

Abstract
Fluorescence recovery after photobleaching (FRAP) is a widely used imaging technique for measuring the mobility of fluorescently tagged proteins in living cells. Although FRAP presumes that high-intensity illumination causes only irreversible photobleaching, reversible photoswitching of many fluorescent molecules, including GFP, can also occur. Here, we show that this photoswitching is likely to contaminate many FRAPs of GFP, and worse, the size of its contribution can be up to 60% under different experimental conditions, making it difficult to compare FRAPs from different studies. We develop a procedure to correct FRAPs for photoswitching and apply it to FRAPs of the GFP-tagged histone H2B, which, depending on the precise photobleaching conditions exhibits apparent fast components ranging from 9–36% before correction and ∼1% after correction. We demonstrate how this ∼1% fast component of H2B-GFP can be used as a benchmark both to estimate the role of photoswitching in previous FRAP studies of TATA binding proteins (TBP) and also as a tool to minimize the contribution of photoswitching to tolerable levels in future FRAP experiments. In sum, we show how the impact of photoswitching on FRAP can be identified, minimized, and corrected.
Introduction
Over the past decade, fluorescence recovery after photobleaching (FRAP) has been used extensively to measure the mobility of fluorescently tagged proteins in living cells (1,2). These measurements have provided many insights into the dynamics of cellular processes, but there remain a number of assumptions in FRAP that have yet to be thoroughly evaluated (3).
One fundamental presumption is that the conversion from a bright state to a dark state is irreversible (4). This means that the measured fluorescence recovery is due exclusively to the influx of fluorescent molecules into the bleach spot, therefore providing a direct assay of molecular mobility. However, light exposure can convert fluorescent molecules to a dark state either irreversibly (photobleaching) or reversibly (photoswitching). Photoswitched molecules can later revert to the bright state, yielding a reversible fraction, a process also known as reversible photobleaching (5–9). An undetected reversible fraction will cause overestimates of protein mobility in FRAP. Many fluorescent molecules, including GFP and its derivatives, exhibit some degree of photoswitching. The photophysical properties of this process have been well characterized (6,7,10,11), but the impact of photoswitching on FRAP, namely how much overestimation of mobility occurs, has only been investigated in a handful of studies (5,9).
By photobleaching fixed cells, Dayel et al. showed that GFP can undergo rapid photoswitching with a reversible fraction of 5–15% (5). Sinnecker et al. (9) also investigated photoswitching, but of the GFP derivatives CFP and YFP in live cells. They performed FRAP on the same membrane protein tagged either with YFP or CFP (9). They found that the YFP fusion was largely immobile, whereas the CFP fusion had a 60% fast component, which they attributed to photoswitching of CFP.
The preceding studies indicate that photoswitching of GFP and its derivatives can influence FRAPs, but the estimated size of the reversible fraction differed markedly. As a result, it remains unclear whether photoswitching has a significant impact on most FRAPs. The primary reason for this is that there is no established procedure to evaluate how much photoswitching occurs in a FRAP experiment. As a consequence, most FRAP studies to date have ignored the possibility of photoswitching.
Here, using typical FRAP conditions, we show that photoswitching occurs with five different fluorescent proteins: GFP, YFP, mCherry, TagRFP, and mTFP1. We find that for GFP, the size of the reversible fraction depends very sensitively on the strength of the intentional photobleach, and we show that this can give rise to very different FRAP recoveries for the histone H2B-GFP and the TATA-binding protein (TBP)-GFP under FRAP conditions commonly used by different laboratories.
We develop a mathematical method to correct for reversible behavior in FRAP and apply it to H2B-GFP FRAPs, obtaining a fast component of 0.8 ± 0.8%. With this knowledge, we show that FRAP of H2B-GFP can be used to help reconcile differences in previous TBP FRAP analyses by estimating the contributions of reversible fractions in these different studies. We also show that FRAP of H2B in live cells can be used to select photobleaching conditions that minimize the reversible effect. Once these optimal photobleaching conditions are selected, we find errors of no more than 5% between quantitative estimates of TBP binding parameters and fast components before and after applying our mathematical method to correct for reversible fractions. This suggests that selection of optimal photobleaching conditions by performing FRAP of H2B in live cells may be sufficient to minimize the effects of photoswitching in many FRAP studies. If the reversible fraction cannot be reduced to tolerable levels, or if more accurate estimation is needed, our correction procedure can be applied.
