

Abstract
Osteonecrosis of the jaw (ONJ), a side-effect of bisphosphonate therapy, is characterized by exposed bone that fails to heal within eight weeks. Healing time of oral epithelial wounds is decreased in the presence of amino-bisphosphonates; however, the mechanism remains unknown. We examined human tissue from individuals with ONJ and non-bisphosphonate-treated controlindividuals to identify changes in oral epithelium and connective tissue. Oral and intravenous bisphosphonate-treated ONJ sites had reduced numbers of basal epithelial progenitor cells, as demonstrated by a 13.8 ± 1.1% and 31.9 ± 5.8% reduction of p63 expression, respectively. No significant differences in proliferation rates, vessel density, or macrophage number were noted. In vitro treatment of clonal and primary oral keratinocytes with zoledronic acid (ZA) inhibited p63, and expression was rescued by the addition of mevalonate pathway intermediates. In addition, both ZA treatment and p63 shRNA knock-down impaired formation of 3D Ex Vivo Produced Oral Mucosa Equivalents (EVPOME) and closure of an in vitro scratch assay. Analysis of our data suggests that bisphosphonate treatment may delay oral epithelial healing by interfering with p63-positive progenitor cells in the basal layer of the oral epithelium in a mevalonate-pathway-dependent manner. This delay in healing may increase the likelihood of osteonecrosis developing in already-compromised bone.
Keywords: osteonecrosis, oral pathology, gingival, epithelia, keratinocytes, zoledronic acid
Introduction
Osteonecrosis of the jaw (ONJ) is a side-effect of bisphosphonate (BP) therapy characterized by dehiscence of oral epithelium and exposure of bone that fails to heal within 8 wks in individuals without history of radiation therapy (Novince et al., 2009). The majority of cases occur in those who are taking therapeutic doses of high-potency intravenous (IV) bisphosphonates such as zoledronic acid (ZA) and pamidronate disodium (Woo et al., 2006). Amino-bisphosphonates suppress bone turnover by inhibiting osteoclast formation and promoting apoptosis. This occurs through inhibition of farnesyl and geranyl–geranyl moiety production, resulting in reduced isoprenylation of the GTPases required for maintenance of cytoskeletal structure and vesicular traffic (Luckman et al., 1998; Fisher et al., 1999; Bergström et al., 2000). There are many hypotheses regarding the development of ONJ, but the exact mechanism has not been determined (Novince et al., 2009). Retrospective studies have associated trauma to the dento-alveolar complex with risk of developing ONJ. However, this does not explain why trauma to the alveolar soft tissue, spontaneously or during periodontal surgery, is the initiating event in a significant percentage of individuals, over 25% of cases in some series (Marx et al., 2005). This suggests the possibility of a direct role of compromised oral mucosal wound healing in the pathogenesis of ONJ.
Following loss of continuity of epithelial coverage, inflammatory cells migrate to the wound site (Warburton et al., 2005). Secreted factors then stimulate proliferation and migration of connective tissue fibroblasts, which coordinate with neo-vascularization and re-epithelialization to result in wound regeneration (Stephens and Genever, 2007). Alterations in any of these pathways would lead to delayed soft-tissue healing at sites of injury, prolonging the time during which the underlying bone is exposed to pathogenic oral flora and increasing the likelihood of osteonecrosis developing in already-compromised bone.
To determine whether bisphosphonate-related inhibition of re-epithelialization plays a role in the pathogenesis of ONJ, we examined archival human tissue harvested from individuals with ONJ and from non-bisphosphonate-treated control individuals to identify changes in the oral epithelium and connective tissue. Since re-epithelialization involves reciprocal interactions between epithelial cells at the border of an ulcer, the underlying connective tissue, surrounding inflammatory mediators, and neo-vascularization, aspects of each of these processes were measured. P63, a selective marker of basal/stem cells of stratified epithelium (Pellegrini et al., 2001), is constitutively expressed in basal cell nuclei of squamous epithelium. P63 has multiple diverse isoforms (Little and Jochemsen, 2002) and is required for initiation of epithelial stratification during the development and maintenance of basal keratinocytes in mature epidermis (Mills et al., 1999; Yang et al., 1999; Koster et al., 2004; Senoo et al., 2007). We hypothesized that alterations in the numbers of p63-positive progenitors may link ONJ to delayed epithelial wound healing, as observed with bisphosphonate treatment (Amagase et al., 2007; Kobayashi et al., 2010).
