of Mycoplasma species possessing the gene) is shown on the right side of clustering result. Abbreviations – mha: M. haemofelis; mss: M. suis Illinois; msk: M. suis KI3806; mpe: M. penetrans; mga: M. gallisepticum; mge: M. genitalium; mpn: M. pneumoniae; mmy: M. mycoides; mcp: M. capricolum; mlc: M. leachii; mat: M. arthritidis; mho: M. hominis; mmo: M. mobile; mhr: M. hyorhinis; mhy: M. hyopneumoniae 232; mhj: M. hyopneumoniae J; mhp: M. hyopneumoniae 7448; mco: M. conjunctivae; mpu: M. pulmonis; mfm: M. fermentans M64; mfr: M. fermentans JER; maa: M. agalactiae PG2; mal: M. agalactiae 5632; mbv: M. bovis PG45; msy: M. synoviae; mcd: M. crocodyli; and poy: Phytoplasma OY.
The analysis of the proteins involved in genetic information processing unambiguously indicated that they are widely distributed among the members of the sequenced Mycoplasmataceae family (Figure 4). Interestingly, the nucleotide metabolism and genetic information processing deservedly exhibited highly conserved profiles of involved genes, whereas M. haemofelis and M. suis seemed to be deficient in some genes. The DNA polymerase III holoenzyme of M. fermentans and the other Mycoplasma species resembles the Gram (+) type and is simpler than that of the Gram (-) model organism [63]. The Mycoplasma species polymerase III holoenzymes are made up of the same subunits as that of the Gram (+) Bacillus subtilis which comprises the α, β, γ, τ, δ, and δ′ subunits. Although the gene encoding the ε subunit with 3′–5′ exonuclease activity could not be found in their genomes, the structure of the Mycoplasmas α subunit protein resembles the PolC-type (Gram (+) type) α subunit, which contains both the DnaE (α subunit) and DnaQ (ε subunit) domains with the DNA polymerase and 3′–5′ exonuclease, respectively, activities suggesting a B. subtilis-like mechanism of DNA replication.
The DNA-directed RNA polymerase α, β, and β′ subunits are sufficient to form a minimal core (ββ′α2) of RNA polymerase for catalyzing the polymerization of nucleoside triphosphates into RNA [64]. The RNA polymerase primary σ factor, which promotes the attachment of RNA polymerase to a specific site in the promoter for transcription initiation, is highly conserved among the sequenced Mycoplasma species. In addition to these subunits, the δ subunit which is ubiquitous among the Gram (+) bacteria were also found to be conserved in some of the Mycoplasma species including M. haemofelis, M. suis, M. leachii, M. capricolum, M. mycoides, M. penetrans, M. pneumoniae, M. genitalium, and M. gallisepticum but not in M. fermentans. Although one gene (MfeM64YM_0186) encoding a protein containing the domain of RNA polymerase δ subunit (pfam05066) is present on the chromosome of M. fermentans M64, the E-value is only 6.3×10−5 and its protein size (89 a.a.) is markedly smaller than those (134∼170 a.a.) of other sequenced Mycoplasmas. Importantly, the sequence similarity analysis of the protein MfeM64YM_0186 demonstrated that all significant hits (E-value<10−5) were annotated as putative uncharacterized protein. Therefore, MfeM64YM_0186 was considered a hypothetical protein and might be degenerated from the typical RNA polymerase δ subunit. The δ subunit is involved in both the initiation and recycling phases of transcription, and may also serve as a virulence factor [65]. The specificity of transcription and the efficiency of RNA synthesis are increased in the presence of the δ subunit because of enhanced recycling. Previous studies reported that mutation of the δ subunit gene would result in extended lag phase growth [66] and a defect in starvation-induced stationary-phase survival or recovery [67]. This subunit does not exist in all Mycoplasma species but is likely responsible for the complex regulation and flexibility of gene expression in those Mycoplasma species bearing the δ subunit genes.
The aminoacy-tRNA synthetases and ribosomal genes also have a high degree of conservation among the sequenced Mycoplasmas. Similar to the Gram (+) B. subtilis, Mycoplasmas do not possess glutaminyl-tRNA synthetase. They may take a similar strategy as B. subtilis and other organisms which lack this enzyme by charging glutamate to both tRNAGlu and tRNAGln with a nondiscriminating glutamyl-tRNA synthetase and then converting glutamyl-tRNAGln to glutaminyl-tRNAGln with glutamyl-tRNAGln amidotransferase (GatABC complex). This is partly supported by observation that M. fermentans and the other 21 Mycoplasma species and strains have a protein homolog (MfeM64YM_0687 in M. fermentans) more similar to the nondiscriminating glutamyl-tRNA synthetase of B. subtilis (61.7% similarity) than the discriminating E. coli enzyme (53% similarity). Mycoplasma species has slightly less ribosomal subunit proteins than those of E. coli (total 55). Furthermore, the genes encoding the GatABC complex are also present in M. fermentans M64 and other sequenced Mycoplasmas. Upon the manually curated annotations from KEGG database (http://www.genome.jp/kegg/pathway/ko/ko03010.html) and validations for smaller proteins (<100 a.a.) by performing TBLASTN, a total of 47 proteins of the ribosome complex are highly conserved among the sequenced Mycoplasma species and strains. The S1, S6, S21, L9, and L32 subunits are absent in some of the species and S22, L25, and L30 subunits could not be found in any of the species. In line with our observations, Kawauchi et al. [68] employed two-dimensional polyacrylamide gel electrophoresis to analyze the M. capricolum ribosomes, and identified 30 proteins of large ribosomal subunit and 21 proteins of small ribosomal subunit. The M. capricolum ribosome lacks S1, S22, L25, and L30 proteins. S1 protein is present in seven species belonging to three close phylogenetic clusters including the M. lipophilum, M. synoviae, and M. pulmonis clusters [58] and two other species. Furthermore, S21 protein restrictively exists in nine species of the hemoplasma, pneumoniae, and spiroplasma groups [57]. Together, these observations suggested that some of these proteins may show the specificity for certain phylogenetic clades and be dispensable in Mycoplasmas as reported previously [12], [69].