Materials and Methods
Cells and constructs
We used a previously described H2B-GFP construct (12). To produce TBP-GFP, the open reading frame encoding human TBP was amplified using appropriate oligonucleotides flanked with XhoI and EcoRI, and then inserted into pEGFP-N1 (Clontech, Mountain View, CA). For FRAPs, HeLa cells were transiently transfected with pEGFP-N1, pEYFP-N1, pmCherry-C1, pTagRFP-N (Axxora, San Diego, CA), pmTFP1-N (Allele Biotech, San Diego, CA), H2B-GFP, or TBP-GFP DNA (12) using Lipofectamine LTX reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. At 24 h after transfection, cells were also prepared for live-cell FRAP experiments as previously described (13), whereas for fixed-cell FRAPs, cells were first incubated in 3.5% paraformaldehyde for 60 min at room temperature on a shaker at 50 rpm, and then washed three times with fresh medium.
FRAP conditions
Data collection and processing
The FRAP experiments with unconjugated fluorescent proteins were performed on a Zeiss LSM 5 LIVE DuoScan confocal microscope using a 63X/1.4 NA oil immersion objective. The temperature of the incubated stage was set to 37°C, and the CO2 content was held at 5%. For each fluorescent protein, imaging size was 500 × 500 pixels (50 × 50 μm2), imaging time was 50 ms, and the optical slice was 2.0 μm. We used different lasers, dichroics, and filter sets depending on the spectral characteristics of each protein (Table S1 in the Supporting Material). For photobleaching, we used the same 100-mW 488-nm laser line, because this laser line yielded the strongest bleach for each of the proteins (better than a 50-mW 405 laser line for mTFP1 or a 40-mW 561 laser line for TagRFP and mCherry—see Table S1 for details of the optical configurations).
The H2B-GFP whole-nucleus FRAP experiments were performed under the same conditions for GFP as described above except that imaging size was set to 250 × 250 pixels (25 × 25 μm2), and imaging time was 33 ms. The intentional photobleaching was performed with the 488-nm line under the different conditions described in the Results. Control measurements for observational photobleaching and subsequent data processing were carried out as described earlier (14). FRAPs on fixed cells were performed in the same medium and under incubation conditions identical to those for live cells. The strip FRAPs for H2B-GFP were performed on a Zeiss LSM 510. The circle FRAPs for H2B-GFP were performed under the same conditions as for the H2B-GFP whole-nucleus bleaches, except that the intentional photobleach was performed with a small circle of 2.0 μm in diameter. See Table S1 for details.
In an attempt to match the conditions used in the previous TBP-GFP FRAP experiments, we performed FRAP on a Zeiss LSM 510, which was the same instrument used in the preceding two TBP FRAP studies in mammalian cells. See Section 5 and Table S2 in the Supporting Material for details of both the published protocols and our protocols designed to approximate them. To correct the reversible fraction of TBP, we performed FRAPs on a Zeiss LSM 5 LIVE with the same conditions used for H2B-GFP, except that the acousto-optic tunable filter was set to 20% to observe dimmer cells, since TBP overexpression had been reported to yield slower FRAPs and lead to underestimation of the fast component (15).
Fitting of the FRAP data
The FRAP model equations were programmed in MATLAB (The MathWorks, Natick, MA), and the routine lsqcurvefit was used to fit the models to experimental data. Details of the fitting procedure can be found in Mueller et al. (14). The MATLAB source code for the newly developed FRAP procedure to correct for photoswitching (Eq. 5 in Results and Discussion) and the modeling protocol are available upon request.
Results and Discussion
GFP exhibits photoswitching after intentional photobleaching
Our objective is to characterize and ultimately mitigate the effects of photoswitching in FRAP experiments. This photoswitching behavior is likely to be sensitive to a variety of environmental conditions, including pH (9), so our experiments have been designed to determine these effects within live cells, or as close as possible to live cells, namely in fixed cells.
Thus, to test for photoswitching of GFP we transfected live cells with unconjugated GFP, photobleached the entire cell, and then measured the recovery over 20 s. Although this procedure should bleach virtually all of the fluorescence to yield no recovery, we consistently measured a fluorescence recovery. We quantified the recovery by renormalizing it such that the intensity before the bleach was 1 and the intensity immediately after the bleach was 0 (Fig. 1 A). One possible explanation for this renormalized recovery (∼9%) was that it reflects the percentage of the fluorescence that returns to the fluorescent state from the dark state.
Figure 1.