Materials & Methods
Immunohistochemistry
Formalin-fixed sections were deparaffinized in xylene and rehydrated. Heat-induced antigen retrieval (p63, Ki67, CD68) was performed in citrate buffer (Citrate pH6.0™, Dako:S1699, Carpinteria, CA, USA). FactorVIII-stained specimens were pre-treated with protease K. Antigen retrieval was not performed for PCNA.
Slides were stained with a Dako AutoStainer™ (Carpinteria, CA, USA) with either the labeled streptavidin-biotin method (p63 and Ki67; DakoCytomation LSAB+ System-HRP™, Dako:K0690) or peroxidase-labeled polymer conjugated to secondary antibody (DakoCytomation EnVision+ System-HRP Labeled Polymer™) anti-mouse (PCNA, CD68; Dako:K4001) or anti-rabbit (von Willebrand factor; Dako:K4003), developed with DAB (Liquid DAB+ Substrate System™, Dako:K3468) and counterstained with hematoxylin. Endogenous peroxidase activity was blocked by five-minute immersion in 3% hydrogen peroxide (Peroxidase Block Solution™, Dako:S200130-2). Primary antibodies and dilutions were: p63 (1:100, Neomarker/LabVision:MS-1081-P1); Ki67 (1:50, Dako:M7240); PCNA (1:100, Dako:M0879); CD68 (1:1600, Dako:M0814); and FactorVIII (1:250, Dako:A0082).
Two blinded researchers examined all slides independently. For p63, Ki67, and PCNA, the percentage of positive-staining nuclei in the basal layer and the 2 layers of epithelial cells immediately superior (suprabasal) were determined in a single representative field (Bortoluzzi et al., 2004). For FactorVIII,the numbers of small, medium, and large vessels in a representative field were counted. For CD68, the number of positive cells in a representative field was counted. Results are reported as the average of the two examiners’ values.
NOK-SI Culture
Spontaneously immortalized oral keratinocyte line ‘NOK-SI’ (a generous gift of Rogerio Castilho, University of Michigan) culture and scratch assay were performed as described previously (Castilho et al., 2010). Cells were maintained in DMEM, 10% FBS, and penicillin/streptomycin. Scratches were generated with a 200-µL pipette tip and “healed” in 5% serum after treatment with ZA (Zometa®, Novartis, Basel, Switzerland). For immunohistochemistry, cells were cultured in Permanox® chamber-slides (Lab-Tek:700400, Rochester, NY, USA), fixed in methanol, permeabilized in 1% Triton-X-100, blocked in 2.5% goat serum/2.5% donkey serum/0.1% Triton-X-100, and dual-stained with 1:400 p63 and 1:200 β-tubulin (AbCam:6046, Cambridge, MA, USA). Secondary antibodies were: 1:400 Cy™2 goat anti-rabbit and 1:500 Cy™3 donkey anti-mouse (Jackson ImmunoResearch, West Grove, PA, USA). We determined positive p63 staining by generating an image composite of 3 random fields per sample, converting to 8-bit image, and then applying a common threshold with ImageJ (Scion Corp., Frederick, MD, USA). The number of p63-positive cells was normalized to the total number of cells per field.
Western Blot
Cells were treated with 5-15 µM trans,trans-Farnesol (FOH) (Sigma:277541, Sigma, St. Louis, MO, USA) or geranylgeraniol (GGOH) (Sigma:G3278) where indicated. Western blot was performed as described previously with minor modification (Scheller et al., 2010). Nuclear extracts were harvested with the Active Motif kit (Cat:40010). Cells were collected in ice-cold PBS and centrifuged, followed by re-suspension in 1x hypotonic buffer, 15 min. Detergent was added for cytoplasmic lysis, and nuclei collected by centrifugation. Nuclear pellet underwent lysis in RIPA buffer (Santa Cruz:24948, Santa Cruz Biotechnology, Santa Cruz, CA, USA) as described previously (Scheller et al., 2010). Whole-cell extracts were made when complete cell pellets underwent lysis with RIPA buffer. A 6- to 8-µg (nuclear extract) or 20- to 25-µg (whole-cell extract) quantity per lane was probed with 1:500 anti-p63, 1:1000 TATA binding protein (AbCam:ab818), or 1:1000 GAPDH (Chemicon® International: MAB374, Billerica, MA, USA), followed by 1:3000 goat-anti mouse (Santa Cruz: 2005).
