

Abstract
The human pathogenic fungus Candida albicans is the predominant cause of both superficial and invasive forms of candidiasis. Clinical observations indicate that mucocutaneous Candida infections are commonly associated with defective cell-mediated immune responses. The importance of the innate immune system as a first-line defense against pathogenic challenge has long been recognized. Over the last decade, many key molecules mediating innate host defense have been identified. Central to these developments is the discovery of pattern recognition receptors such as Toll-like receptors and C-type lectin-receptors that induce innate immune responses and also modulate cellular and humoral adaptive immunity during Candida infections. Although a large amount of information is now available in systemic infections, little is known about localized infections. We address the most relevant pattern recognition receptors and their signaling mechanisms in oral epithelial cells, to gain a better understanding of their contributions to antifungal innate immunity.
Keywords: oral epithelium, innate immunity, Toll-like receptors, C-type lectin-receptors, Candida albicans
Introduction
The mucosal epithelium has immense importance in host defense and immune surveillance, since it is the primary cell layer that initially encounters the majority of micro-organisms. The most important function of the immune system is to discriminate between friend and foe, a property that is essential for maintaining immune homeostasis. This specialized interaction will result in either passive coexistence between microbe and host, as in the case of commensal microbes, or in a violation of the mucosal barrier and subsequent cell injury, as in the case of microbial pathogens. The cells that comprise the innate immune response are primarily phagocytes, including neutrophils and macrophages, and the cells that line the epithelial mucosa. Originally, it was thought that the epithelium serves only as a passive barrier against invading pathogens. Barrier function alone is usually adequate to restrain commensal microbes, but is often insufficient to protect against microbial pathogens. However, recently it has become apparent that epithelial cells are capable of triggering an immune response similar to that of cells of the myeloid lineage, thus playing a crucial role in the active recognition of microbes. Accordingly, the oral epithelium is able to secrete a variety of defense effector molecules (Diamond et al., 2008) and to orchestrate an immune inflammatory response to activate myeloid cells in the submucosal layers to clear the invading pathogens (Cutler and Jotwani, 2006).
The frequency of mucosal and cutaneous fungal infections is increasing worldwide, with oral candidiasis being the most common human fungal infection, especially in early and later life (Samaranayake et al., 2009). Oral candidiasis is a common opportunistic infection of the oral cavity and presents a challenge for immunologically competent and immunodeficient patients alike. Various clinical presentations are traditionally divided into acute and chronic forms. Acute pseudomembranous candidiasis (mucosal candidiasis, oral thrush) presents with stippled (later confluent) white plaque (that can be wiped off) on bright red and lightly bleeding mucosa. Chronic atrophic candidiasis (denture-related stomatitis) is associated with erythema and edema of the oral mucosa, often found on the fitting surfaces of dentures. Also belonging to the category of oral candidiasis are perlèche, candidal leukoplakia (chronic hyperplastic candidiasis), candidal cheilitis, and chronic mucocutaneous candidiasis (CMC), a rare form that is associated with immune deficiency. Life-threatening systemic infection is generally limited to severely immunocompromised patients, such as neutropenic patients, often after nosocomial infection. The number of fungal infections as a proportion of all nosocomial infections doubled during a 10-year period in the United States. In immunocompetent patients, predisposing factors are responsible for infection or even chronic recurrent mucocutaneous candidiasis. Oropharyngeal and vaginal infections are the most common manifestation; predisposing factors include antibiotic, glucocorticosteroid, and hormone therapies, as well as diabetes mellitus and infections such as HIV and AIDS. Around 80% of all fungal infections are caused by Candida, typically Candida albicans (C. albicans) (Ruhnke, 2006). However, non-albicans Candida spp., such as Candida glabrata, Candida tropicalis, Candida parapsilosis, Candida guilliermondii, and Candida krusei are also pathogenic to humans and have emerged as important opportunistic pathogens in the oral mucosa (Li et al., 2007; Samaranayake et al., 2009).
C. albicans interacts with epithelial cells in terms of adherence, invasion, and induction of cell damage (Fig. 1). Virulence factors are crucial in determining the role of opportunistic pathogens in infections. Important virulence factors expressed by C. albicans include dimorphism (ability to grow in either yeast or mycelial form), adhesion factors, phenotypic switching, thigmotropism (ability to identify intercellular junctions at the mucosal surface by contact sensing and their targeted penetration), and secretion of hydrolytic enzymes such as lipase, phospholipase, and proteinase (reviewed in Calderone and Fonzi, 2001; Schaller et al., 2005). The interaction between virulence factors of C. albicans and host defense mechanisms plays a central role in determining whether colonization remains harmless or leads to infection of the epithelium and possibly systemic infection. Recognition of C. albicans by the innate host defense system is mediated by pattern-recognition receptors (PRRs) from the Toll-like receptor (TLR), C-type lectin-receptor (CLR), and NOD-like receptor (NLR) families (Netea et al., 2008a; Bryant and Fitzgerald, 2009). To date, most investigations have focused on the interaction of C. albicans with macrophages, and on systemic infections. At present, we understand little about how the oral mucosa regulates itself in the context of fungal infections, although recent studies have furthered our understanding of pathogen recognition and signaling mechanisms in oral epithelial cells. This review will discuss recent advances in our understanding of the role PRRs play in signaling or regulating the immune response against fungal pathogens in the oral mucosa. In addition, since PRRs form a crucial link between innate and adaptive immunity, adaptive cell responses will also be discussed.
Figure 1.
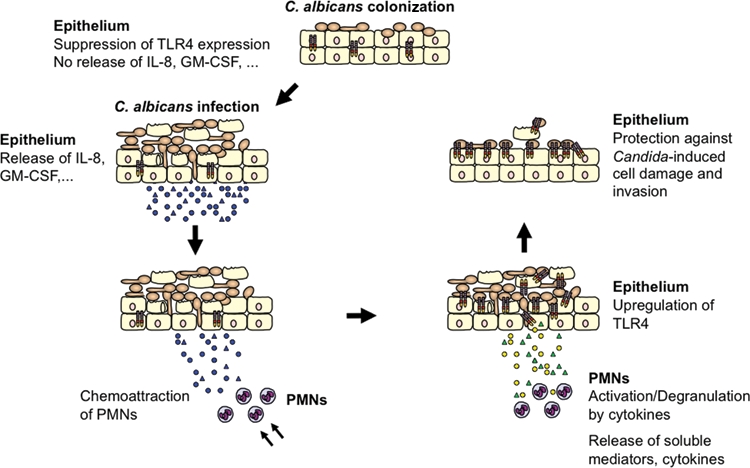
Model of TLR4-mediated and PMN-dependent antifungal defense by the oral epithelium. In general, experimental oral infection can be divided into 3 phases: an attachment phase, an invasion phase, and a tissue destruction phase. Although C. albicans normally exists as a yeast cell, adherent yeast cells rapidly form germ tubes after contact with epithelial cells, and hyphae penetrate the epithelium. Tissue damage is increased dramatically over time, when hyphae penetrate the tissue not only in the top layer, but also in deeper epithelial cell layers. In contrast, epithelial cells control fungal cell growth and invasion. During colonization of the oral epithelium, C. albicans suppresses TLR4 expression and does not induce cytokine production. Infection, particularly in predisposed patients, leads to increased cytokine secretion that recruits and stimulates PMNs at the site of infection. After recruitment, several cytokines, especially TNF, are directly involved in initiating the subsequent PMN-mediated up-regulation of epithelial TLR4 via a process that does not require PMN infiltration of the mucosal tissues. Finally, epithelial TLR4 directly protects the oral mucosa from fungal invasion and cell injury, possibly by production of antimicrobial peptides.
Pattern-Recognition Receptors in Fungal Recognition
The innate immune system recognizes conserved pathogen-associated molecular patterns (PAMPs), which represent broad groups of microbial species rather than a single specific species, through germline-encoded proteins, such as PRRs (Janeway and Medzhitov, 2002).