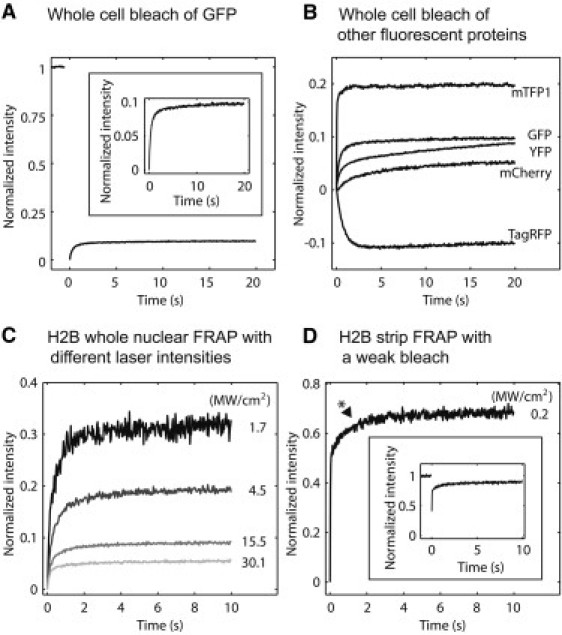
Fluorescent proteins exhibit markedly different reversible fractions. (A) Photoswitching behaviors of GFP, with a zoomed-in view (inset). To eliminate gradients in fluorescence, whole cells were photobleached, and then whole-cell intensity was measured over time. The resulting recovery curves were renormalized such that prebleach intensity was 1 and postbleach intensity was 0. Thus, the renormalized curve shows the relative contribution of photoswitching to FRAP. (B) Photoswitching behaviors of four other fluorescent proteins. In this assay, an ideal probe for FRAP would show rapid photobleaching and then no recovery. (C) Live cells expressing H2B-GFP were subjected to whole-nuclear bleaches using a single bleach iteration at different laser intensities. Stronger laser intensities reduced the reversible fraction. (D) In a real FRAP experiment (here using a strip photobleach), the fast component of H2B can reach 60% even with a reasonable bleach depth (inset; the bleach depth was not normalized to zero). The asterisk next to the curve indicates the point on the measured curve where the change in slope is minimized, which is how we estimated fast components.
An alternative explanation for this 9% recovery was detector photoblinding (14), which is a transient loss in sensitivity that can occur on a photomultiplier tube after it has been exposed to the intense fluorescence produced by a photobleach. When present, detector photoblinding produces an artifactual increase in measured fluorescence as the detector regains sensitivity. However, we found that the CCD camera used for the measurement in Fig. 1 A did not exhibit detector photoblinding (Fig. S1 A), thereby excluding this possibility.
A second alternative explanation for the 9% recovery was that the photobleach was nonuniform, particularly along the z axis, where a conical photobleach profile is expected (16). If so, then small spatial gradients in fluorescence could exist and their relaxation by diffusion could conceivably produce some influx of fluorescence into the focal plane. To exclude this, we performed the same whole-cell photobleach on fixed cells, measuring a fluorescence recovery (∼10%) comparable to that in live cells (Fig. S1 B). This suggests that diffusion in the live cells did not contribute to the measured recovery.
However, in principle, these very similar recoveries in live and fixed cells could have arisen by chance if the 60 min paraformaldehyde fixation procedure failed to cross-link a 10% fraction of the GFP. To test for this possibility, we also fixed cells containing GFP-H2B, most of which is thought to be tightly associated to chromatin inside of cells with only a small diffusible component (17,18). Despite this small free fraction, the fluorescence recovery in the fixed H2B cells was very similar to the recovery in the fixed GFP cells (Fig. S1 C), indicating that these recoveries were not due to diffusion of unfixed molecules.
We conclude that the 9% recovery seen in a whole-cell photobleach primarily reflects the photoswitching of GFP molecules within live cells.
Four other fluorescent proteins also exhibit far-from-optimal photobleaching
Any sort of reversible behavior can complicate FRAP experiments, so we tested four alternative tags, namely YFP, mCherry, TagRFP, and mTFP1 to see if any would provide a better label for FRAP. We estimated the reversible fraction for each tag by using the same whole-live-cell photobleach procedure described above. An optimal tag for FRAP would show a very small reversible fraction that recovered very quickly.
mTFP1 yielded a much larger reversible fraction than GFP (20% vs. 9%) but a somewhat faster reversion rate (2–3 s vs. 3–4 s) (Fig. 1 B). YFP yielded a similar reversible fraction to GFP (∼9%) but a slower reversion rate (∼20 s) (Fig. 1 B). mCherry also displayed a slower reversion rate (∼20 s), but with a smaller reversible fraction (5%) (Fig. 1 B).