shRNA
Lentiviral shRNA clones in a pGIPZ vector from Open Biosystems (Thermo Scientific, Huntsville, AL, USA) were screened for ability to knock down p63. Virus was produced by the University of Michigan Vector Core with University of Michigan IBC approval. Clone V2LHS_24246 targeted against GenBank accession NM_003722 was selected. Cells were transduced at ~3-6 MOI in complete media with 8 µg/mL polybrene (Santa Cruz:134220) overnight. Control cells were transduced with scrambled pGIPZ vector. 1°OK were assayed after 4 days, and NOK-SI were selected in 1 µg/mL puromycin (Santa Cruz:108071) for 3 passages before utilization.
Primary Keratinocytes and EVPOME
Human oral mucosal tissue harvesting was approved by the University of Michigan IRB (Appendix Table 2). Primary human oral keratinocyte (1°OK) culture and Ex Vivo Produced Oral Mucosa Equivalents (EVPOMEs) have been described previously (Izumi et al., 1999, 2003). Briefly, 1°OK were enzymatically dissociated from tissue samples, and cells were cultured in chemically defined medium (EpiLife+EDGS; 0.06 mM calcium; 25 µg/mL gentamicin; 0.375 µg/mL fungizone) (Invitrogen, Carlsbad, CA, USA). For EVPOMEs, 200 K cells/cm2 were seeded on 1 cm2 acellular cadaver skin (AlloDerm®) that was pre-soaked in 5 µg/cm2 human type IV collagen at 4°C overnight. Cells+AlloDerm® were submerged in medium containing 1.2 mM calcium for 4 days before being raised to an air-liquid phase for 7 days. A 10−5 M quantity of ZA was added on day 3. Samples were fixed in 10% formalin with immunohistochemistry of 1°OK as above; immunohistochemistry for paraffin-sectioned EVPOMEs was as above with minor modification. Slides were processed manually, and p63 signal was amplified with ImmPRESS reagent (VectorLabs: MP-7500, Burlingame, CA, USA) and imaged with ImmPACT DAB substrate (VectorLabs:SK-4105).
Statistical Analysis
One-way ANOVA for 3 independent samples was performed with VassarStats (R. Lowry, Vassar College, Poughkeepsie, NY, USA). To compare individual groups, we used a two-tailed, homoscedastic t test. Differences were statistically significant when p < 0.05 and of non-significant trend when p < 0.100.
Results
Human Tissue Analysis
The number of p63-positive cells in the basal and parabasal layers was higher in the control tissue (86.1 ± 3.4) as compared with the Oral-BP (74.2 ± 6.1, p = 0.087) and IV-BP (58.6 ± 10.6, p = 0.010) groups (Appendix, Figs. 1A, 1B). No differences in epithelial or stromal proliferation rates were noted, as measured by Ki67 and PCNA (Fig. 1A). Likewise, no significant differences were noted in concentration of macrophages or blood vessel density, as determined by CD68 and FactorVIII expression, respectively (Fig. 1A). A non-significant trend toward increased numbers of CD68-positive macrophages was observed in the IV-BP group (Fig. 1A).
Figure 1.
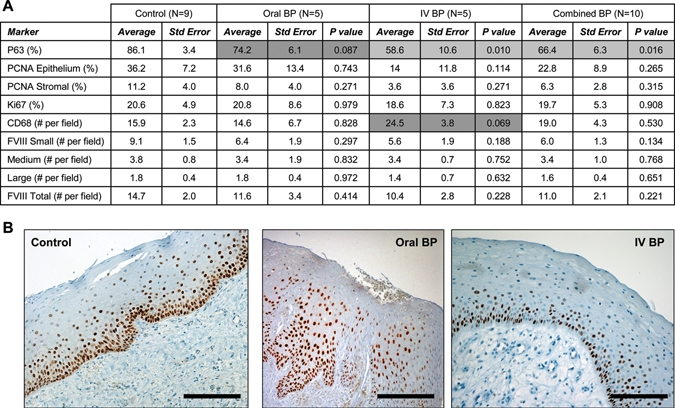
Bisphosphonates inhibit expression of p63 in basal oral keratinocytes. (A) Tabulated immunohistochemistry results. (B) Representative images of control, Oral-BP, and IV-BP epithelial staining for p63. Results are reported as average ± standard error of the mean.