Potential Essential Genes in M. Fermentans
The essential genes from the closely related M. pulmonis, M. arthritidis, and M. genitalium and the distantly related B. subtilis and E. coli identified in previous studies were compared with the proteins predicted from the M. fermentans M64 genome to shed light on M. fermentans likely essential genes necessary for a minimal self-replicating cell (Figure 5) [12], [69], [70], [71], [72]. It is notable that more essential genes in M. pulmonis, M. arthritidis, and M. genitalium than the other two species have homologs in M. fermentans. Approximately 303 (97.7%) of the 310 M. pulmonis, 315 (75.7%) of the 416 M. genitalium, 282 (73.8%) of the 382 M. genitalium, 150 (55.3%) of the 271 B. subtilis, and 168 (55.4%) of the 303 E. coli essential gene homologs were found in the M. fermentans M64 genome. Although the matched essential genes in M. fermentans have yet to be validated, a subset of essential genes in Mycoplasma species that seem to be distinct from the two distantly related bacteria were identified. The higher similarity of M. fermentans with M. pulmonis than with M. arthritidis and M. genitalium is in accordance with their phylogenetic distance (Figure S6). A relatively higher degree of conservation is observed for the essential genes participating in translation and transcription. In addition, a significant fraction of the essential genes involved in replication and repair, nucleotide metabolism, amino acid metabolism, and membrane transport also appeared to be highly conserved in the analyzed species. On the other hand, some of the proteins only seemed to be essential to one or more but not all species. It is conceivable that the genome reduction was accompanied by a shift of the essential gene sets during the evolution as shown in carbohydrate metabolism and membrane transport (Figure 5).
Figure 5. Comparison of predicted genes of M. fermentans M64 with the reported essential genes of five bacteria species.

BLASTP was implemented to compare the proteome of M. fermentans M64 (mfm) against the proteomes of M. pulmonis (mpu), M. arthritidis (mat), M. genitalium (mge), B. subtilis (bsu), and E. coli (eco). The gene names (or KEGG Orthology numbers) and KEGG functional categories are indicated. Black rectangles indicate the essential genes in mpu, mat, mge, bsu, and eco. Light grey rectangles indicate the genes which had not been shown to be essential. Dark gray rectangles indicate the potential essential genes in M. fermentans. White rectangles indicate the missing genes. The genes involved in more than one functional category are indicated by asterisk.
Conservation of Residual Metabolism and Essential Genes among the Mycoplasmas
Analysis of the conservation of the genes among the Mycoplasmas indicated that some of the residual or partial metabolic capabilities in M. fermentans are highly conserved among the Mycoplasma species. A cross examination of the essential genes in M. pulmonis [69], M. arthritidis [12], and M. genitalium [70] suggested that many of the conserved genes in the residual metabolic functions are likely essential in Mycoplasmas. The incomplete nucleotide and carbohydrate metabolism were chosen as the examples to illustrate this property. Notwithstanding Mycoplasmas lack the full capability of de novo nucleotide synthesis, a total of 21 and 22 genes in purine and pyrimidine metabolic pathways, respectively, were found in the M. fermentans genome. Phylogenetic comparison of the nucleotide metabolic networks indicated that the genes present in M. fermentans are more conserved in purine metabolism than those in the pyrimidine pathway among the 21 sequenced Mycoplasma species (Figure 6A). The enzymes which catalyzed clusters of connected reactions in purine metabolic pathway, except for Dgk and DeoB only present in six and twelve other species, respectively, are highly conserved among the Mycoplasmas. Most species have the enzymes for the synthesis of ATP from adenine or adenosine, and GTP and dGTP from guanine or guanosine. However, three phylogenetically related species - M. fermentans, M. agalactiae, and M. bovis in the M. lipophilum cluster and two other related species - M. arthritidis and M. hominis in the M. hominis cluster [58] lack the ribonucleotide diphosphate reductase complex for the conversion of ADP to dADP and thus dATP might be synthesized from dADP or dAMP or uptake from an extracellular source. On the other hand, the enzymes in the pyrimidine metabolic pathway are relatively less conserved among the Mycoplasmas. Due to the lack of at least one key enzyme, all sequenced species may rely on external supply of UTP, dCTP, and dTTP from the hosts or culture media. For instance, M. fermentans is short of genes encoding dCTP deaminase involved in the conversion of CTP to UTP and nucleoside-diphosphate kinase responsible for the conversion of UDP to UTP, dCDP to dCTP, and dTDP to dTTP. Moreover, ribonucleoside-triphosphate reductase catalyzing the conversion of CTP to dCTP is also absent in it. Some of the species such as M. fermentans carry the pyrG gene and thus may be able to convert UTP to CTP.
Figure 6. Phylogenetic conservation analysis of nucleotide (A) and carbohydrate (B) metabolic networks of M. fermentans M64 and 20 other Mycoplasma species.

M. fermentans M64 metabolic networks were served as the references in the comparisons. The small open squares represent the compounds and the colored circles represent the proteins (enzymes) participating in the metabolisms. Circles with thick and light blue circumference denote the potential essential genes inferred from the comparison with M. pulmonis, M. arthritidis, and M. genitalium (Figure 5). The arrows and lines linking the enzymes and substrates indicate the direction of the reactions. Enzymes involved in more than one metabolism are annotated with a single letter code of the other metabolisms (A: Amino acids; C: Carbohydrate; N: Nucleotide; V: cofactors and Vitamins) next to the circles. The color scale (bottom) indicates the phylogenetic conservation of proteins in the analyzed Mycoplasma species.
Of the carbohydrate metabolism, M. fermentans M64 and the other Mycoplasma species seemed to possess a highly conserved glycolysis pathway as well as the incomplete and partially conserved pentose phosphate and pyruvate metabolic networks (Figure 6B). Upstream of glycolysis pathway, α-D-glucose and β-D-glucose may enter the pathway through the enzyme GlcK which was only found in M. fermentans M64 and seven other species. The joint presence of AmyC and MalL in a few species including M. fermentans, M. pulmonis, M. hyorhinis, and M. crocodyli suggested that starch may be converted to dextran and then to α-D-glucose and subsequently enter the glycolysis pathway in these species. In addition, the presence of the FruA, FruK, and Fba, which have different degree of conservation, suggested that fructose may also be utilized by M. fermentans M64 and some other Mycoplasma species. In contrast to the highly conserved Fba which interconverts D-fructose 1,6-bisphosphate and glycerone phosphate plus D-glyceraldehyde 3-phosphate, FruA does not exist in all of two hemoplasma species and six species in the M. hominis group including M. lipophilum, M. hominis, and M. synoviae clusters. FruK is present in all of three species belonging to the spiroplasma group, two species in the pneumoniae group, and two species in the hominis group including M. hyorhinis and M. fermentans. Within the pentose phosphate pathway, the enzymes Tkt, Rpe, and RpiB for the conversion of D-glyceraldehyde 3-phosphate to D-ribulose 5-phosphate are conserved in all species except for the hemoplasma. The enzyme PrsA, which converts D-ribulose 5-phosphate to 5-Phospho-alpha-D-ribose 1-diphosphate (PRPP) could be found in all Mycoplasma species. Many Mycoplasma species seemed to be able to catabolize pyruvate into acetyl-CoA and acetate through a conserved pathway, whereas only some have a complete set of enzymes to further metabolize pyruvate to lactate or ethanol. M. fermentans, M. agalactiae, M. bovis, and M. crocodyli in the hominis group, M. mycoides and M. leachii in the spiroplasma group, and M. penetrans have lactate dehydrogenase catalyzing the interconversion between lactate and pyruvate. Of these species, M. fermentans, M. crocodyli, and M. penetrans are also capable of converting acetate to ethanol by exerting aldehyde dehydrogenase and alcohol dehydrogenase which only exist in a couple of species. This implies that M. fermentans M64 and several other Mycoplasma species may be able to undergo lactic or alcoholic fermentation. Similarly, when the enzymes in the incomplete amino acid and vitamin and cofactor metabolic networks were examined, some of the enzymes also found to be conserved, suggesting that at least some of the residual metabolic activities may be related to their survival (figure S7).