TagRFP was unique in that it showed very slow conversion to the dark state, such that measurements of the recovery phase still contained a fluorescence loss over the first few seconds (Fig. 1 B). Only by 4–5 s after the photobleach had TagRFP finally decayed to a steady-state value, which then increased only slightly, suggesting a small reversible fraction. Despite the negligible reversible fraction for TagRFP, like GFP, it required ∼4 s to achieve an equilibrated state (Fig. 1 B), so it is also unsuited for most FRAP experiments. Thus, none of these fluorescent proteins yielded satisfactory behavior for FRAP.
The reversible fraction of GFP varies considerably depending on the bleaching conditions
The preceding data indicate that complicated photoswitching behaviors are a common property of fluorescent proteins. GFP has been the most widely used fluorescent protein for FRAP, and our tests of other fluorescent proteins suggest that there is not a simple alternative. Thus, we proceeded to further investigate the consequences of GFP's reversible behavior for FRAP. To do so, we again used H2B-GFP, since it is mostly bound to chromatin and is expected to have at best a small free component (17,18). We subjected live cells transfected with H2B-GFP to a whole-nucleus photobleach, and then to varied photobleaching parameters typically used in FRAP experiments. First, we gradually decreased the laser power during the photobleach to replicate the effects of weaker photobleaches that can arise in real FRAPs due to older or misaligned lasers. Second, we also changed the number of iterations of the photobleach.
We found that the laser intensity of the intentional photobleach had a striking effect on the reversible fraction, which increased from 5% to 30% as the laser intensity decreased (Fig. 1 C). Consistent with this, the number of bleach iterations also had a marked effect on the reversible fraction, which could be reduced to as low as 1% when using five bleach iterations (data not shown). These effects in whole nuclear FRAPs directly translated to real FRAPs. Bleaching of a 0.5-μm-wide strip across the nucleus produced H2B recoveries with fast components ranging from 5% to 60% that depended on the strength of the photobleach (the worst case (60%) is shown in Fig. 1 D). It is important to note that even this weakest photobleach (with a 60% H2B fast component) still produced FRAP curves with a substantial bleach depth (Fig. 1 D, inset) demonstrating that large reversible fractions can arise under standard FRAP conditions.
A mathematical model to correct for photoswitching in FRAP
The preceding results show that FRAPs of GFP-H2B in live cells under different photobleaching conditions can give rise to a wide range of recoveries. Our data strongly suggest that the differences are due to different photoswitchable fractions induced by different photobleaching conditions, since we could obtain very different fast components simply by changing the intensity or duration of the photobleach. Our smallest fast component for H2B was 5%, which provides an upper bound on the size of the true H2B fast component, but it is not clear how much of this 5% fast component is still due to a reversible fraction. This will be a general problem in any FRAP experiment.
To address this, we developed a correction procedure for photoswitching in FRAP. A first approach might be to estimate the size of the photoswitchable fraction for the photobleaching conditions in use and then subtract this from the initial part of the FRAP. However, this neglects the fact that a substantial fraction of the photoswitchable molecules may diffuse out of the bleach spot if the protein under study has a large fast component. The subtraction procedure will then lead to underestimation of the fast component. Alternatively, the time required for the photoswitchable fraction to recover could be measured, and then this part of the initial FRAP curve could be ignored. This, however, will also lead to underestimation of the true fast component, since some of the discarded recovery can reflect the true recovery of the protein under study. Thus, proper correction of photoswitching in FRAP requires a more rigorous mathematical model that accounts for the movements of fluorescent molecules into the bleach spot, as well as the appearance of molecules that revert to the bright state, some of which may diffuse out of the bleach spot.
Let be the measured FRAP recovery that should already be corrected for observational photobleaching, if it occurs. Note that in some cases, the observational photobleaching correction is itself also complicated by photoswitching (see Section 2 in the Supporting Material for details).