ZA Suppression of p63 Is Mevalonate-pathway-dependent
NOK-SI cells were treated for 24 hrs with increasing concentrations of ZA. Decreases in numbers of p63-positive cells were observed at 10−6 M (46.8 ± 11.8%, p = 0.030) and 10−5 M ZA (25.2 ± 14.4%, p = 0.014) when compared with controls (86.5 ± 2.66%) (Figs. 2A, 2B). The addition of mevalonate pathway intermediates restored p63 expression. FOH increased p63 in nuclear extracts (Fig. 2C) and immunohistochemically (Fig. 2D) in a dose-dependent manner, from 5-15 µM. Regulation of p63 expression by GGOH occurred at 5 µM in the nuclear extract but not during immunohistochemical analysis (Figs. 2C, 2D). A similar decrease in numbers of p63-positive cells after ZA treatment was observed with 1°OK after 10−6 M (43.3 ± 1.6%, p = 0.008) and 10−5 M (46.1 ± 6.3%, p = 0.018) ZA treatment compared with controls (74.4 ± 11%) (Fig. 3A).
Figure 2.
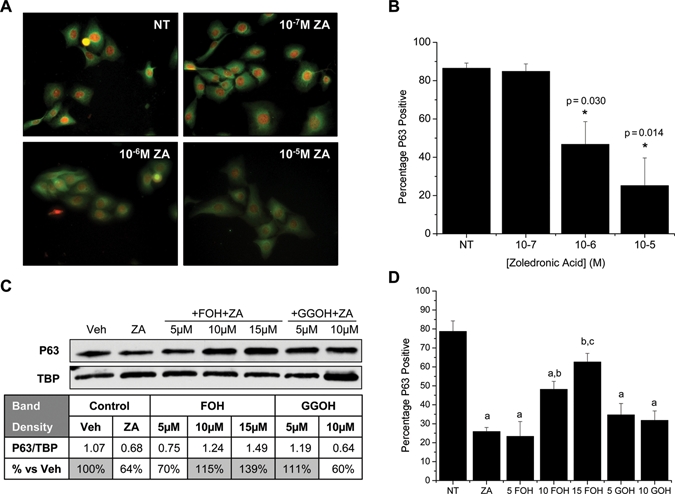
Zoledronic acid inhibits p63 in clonal oral keratinocytes. (A) Immunocytochemistry depicting p63 nuclear stain (red) and B-tubulin cytoplasmic stain (green). (B) Percentage p63-positive cells as calculated by applying a common threshold to matched p63-stained images and comparison with total number of cells per field. Results are reported as the average of 3 independent experiments (N = 3), with 3 random fields analyzed per treatment regimen per experiment. (C) Representative Western blot of nuclear extracts from NOK-SI treated with 10-5 M ZA, vehicle control, FOH, or GGOH. Band densitometry was used to normalize p63 to the TATA binding protein (TBP) loading standard. (D) Percentage p63-positive cells calculated as in (B) after treatment with vehicle (NT), 10-5 M ZA, ZA+5-15 µM FOH, or ZA+5-10 µM GGOH. (a) Significantly less than NT control; (b) significantly higher than the ZA group; (c) significantly higher than the 10 FOH group. Results are reported as mean ± standard deviation.
Figure 3.