Toll-like Receptors
TLRs are a family of evolutionarily conserved receptors that react to bacterial, viral, or fungal antigens or to endogenous factors released during cell injury. The capacity to recognize a variety of common microbial antigens and endogenous factors indicates that a primary function of TLRs is to act as sentinel receptors to alert the innate immune system to infection or tissue damage (Takeda et al., 2003). To date, the TLR family is composed of 10 members in humans (TLR1–TLR10) and 12 in the mouse (TLR1-TLR9 and TLR11-TLR13). All TLRs are characterized as type I transmembrane receptors with an extracellular leucine-rich repeat domain and a cytoplasmic tail with high similarity to the type 1 interleukin-1 (IL-1) receptor. The leucine-rich repeat domains of TLRs bind different microbial components (PAMPs), including bacterial cell wall molecules such as lipopolysaccharide and peptidoglycan, proteins (e.g., flagellin), as well as double- or single-stranded RNA of viruses or unmethylated CpG DNA. Ligation of TLRs leads to activation of a protease cascade inducing transcription factors such as nuclear factor (NF)-κB and interferon regulatory factors 3/7, followed by enhanced transcription of antimicrobial peptides, cytokines, chemokines, and co-stimulatory molecules. As such, TLRs function as critical mediators between innate and adaptive immune responses.
The recognition of C. albicans and other medically important fungi by cells of the immune system have been recently reviewed extensively (Roeder et al., 2004; Netea et al., 2006a); thus, only a short overview will be presented here. The major PRRs and their putative ligands derived from C. albicans are illustrated in Fig. 2. Most studies so far have addressed the 3 major fungal pathogens, C. albicans, Aspergillus fumigatus, and Cryptococcus neoformans, and only few reports dealt with specific fungal PAMPs and their involvement in innate immunity (Roeder et al., 2004). While the major focus of antifungal innate immunity has been on systemic Candida infections, less is known about the function of TLRs during localized fungal infections. From the study of fungal infections in knock-out mice deficient in either TLRs or TLR-associated adaptor molecules, it appears that specific TLRs such as TLR2, TLR4 (Roeder et al., 2004; Netea et al., 2008a; Gil and Gozalbo, 2009), TLR6 (Netea et al., 2008b), and TLR9 (Bellocchio et al., 2004; van de Veerdonk et al., 2008; Miyazato et al., 2009) play differential roles in the activation of the various arms of the innate immune response. Deletion of the intracellular adaptor MyD88 renders mice highly susceptible to infections with fungi, although the role of the individual TLRs has not been established decisively in all cases and is often controversial. Different research groups have demonstrated divergent roles for TLR2 and TLR4, and their importance in the control of C. albicans and C. neoformans infections is still unclear.
Figure 2.
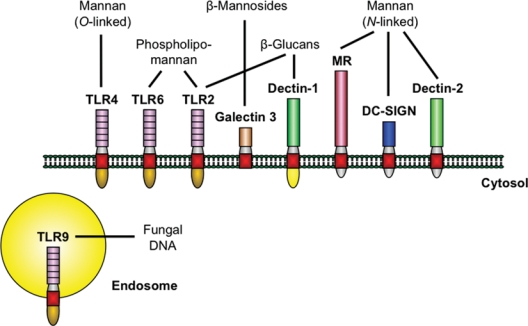
The major PRRs involved in the recognition of specific C. albicans PAMPs. Stimulation of host response by C. albicans at the cell membrane is mediated by a limited number of PRRs from the TLR and CLR families. Upon activation, the receptors trigger common adaptor molecules, intracellular pathways, and transcription factors (not shown). However, the specificity of the host response is maintained by the different repertoire of receptors stimulated by certain fungal PAMPs, as well as by the complex interactions between pathways. The depicted PRRs are predominantly expressed on cells of the myeloid lineage. Oral epithelial cells have been shown to express functional TLR2, TLR4, TLR6, and TLR9, emphasizing the importance of TLRs in the interaction of C. albicans and oral epithelium.
C-type Lectin-receptor Receptors
The CLRs are a large superfamily of proteins characterized by C-type lectin-like domains (Zelensky and Gready, 2005). Several extracellular and transmembrane CLRs— including the mannose receptor (MR), Dectin-1, Dectin-2, dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN), and macrophage-inducible C-type lectin (Mincle)—are involved in antifungal immunity, although their roles have not been completely understood. Importantly, these receptors mediate fungal binding, uptake, and killing and also contribute to the initiation and/or modulation of the immune response to these organisms (Netea et al., 2008a; Willment and Brown, 2008).
The MR (CD206) is a prototypical type I (group VI) transmembrane protein and is mainly expressed by macrophages, as well as by DCs (Taylor et al., 2005b). Several organisms, including C. albicans, C. neoformans, Pneumocystis carinii, and other pathogens such as bacteria and viruses are recognized by the MR (Taylor et al., 2005a; McKenzie et al., 2007). The receptor binds mannose, fucose, N-acetylglucosamine, and glucose. After carbohydrate recognition, the receptor mediates internalization of pathogens by phagocytosis, thereby leading to intracellular killing. Although the MR lacks classic signaling motifs, it mediates a variety of cellular responses, including induction of NF-κB activation and the production of numerous defensive cytokines, including IL-12, IL-8, IL-1ß, IL-6, IL-17 and granulocyte-macrophage colony-stimulating factor (GM-CSF), and can also enhance MR shedding (Zhang et al., 2004; Taylor et al., 2005a; Netea et al., 2006b; Tachado et al., 2007; van de Veerdonk et al., 2009b). The receptor has also been implicated in the phagocytosis of fungi, although its exact role in this process has not been fully defined (Le Cabec et al., 2005; Taylor et al., 2005a). Furthermore, the MR may perform an immunosuppressive function in its ability to inhibit the production of inflammatory cytokines when certain fungal pathogens are recognized (Zhang et al., 2005). In MR-deficient mice, fungal infection did not result in increased susceptibility, although minor changes were observed in fungal burdens on infection with C. albicans (Lee et al., 2003). More recently, it has been shown that the MR induces IL-17 production by Candida mannan in the absence of mitogenic stimulation, even more potently than Gram-negative bacteria (van de Veerdonk et al., 2009b).
Dectin-1 is a type II transmembrane receptor and belongs to the natural killer cell receptor-like CLRs (Brown, 2006). The extracellular carbohydrate recognition domain (CRD) selectively binds β-glucan polymers, a major component of yeast and mycobacterial cell walls, and mediates the phagocytosis of zymosan particles and intact yeast (Herre et al., 2004; Underhill et al., 2005). Dectin-1 also synergizes with TLR2 and TLR4-induced signals, inducing tumor necrosis factor (TNF), IL-10, transforming growth factor-β, and maturation of DCs (Brown et al., 2003; Gantner et al., 2003; Dillon et al., 2006). The cell wall of C. albicans consists of large amounts of β-glucan covered by a layer of mannans that prevent direct exposure to dectin-1. β-Glucan is exposed and can bind dectin-1 only at the level of the budding scars of blastoconidia, where the integrity of the cell wall is disrupted. Hyphae also have a thin layer of β-glucan in their cell wall that can be detected by antibodies against β-glucan (Torosantucci et al., 2009); however, this type of β-glucan does not seem to bind dectin-1 (Gantner et al., 2005; Netea et al., 2008a). Dectin-1−/− mice are more susceptible to disseminated candidiasis (Taylor et al., 2007), and this observation is strengthened by higher susceptibility to disseminated candidiasis in mice deficient for caspase recruitment domain (CARD) 9, a key transducer of dectin-1 signaling. (Gross et al., 2006). However, a different study could not confirm the nonredundant role of dectin-1 in C. albicans infection (Saijo et al., 2007).
Dectin-2 is a type II transmembrane receptor with a single CRD and a stalk region, but without an intracellular signaling motif (Willment and Brown, 2008; Graham and Brown, 2009). Dectin-2 is expressed on macrophages and dendritic cells and is up-regulated when these cells are stimulated with particles containing high-mannose structures such as C. albicans hyphae (Taylor et al., 2005c; McGreal et al., 2006). Dectin-2 signaling occurs in collaboration with the Fc receptor γ chain, activates NF-κB, and can induce IL-1Ra and TNF production (Sato et al., 2006).