This measured recovery, , reflects two components: the entry of fluorescent molecules into the bleach spot described by the conventional FRAP equations, , plus a new component arising from a fraction of the molecules, , that revert to the bright state from the dark state. Thus,
| (1) |
To find equations that describe the reversible fraction , we note that this contribution to the measured recovery curve is effectively a time-dependent reactivation of the photoswitched GFP. This is mathematically equivalent to a photoactivation or FLAP (fluorescence loss after photoactivation) experiment, except that in these experiments all of the molecules are instantly photoactivated, whereas in photoswitching, molecules revert to the bright state over a time course. Thus, to describe the reactivation occurring in photoswitching, we must modulate the equations to describe a FLAP by a function accounting for the fraction of molecules that have reverted to the bright state, . Note in fact that depends on the history of the FLAP up to time t rather than just the current FLAP at time t; however, the following equation is assumed for theoretical and computational simplicity:
| (2) |
Like FRAP, FLAP depends on space and time. FLAP has been quantified before (19), but to our knowledge the connection between FRAP and FLAP has not been directly demonstrated. In Section 3 of the Supporting Material, we show that
| (3) |
To implement this equation, the initial condition for the FLAP model, namely, the photoactivation profile , must be converted to an equivalent photobleaching profile , where the quotes on the subscript “ble” indicate that this is not a true photobleach, but instead the photobleach that would occur by transforming the FLAP experiment into the corresponding FRAP experiment. We show in the Section 3 of the Supporting Material that the equivalent photobleaching profile is given by .
Substitution of Eqs. 2 and 3 in Eq. 1 yields the full equation to describe the recovery profile after the photobleach:
| (4) |
where the terms and have been added to the functional dependence of the FRAP terms to indicate that they have fundamentally different initial conditions. This is because the number of molecules that undergo reactivation is only a fraction of the number of molecules in the dark state.
To use Eq. 4 to fit FRAP data, , , and R(t) must be measured. can be obtained directly from the live-cell FRAP data, but to isolate the reversible behavior characterized by and R(t), measurements must be made in fixed cells expressing H2B-GFP. For these measurements, we estimated that the fraction of freely diffusible H2B-GFP after fixation is at most 0.08% (Section 1 in the Supporting Material), and so the observed recovery can be attributed solely to photoswitching of GFP in the fixed cells. As noted above, photoswitching in fixed cells is similar to photoswitching in live cells, but they are not exactly the same. As described in the next section, we show that fixed-cell reversible fractions can be converted into live-cell reversible fractions by use of a rescaling factor, α. Thus, the final equation to describe the recovery profile after the photobleach is
| (5) |
Measurements required for the photoswitching model
To determine the equivalent photobleaching profile for the reactivation , we performed a circular photobleach in fixed cells with the same conditions used for live cells and then measured the radial-intensity profile as a function of time (Fig. 2 A). After a few seconds, the reversible fraction equilibrated, as indicated by no further change in the profile. This stationary profile reflects only the irreversibly photobleached molecules that remain. The difference between the first profile after the photobleach and this stationary profile therefore reflects the total profile of reactivated molecules arising from photoswitching. We found that this profile could be fit with a piecewise constant function with Gaussian flanks (Fig. 2 B), and so we used one minus this function as the initial condition for the second FRAP term in Eq. 5.
Figure 2.
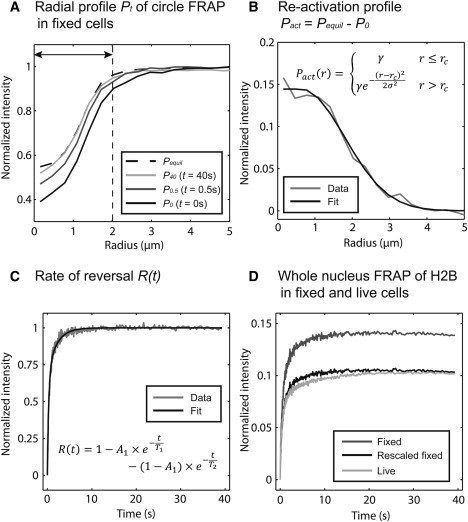
Quantification of photoswitching in a FRAP experiment. (A) FRAP was performed in a small circular region (diameter 2.0 μm) in fixed cells expressing H2B-GFP. The radial-intensity profile, Pt, was measured at different time points, as described earlier (14). The equilibrated profile was measured by averaging the profiles after no measurable increase of fluorescence was detected (Pequil). (B) The reactivation profile was measured by subtracting the profile at t = 0 s (P0) from the equilibrium profile (Pequil). These data were fit (equation shown in the plot) to yield the initial conditions for the reactivation. (C) The rate of reversal was measured in the center of the bleach region (dashed line in A) by computing the area under the curve for Pt – P0 at all time points t. After renormalization, the resultant curve was fit with a double exponential. (D) The reversible behaviors in fixed and live cells were different, as demonstrated by whole-nuclear FRAPs of H2B-GFP. However, for these bleach conditions, the entire fixed-cell recovery can be rescaled by a factor of 0.75 to closely (but not perfectly) match the live-cell recovery.