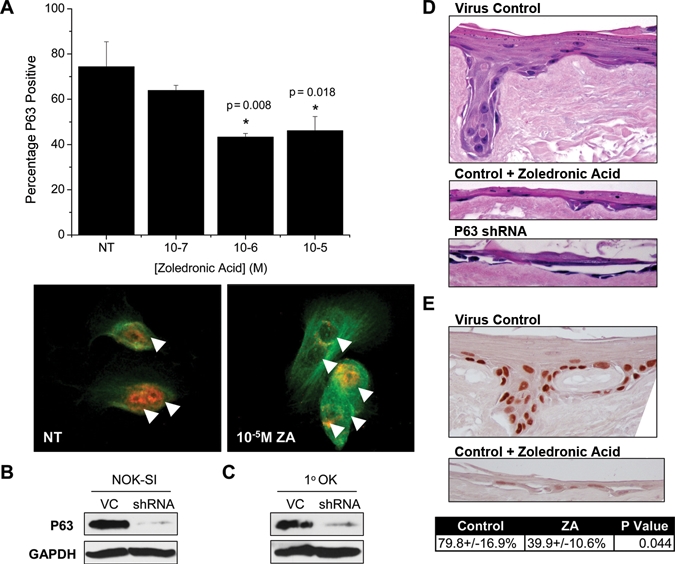
Zoledronic acid (ZA) inhibits p63 in primary oral keratinocytes (1°OK), and both ZA and p63 regulate Ex Vivo Produced Oral Mucosa Equivalents (EVPOME) formation. (A) Percentage p63-positive 1°OK calculated as in Fig. 2B. Red = p63, Green = β-tubulin, arrows = cell nuclei. (B,C) Western blot of p63 in whole-cell extract after shRNA knock-down in clonal NOK-SI keratinocytes or 1°OK, VC: scrambled shRNA virus control. (D) Representative images from 3D EVPOMEs formed after 200 K 1°OK/cm2 seeding onto type IV collagen-coated AlloDerm® (N = 3). (E) Representative images and quantification of p63 staining in the basal/parabasal layers of EVPOMEs (N = 3). Results are reported as mean ± standard deviation.
ZA and p63 Regulate Formation of Stratified Epithelium in 3D Culture
Lentiviral shRNA was used to knock down p63 expression in NOK-SI (Fig. 3B) and 1°OK (Fig. 3C). After 4 days, 1°OK were harvested and seeded on type IV collagen-coated AlloDerm® for EVPOME formation. Upon EVPOME analysis, significant qualitative differences were noted between the groups. Control 1°OK transduced with scrambled shRNA showed robust formation of keratinized, stratified squamous epithelium, 3 to 8 cells thick, with generalized rete ridge formation (Fig. 3D). Treatment with ZA reduced the thickness to 1 to 3 cells with few or no rete ridges (Fig. 3D). Knock-out of p63 nearly ablated the ability of the cells to form an epithelial layer, with > 90% of the surface lacking any epithelial attachment. In places where cells persisted, the acantholytic epithelium was 1 to 2 cells thick (Fig. 3D). Immunohistochemistry for p63 revealed robust expression in the control group basal/parabasal layer (79.8 ± 16.9%), with significant inhibition after ZA treatment (39.9 ± 10.6%, p = 0.044) (Fig. 3E). No p63-positive cells were noted on the p63 knock-out EVPOMEs.
ZA and p63 Regulate Scratch-healing and Proliferation
NOK-SI were plated at high density before being uniformly scratched, and ZA was added directly before scratch generation. After 20 hrs, both 10−6 M and 10−5 M ZA groups showed a significant delay in healing that continued up to 36 hrs (Figs. 4A, 4B). Similarly, NOK-SI with stable knock-down of p63 had significantly impaired scratch closure after 24 hrs (Fig. 4C). In addition, the cell morphology was altered, with more spreading and decreased density (Fig. 4D). Proliferation defects at all time-points from 30 hrs on appeared only in the 10−5 M ZA group (Appendix Fig. A). However, proliferation decreases were also noted with 10−6 M ZA at 30 hrs and 10−7 M ZA at 56 hrs (Appendix Fig. A). Proliferation of p63 knock-out cells was also impaired, as shown by a significant 22% decrease in the slope of their exponential growth phase when compared with that of scrambled controls (Appendix Fig. B).
Figure 4.