DC-SIGN (CD209) is a type II transmembrane receptor with a single CRD and is expressed on dendritic cells and endothelium (Willment and Brown, 2008). The role of DC-SIGN in phagocytosis is questionable, whereas induction of endocytosis and uptake of pathogens has been well-documented (Taylor et al., 2004). The uptake of C. albicans by human DCs depends on the binding of N-linked mannan to DC-SIGN and induces IL-6 production (Cambi et al., 2003, 2008).
Mincle (also called Clec4e and Clecsf9) is a type II transmembrane protein with a single CRD, a short stalk region, and an intracellular region that signals by association with Fc receptor γ adaptor (Graham and Brown, 2009). Both human and mouse Mincle have been recently shown to bind C. albicans and contribute to cytokine stimulation (Bugarcic et al., 2008), and Mincle-deficient mice were more susceptible to systemic candidiasis (Wells et al., 2008).
NOD-like Receptors
NLRs are a family of intracellular immune receptors characterized by leucine-rich repeats and a nucleotide-binding domain. Like TLRs, NLRs recognize microbial products, as well as other intracellular danger signals, thus triggering host defense pathways through the activation of the NF-κB response and inflammatory caspases (Martinon et al., 2009). Several members of the NLR family, including NLRP3 (also known as NALP3 and cryopyrin), form large multiprotein complexes, termed inflammasome, which in turn activate caspase-1, leading to the processing and secretion of IL-1β and IL-18 (Bryant and Fitzgerald, 2009). Recent reports link IL-1β production induced by C. albicans to the NLRP3 inflammasome (Gross et al., 2009; Hise et al., 2009; Joly et al., 2009; Kumar et al., 2009; van de Veerdonk et al., 2009a). Noteworthy, TLR2, Dectin-1, and NLRP3 were crucial for the protection from dissemination of C. albicans in a murine model of oral mucosal infection (Hise et al., 2009). However, at present, the extent to which oral epithelial cells contribute to the observed defense mechanism is unknown.
Expression of PRRs in Oral Epithelial Cells
Most TLRs are expressed constitutively in oral epithelial cells, healthy epithelial tissue (Mahanonda and Pichyangkul, 2007; Beklen et al., 2008), and oral mucosa biopsies from patients with oral candidiasis (Ali et al., 2008). Previously, using a model of oral reconstituted human epithelium (RHE), we studied several different aspects of host-Candida interactions (Schaller et al., 1998, 1999, 2002, 2004, 2006; Weindl et al., 2007; Schaller and Weindl, 2009). Although the model consists of transformed cells (cell line TR146 derived from a carcinoma of the oral epithelium) (Rupniak et al., 1985), all natural major markers of the epithelial basement membrane and of epithelial differentiation are expressed. More important, despite the artificiality of the model, it behaves like human in vivo epithelium when treated with pathogens and pharmacologically active agents, respectively (Schaller and Weindl, 2009), and it mimics the clinical setting of C. albicans infections in the oral cavity (Wilson et al., 2009). Analysis of the oral RHE by real-time RT-PCR demonstrated a high degree of similarity in TLR expression profiles between the oral RHE and buccal epithelial samples isolated from healthy individuals (Weindl et al., 2007). In the oral RHE model, all TLRs except TLR7 at a low level are constitutively expressed. Similarly, in samples from healthy individuals, all TLRs except TLR5 and TLR7 are detected. The most commonly expressed TLR genes in vivo are TLR1, TLR2, TLR4, and TLR8, with TLR1 being the most highly expressed gene. Increased expression of TLR2 and TLR4 has previously been observed in inflamed gingival epithelial tissues (Sugawara et al., 2006). The immunohistochemical expression of 9 classes of TLRs (TLR1 to TLR9) was demonstrated in a series of sections from chronic hyperplastic candidiasis, leukoplakia, and healthy tissue (Ali et al., 2008).
In contrast, much less is known about the expression of CLRs in the oral cavity. Currently, there are no data published on the role of MR in localized Candida infections. The receptor is expressed in keratinocytes (Szolnoky et al., 2001) and oral epithelial cells (Wagener et al. and Weig et al., unpublished observations). However, gene expression analysis in the oral RHE model showed no significant differences upon infection with C. albicans (Weig et al., unpublished observations). In oral epithelial cells, MR blocking did not alter cytokine secretion levels of IL-6, IL-8, and GM-CSF upon stimulation with Candida cell wall components (Wagener et al., unpublished observations).
The function of dectin-1 in mucosal candidiasis has not yet been established, but several facts suggest that dectin-1 might play a crucial role in the mucosal immunity against C. albicans, at least in the intestine. Myeloid lineage cells in the intestinal tract express dectin-1, and the outgrowth of C. albicans in the digestive tract from dectin-1−/− mice was disproportionately high, leading to occlusion and contributing to increased mortality (Reid et al., 2004; Taylor et al., 2002, 2007). Furthermore, dectin-1 is paramount for IL-17 induction by C. albicans (Leibundgut-Landmann et al., 2007; Osorio et al., 2008), and patients with impaired IL-17 production caused by STAT3 (signal transducer and activator of transcription 3) mutations (hyper-IgE syndrome) and CMC have recurrent Candida infections (Eyerich et al., 2008; Ma et al., 2008; Milner et al., 2008). Previous studies have failed to demonstrate Dectin-1 expression in epithelial cells from the gastrointestinal tract (Rice et al., 2005), lung (Evans et al., 2005; Lee et al., 2009b), and gingiva (Laube et al., 2008). However, epidermal keratinocytes appear to express functional Dectin-1 (Lee et al., 2009a), and its expression can also be induced by mycobacteria in airway epithelial cells (Lee et al., 2009b). We observed that Dectin-1 is expressed in the oral RHE, but gene expression is not inducible by C. albicans, and Dectin-1 ligands did not stimulate cytokine secretion (Wagener et al. and Weig et al., unpublished observations). This suggests that Dectin-1 plays only a minor role in oral epithelial cells, and that other PRRs might contribute to the interaction between the epithelial cells and C. albicans PAMPs. As for Dectin-2, DC-SIGN, and Mincle, these receptors seem not to be expressed in oral epithelial cells (Weindl and Schaller, unpublished observations).
Oral epithelial cells express the members of the NLR family NLRC1 (NOD1) and NLRC2 (NOD2), and stimulation with synthetic ligands strongly increased the expression of antimicrobial molecules, while pro-inflammatory cytokines were not induced (Uehara et al., 2005, 2007; Sugawara et al., 2006; Uehara and Takada, 2008). Although C. albicans is not recognized by NLRC1 and NLRC2 (van der Graaf et al., 2006), another NLR member, NLRP3, might have an important function in the host defense against mucosal Candida infections (Hise et al., 2009). Interestingly, NLRP3 is strongly expressed by keratinocytes in non-keratinizing epithelia such as the oral cavity and esophagus (Kummer et al., 2007). The potential role of NLRP3 in oral epithelial cells is further supported by studies showing increased IL-1β and IL-18 levels upon stimulation with C. albicans (Rouabhia et al., 2002; Mostefaoui et al., 2004; Schaller et al., 2004; Tardif et al., 2004; Weindl et al., 2007).
Regulation of Innate Immune Pathways BY C. albicans
C. albicans is a harmless colonizer of mucosal surfaces in healthy individuals. During the period of colonization, extensive fungal growth is limited through the release of antimicrobial peptides from epithelial cells, or due to the existence of other bacteria of the microbial flora. In this stage of colonization, without clinical symptoms and signs of inflammation, neither the facultative pathogen nor the host might induce a (TLR-mediated) inflammatory cytokine response. However, when these conditions become unbalanced—for instance, due to antibiotic therapy or immunosuppression—superficial or even systemic infections may occur.