Genes Outside of the Mycoplasma Clade
Sequence analysis identified 15 genes in M. fermentans M64 with no homolog among all of significant BLASTP hits coming from Mycoplasma species, suggesting that they are not conserved in the Mycoplasma clade (See comment on the Table S2). These genes might be the vestiges of the genome reduction from the last common Gram-positive ancestor, or be the horizontally transferred genes. The genes were dispersedly distributed on the chromosome. Among these, 7 genes were closest to the Gram-positive bacteria, and 8 genes were closest to Gram-negative bacteria. All of 7 Gram-positive bacteria belonged to Firmicutes, of which 6 belonged to Clostridia. Those 7 genes with highest homologies to Firmicutes were regarded as the possible preserved genes after the onset of the evolution of genome minimization. In addition, the 8 genes closest to Gram-negative bacteria, including 4 species belonging to Bacteroidetes/Chlorobi group, 3 to Cyanobacteria, and 1 to Spirochaetes, might have been transferred laterally to M. fermentans M64. No UGA codon was detected in these 8 putative horizontally transferred gene candidates, suggesting that these genes might not have been transferred for a long period and thus was insufficient to induce the codon bias. The proteins encoded by the 15 genes consisted of 5 conserved hypothetical proteins, 3 AAA+superfamily ATPases, and 7 different proteins. During the evolution of M. fermentans M64, AAA+superfamily ATPase might play an important role in the compensation for genome minimization because this protein is accountable for a large variety of diverse functions via molecular remodeling dependent of the energy from ATP hydrolysis, such as protein degradation, DNA recombination, replication, repair, and so on [73]. Moreover, the hsdS among the 15 genes encodes a subunit of type I restriction enzyme which could protect the hosts against foreign DNA such as those from bacteriophages. For those 15 gene candidates, the protein phylogeny was reconstructed for M. fermentans M64 homolog and the twelve most similar homologs of different organisms in the BLASTP results, and further, M. fermentans M64 and those twelve organisms were analyzed for the 23S rRNA phylogeny to elucidate their evolutionary relationship. Regarding the ORF MfeM64YM_1027, M. fermentans M64 occupied a distinct, individual branch with strong branch support and was distant from Clostridium perfringens in the 23S rRNA phylogenetic tree (Figure S8A). However, M. fermentans M64 MfeM64YM_1027 was closest to the homolog of C. perfringens, raising the possibility that this gene might be a remnant after genome minimization (Figure S8B). Similar result was also observed for another gene (MfeM64YM_0060) which product was closest to the homolog of a Gram-negative bacterium (Figure S8C and D). Taken together, the preserved genes after the onset of genome reduction and horizontally transferred gene candidates appeared to play some critical roles in the versatility of a small genome and shaping of a limited metabolism.
Conclusion
The genome of M. fermentans M64, despite the interference of a large proportion of repetitive sequences when assembling the whole genome shotgun sequences, has been successfully determined in our previous study [52]. Rare conservation of transposable elements among the Mycoplasmas suggests that most elements were integrated in the genomes after speciation. Clustering analysis of Mycoplasma genes indicated the conservations of degenerated metabolisms and cellular processes, suggesting that some of the remaining metabolic functions might be indispensable. On account of the lack of complete amino acid, nucleotide, and vitamin and cofactor synthesis pathways, these compounds or their intermediate metabolites must be obtained from external sources and entered the cells in the forms such as peptides, amino acids, and nucleotides with poorly understood transport systems. The completion of the M64 and JER strains of M. fermentans and the other Mycoplasma genomes would facilitate the revelation of these systems and the biology of this interesting group of microorganisms with reduced genomes.
Materials and Methods
Comparative Metabolomics
The metabolism of M. fermentans M64 and the other Mycoplasma species were compared by clustering analysis of the homologous genes. The clustering analysis and comparison of the presence and absence of genes in different functional groups was based on the KO (KEGG Orthology) assignments of the homologous proteins in KEGG database. M. fermentans M64 protein sequences were submitted to the KEGG Automatic Annotation Server (KAAS) [74] to obtain the KO number and pathway assignments. While similar information of the other Mycoplasma species and Phytoplasma OY were extracted from the organism-specific files in the KEGG database.
Conservation of Metabolisms
A new method for evaluating the phylogenetic conservation of metabolism in a genus/group of organisms was developed to predict the most conserved metabolism among the Mycoplasma species. This analysis is based on the hypothesis that the probability of the preservation of an enzymatic reaction in two closely related species should be different from that of two distantly related species. The M. fermentans M64 metabolic networks constructed with the KO number and KEGG pathway information served as the reference networks in this analysis. The conservation of enzymes, reactions, and metabolic pathway between M. fermentans M64 and the 19 Mycoplasma species and Phytoplasma OY were calculated with the following equations:
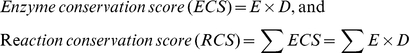 |
in which, ECS calculates the conservation of the enzyme of the compared species; E is the presence (1) or absence (0) of the enzyme; D is the “Phylogenetic Distance” between M. fermentans and the compared species which is expressed as the substitution rate (D) indicated in the 23S rRNA phylogenetic tree of the 21 Mycoplasma species and one outgroup species. The phylogenetic tree was constructed with PHYML 3.0 [75] and evaluated with the aLRT method [76]. The phylogenetic distance was considered as a weight for the enzyme conservation in this analysis. If an enzyme exists in two distantly related species, the enzyme likely has higher conservation in this genus/group of organisms, and vice versa. Each RCS represents the conservation of the enzyme orthologs in all of compared species which catalyze the same reaction.
Essential Genes
The essential genes of M. pulmonis, M. arthritidis, M. genitalium, B. subtilis, and E. coli identified in previous studies [12], [69], [70], [71], [72] and the other proteins predicted from the genomes were submitted to KAAS to obtain their KO numbers via sequence homology searches. The M. fermentans M64 essential genes were predicted by clustering analysis with the essential and putative non-essential proteins according to the KO number and through BLASTP [77] analysis of each protein against a consolidated database of the proteomes of the six analyzed species.
Genes Outside of the Mycoplasma Clade
The M. fermentans M64 genes outside of the Mycoplasma clade were identified by examining all of significant hits of the BLASTP [77] search results against the proteomes of sequenced prokaryotic organisms retrieved from the UniProt database (http://www.uniprot.org/downloads). Genes were considered to be outside of the Mycoplasma clade if no hit among all significant matches belonged to Mycoplasma species as described previously [15]. Then the closest organisms for genes outside of the Mycoplasma clade were assigned as the top non-Mycoplasma hit which had more than 70% sequence coverage in the alignment with M. fermentans sequence.