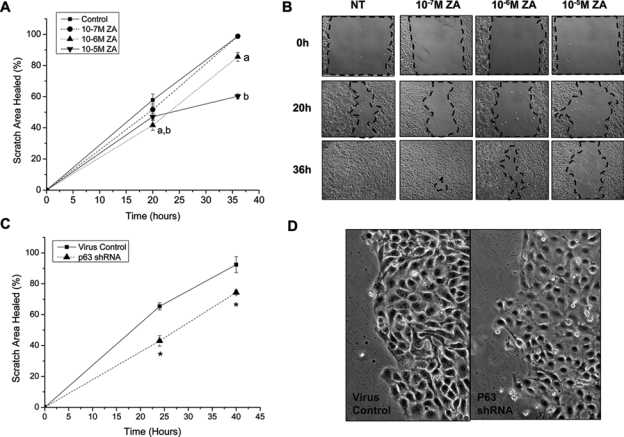
Zoledronic acid and p63 delay scratch closure in vitro. (A,B) Confluent layers of NOK-SI cells were treated with increasing concentrations of ZA and uniformly scratched with a 200-µL pipette tip. Closure was measured at 20 and 36 hrs. N = 6-8 unique scratches per group. (a) 10−6 M ZA significance; (b) 10−5 M ZA significance. (C) Confluent NOK-SI maintained in 1 µg/mL puromycin with stable knock-down of p63 were uniformly scratched. Closure was measured at 24 and 40 hrs (N = 5-7). (D) Morphology of cells at 24 hrs showed decreased cell density in the shRNA p63 group. Results are reported as mean ± standard deviation.
Discussion
Inhibition of mucosal wound healing in vivo and in vitro has been demonstrated after bisphosphonate treatment (Amagase et al., 2007; Landesberg et al., 2008; Kobayashi et al., 2010), yet the mechanism and its role in the pathogenesis of ONJ remain unknown. We present evidence, both in vivo and in vitro, that bisphosphonates can decrease the number of p63-positive basal epithelial progenitor cells. We hypothesize that this contributes to reduced epithelial healing and speculate that this may potentiate insults to the oral cavity, increasing the risk of ONJ development in already-compromised bone.
Since re-epithelialization involves reciprocal interactions among epithelial cells, underlying connective tissue, the surrounding inflammatory mediators, and neo-vascularization, we examined aspects of several of these components. Amino-bisphosphonates block bone resorption through inhibition of osteoclast formation and function. Although the macrophage originates from a common precursor, much less is known about the effects of bisphosphonates on macrophage activity. We found a non-significant trend (p = 0.069) for increased macrophage numbers in the IV bisphosphonate group. Amino-bisphosphonates have also been shown to be inhibitors of angiogenesis (Giraudo et al., 2004; Amagase et al., 2007). However, we failed to identify changes in blood vessel density between tissue samples from bisphosphonate-treated and control individuals.
Adverse effects of amino-bisphosphonates on mucosal tissues in the clinical setting range from esophageal erosions and gastrointestinal disturbances to oral ulcerations following direct epithelial contact (Gonzalez-Moles and Bagan-Sebastian, 2000; Monkkonen et al., 2003; Rubegni and Fimiani, 2006). The adverse effects on the gastric mucosa appear to be related to the mevalonate pathway, resulting in inhibition of proliferation and increased apoptosis of gastric epithelial cell lines at concentrations of bisphosphonates around 10−5 M (Suri et al., 2001). In our study, both treatment with ZA and knock-down of p63 in NOK-SI cells showed reduction in proliferation. However, this proliferation defect was not observed in human ONJ biopsies with PCNA or Ki67 staining. This is likely because our biopsy sample size was not large enough for a significant difference to be detected.
Previous literature, our in vitro scratch assay, and 3D EVPOME formation support the notion that p63 expression is necessary for formation of a mature stratified squamous epithelium (Yang et al., 1999). Treatment of both NOK-SI clonal and 1°OK with 10−5 M ZA mimicked the delayed scratch healing, decrease in proliferation, and poor 3D EVPOME formation observed upon knock-down of p63. This strongly suggests that inhibition of p63 expression by amino-bisphosphonates, as was observed in ONJ biopsy specimens, in vitro cell cultures, and 3D EVPOMEs, may contribute to decreased stratification and delayed wound healing. Partial percent p63 inhibition by ZA, as opposed to near-complete inhibition with shRNA, is likely responsible for the decreased severity of the ZA-treated phenotype when compared with p63 shRNA in our scratch and EVPOME assays. This may also be one reason bisphosphonates delay but do not completely inhibit epithelial healing in vivo.
Last, analysis of our data implies that bisphosphonate regulation of p63 likely occurs via the inhibition of farnesylation and modulation of the mevalonate pathway. Inhibition of p63 expression by oral keratinocytes was rescued dose-dependently by the addition of FOH and, less consistently, by GGOH. There are relatively few known farnesylated proteins; one study identified 17, including several members of the Ras protein family (Khoet al., 2004). Though we do not yet understand how farnesylated proteins regulate p63 expression, a recent publication found that overexpression of H-Ras in immortalized keratinocytes increased vimentin and p63 co-localization in vitro (Vaughanet al., 2009). This suggests that inhibition of H-Ras farnesylation by ZA may have an opposing effect and reduce nuclear p63 accumulation, as observed in this study. Future work will be necessary to explore isoprenylated intermediates and their ability to regulate p63 expression in keratinocytes.