To determine the function accounting for the fraction of molecules that have reverted to the bright state, R(t), we measured the average intensity across the central part of the reversible recovery profiles in Fig. 2 A as a function of time. After renormalizing from 0 to 1, this yielded a curve that could be fit with a double exponential (Eq. 6, Fig. 2 C).
| (6) |
To obtain the rescaling factor, α, between fixed and live cells, we performed whole-nuclear bleaches in fixed and live cells, and compared reversible behaviors. We found that the fixed reversible recovery could be converted to the live reversible recovery by a one-parameter rescaling of the fixed curve (Fig. 2 D). By measuring this rescaling factor at a series of photobleaches with increasing intensity, we generated a calibration curve relating the scale factor to the size of the reversible fraction in fixed cells (Fig. S4). We then measured the reversible fraction produced by our FRAP conditions in fixed cells and used the calibration curve to find the appropriate rescaling factor, α.
With measurements of , , R(t), and α, Eq. 5 can be used to fit the measured FRAP recovery. This requires a FRAP model that describes the mobility of the protein under study. In the next section for H2B correction, we used our reaction diffusion model (13) that presumes diffusion and binding of the protein to immobile chromatin sites. This model has three unknown parameters, the molecule's diffusion constant and its association and dissociation rates with chromatin. Once the parameters are estimated, the true FRAP recovery is obtained by substituting these estimated parameters into the reaction-diffusion term for FRAP in Eq. 5, namely .
Note that the preceding general approach is suitable for any FRAP bleach geometry. In Section 3 of the Supporting Material, we develop the procedure for a circular FRAP. Also note that the new model is an extension of the basic FRAP model described in Mueller et al. (14) and is subject to all of the assumptions described for that model. This includes the assumption of no variation in the photobleach profile with z, which means in practice that bleach diameters should be on the order of 1 μm or larger. It also includes the assumption that the first measurement is made immediately after the photobleach, which in practice means the delay between the bleach and first measurement should be minimized (our delay is 45 ms).
Tests of the photobleaching correction procedure
We performed two tests of the effectiveness and accuracy of the correction procedure for photoswitching. First, we asked how reproducible our measurement was of the reversible fraction in fixed cells, since this is the primary component in the correction procedure. We measured this fraction under seven different photobleaching conditions and performed each measurement five times. From this, we calculated the standard deviation of the estimated reversible fraction under each condition. These standard deviations ranged from 0.4% to 3.7%, indicating that the reversible fraction can be accurately estimated. (Table S3).
Second, we asked how much variation there would be in estimating the true fast component of H2B. Using Eq. 5 as implemented for a circle FRAP, we corrected the seven FRAP curves of H2B, which showed fast components that ranged from 8.5% to 35.8% (Fig. 3, A and C). After correction, the fast components ranged from 0.0% to 2.2% (Fig. 3, B and C), with a mean value of 0.8 ± 0.8%. These results also suggest that the correction procedure is not subject to large variations, since we could obtain a very similar fast component for H2B regardless of the size of the initial fast component. Our estimate of an ∼1% fast component of H2B is also consistent with the expectation that virtually all of H2B is tightly bound to chromatin (17,18).
Figure 3.
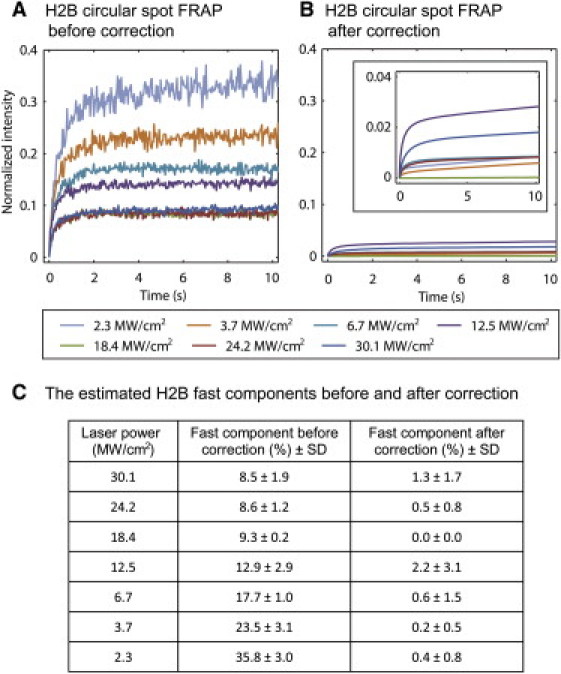
Photoswitching correction. (A) We performed FRAP on nuclei of live cells expressing H2B-GFP bleaching a small circular spot (diameter 2.0 μm) with different laser intensities (see legend below A and B). (B) After application of our correction procedure for photoswitching, the fast components of all H2B curves were reduced to 0–2% with a mean fast component of 1%. Note that the corrected curves are smooth, since they reflect a fit of the data using the photoswitching model. (C) We estimated the fast component of H2B under each photobleaching condition before and after the correction. The fast components ranged from 8.5–35.8% before the photoswitching correction and were reduced to a mean value of 0.8 ± 0.8% after the photoswitching correction.