Although oral epithelial cells express TLRs, no studies have yet demonstrated TLR up-regulation upon stimulation with C. albicans. Heat-killed C. albicans cells failed to modulate epithelial TLR expression (Pivarcsi et al., 2003). Similarly, in an infection model of oral candidiasis, both heat-killed and viable C. albicans cells were unable to up-regulate epithelial TLR expression, despite the fungus causing clear signs of mucosal damage (Weindl et al., 2007). However, C. famata, which accounts for 1-3% of candidiasis, has been shown to induce slight TLR mRNA expression in human gingival epithelial cells (Bahri et al., 2010). Specifically, the expression of TLR2, 4, and 6 was up-regulated, although only minimally. With regard to commensal organisms, it has been suggested that rapid responsiveness by epithelial TLRs may create the danger of an immune overreaction (Strober, 2004). Thus, one possible explanation for the lack of direct TLR up-regulation by C. albicans may be because the fungus is usually a harmless colonizer of oral mucosal surfaces in approximately 40% of healthy individuals (Arendorf and Walker, 1979) and may even actively down-regulate epithelial responses by unknown mechanisms (Weindl and Schaller, unpublished data). In addition, during the carrier state, it would serve little purpose for the host to activate a TLR-mediated inflammatory response when it is not required. However, during oral infection with Candida, many cytokines are secreted by oral epithelial cells, which maintain a central role in the protection against fungal organisms (Dongari-Bagtzoglou and Fidel, 2005). In general, pro-inflammatory cytokines (IL-1α, IL-1β, IL-6, IL-8, TNF, GM-CSF, and others) regulate leukocyte trafficking (Eversole et al., 1997) and/or activate a strong antifungal response by these cells (Dongari-Bagtzoglou et al., 2005). In addition, oral epithelial cells are capable of inducing antimicrobial peptides, such as defensins, cathelicidins, and histatins (reviewed in Diamond et al., 2008), which control C. albicans growth and infection. Among these peptides, human β-defensin-2 (hBD-2), hBD-3 (Joly et al., 2004; Feng et al., 2005; Schneider et al., 2005), LL-37 (Turner et al., 1998), and histatin-5 (Oppenheim et al., 1988) exhibit potent anti-candidal properties.
The addition of PMNs to an in vitro model of oral candidiasis enhanced a T-helper (Th)1-type immune response (interferon-γ, TNF), down-regulated the expression of the Th2-type cytokine IL-10, and was associated with protection against Candida-induced tissue damage (Schaller et al., 2004). PMNs could protect the epithelium from C. albicans–induced cell injury via a process that was independent of phagocytosis, PMN transmigration, or physical PMN-epithelial cell contact. Interestingly, the immunological cross-talk between C. albicans-infected oral epithelium and PMNs causes PMN-mediated up-regulation of epithelial TLR4 (Weindl et al., 2007). Furthermore, epithelial TLR4 is directly responsible for protecting the mucosal surface from fungal invasion and cell injury. Cytokines seem not to have an essential function in direct host defense against invading fungi, even in the presence of PMNs. Rather, cytokines are crucial for the activation of PMNs and/or are released from the PMNs, which, in turn, results in up-regulation of epithelial TLR4 and protection from fungal invasion (see Fig. 1). Among the cytokines, TNF showed the most potent effect, which confirms the important role of this cytokine in host defense against opportunistic fungal infections (Filler et al., 2005). Absence of this cytokine strongly impairs neutrophil recruitment and effective phagocytosis of C. albicans (Netea et al., 1999). In an in vitro model of esophageal candidiasis, co-incubation of PMNs with C. albicans leads to a significant up-regulation of hBD-2 and hBD-3 in esophageal cells compared with effects of PMNs or C. albicans alone (Steubesand et al., 2009). Thus, increased PMN-dependent production of antimicrobial peptides by epithelial cells could contribute to the protective effect and further underlines the important role for PMNs in clearance of experimental oral candidiasis.
At present, it is unknown which fungal cell wall structures are recognized in oral epithelial cells. Both TLR2 and TLR4 have been implicated in the recognition of C. albicans by immune cells (Roeder et al., 2004). Recognition of surface N-linked mannosyl residues is mediated by mannose receptor and O-linked mannosyl residues by TLR4, phospholipomannan is detected by TLR2, and β-glucan structures are recognized by TLR2 in collaboration with Dectin-1 (Gantner et al., 2003; Jouault et al., 2003; Netea et al., 2006b). In epidermal keratinocytes, C. albicans-native phospholipomannan may contribute to the inflammatory responses of cutaneous candidiasis via TLR2-NF-κB and p38 mitogen-activated protein kinase signaling pathways (Li et al., 2009). However, studies on oral epithelial cells are needed to address the recognition of fungal cell wall structures by PRRs.
Similarly, epithelial cytokine production or expression of antimicrobial peptides, induced by C. albicans, has not yet been associated with specific PRRs. Various Candida strains strongly induce GM-CSF in human oral epithelial cells and three-dimensional models (Dongari-Bagtzoglou and Kashleva, 2003; Weindl et al., 2007; Li and Dongari-Bagtzoglou, 2009). GM-CSF is a potent cytokine involved in the enhancement of proliferation, activation, and fungicidal activity of immune cells. Analysis of the data indicates that TLR4 is not involved in Candida-induced GM-CSF and IL-8 production in epithelial cells (Weindl et al., 2007; Li and Dongari-Bagtzoglou, 2009). In this regard, the adhesion receptor CDw17 (lactosylceramide) might be responsible, at least partially, for GM-CSF activation mediated by NF-κB (Li and Dongari-Bagtzoglou, 2009). Studies indicate that CDw17 may function as an alternate β-glucan receptor in Pneumocystis carinii; however, it has been suggested that secretion of GM-CSF is independent of the cell wall component ß-glucan in C. glabrata.
Recently, specific human gene mutations and polymorphisms have been linked to signal pathways resulting in susceptibility for C. albicans. STAT3 (signal transducer and activator of transcription 3) mutations identified in patients with hyper-IgE syndrome have been attributed to a defective IL-17 production and a diminished Th17 response, resulting in recurrent mucosal Candida infections (Eyerich et al., 2008; Ma et al., 2008; Milner et al., 2008). Similarly, deficiency in Dectin-1 signaling pathways has been linked to mucocutaneous candidiasis (Ferwerda et al., 2009; Glocker et al., 2009).
A recent study reported a Dutch family with impaired in vitro responses to β-glucan, leading to infections of nails and mucosa (Ferwerda et al., 2009). The study demonstrated that the production of IL-6 and IL-17 was impaired only in response to the dectin-1 ligand, β-glucan. The specific stop codon the authors identified in dectin-1 is remarkably common in some parts of Africa and Europe (allele frequency, 3 to 7%), suggesting that unknown factors maintain it in populations. Another study identified an extended Iranian family with predominantly mucocutaneous, but also fatal, candidiasis of the central nervous system, caused by mutations in the adaptor protein CARD9, impairing both dectin-1 signaling and Th17 production (Glocker et al., 2009). CARD9 also directs a variety of other cell-surface and intracellular signals, including the p38 mitogen-activated protein kinase and Jun N-terminal kinase pathways, possibly accounting for its greater clinical severity as compared with isolated dectin-1 deficiency (Ferwerda et al., 2009). The CARD9 mutation appears to be rare, and its rarity is commensurate with its severity. Both studies, however, need to be interpreted with caution, since both patient groups show mucocutaneous manifestations, whereas the functional studies were performed on leukocytes. It remains to be proven whether the key mechanisms in these cases of severe candidiasis consist of impaired dectin-1 signaling at the epithelial level or impaired leukocyte activation, mediated through cytokines such as IL-17.
Oral Mucosal T-Cell Responses to C. albicans
Activation of the innate immune system by C. albicans induces the secretion of a variety of pro-inflammatory cytokines and the expression of co-stimulatory molecules. It is generally accepted that induction of a Th1-type cellular response is crucial for the defense against C. albicans (Fidel et al., 1997; Romani, 1999; Schaller et al., 2004). In contrast, a Th2 cellular response is considered non-protective, since it induces class-switch to non-opsonizing antibody subclasses and IgE (Savolainen et al., 1996; Clemons and Stevens, 2001). Investigation of the role of Th17 in mediating the immune response has shown that Th17 memory cells are induced by Candida hyphae (Acosta-Rodriguez et al., 2007; Zhou et al., 2008), and in a murine model, IL-17AR knock-out mice had an increased susceptibility to systemic (Huang et al., 2004) and oropharyngeal candidiasis (OPC) (Conti et al., 2009). In contrast, deleterious effects of IL-17 inflammatory activities have also been demonstrated (De Luca et al., 2007; Zelante et al., 2007; Bozza et al., 2008). Patients with impaired IL-17 production suffer from mucosal C. albicans infections in hyper-IgE syndrome and CMC (Eyerich et al., 2008; Ma et al., 2008; Milner et al., 2008). In contrast to Th cells, regulatory T-cells suppress inflammatory responses in disseminated C. albicans, resulting in higher susceptibility in mice (Netea et al., 2004; Sutmuller et al., 2006). However, the tolerance-inducing effects of regulatory T-cells seem to be beneficial at mucosal sites (De Luca et al., 2007; Vignali et al., 2008).