In summary, our results suggest that bisphosphonates may delay mucosal wound-healing and potentially predispose to the development of ONJ by reducing the number of p63-positive epithelial progenitor cells in a mevalonate-pathway-dependent manner. Our complementary studies have indicated that similar effects on p63 expression, wound closure, and epithelial stratification are observed when human oral keratinocytes are treated with 10−6 M to 10−5 M ZA in vitro, suggesting that this may be a relevant test concentration for future studies. Future work will include exploration of regulation of p63 by isoprenylated intermediates, as well as expansion of our analysis to determine if an individual’s susceptibility plays a role in bisphosphonate regulation of epithelial p63 expression.
Supplementary Material
Acknowledgments
Many thanks to Chris Strayhorn, Nancy McAnsh, Tom Lannigan, and Rogerio Castilho for their assistance.
Footnotes
This work was supported by the University of Michigan Department of Periodontics and Oral Medicine (PCE), R01-DE13835 (PHK), F30-DE019577 (ELS), and R01-DE019431 (SEF).
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/supplemental.
References
- Amagase K, Hayashi S, Nishikawa K, Aihara E, Takeuchi K. (2007). Impairment of gastric ulcer healing by alendronate, a nitrogen-containing bisphosphonate, in rats. Dig Dis Sci 52:1879-1889 [DOI] [PubMed] [Google Scholar]
- Bergström JD, Bostedor RG, Masarachia PJ, Reszka AA, Rodan G. (2000). Alendronate is a specific, nanomolar inhibitor of farnesyl diphosphate synthase. Arch Biochem Biophys 373:231-241 [DOI] [PubMed] [Google Scholar]
- Bortoluzzi MC, Yurgel LS, Dekker NP, Jordan RC, Regezi JA. (2004). Assessment of p63 expression in oral squamous cell carcinomas and dysplasias. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 98:698-704 [DOI] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Leelahavanichkul K, Zheng Y, Bugge T, Gutkind JS. (2010). Rac1 is required for epithelial stem cell function during dermal and oral mucosal wound healing but not for tissue homeostasis in mice. PLoS One 5:e10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JE, Rogers MJ, Halasy JM, Luckman SP, Hughes DE, Masarachia PJ, et al. (1999). Alendronate mechanism of action: geranylgeraniol, an intermediate in the mevalonate pathway, prevents inhibition of osteoclast formation, bone resorption, and kinase activation in vitro. Proc Natl Acad Sci USA 96:133-138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudo E, Inoue M, Hanahan D. (2004). An amino-bisphosphonate targets MMP-9-expressing macrophages and angiogenesis to impair cervical carcinogenesis. J Clin Invest 114:623-633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Moles MA, Bagan-Sebastian JV. (2000). Alendronate-related oral mucosa ulcerations. J Oral Pathol Med 29:514-518 [DOI] [PubMed] [Google Scholar]
- Izumi K, Takacs G, Terashi H, Feinberg SE. (1999). Ex vivo development of a composite human oral mucosal equivalent. J Oral Maxillofac Surg 57:571-577 [DOI] [PubMed] [Google Scholar]
- Izumi K, Feinberg SE, Iida A, Yoshizawa M. (2003). Intraoral grafting of an ex vivo produced oral mucosa equivalent: a preliminary report. Int J Oral Maxillofac Surg 32:188-197 [DOI] [PubMed] [Google Scholar]
- Kho Y, Kim SC, Jiang C, Barma D, Kwon SW, Cheng J, et al. (2004). A tagging-via-substrate technology for detection and proteomics of farnesylated proteins. Proc Natl Acad Sci USA 101:12479-12484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Hiraga T, Ueda A, Wang L, Matsumoto-Nakano M, Hata K, et al. (2010). Zoledronic acid delays wound healing of the tooth extraction socket, inhibits oral epithelial cell migration, and promotes proliferation and adhesion to hydroxyapatite of oral bacteria, without causing osteonecrosis of the jaw, in mice. J Bone Miner Metab 28:165-175 [DOI] [PubMed] [Google Scholar]