Genome Sequence Accession Numbers
The accession numbers of the M. fermentans M64 genome sequence is NC_014921. The species for comparative analyses included M. fermentans strain JER (NC_014552), M. haemofelis (NC_014970), M. suis Illinois (NC_015155), M. suis KI3806 (NC_015153), M. penetrans (NC_004432), M. gallisepticum (NC_004829), M. genitalium (NC_000908), M. pneumoniae (NC_000912), M. mycoides (NC_005364), M. capricolum (NC_007633), M. leachii (NC_014751), M. arthritidis (NC_011025), M. hominis (NC_013511), M. mobile (NC_006908), M. hyorhinis (NC_014448), M. hyopneumoniae 232 (NC_006360), M. hyopneumoniae J (NC_007295), M. hyopneumoniae 7448 (NC_007332), M. conjunctivae (NC_012806), M. pulmonis (NC_002771), M. agalactiae PG2 (NC_009497), M. agalactiae 5632 (NC_013948), M. bovis PG45 (NC_014760), M. synoviae (NC_007294), M. crocodyli (NC_014014), Phytoplasma OY (NC_005303), B. subtilis subsp. subtilis str. 168 (NC_000964), and E. coli str. K-12 substr. MG1655 (NC_000913).
Supporting Information
The circular representation of the M. fermentans M64 genome. The ticks on the outermost concentric circle indicate the relative genomic positions in base pairs, where position one is the first base of the upstream intergenic region of dnaA gene. Second and third concentric circles: predicted protein coding genes on the plus and minus strands, respectively. The various functional groups of the predicted genes (color coded) are categorized according to the COG classification. Fourth concentric circle: locations of rRNA (gray) and tRNAs (green) genes. Fifth concentric circle: ICEF-I and IIs (red arrows), ICEF-III (green arrows), ΦMFV1 prophage (blue arrows), IS elements [IS1550 (dark brown), IS1630 (brown), ISMf1 (orange)], 16S-23S rRNA operons (blue), and non-IS repeats (yellow). Sixth and seventh concentric circles: GC content and GC skew calculated in 5,000 bp of sliding window with 100 bp of step window, respectively.
(TIF)
Validation of the large repeats and genome assembly of M. fermentans M64. A. and C. EtBr-stained gel images of restriction enzymes (BglI, MluI, KpnI, and AdhI)-digested M64 genomic DNA. These fragments were resolved by CHEF gel electrophoresis in 1% agarose and 0.5X TBE at 6.0 v/cm for approximately 21.5 hr with pulsing times of 0.92 to 19.89 s and 0.92 to 37.34 s, respectively. B. and D. Southern blot analyses of DNA fragments in A and C, respectively. The BglI and MluI, KpnI, and AhdI-digested fragments were hybridized with the ICEF-I and ICEF-II (926 bp), ICEF-III (848 bp), prophage ΦMFV1 (920 bp) specific probes, respectively. The Southern blotting was conducted with DIG High Prime DNA Labeling and Detection Starter Kit (Roche, Basel, Switzerland). Lane M: MidRange I PFG Marker (New England Biolabs, Ipswich, Massachusetts); Lane U: Uncut genomic DNA; The size of the marker bands are indicated on the right.
(TIF)
Comparison among ICEF-IA, ICEF-IIA, and ICEF-IIIA of M. fermentans M64 indicated ICEF-III is a new family of ICE. A., B., and C. Results of sequence comparison of ICEF-IIIA and ICEF-IA, ICEF-IA and ICEF-IIA, and ICEF-IIA and ICEF-IIIA, respectively. The sequence similarities between ICEF-IIIA and ICEF-IA, ICEF-IA and ICEF-IIA, and ICEF-IIA and ICEF-IIIA are, 45.6%, 91.9%, and 45.4%, respectively. The GC content (sliding window: 120 bp) of each element was plotted on top or bottom of each panel. The arrows indicate the ORFs and the four-digit numbers represent the locus name (i.e., the number follow “MfeM64YM_” in locus tag). The two strips in dark gray stand for the forward and reverse strands of DNA. The direct and complementary matched regions between 2 elements are linked by blue and red lines, respectively. D. Phylogenetic tree of the 11 intact Integrative Conjugal Element (ICE) of Mycoplasmas. ICEB, ICEA, ICEF, ICEH, ICEM, and ICEC represent the elements in M. bovis, M. agalactiae, M. fermentans, M. hyopneumoniae, M. mycoides, and M. capricolum genomes, respectively. The scale bar stands for the estimated number of nucleotide substitutions per site.
(TIF)
A global view of M. fermentans metabolic networks showing the connected reactions in carbohydrate (green), nucleotide (blue), amino acids (red), cofactors and vitamins (orange), and lipid (pink) metabolisms. The map is produced by matching the M64 predicted proteins to metabolic pathways with the “color pathway” tool in KEGG (http://www.genome.jp/kegg/tool/color_pathway.html).
(TIF)
M. fermentans M64 purine (A) and pyrimidine (B) metabolic pathways contain many small clusters of connected reactions. Small circles represent the metabolic intermediates and green rectangles with enzyme catalog numbers denote the proteins in M. fermentans.
(TIF)
Phylogenetic tree of the 21 Mycoplasma species analyzed in Figure 4 . Maximum-likelihood method was performed on the 23S rRNA to reconstruct this tree using PHYML 3.0. The branch reliability was evaluated by implementing the aLRT method. Phytoplasma OY was set as the outgroup. The scale bar stands for the estimated number of nucleotide substitutions per site.
(TIF)
Phylogenetic comparative analysis of cofactors and vitamins (A) and amino acid (B) metabolic networks of M. fermentans M64 and 20 other Mycoplasma species. The small open squares represent the compounds and the colored circles represent the proteins (enzymes) participated in the metabolism. Circles with thick and light blue circumference represent the predicted essential genes. The arrows and lines linking the enzymes and substrates indicate the direction of the reactions. Small rectangle in B represents a putative horizontally transferred gene candidate. Enzymes involved in more than one metabolism are indicated with a one letter code (A: Amino acids metabolism; C: Carbohydrate metabolism; N: Nucleotide metabolism; V: metabolism and cofactors and Vitamins) next to the circles. The color scale (bottom) indicates the phylogenetic conservation of the proteins in 21 Mycoplasma species.
(TIF)
Phylogenetic relationships of MfeM64YM_1027 and MfeM64YM_0060 outside of the Mycoplasma clade. A. and C. The 23S rRNA trees of the species shown in B and D, respectively. B. and D. The phylogenetic trees of MfeM64YM_1027 and MfeM64YM_0060, respectively, and their closest homologs present in 12 other species. Maximum-likelihood method was used to reconstruct the trees. The branch reliability was evaluated by implementing the aLRT method. The scale bar stands for the estimated number of nucleotide (A and C) or amino acid (B and D) substitutions per site.