Based on these results, we propose that FRAP of H2B-GFP in live cells can be used as a standard for evaluating the presence of photoswitching under a certain set of photobleaching conditions. The cell line and the plasmid encoding H2B-GFP in use here are readily available and so could be obtained by any laboratory performing FRAP experiments. The photobleaching conditions selected for an experiment can then be applied to live cells transfected with H2B-GFP and the fast component estimated. If this fast component exceeds a few percent, then the photobleaching conditions give rise to photoswitching, and so the conditions should be changed until the H2B-GFP fast component is reduced to tolerable levels. Better bleaching conditions can be determined empirically by increasing the bleaching laser power, number of bleach iterations, or pixel dwell time. If the reversible fraction cannot be reduced to tolerable levels, or more accurate quantitative estimation is needed, then the correction procedure described above can be applied. We apply this general strategy in the next section to FRAP analysis of TBP, which is a key component of the polymerase complex.
A potential contribution of photoswitching to discrepant TBP FRAP curves
A large body of in vitro data suggests that TBP is stably bound at transcriptionally active promoters. However, in mammalian cells, two different studies have produced very different TBP FRAP curves, leading to potentially different conclusions about TBP mobility (15,20). An especially puzzling question raised by these two studies has been how FRAP data could be so different between two groups studying the same protein.
The differences between these two FRAP curves are clear when the data are extracted from each study and plotted on the same graph (Fig. 4 A). de Graaf et al. (15) argued that this difference could have been due to overexpression of TBP in Chen et al. (20). This is plausible, since de Graaf et al. showed that the protein BTAF1 reduces TBP's residence time on DNA. Thus, higher TBP expression levels could eventually titrate out the protein BTAF1, leading to slower FRAPs. Consistent with this, de Graaf et al. showed that brighter cells yielded slower TBP FRAPs, and we confirmed this finding when we performed FRAPs of TBP in dim and bright cells (Fig. S5 C). However, under our conditions, the change in the FRAP curves due to TBP concentration was ∼10%, which was not large enough to explain the ∼30% difference between the published curves.
Figure 4.
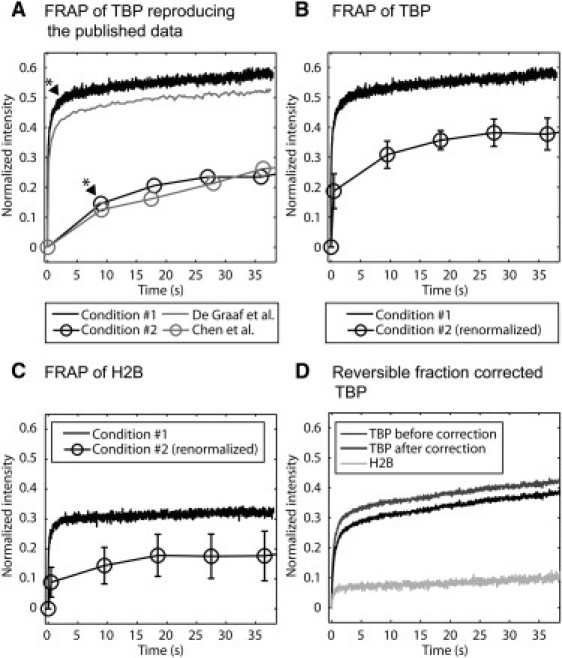
Reconciling discrepant predictions of TBP fast components. (A) We were able to roughly match two published FRAP curves by approximating the different imaging conditions used in the two studies (15,20). Our two FRAP curves were obtained on the same cells expressing similar levels of TBP. The difference in fast component between conditions 1 and 2 is ∼30%. (See Fig. 1 legend for the procedure to estimate fast components). (B) We renormalized the curve from condition 2 with the true bleach depth by measuring the intensity in the bleach region as quickly as possible after the condition 2 photobleach. This reduced the difference in fast component between conditions 1 and 2 to ∼20%. (C) We applied the photobleaches of conditions 1 and 2 to H2B-GFP in live cells and found an ∼20% difference in reversible fractions, roughly accounting for the remaining difference from B. (D) We performed FRAP using a circular photobleach on cells expressing low levels of TBP and then applied our correction procedure for photoswitching to the TBP curve, yielding an estimated fast component of 26%. The uncorrected H2B curve shows the level of photoswitching present under these conditions.