Th17 cells are a distinct lineage from Th1 and Th2 cells, characterized by the release of IL-17A and IL-17F, IL-22 and IL-26. Receptors for IL-17A and IL-17F (Il-17Ra and IL-17Rc) are present in several cell types, including antigen-presenting cells and epithelial cells (Xie et al., 2000; Gaffen, 2009). In contrast, receptors for IL-22 and IL-26 appear to be localized to the epithelium (Xie et al., 2000; Sheikh et al., 2004; Wolk et al., 2004). Very little is known about the role of IL-26 during mucosal infection, because rodents do not express this cytokine. During colonization of the oral cavity with C. albicans, IL-17 receptor signaling is essential for defense (Conti et al., 2009). Interestingly, Th17-deficient (IL-23p19-/-) and IL-17R-deficient (IL-17RA-/-) mice experienced severe OPC, whereas Th1-deficient (IL-12p35-/-) mice showed low fungal burdens and no apparent sign of disease. Furthermore, neutrophil recruitment was impaired in IL-23p19-/- and IL-17RA-/-, but not IL-12-/-, mice, and T-cell receptor αβ cells were more important than γδ cells. In contrast, mice deficient in the Th17 cytokine IL-22 were only mildly susceptible to OPC, indicating that IL-17 rather than IL-22 is crucial in defense against oral candidiasis. Gene profiling of oral mucosal tissue showed strong induction of Th17 signature genes, including beta defensin-3 and CXC chemokines. hBD-3 has candidacidal activity in vitro (Vylkova et al., 2006), and saliva from wild-type mice, but not IL-17Ra-/- mice, has candidacidal activity, indicating that IL-17 also controls C. albicans proliferation by promoting secretion of antimicrobial peptides (Conti et al., 2009). However, more work is needed to understand whether Th17 responses also govern the response to oral candidiasis in humans, because it is unclear whether mouse and human diseases have the same etiology. In humans, OPC has diverse etiologies ranging from antimicrobial use, to immune dysregulation associated with advanced HIV infection, to mutations in autoimmune regulator genes. Patients with CMC show differences in cytokine production, including IL-23, depending on the etiology of the disorder (Ryan et al., 2008). Although HIV infection and host genetics are likely to be important variables in Th17 expression, environmental factors, particularly those that affect the microbiota, could also influence Th cell polarization. Although microbe-specific motifs could induce Th polarization, common microbial PAMPs that induce Th1 or Th17 may also be present. Thus, a diverse group of microbial ligands could induce different responses, and immunity toward pathogens could be less specific than previously thought.
Analysis of the data together indicates optimal protection against (chronic) mucosal Candida infections by Th1, Th17, and regulatory T-cells. An effective Th1 and antibody (humoral) response are crucial for defense against disseminated Candida infections. In the case of localized Candida infections, however, more work is needed to decipher their relative contributions.
Conclusions
The above-mentioned studies clearly demonstrate that PRRs contribute to the signal transduction induced by C. albicans, to the induction of inflammation, and to the activation of adaptive immunity. The simultaneous activation of multiple PRRs by one fungal pathogen endows the immune system with a broad range of possibilities for a specific and effective immune response. However, the contribution of PRRs in mucosal tissues has only recently been studied. Among PRRs, TLRs play a crucial role in detecting danger signals and triggering innate immune responses that prevent pathogens from invading the host and spreading systemically. Other innate immune receptors expressed at the mucosal surfaces are likely to participate in such defense mechanisms as shown for dectin-1. PRRs are also involved in mediating epithelial homeostatic functions facilitating mucosal tissue repair and remodeling following inflammation. How pathogens and commensals stimulate distinct mucosal responses while expressing similar molecular patterns is only now beginning to be understood. On other tissues, such as the intestine, it has been shown that commensals are able to modulate pro-inflammatory responses by interfering with TLR signaling cascades. Pathogens use additional virulence factors that participate in the onset of pro-inflammatory responses. Thus, the wide variety of responses to micro-organisms may be explained by the diversity of co-receptors that can be engaged to induce functional innate immune signaling.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Sch 897/1-3, Sch 897/3-1, Sch 897/3-2, graduate college 685) and by NIDCR grant R01 DE017514-01.
References
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, et al. (2007). Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639-646 [DOI] [PubMed] [Google Scholar]
- Ali A, Natah S, Konttinen Y. (2008). Differential expression of Toll-like receptors in chronic hyperplastic candidosis. Oral Microbiol Immunol 23:299-307 [DOI] [PubMed] [Google Scholar]
- Arendorf TM, Walker DM. (1979). Oral candidal populations in health and disease. Br Dent J 147:267-272 [DOI] [PubMed] [Google Scholar]
- Bahri R, Saidane-Mosbahi D, Rouabhia M. (2010). Candida famata modulates Toll-like receptor, beta-defensin, and proinflammatory cytokine expression by normal human epithelial cells. J Cell Physiol 222:209-218 [DOI] [PubMed] [Google Scholar]
- Beklen A, Hukkanen M, Richardson R, Konttinen YT. (2008). Immuno histochemical localization of Toll-like receptors 1-10 in periodontitis. Oral Microbiol Immunol 23:425-431 [DOI] [PubMed] [Google Scholar]
- Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, et al. (2004). The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172:3059-3069 [DOI] [PubMed] [Google Scholar]
- Bozza S, Zelante T, Moretti S, Bonifazi P, DeLuca A, D’Angelo C, et al. (2008). Lack of Toll IL-1R8 exacerbates Th17 cell responses in fungal infection. J Immunol 180:4022-4031 [DOI] [PubMed] [Google Scholar]
- Brown GD. (2006). Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol 6:33-43 [DOI] [PubMed] [Google Scholar]
- Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. (2003). Dectin-1 mediates the biological effects of beta-glucans. J Exp Med 197:1119-1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant C, Fitzgerald KA. (2009). Molecular mechanisms involved in inflammasome activation. Trends Cell Biol 19:455-464 [DOI] [PubMed] [Google Scholar]
- Bugarcic A, Hitchens K, Beckhouse AG, Wells CA, Ashman RB, Blanchard H. (2008). Human and mouse macrophage-inducible C-type lectin (Mincle) bind Candida albicans. Glycobiology 18:679-685 [DOI] [PubMed] [Google Scholar]
- Calderone RA, Fonzi WA. (2001). Virulence factors of Candida albicans. Trends Microbiol 9:327-335 [DOI] [PubMed] [Google Scholar]
- Cambi A, Gijzen K, de Vries JM, Torensma R, Joosten B, Adema GJ, et al. (2003). The C-type lectin DC-SIGN (CD209) is an antigen-uptake receptor for Candida albicans on dendritic cells. Eur J Immunol 33:532-538 [DOI] [PubMed] [Google Scholar]
- Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, et al. (2008). Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J Biol Chem 283:20590-20599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons KV, Stevens DA. (2001). Overview of host defense mechanisms in systemic mycoses and the basis for immunotherapy. Semin Respir Infect 16:60-66 [DOI] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206:299-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler CW, Jotwani R. (2006). Dendritic cells at the oral mucosal interface [review]. J Dent Res 85:678-689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Montagnoli C, Zelante T, Bonifazi P, Bozza S, Moretti S, et al. (2007). Functional yet balanced reactivity to Candida albicans requires TRIF, MyD88, and IDO-dependent inhibition of Rorc. J Immunol 179:5999-6008 [DOI] [PubMed] [Google Scholar]