(TIF)
Unique and common genes of the M. fermentans strains M64, JER, and PG18.
(XLSX)
Genes of M. fermentans M64 outside of the Mycoplasma clade.
(XLSX)
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by Grant 96-2628-B-010-003-MY3 from the National Science Council and intramural funding from the Aim for the Top University Grant awarded to National Yang Ming University from the Ministry of Education in Taiwan, Republic of China. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Yus E, Maier T, Michalodimitrakis K, van Noort V, Yamada T, et al. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009;326:1263–1268. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- 2.Guell M, van Noort V, Yus E, Chen WH, Leigh-Bell J, et al. Transcriptome complexity in a genome-reduced bacterium. Science. 2009;326:1268–1271. doi: 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- 3.Fraser CM, Gocayne JD, White O, Adams MD, Clayton RA, et al. The minimal gene complement of Mycoplasma genitalium. Science. 1995;270:397–403. doi: 10.1126/science.270.5235.397. [DOI] [PubMed] [Google Scholar]
- 4.Peterson SN, Hu PC, Bott KF, Hutchison CA A survey of the Mycoplasma genitalium genome by using random sequencing. J Bacteriol. 1993;175:7918–7930. doi: 10.1128/jb.175.24.7918-7930.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Papazisi L, Gorton TS, Kutish G, Markham PF, Browning GF, et al. The complete genome sequence of the avian pathogen Mycoplasma gallisepticum strain R(low). Microbiology. 2003;149:2307–2316. doi: 10.1099/mic.0.26427-0. [DOI] [PubMed] [Google Scholar]
- 6.Sasaki Y, Ishikawa J, Yamashita A, Oshima K, Kenri T, et al. The complete genomic sequence of Mycoplasma penetrans, an intracellular bacterial pathogen in humans. Nucleic Acids Res. 2002;30:5293–5300. doi: 10.1093/nar/gkf667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pereyre S, Sirand-Pugnet P, Beven L, Charron A, Renaudin H, et al. Life on arginine for Mycoplasma hominis: clues from its minimal genome and comparison with other human urogenital mycoplasmas. PLoS Genet. 2009;5:e1000677. doi: 10.1371/journal.pgen.1000677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kitzerow A, Hadding U, Henrich B. Cyto-adherence studies of the adhesin P50 of Mycoplasma hominis. J Med Microbiol. 1999;48:485–493. doi: 10.1099/00222615-48-5-485. [DOI] [PubMed] [Google Scholar]
- 9.Baseman JB, Cole RM, Krause DC, Leith DK. Molecular basis for cytadsorption of Mycoplasma pneumoniae. J Bacteriol. 1982;151:1514–1522. doi: 10.1128/jb.151.3.1514-1522.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Himmelreich R, Hilbert H, Plagens H, Pirkl E, Li BC, et al. Complete sequence analysis of the genome of the bacterium Mycoplasma pneumoniae. Nucleic Acids Res. 1996;24:4420–4449. doi: 10.1093/nar/24.22.4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calderon-Copete SP, Wigger G, Wunderlin C, Schmidheini T, Frey J, et al. The Mycoplasma conjunctivae genome sequencing, annotation and analysis. BMC Bioinformatics. 2009;10(Suppl 6):S7. doi: 10.1186/1471-2105-10-S6-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dybvig K, Zuhua C, Lao P, Jordan DS, French CT, et al. Genome of Mycoplasma arthritidis. Infect Immun. 2008;76:4000–4008. doi: 10.1128/IAI.00516-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sirand-Pugnet P, Lartigue C, Marenda M, Jacob D, Barre A, et al. Being pathogenic, plastic, and sexual while living with a nearly minimal bacterial genome. PLoS Genet. 2007;3:e75. doi: 10.1371/journal.pgen.0030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minion FC, Lefkowitz EJ, Madsen ML, Cleary BJ, Swartzell SM, et al. The genome sequence of Mycoplasma hyopneumoniae strain 232, the agent of swine mycoplasmosis. J Bacteriol. 2004;186:7123–7133. doi: 10.1128/JB.186.21.7123-7133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaffe JD, Stange-Thomann N, Smith C, DeCaprio D, Fisher S, et al. The complete genome and proteome of Mycoplasma mobile. Genome Res. 2004;14:1447–1461. doi: 10.1101/gr.2674004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Westberg J, Persson A, Holmberg A, Goesmann A, Lundeberg J, et al. The genome sequence of Mycoplasma mycoides subsp. mycoides SC type strain PG1T, the causative agent of contagious bovine pleuropneumonia (CBPP). Genome Res. 2004;14:221–227. doi: 10.1101/gr.1673304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasconcelos AT, Ferreira HB, Bizarro CV, Bonatto SL, Carvalho MO, et al. Swine and poultry pathogens: the complete genome sequences of two strains of Mycoplasma hyopneumoniae and a strain of Mycoplasma synoviae. J Bacteriol. 2005;187:5568–5577. doi: 10.1128/JB.187.16.5568-5577.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chambaud I, Heilig R, Ferris S, Barbe V, Samson D, et al. The complete genome sequence of the murine respiratory pathogen Mycoplasma pulmonis. Nucleic Acids Res. 2001;29:2145–2153. doi: 10.1093/nar/29.10.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown DR, Farmerie WG, May M, Benders GA, Durkin AS, et al. Genome sequences of Mycoplasma alligatoris A21JP2T and Mycoplasma crocodyli MP145T. J Bacteriol. 2011;193:2892–2893. doi: 10.1128/JB.00309-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, Fang L, Li S, Li Q, Zhou Z, et al. Complete genome sequence of Mycoplasma hyorhinis strain HUB-1. J Bacteriol. 2010;192:5844–5845. doi: 10.1128/JB.00946-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rechnitzer H, Brzuszkiewicz E, Strittmatter A, Liesegang H, Lysnyansky I, et al. Genomic features and insights into the biology of Mycoplasma fermentans. Microbiology. 2011;157:760–773. doi: 10.1099/mic.0.043208-0. [DOI] [PubMed] [Google Scholar]
- 22.Wise KS, Calcutt MJ, Foecking MF, Roske K, Madupu R, et al. Complete genome sequence of Mycoplasma bovis type strain PG45 (ATCC 25523). Infect Immun. 2011;79:982–983. doi: 10.1128/IAI.00726-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barker EN, Helps CR, Peters IR, Darby AC, Radford AD, et al. Complete genome sequence of Mycoplasma haemofelis, a hemotropic mycoplasma. J Bacteriol. 2011;193:2060–2061. doi: 10.1128/JB.00076-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Messick JB, Santos AP, Guimaraes AM. Complete genome sequences of two hemotropic mycoplasmas, Mycoplasma haemofelis strain Ohio2 and Mycoplasma suis Strain Illinois. J Bacteriol. 2011;193:2068–2069. doi: 10.1128/JB.00133-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watanabe T, Shibata K, Yoshikawa T, Dong L, Hasebe A, et al. Detection of Mycoplasma salivarium and Mycoplasma fermentans in synovial fluids of temporomandibular joints of patients with disorders in the joints. FEMS Immunol Med Microbiol. 1998;22:241–246. doi: 10.1111/j.1574-695X.1998.tb01212.x. [DOI] [PubMed] [Google Scholar]