- Koster MI, Kim S, Mills AA, DeMayo FJ, Roop DR. (2004). p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev 18:126-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landesberg R, Cozin M, Cremers S, Woo V, Kousteni S, Sinha S, et al. (2008). Inhibition of oral mucosal cell wound healing by bisphosphonates. J Oral Maxillofac Surg 66:839-847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little NA, Jochemsen AG. (2002). p63. Int J Biochem Cell Biol 34:6-9 [DOI] [PubMed] [Google Scholar]
- Luckman SP, Coxon FP, Ebetino FH, Russell RG, Rogers MJ. (1998). Heterocycle-containing bisphosphonates cause apoptosis and inhibit bone resorption by preventing protein prenylation: evidence from structure-activity relationships in J774 macrophages. J Bone Miner Res 13:1668-1678 [DOI] [PubMed] [Google Scholar]
- Marx RE, Sawatari Y, Fortin M, Broumand V. (2005). Bisphosphonate-induced exposed bone (osteonecrosis/osteopetrosis) of the jaws: risk factors, recognition, prevention, and treatment. J Oral Maxillofac Surg 63:1567-1575 [DOI] [PubMed] [Google Scholar]
- Mills AA, Zheng B, Wang XJ, Vogel H, Roop DR, Bradley A. (1999). p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398:708-713 [DOI] [PubMed] [Google Scholar]
- Monkkonen H, Tormalehto S, Asunmaa K, Niemi R, Auriola S, Vepsalainen J, et al. (2003). Cellular uptake and metabolism of clodronate and its derivatives in Caco-2 cells: a possible correlation with bisphosphonate-induced gastrointestinal side-effects. Eur J Pharm Sci 19:23-29 [DOI] [PubMed] [Google Scholar]
- Novince CM, Ward BB, McCauley LK. (2009). Osteonecrosis of the jaw: an update and review of recommendations. Cells Tissues Organs 189:275-283 [DOI] [PubMed] [Google Scholar]
- Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, et al. (2001). p63 identifies keratinocyte stem cells. Proc Natl Acad Sci USA 98:3156-3161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubegni P, Fimiani M. (2006). Images in clinical medicine. Bisphosphonate-associated contact stomatitis. N Engl J Med 355:e25. [DOI] [PubMed] [Google Scholar]
- Scheller EL, Song J, Dishowitz MI, Soki FN, Hankenson KD, Krebsbach PH. (2010). Leptin functions peripherally to regulate differentiation of mesenchymal progenitor cells. Stem Cells 28:1071-1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo M, Pinto F, Crum CP, McKeon F. (2007). p63 is essential for the proliferative potential of stem cells in stratified epithelia. Cell 129:523-536 [DOI] [PubMed] [Google Scholar]
- Stephens P, Genever P. (2007). Non-epithelial oral mucosal progenitor cell populations. Oral Dis 13:1-10 [DOI] [PubMed] [Google Scholar]
- Suri S, Monkkonen J, Taskinen M, Pesonen J, Blank MA, Phipps RJ, et al. (2001). Nitrogen-containing bisphosphonates induce apoptosis of Caco-2 cells in vitro by inhibiting the mevalonate pathway: a model of bisphosphonate-induced gastrointestinal toxicity. Bone 29:336-343 [DOI] [PubMed] [Google Scholar]
- Vaughan MB, Ramirez RD, Andrews CM, Wright WE, Shay JW. (2009). H-ras expression in immortalized keratinocytes produces an invasive epithelium in cultured skin equivalents. PLoS One 4:e7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburton G, Nares S, Angelov N, Brahim JS, Dionne RA, Wahl SM. (2005). Transcriptional events in a clinical model of oral mucosal tissue injury and repair. Wound Repair Regen 13:19-26 [DOI] [PubMed] [Google Scholar]
- Woo SB, Hellstein JW, Kalmar JR. (2006). Narrative [corrected] review: bisphosphonates and osteonecrosis of the jaws. Ann Intern Med 144:753-761; erratum in Ann Intern Med 145:325, 2006 [DOI] [PubMed] [Google Scholar]
- Yang A, Schweitzer R, Sun D, Kaghad M, Walker N, Bronson RT, et al. (1999). p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 398:714-718 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.