- Diamond G, Beckloff N, Ryan LK. (2008). Host defense peptides in the oral cavity and the lung: similarities and differences [review]. J Dent Res 87:915-927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon S, Agrawal S, Banerjee K, Letterio J, Denning TL, Oswald-Richter K, et al. (2006). Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest 116:916-928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Kashleva H. (2003). Granulocyte-macrophage colony-stimulating factor responses of oral epithelial cells to Candida albicans. Oral Microbiol Immunol 18:165-170 [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Fidel PL., Jr (2005). The host cytokine responses and protective immunity in oropharyngeal candidiasis [review]. J Dent Res 84:966-977 [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Villar CC, Kashleva H. (2005). Candida albicans-infected oral epithelial cells augment the anti-fungal activity of human neutrophils in vitro. Med Mycol 43:545-549 [DOI] [PubMed] [Google Scholar]
- Evans SE, Hahn PY, McCann F, Kottom TJ, Pavlovic ZV, Limper AH. (2005). Pneumocystis cell wall beta-glucans stimulate alveolar epithelial cell chemokine generation through nuclear factor-kappaB-dependent mechanisms. Am J Respir Cell Mol Biol 32:490-497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eversole LR, Reichart PA, Ficarra G, Schmidt-Westhausen A, Romagnoli P, Pimpinelli N. (1997). Oral keratinocyte immune responses in HIV-associated candidiasis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 84:372-380 [DOI] [PubMed] [Google Scholar]
- Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H, et al. (2008). Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol 128:2640-2645 [DOI] [PubMed] [Google Scholar]
- Feng Z, Jiang B, Chandra J, Ghannoum M, Nelson S, Weinberg A. (2005). Human beta-defensins: differential activity against candidal species and regulation by Candida albicans. J Dent Res 84:445-450 [DOI] [PubMed] [Google Scholar]
- Ferwerda B, Ferwerda G, Plantinga TS, Willment JA, van Spriel AB, Venselaar H, et al. (2009). Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med 361:1760-1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidel PL, Jr, Ginsburg KA, Cutright JL, Wolf NA, Leaman D, Dunlap K, et al. (1997). Vaginal-associated immunity in women with recurrent vulvovaginal candidiasis: evidence for vaginal Th1-type responses following intravaginal challenge with Candida antigen. J Infect Dis 176:728-739 [DOI] [PubMed] [Google Scholar]
- Filler SG, Yeaman MR, Sheppard D. (2005). Tumor necrosis factor inhibition and invasive fungal infections. Clin Infect Dis 41(Suppl 3):208-212 [DOI] [PubMed] [Google Scholar]
- Gaffen SL. (2009). Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 9:556-567; erratum in Nat Rev Immunol 9:747, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. (2003). Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med 197:1107-1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, Underhill DM. (2005). Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J 24:1277-1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil ML, Gozalbo D. (2009). Role of Toll-like receptors in systemic Candida albicans infections. Front Biosci 14:570-582 [DOI] [PubMed] [Google Scholar]
- Glocker EO, Hennigs A, Nabavi M, Schaffer AA, Woellner C, Salzer U, et al. (2009). A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med 361:1727-1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham LM, Brown GD. (2009). The Dectin-2 family of C-type lectins in immunity and homeostasis. Cytokine 48:148-155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, et al. (2006). Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442:651-656 [DOI] [PubMed] [Google Scholar]
- Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, et al. (2009). Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459:433-436 [DOI] [PubMed] [Google Scholar]
- Herre J, Marshall AS, Caron E, Edwards AD, Williams DL, Schweighoffer E, et al. (2004). Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 104:4038-4045 [DOI] [PubMed] [Google Scholar]
- Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, et al. (2009). An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host Microbe 5:487-497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Na L, Fidel PL, Schwarzenberger P. (2004). Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190:624-631 [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. (2002). Innate immune recognition. Annu Rev Immunol 20:197-216 [DOI] [PubMed] [Google Scholar]
- Joly S, Maze C, McCray PB, Jr, Guthmiller JM. (2004). Human beta-defensins 2 and 3 demonstrate strain-selective activity against oral microorganisms. J Clin Microbiol 42:1024-1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly S, Ma N, Sadler JJ, Soll DR, Cassel SL, Sutterwala FS. (2009). Cutting edge: Candida albicans hyphae formation triggers activation of the Nlrp3 inflammasome. J Immunol 183:3578-3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouault T, Ibata-Ombetta S, Takeuchi O, Trinel PA, Sacchetti P, Lefebvre P, et al. (2003). Candida albicans phospholipomannan is sensed through Toll-like receptors. J Infect Dis 188:165-172 [DOI] [PubMed] [Google Scholar]
- Kumar H, Kumagai Y, Tsuchida T, Koenig PA, Satoh T, Guo Z, et al. (2009). Involvement of the NLRP3 inflammasome in innate and humoral adaptive immune responses to fungal beta-glucan. J Immunol 183:8061-8067 [DOI] [PubMed] [Google Scholar]
- Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, et al. (2007). Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem 55:443-452 [DOI] [PubMed] [Google Scholar]
- Laube DM, Dongari-Bagtzoglou A, Kashleva H, Eskdale J, Gallagher G, Diamond G. (2008). Differential regulation of innate immune response genes in gingival epithelial cells stimulated with Aggregatibacter actinomycetemcomitans. J Periodontal Res 43:116-123 [DOI] [PubMed] [Google Scholar]
- Le Cabec V, Emorine LJ, Toesca I, Cougoule C, Maridonneau-Parini I. (2005). The human macrophage mannose receptor is not a professional phagocytic receptor. J Leukoc Biol 77:934-943 [DOI] [PubMed] [Google Scholar]
- Lee HM, Shin DM, Choi DK, Lee ZW, Kim KH, Yuk JM, et al. (2009a). Innate immune responses to Mycobacterium ulcerans via Toll-like receptors and dectin-1 in human keratinocytes. Cell Microbiol 11:678-692 [DOI] [PubMed] [Google Scholar]
- Lee HM, Yuk JM, Shin DM, Jo EK. (2009b). Dectin-1 is inducible and plays an essential role for mycobacteria-induced innate immune responses in airway epithelial cells. J Clin Immunol 29:795-805 [DOI] [PubMed] [Google Scholar]
- Lee SJ, Zheng NY, Clavijo M, Nussenzweig MC. (2003). Normal host defense during systemic candidiasis in mannose receptor-deficient mice. Infect Immun 71:437-445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibundgut-Landmann S, Gross O, Robinson MJ, Osorio F, Slack EC, Tsoni SV, et al. (2007). Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 8:630-638 [DOI] [PubMed] [Google Scholar]
- Li L, Dongari-Bagtzoglou A. (2009). Epithelial GM-CSF induction by Candida glabrata. J Dent Res 88:746-751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Redding S, Dongari-Bagtzoglou A. (2007). Candida glabrata: an emerging oral opportunistic pathogen [review]. J Dent Res 86:204-215 [DOI] [PubMed] [Google Scholar]
- Li M, Chen Q, Shen Y, Liu W. (2009). Candida albicans phospholipomannan triggers inflammatory responses of human keratinocytes through Toll-like receptor 2. Exp Dermatol 18:603-610 [DOI] [PubMed] [Google Scholar]
- Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, et al. (2008). Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 205:1551-1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanonda R, Pichyangkul S. (2007). Toll-like receptors and their role in periodontal health and disease. Periodontol 2000 43:41-55 [DOI] [PubMed] [Google Scholar]