- 26.Haier J, Nasralla M, Franco AR, Nicolson GL. Detection of mycoplasmal infections in blood of patients with rheumatoid arthritis. Rheumatology (Oxford) 1999;38:504–509. doi: 10.1093/rheumatology/38.6.504. [DOI] [PubMed] [Google Scholar]
- 27.Gilroy CB, Keat A, Taylor-Robinson D. The prevalence of Mycoplasma fermentans in patients with inflammatory arthritides. Rheumatology (Oxford) 2001;40:1355–1358. doi: 10.1093/rheumatology/40.12.1355. [DOI] [PubMed] [Google Scholar]
- 28.Kraft M, Cassell GH, Henson JE, Watson H, Williamson J, et al. Detection of Mycoplasma pneumoniae in the airways of adults with chronic asthma. Am J Respir Crit Care Med. 1998;158:998–1001. doi: 10.1164/ajrccm.158.3.9711092. [DOI] [PubMed] [Google Scholar]
- 29.Vojdani A, Choppa PC, Tagle C, Andrin R, Samimi B, et al. Detection of Mycoplasma genus and Mycoplasma fermentans by PCR in patients with Chronic Fatigue Syndrome. FEMS Immunol Med Microbiol. 1998;22:355–365. doi: 10.1111/j.1574-695X.1998.tb01226.x. [DOI] [PubMed] [Google Scholar]
- 30.Cultrera R, Roulland-Dussoix D, Romani R, Contini C. Use of PCR to detect mycoplasma DNA in respiratory tract specimens from adult HIV-positive patients. J Med Microbiol. 1998;47:983–986. doi: 10.1099/00222615-47-11-983. [DOI] [PubMed] [Google Scholar]
- 31.Sasaki Y, Honda M, Naitou M, Sasaki T. Detection of Mycoplasma fermentans DNA from lymph nodes of acquired immunodeficiency syndrome patients. Microb Pathog. 1994;17:131–135. doi: 10.1006/mpat.1994.1059. [DOI] [PubMed] [Google Scholar]
- 32.Dawson MS, Hayes MM, Wang RY, Armstrong D, Kundsin RB, et al. Detection and isolation of Mycoplasma fermentans from urine of human immunodeficiency virus type 1-infected patients. Arch Pathol Lab Med. 1993;117:511–514. [PubMed] [Google Scholar]
- 33.Hawkins RE, Rickman LS, Vermund SH, Carl M. Association of mycoplasma and human immunodeficiency virus infection: detection of amplified Mycoplasma fermentans DNA in blood. J Infect Dis. 1992;165:581–585. doi: 10.1093/infdis/165.3.581. [DOI] [PubMed] [Google Scholar]
- 34.Ainsworth JG, Clarke J, Lipman M, Mitchell D, Taylor-Robinson D. Detection of Mycoplasma fermentans in broncho-alveolar lavage fluid specimens from AIDS patients with lower respiratory tract infection. HIV Med. 2000;1:219–223. doi: 10.1046/j.1468-1293.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- 35.Ainsworth JG, Hourshid S, Webster AD, Gilroy CB, Taylor-Robinson D. Detection of Mycoplasma fermentans in healthy students and patients with congenital immunodeficiency. J Infect. 2000;40:138–140. doi: 10.1016/s0163-4453(00)80005-5. [DOI] [PubMed] [Google Scholar]
- 36.Chingbingyong MI, Hughes CV. Detection of Mycoplasma fermentans in human saliva with a polymerase chain reaction-based assay. Arch Oral Biol. 1996;41:311–314. doi: 10.1016/0003-9969(96)84556-0. [DOI] [PubMed] [Google Scholar]
- 37.Shibata K, Kaga M, Kudo M, Dong L, Hasebe A, et al. Detection of Mycoplasma fermentans in saliva sampled from infants, preschool and school children, adolescents and adults by a polymerase chain reaction-based assay. Microbiol Immunol. 1999;43:521–525. doi: 10.1111/j.1348-0421.1999.tb02437.x. [DOI] [PubMed] [Google Scholar]
- 38.Montagnier L, Blanchard A. Mycoplasmas as cofactors in infection due to the human immunodeficiency virus. Clin Infect Dis. 1993;17(Suppl 1):S309–315. [PubMed] [Google Scholar]
- 39.Iyama K, Ono S, Kuwano K, Ohishi M, Shigematsu H, et al. Induction of tumor necrosis factor alpha (TNF alpha) and enhancement of HIV-1 replication in the J22HL60 cell line by Mycoplasma penetrans. Microbiol Immunol. 1996;40:907–914. doi: 10.1111/j.1348-0421.1996.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 40.Beecham HJ, Lo SC, Lewis DE, Comer SW, Riley KJ, et al. Recovery from fulminant infection with Mycoplasma fermentans (incognitus strain) in non-immunocompromised host. Lancet. 1991;338:1014–1015. doi: 10.1016/0140-6736(91)91874-t. [DOI] [PubMed] [Google Scholar]
- 41.Dallo SF, Baseman JB. Intracellular DNA replication and long-term survival of pathogenic mycoplasmas. Microb Pathog. 2000;29:301–309. doi: 10.1006/mpat.2000.0395. [DOI] [PubMed] [Google Scholar]
- 42.Taylor-Robinson D, Sarathchandra P, Furr PM. Mycoplasma fermentans--HeLa cell interactions. Clin Infect Dis. 1993;17(Suppl 1):S302–304. doi: 10.1093/clinids/17.supplement_1.s302. [DOI] [PubMed] [Google Scholar]
- 43.Blanchard A, Hamrick W, Duffy L, Baldus K, Cassell GH. Use of the polymerase chain reaction for detection of Mycoplasma fermentans and Mycoplasma genitalium in the urogenital tract and amniotic fluid. Clin Infect Dis. 1993;17(Suppl 1):S272–279. doi: 10.1093/clinids/17.supplement_1.s272. [DOI] [PubMed] [Google Scholar]
- 44.Johnson S, Sidebottom D, Bruckner F, Collins D. Identification of Mycoplasma fermentans in synovial fluid samples from arthritis patients with inflammatory disease. J Clin Microbiol. 2000;38:90–93. doi: 10.1128/jcm.38.1.90-93.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choppa PC, Vojdani A, Tagle C, Andrin R, Magtoto L. Multiplex PCR for the detection of Mycoplasma fermentans, M. hominis and M. penetrans in cell cultures and blood samples of patients with chronic fatigue syndrome. Mol Cell Probes. 1998;12:301–308. doi: 10.1006/mcpr.1998.0186. [DOI] [PubMed] [Google Scholar]
- 46.Hu WS, Yang CC. IS1550 from Mycoplasma fermentans is transposable in Escherichia coli. FEMS Microbiol Lett. 2001;198:159–164. doi: 10.1111/j.1574-6968.2001.tb10636.x. [DOI] [PubMed] [Google Scholar]
- 47.Calcutt MJ, Lavrrar JL, Wise KS. IS1630 of Mycoplasma fermentans, a novel IS30-type insertion element that targets and duplicates inverted repeats of variable length and sequence during insertion. J Bacteriol. 1999;181:7597–7607. doi: 10.1128/jb.181.24.7597-7607.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Calcutt MJ, Lewis MS, Wise KS. Molecular genetic analysis of ICEF, an integrative conjugal element that is present as a repetitive sequence in the chromosome of Mycoplasma fermentans PG18. J Bacteriol. 2002;184:6929–6941. doi: 10.1128/JB.184.24.6929-6941.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roske K, Calcutt MJ, Wise KS. The Mycoplasma fermentans prophage phiMFV1: genome organization, mobility and variable expression of an encoded surface protein. Mol Microbiol. 2004;52:1703–1720. doi: 10.1111/j.1365-2958.2004.04087.x. [DOI] [PubMed] [Google Scholar]
- 50.Pitcher D, Hilbocus J. Variability in the distribution and composition of insertion sequence-like elements in strains of Mycoplasma fermentans. FEMS Microbiol Lett. 1998;160:101–109. doi: 10.1111/j.1574-6968.1998.tb12898.x. [DOI] [PubMed] [Google Scholar]