- Martinon F, Mayor A, Tschopp J. (2009). The inflammasomes: guardians of the body. Annu Rev Immunol 27:229-265 [DOI] [PubMed] [Google Scholar]
- McGreal EP, Rosas M, Brown GD, Zamze S, Wong SY, Gordon S, et al. (2006). The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology 16:422-430 [DOI] [PubMed] [Google Scholar]
- McKenzie EJ, Taylor PR, Stillion RJ, Lucas AD, Harris J, Gordon S, et al. (2007). Mannose receptor expression and function define a new population of murine dendritic cells. J Immunol 178:4975-4983 [DOI] [PubMed] [Google Scholar]
- Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, et al. (2008). Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452:773-776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazato A, Nakamura K, Yamamoto N, Mora-Montes HM, Tanaka M, Abe Y, et al. (2009). Toll-like receptor 9-dependent activation of myeloid dendritic cells by deoxynucleic acids from Candida albicans. Infect Immun 77:3056-3064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mostefaoui Y, Claveau I, Rouabhia M. (2004). In vitro analyses of tissue structure and interleukin-1beta expression and production by human oral mucosa in response to Candida albicans infections. Cytokine 25:162-171 [DOI] [PubMed] [Google Scholar]
- Netea MG, van Tits LJ, Curfs JH, Amiot F, Meis JF, van der Meer JW, et al. (1999). Increased susceptibility of TNF-alpha lymphotoxin-alpha double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J Immunol 163:1498-1505 [PubMed] [Google Scholar]
- Netea MG, Sutmuller R, Hermann C, Van der Graaf CA, Van der Meer JW, van Krieken JH, et al. (2004). Toll-like receptor 2 suppresses immunity against Candida albicans through induction of IL-10 and regulatory T cells. J Immunol 172:3712-3718 [DOI] [PubMed] [Google Scholar]
- Netea MG, Ferwerda G, van der Graaf CA, Van der Meer JW, Kullberg BJ. (2006a). Recognition of fungal pathogens by Toll-like receptors. Curr Pharm Des 12:4195-4201 [DOI] [PubMed] [Google Scholar]
- Netea MG, Gow NA, Munro CA, Bates S, Collins C, Ferwerda G, et al. (2006b). Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest 116:1642-1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Brown GD, Kullberg BJ, Gow NA. (2008a). An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 6:67-78 [DOI] [PubMed] [Google Scholar]
- Netea MG, van de Veerdonk F, Verschueren I, van der Meer JW, Kullberg BJ. (2008b). Role of TLR1 and TLR6 in the host defense against disseminated candidiasis. FEMS Immunol Med Microbiol 52:118-123 [DOI] [PubMed] [Google Scholar]
- Oppenheim FG, Xu T, McMillian FM, Levitz SM, Diamond RD, Offner GD, et al. (1988). Histatins, a novel family of histidine-rich proteins in human parotid secretion. Isolation, characterization, primary structure, and fungistatic effects on Candida albicans. J Biol Chem 263:7472-7477 [PubMed] [Google Scholar]
- Osorio F, Leibundgut-Landmann S, Lochner M, Lahl K, Sparwasser T, Eberl G, et al. (2008). DC activated via dectin-1 convert Treg into IL-17 producers. Eur J Immunol 38:3274-3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivarcsi A, Bodai L, Rethi B, Kenderessy-Szabo A, Koreck A, Szell M, et al. (2003). Expression and function of Toll-like receptors 2 and 4 in human keratinocytes. Int Immunol 15:721-730 [DOI] [PubMed] [Google Scholar]
- Reid DM, Montoya M, Taylor PR, Borrow P, Gordon S, Brown GD, et al. (2004). Expression of the beta-glucan receptor, Dectin-1, on murine leukocytes in situ correlates with its function in pathogen recognition and reveals potential roles in leukocyte interactions. J Leukoc Biol 76:86-94 [DOI] [PubMed] [Google Scholar]
- Rice PJ, Adams EL, Ozment-Skelton T, Gonzalez AJ, Goldman MP, Lockhart BE, et al. (2005). Oral delivery and gastrointestinal absorption of soluble glucans stimulate increased resistance to infectious challenge. J Pharmacol Exp Ther 314:1079-1086 [DOI] [PubMed] [Google Scholar]
- Roeder A, Kirschning CJ, Rupec RA, Schaller M, Weindl G, Korting HC. (2004). Toll-like receptors as key mediators in innate antifungal immunity. Med Mycol 42:485-498 [DOI] [PubMed] [Google Scholar]
- Romani L. (1999). Immunity to Candida albicans: Th1, Th2 cells and beyond. Curr Opin Microbiol 2:363-367 [DOI] [PubMed] [Google Scholar]
- Rouabhia M, Ross G, Pagé N, Chakir J. (2002). Interleukin-18 and gamma interferon production by oral epithelial cells in response to exposure to Candida albicans or lipopolysaccharide stimulation. Infect Immun 70:7073-7080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhnke M. (2006). Epidemiology of Candida albicans infections and role of non-Candida-albicans yeasts. Current drug targets 7:495-504 [DOI] [PubMed] [Google Scholar]
- Rupniak HT, Rowlatt C, Lane EB, Steele JG, Trejdosiewicz LK, Laskiewicz B, et al. (1985). Characteristics of four new human cell lines derived from squamous cell carcinomas of the head and neck. J Natl Cancer Inst 75:621-635 [PubMed] [Google Scholar]
- Ryan KR, Hong M, Arkwright PD, Gennery AR, Costigan C, Dominguez M, et al. (2008). Impaired dendritic cell maturation and cytokine production in patients with chronic mucocutanous candidiasis with or without APECED. Clin Exp Immunol 154:406-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo S, Fujikado N, Furuta T, Chung S, Kotaki H, Seki K, et al. (2007). Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol 8:39-46 [DOI] [PubMed] [Google Scholar]
- Samaranayake LP, Keung Leung W, Jin L. (2009). Oral mucosal fungal infections. Periodontol 2000 49:39-59 [DOI] [PubMed] [Google Scholar]
- Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, et al. (2006). Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J Biol Chem 281:38854-38866 [DOI] [PubMed] [Google Scholar]
- Savolainen J, Rantala A, Nermes M, Lehtonen L, Viander M. (1996). Enhanced IgE response to Candida albicans in postoperative invasive candidiasis. Clin Exp Allergy 26:452-460 [PubMed] [Google Scholar]
- Schaller M, Weindl G. (2009). Models of oral and vaginal candidiasis based on in vitro reconstituted human epithelia for the study of host-pathogen interactions. Methods Mol Biol 470:327-345 [DOI] [PubMed] [Google Scholar]
- Schaller M, Schäfer W, Korting HC, Hube B. (1998). Differential expression of secreted aspartyl proteinases in a model of human oral candidosis and in patient samples from the oral cavity. Mol Microbiol 29:605-615 [DOI] [PubMed] [Google Scholar]
- Schaller M, Korting HC, Schäfer W, Bastert J, Chen W, Hube B. (1999). Secreted aspartic proteinase (Sap) activity contributes to tissue damage in a model of human oral candidosis. Mol Microbiol 34:169-180 [DOI] [PubMed] [Google Scholar]
- Schaller M, Mailhammer R, Grassl G, Sander CA, Hube B, Korting HC. (2002). Infection of human oral epithelia with Candida species induces cytokine expression correlated to the degree of virulence. J Invest Dermatol 118:652-657 [DOI] [PubMed] [Google Scholar]
- Schaller M, Boeld U, Oberbauer S, Hamm G, Hube B, Korting HC. (2004). Polymorphonuclear leukocytes (PMNs) induce protective Th1-type cytokine epithelial responses in an in vitro model of oral candidosis. Microbiology 150(Pt 9):2807-2813 [DOI] [PubMed] [Google Scholar]
- Schaller M, Borelli C, Korting HC, Hube B. (2005). Hydrolytic enzymes as virulence factors of Candida albicans. Mycoses 48:365-377 [DOI] [PubMed] [Google Scholar]
- Schaller M, Zakikhany K, Naglik JR, Weindl G, Hube B. (2006). Models of oral and vaginal candidiasis based on in vitro reconstituted human epithelia. Nat Protoc 1:2767-2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JJ, Unholzer A, Schaller M, Schäfer-Korting M, Korting HC. (2005). Human defensins. J Mol Med 83:587-595 [DOI] [PubMed] [Google Scholar]
- Sheikh F, Baurin VV, Lewis-Antes A, Shah NK, Smirnov SV, Anantha S, et al. (2004). Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J Immunol 172:2006-2010 [DOI] [PubMed] [Google Scholar]