- 51.Hu WS, Hayes MM, Wang RY, Shih JW, Lo SC. High-frequency DNA rearrangements in the chromosomes of clinically isolated Mycoplasma fermentans. Curr Microbiol. 1998;37:1–5. doi: 10.1007/s002849900327. [DOI] [PubMed] [Google Scholar]
- 52.Shu HW, Liu TT, Chan HI, Liu YM, Wu KM, et al. Genome sequence of the repetitive-sequence-rich Mycoplasma fermentans strain M64. J Bacteriol. 2011;193:4302–4303. doi: 10.1128/JB.05228-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ishida N, Irikura D, Matsuda K, Sato S, Sone T, et al. Molecular cloning and expression of a novel cholinephosphotransferase involved in glycoglycerophospholipid biosynthesis of Mycoplasma fermentans. Curr Microbiol. 2009;58:535–540. doi: 10.1007/s00284-009-9362-6. [DOI] [PubMed] [Google Scholar]
- 54.Chu LJ, Yang H, Shih P, Kao Y, Tsai YS, et al. J Proteome Res; 2011. Metabolic capabilities and systems fluctuations in Haloarcula marismortui revealed by integrative genomics and proteomics analyses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neimark H, Johansson KE, Rikihisa Y, Tully JG. Proposal to transfer some members of the genera Haemobartonella and Eperythrozoon to the genus Mycoplasma with descriptions of ‘Candidatus Mycoplasma haemofelis’, ‘Candidatus Mycoplasma haemomuris’, ‘Candidatus Mycoplasma haemosuis’ and ‘Candidatus Mycoplasma wenyonii’. Int J Syst Evol Microbiol. 2001;51:891–899. doi: 10.1099/00207713-51-3-891. [DOI] [PubMed] [Google Scholar]
- 56.Rikihisa Y, Kawahara M, Wen B, Kociba G, Fuerst P, et al. Western immunoblot analysis of Haemobartonella muris and comparison of 16S rRNA gene sequences of H. muris, H. felis, and Eperythrozoon suis. J Clin Microbiol. 1997;35:823–829. doi: 10.1128/jcm.35.4.823-829.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weisburg WG, Tully JG, Rose DL, Petzel JP, Oyaizu H, et al. A phylogenetic analysis of the mycoplasmas: basis for their classification. J Bacteriol. 1989;171:6455–6467. doi: 10.1128/jb.171.12.6455-6467.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pettersson B, Tully JG, Bolske G, Johansson KE. Updated phylogenetic description of the Mycoplasma hominis cluster (Weisburg, et al. 1989) based on 16S rDNA sequences. Int J Syst Evol Microbiol 50 Pt. 2000;1:291–301. doi: 10.1099/00207713-50-1-291. [DOI] [PubMed] [Google Scholar]
- 59.Van Demark PJ. Respiratory pathways in the mycoplasma. Ann N Y Acad Sci. 1967;143:77–84. doi: 10.1111/j.1749-6632.1967.tb27647.x. [DOI] [PubMed] [Google Scholar]
- 60.Smith SL, Vandemark PJ, Fabricant J. Respiratory Pathways in the Mycoplasma. I. Lactate Oxidation by Mycoplasma Gallisepticum. J Bacteriol. 1963;86:893–897. doi: 10.1128/jb.86.5.893-897.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev. 1998;62:1094–1156. doi: 10.1128/mmbr.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pollack JD. The necessity of combining genomic and enzymatic data to infer metabolic function and pathways in the smallest bacteria: amino acid, purine and pyrimidine metabolism in Mollicutes. Front Biosci. 2002;7:d1762–1781. doi: 10.2741/A878. [DOI] [PubMed] [Google Scholar]
- 63.Baker TA, Wickner SH. Genetics and enzymology of DNA replication in Escherichia coli. Annu Rev Genet. 1992;26:447–477. doi: 10.1146/annurev.ge.26.120192.002311. [DOI] [PubMed] [Google Scholar]
- 64.Lampe M, Binnie C, Schmidt R, Losick R. Cloned gene encoding the delta subunit of Bacillus subtilis RNA polymerase. Gene. 1988;67:13–19. doi: 10.1016/0378-1119(88)90003-0. [DOI] [PubMed] [Google Scholar]
- 65.Jones AL, Needham RH, Rubens CE. The Delta subunit of RNA polymerase is required for virulence of Streptococcus agalactiae. Infect Immun. 2003;71:4011–4017. doi: 10.1128/IAI.71.7.4011-4017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lopez de Saro FJ, Yoshikawa N, Helmann JD. Expression, abundance, and RNA polymerase binding properties of the delta factor of Bacillus subtilis. J Biol Chem. 1999;274:15953–15958. doi: 10.1074/jbc.274.22.15953. [DOI] [PubMed] [Google Scholar]
- 67.Watson SP, Antonio M, Foster SJ. Isolation and characterization of Staphylococcus aureus starvation-induced, stationary-phase mutants defective in survival or recovery. Microbiology 144 (Pt. 1998;11):3159–3169. doi: 10.1099/00221287-144-11-3159. [DOI] [PubMed] [Google Scholar]
- 68.Kawauchi Y, Muto A, Osawa S. The protein composition of Mycoplasma capricolum. Mol Gen Genet. 1982;188:7–11. doi: 10.1007/BF00332989. [DOI] [PubMed] [Google Scholar]
- 69.French CT, Lao P, Loraine AE, Matthews BT, Yu H, et al. Large-scale transposon mutagenesis of Mycoplasma pulmonis. Mol Microbiol. 2008;69:67–76. doi: 10.1111/j.1365-2958.2008.06262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Glass JI, Assad-Garcia N, Alperovich N, Yooseph S, Lewis MR, et al. Essential genes of a minimal bacterium. Proc Natl Acad Sci U S A. 2006;103:425–430. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, et al. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100:4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2: 2006. 2006;0008 doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Snider J, Thibault G, Houry WA. The AAA+superfamily of functionally diverse proteins. Genome Biol. 2008;9:216. doi: 10.1186/gb-2008-9-4-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 76.Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol. 2006;55:539–552. doi: 10.1080/10635150600755453. [DOI] [PubMed] [Google Scholar]
- 77.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 78.Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics. 2007;23:1026–1028. doi: 10.1093/bioinformatics/btm039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The circular representation of the M. fermentans M64 genome. The ticks on the outermost concentric circle indicate the relative genomic positions in base pairs, where position one is the first base of the upstream intergenic region of dnaA gene. Second and third concentric circles: predicted protein coding genes on the plus and minus strands, respectively. The various functional groups of the predicted genes (color coded) are categorized according to the COG classification. Fourth concentric circle: locations of rRNA (gray) and tRNAs (green) genes. Fifth concentric circle: ICEF-I and IIs (red arrows), ICEF-III (green arrows), ΦMFV1 prophage (blue arrows), IS elements [IS1550 (dark brown), IS1630 (brown), ISMf1 (orange)], 16S-23S rRNA operons (blue), and non-IS repeats (yellow). Sixth and seventh concentric circles: GC content and GC skew calculated in 5,000 bp of sliding window with 100 bp of step window, respectively.
(TIF)
Validation of the large repeats and genome assembly of M. fermentans M64. A. and C. EtBr-stained gel images of restriction enzymes (BglI, MluI, KpnI, and AdhI)-digested M64 genomic DNA. These fragments were resolved by CHEF gel electrophoresis in 1% agarose and 0.5X TBE at 6.0 v/cm for approximately 21.5 hr with pulsing times of 0.92 to 19.89 s and 0.92 to 37.34 s, respectively. B. and D. Southern blot analyses of DNA fragments in A and C, respectively. The BglI and MluI, KpnI, and AhdI-digested fragments were hybridized with the ICEF-I and ICEF-II (926 bp), ICEF-III (848 bp), prophage ΦMFV1 (920 bp) specific probes, respectively. The Southern blotting was conducted with DIG High Prime DNA Labeling and Detection Starter Kit (Roche, Basel, Switzerland). Lane M: MidRange I PFG Marker (New England Biolabs, Ipswich, Massachusetts); Lane U: Uncut genomic DNA; The size of the marker bands are indicated on the right.
(TIF)
Comparison among ICEF-IA, ICEF-IIA, and ICEF-IIIA of M. fermentans M64 indicated ICEF-III is a new family of ICE. A., B., and C. Results of sequence comparison of ICEF-IIIA and ICEF-IA, ICEF-IA and ICEF-IIA, and ICEF-IIA and ICEF-IIIA, respectively. The sequence similarities between ICEF-IIIA and ICEF-IA, ICEF-IA and ICEF-IIA, and ICEF-IIA and ICEF-IIIA are, 45.6%, 91.9%, and 45.4%, respectively. The GC content (sliding window: 120 bp) of each element was plotted on top or bottom of each panel. The arrows indicate the ORFs and the four-digit numbers represent the locus name (i.e., the number follow “MfeM64YM_” in locus tag). The two strips in dark gray stand for the forward and reverse strands of DNA. The direct and complementary matched regions between 2 elements are linked by blue and red lines, respectively. D. Phylogenetic tree of the 11 intact Integrative Conjugal Element (ICE) of Mycoplasmas. ICEB, ICEA, ICEF, ICEH, ICEM, and ICEC represent the elements in M. bovis, M. agalactiae, M. fermentans, M. hyopneumoniae, M. mycoides, and M. capricolum genomes, respectively. The scale bar stands for the estimated number of nucleotide substitutions per site.
(TIF)
A global view of M. fermentans metabolic networks showing the connected reactions in carbohydrate (green), nucleotide (blue), amino acids (red), cofactors and vitamins (orange), and lipid (pink) metabolisms. The map is produced by matching the M64 predicted proteins to metabolic pathways with the “color pathway” tool in KEGG (http://www.genome.jp/kegg/tool/color_pathway.html).
(TIF)
M. fermentans M64 purine (A) and pyrimidine (B) metabolic pathways contain many small clusters of connected reactions. Small circles represent the metabolic intermediates and green rectangles with enzyme catalog numbers denote the proteins in M. fermentans.
(TIF)
Phylogenetic tree of the 21 Mycoplasma species analyzed in Figure 4 . Maximum-likelihood method was performed on the 23S rRNA to reconstruct this tree using PHYML 3.0. The branch reliability was evaluated by implementing the aLRT method. Phytoplasma OY was set as the outgroup. The scale bar stands for the estimated number of nucleotide substitutions per site.
(TIF)
Phylogenetic comparative analysis of cofactors and vitamins (A) and amino acid (B) metabolic networks of M. fermentans M64 and 20 other Mycoplasma species. The small open squares represent the compounds and the colored circles represent the proteins (enzymes) participated in the metabolism. Circles with thick and light blue circumference represent the predicted essential genes. The arrows and lines linking the enzymes and substrates indicate the direction of the reactions. Small rectangle in B represents a putative horizontally transferred gene candidate. Enzymes involved in more than one metabolism are indicated with a one letter code (A: Amino acids metabolism; C: Carbohydrate metabolism; N: Nucleotide metabolism; V: metabolism and cofactors and Vitamins) next to the circles. The color scale (bottom) indicates the phylogenetic conservation of the proteins in 21 Mycoplasma species.
(TIF)
Phylogenetic relationships of MfeM64YM_1027 and MfeM64YM_0060 outside of the Mycoplasma clade. A. and C. The 23S rRNA trees of the species shown in B and D, respectively. B. and D. The phylogenetic trees of MfeM64YM_1027 and MfeM64YM_0060, respectively, and their closest homologs present in 12 other species. Maximum-likelihood method was used to reconstruct the trees. The branch reliability was evaluated by implementing the aLRT method. The scale bar stands for the estimated number of nucleotide (A and C) or amino acid (B and D) substitutions per site.
(TIF)
Unique and common genes of the M. fermentans strains M64, JER, and PG18.
(XLSX)
Genes of M. fermentans M64 outside of the Mycoplasma clade.
(XLSX)