- Steubesand N, Kiehne K, Brunke G, Pahl R, Reiss K, Herzig KH, et al. (2009). The expression of the beta-defensins hBD-2 and hBD-3 is differentially regulated by NF-kappaB and MAPK/AP-1 pathways in an in vitro model of Candida esophagitis. BMC Immunol 10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober W. (2004). Epithelial cells pay a Toll for protection. Nat Med 10:898-900 [DOI] [PubMed] [Google Scholar]
- Sugawara Y, Uehara A, Fujimoto Y, Kusumoto S, Fukase K, Shibata K, et al. (2006). Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res 85:524-529 [DOI] [PubMed] [Google Scholar]
- Sutmuller RP, den Brok MH, Kramer M, Bennink EJ, Toonen LW, Kullberg BJ, et al. (2006). Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest 116:485-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szolnoky G, Bata-Csorgo Z, Kenderessy AS, Kiss M, Pivarcsi A, Novak Z, et al. (2001). A mannose-binding receptor is expressed on human keratinocytes and mediates killing of Candida albicans. J Invest Dermatol 117:205-213 [DOI] [PubMed] [Google Scholar]
- Tachado SD, Zhang J, Zhu J, Patel N, Cushion M, Koziel H. (2007). Pneumocystis-mediated IL-8 release by macrophages requires coexpression of mannose receptors and TLR2. J Leukoc Biol 81:205-211 [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. (2003). Toll-like receptors. Annu Rev Immunol 21:335-376 [DOI] [PubMed] [Google Scholar]
- Tardif F, Goulet JP, Zakrazewski A, Chauvin P, Rouabhia M. (2004). Involvement of interleukin-18 in the inflammatory response against oropharyngeal candidiasis. Med Sci Monit 10:BR239-BR249 [PubMed] [Google Scholar]
- Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, et al. (2002). The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol 169:3876-3882 [DOI] [PubMed] [Google Scholar]
- Taylor PR, Brown GD, Herre J, Williams DL, Willment JA, Gordon S. (2004). The role of SIGNR1 and the beta-glucan receptor (dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J Immunol 172:1157-1162 [DOI] [PubMed] [Google Scholar]
- Taylor PR, Gordon S, Martinez-Pomares L. (2005a). The mannose receptor: linking homeostasis and immunity through sugar recognition. Trends Immunol 26:104-110 [DOI] [PubMed] [Google Scholar]
- Taylor PR, Martinez-Pomares L, Stacey M, Lin H, Brown GD, Gordon S. (2005b). Macrophage receptors and immune recognition. Annu Rev Immunol 23:901-944 [DOI] [PubMed] [Google Scholar]
- Taylor PR, Reid DM, Heinsbroek SE, Brown GD, Gordon S, Wong SY. (2005c). Dectin-2 is predominantly myeloid restricted and exhibits unique activation-dependent expression on maturing inflammatory monocytes elicited in vivo. Eur J Immunol 35:2163-2174 [DOI] [PubMed] [Google Scholar]
- Taylor PR, Tsoni S, Willment J, Dennehy KM, Rosas M, Findon H, et al. (2007). Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol 8:31-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torosantucci A, Chiani P, Bromuro C, De Bernardis F, Palma AS, Liu Y, et al. (2009). Protection by anti-beta-glucan antibodies is associated with restricted beta-1,3 glucan binding specificity and inhibition of fungal growth and adherence. PLoS ONE 4:e5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner J, Cho Y, Dinh NN, Waring AJ, Lehrer RI. (1998). Activities of LL-37, a cathelin-associated antimicrobial peptide of human neutrophils. Antimicrob Agents Chemother 42:2206-2214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara A, Takada H. (2008). Synergism between TLRs and NOD1/2 in oral epithelial cells. J Dent Res 87:682-686 [DOI] [PubMed] [Google Scholar]
- Uehara A, Sugawara Y, Kurata S, Fujimoto Y, Fukase K, Kusumoto S, et al. (2005). Chemically synthesized pathogen-associated molecular patterns increase the expression of peptidoglycan recognition proteins via Toll-like receptors, NOD1 and NOD2 in human oral epithelial cells. Cell Microbiol 7:675-686 [DOI] [PubMed] [Google Scholar]
- Uehara A, Fujimoto Y, Fukase K, Takada H. (2007). Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol 44:3100-3111 [DOI] [PubMed] [Google Scholar]
- Underhill DM, Rossnagle E, Lowell CA, Simmons RM. (2005). Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 106:2543-2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Veerdonk FL, Netea MG, Jansen TJ, Jacobs L, Verschueren I, Van der Meer JW, et al. (2008). Redundant role of TLR9 for anti-Candida host defense. Immunobiology 213:613-620 [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Joosten LA, Devesa I, Mora-Montes HM, Kanneganti TD, Dinarello CA, et al. (2009a). Bypassing pathogen-induced inflammasome activation for the regulation of interleukin-1beta production by the fungal pathogen Candida albicans. J Infect Dis 199:1087-1096; erratum in J Infect Dis 199:1716, 2009 [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Marijnissen RJ, Kullberg BJ, Koenen HJ, Cheng SC, Joosten I, et al. (2009b). The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 5:329-340 [DOI] [PubMed] [Google Scholar]
- van der Graaf CA, Netea MG, Franke B, Girardin SE, Van der Meer JW, Kullberg BJ. (2006). Nucleotide oligomerization domain 2 (Nod2) is not involved in the pattern recognition of Candida albicans. Clin Vaccine Immunol 13:423-425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali DA, Collison L, Workman C. (2008). How regulatory T cells work. Nat Rev Immunol 8:523-532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vylkova S, Li XS, Berner JC, Edgerton M. (2006). Distinct antifungal mechanisms: beta-defensins require Candida albicans Ssa1 protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob Agents Chemother 50:324-331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weindl G, Naglik JR, Kaesler S, Biedermann T, Hube B, Korting HC, et al. (2007). Human epithelial cells establish direct antifungal defense through TLR4-mediated signaling. J Clin Invest 117:3664-3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells CA, Salvage-Jones JA, Li X, Hitchens K, Butcher S, Murray RZ, et al. (2008). The macrophage-inducible C-type lectin, mincle, is an essential component of the innate immune response to Candida albicans. J Immunol 180:7404-7413 [DOI] [PubMed] [Google Scholar]
- Willment JA, Brown GD. (2008). C-type lectin receptors in antifungal immunity. Trends Microbiol 16:27-32 [DOI] [PubMed] [Google Scholar]
- Wilson D, Thewes S, Zakikhany K, Fradin C, Albrecht A, Almeida R, et al. (2009). Identifying infection-associated genes of Candida albicans in the postgenomic era. FEMS Yeast Res 9:688-700 [DOI] [PubMed] [Google Scholar]
- Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R. (2004). IL-22 increases the innate immunity of tissues. Immunity 21:241-254 [DOI] [PubMed] [Google Scholar]
- Xie MH, Aggarwal S, Ho WH, Foster J, Zhang Z, Stinson J, et al. (2000). Interleukin (IL)-22, a novel human cytokine that signals through the interferon receptor-related proteins CRF2-4 and IL-22R. J Biol Chem 275:31335-31339 [DOI] [PubMed] [Google Scholar]
- Zelante T, De Luca A, Bonifazi P, Montagnoli C, Bozza S, Moretti S, et al. (2007). IL-23 and the Th17 pathway promote inflammation and impair antifungal immune resistance. Eur J Immunol 37:2695-2706 [DOI] [PubMed] [Google Scholar]
- Zelensky AN, Gready JE. (2005). The C-type lectin-like domain superfamily. Febs J 272:6179-6217 [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhu J, Imrich A, Cushion M, Kinane TB, Koziel H. (2004). Pneumocystis activates human alveolar macrophage NF-kappaB signaling through mannose receptors. Infect Immun 72:3147-3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tachado SD, Patel N, Zhu J, Imrich A, Manfruelli P, et al. (2005). Negative regulatory role of mannose receptors on human alveolar macrophage proinflammatory cytokine release in vitro. J Leukoc Biol 78:665-674 [DOI] [PubMed] [Google Scholar]
- Zhou M, Yang B, Ma R, Wu C. (2008). Memory Th-17 cells specific for C. albicans are persistent in human peripheral blood. Immunol Lett 118:72-81 [DOI] [PubMed] [Google Scholar]