To investigate other possible explanations for the difference between the published TBP FRAP curves, we attempted to reproduce the published data by approximating the imaging and photobleaching conditions reported (see Table S2). We could not do this exactly, because not all the parameter settings were reported, and furthermore, a key parameter, the laser power, will depend on the age and alignment of the laser in use. Nevertheless, we were able to find conditions (referred to as conditions 1 and 2) that allowed us to nearly replicate the published curves, but now, it is important to note, using the same cells with the same expression levels of TBP (Fig. 4 A and Table 1 for the estimated fast components). The similarities between our curves and the published curves include a good match of the actual bleach depths (see Fig. S5, A and B, in which bleach depths were not normalized to zero).
Table 1.
Estimated TBP fast components
| Condition 1 | Condition 2 | Optimal condition before correction | Optimal condition after correction | |
|---|---|---|---|---|
| Fast component (%) ± SD | 49.1 ± 8.4 | 14.6 ± 9.1 | 30.6 ± 2.4 | 26.4 ± 2.6 |
Although it is impossible to exactly replicate the original experiments, we can use the two conditions we identified to investigate how different FRAPs can arise purely due to changes in imaging and bleaching conditions. As we show below, two factors account for the differences in our experiments: the time to acquire the first image of the bleached region and the contribution of photoswitching.
The time to acquire the first image of the bleached region was 482 ms longer for condition 2 compared to condition 1. This difference arose because condition 2 used a larger image that introduced a longer interval between the end of the bleach and the first image of the bleached region. The reason for this is that the bleached square is in the middle of the image in condition 2, so the top half of the image must first be scanned, requiring 482 ms before the bleached region is reached. This does not occur in condition 1, which bleaches and then immediately images the same strip. Due to this poor temporal sampling in condition 2, some of the fast component of TBP is not detected. To correct for this, we estimated the full extent of the bleach depth in condition 2 by performing a photobleach with the same laser intensity and bleach pattern but then measuring the fluorescent intensity in the bleached region as quickly as possible (with no delay rather than 482 ms). We then used this value to normalize the bleach depth to zero for the FRAP data from condition 2. This correction reduced the difference between the fast components observed in conditions 1 and 2 from ∼30% to ∼20% (Fig. 4 B).
We then asked whether photoswitching could explain the remaining difference between the two curves. We cannot directly correct the FRAPs from either condition 1 or condition 2, since our current implementation of the correction method is not designed for FRAPs where only the bleached region is imaged (condition 1) or for FRAPs with square bleach profiles (condition 2). Instead, we used H2B-GFP in live cells as a benchmark to estimate how much photoswitching occurred under conditions 1 and 2. The difference between the resultant H2B-GFP fast components was ∼20% (Fig. 4 C), which roughly accounts for the remaining difference in the fast components estimated from the delay-time-corrected TBP FRAP curves (Fig. 4 B). Note that these fast-component estimates are only approximate, particularly since the temporal sampling of condition 2 is poor. Note also that this simple approach of subtracting out reversible fractions is crude, since the actual contribution to the FRAP will also be influenced by the rate of reversion balanced by the rate at which these molecules leave the bleached region. Nevertheless, our analysis suggests that by accounting for the temporal sampling rate and the amount of photoswitching, the FRAP curves from the two previous studies are in fact very similar. This is reassuring, because it demonstrates that there are not fundamental differences in the behavior of TBP in these two published studies.
Finally, we tested our suggested procedure of using FRAP of H2B to select photobleaching conditions that would minimize the reversible fraction. We used a 45-ms circular photobleach and then varied the laser intensity, pixel dwell time, and number of iterations during the photobleach. The smallest fast component of H2B that we could obtain was 6%, suggesting a residual 5% reversible fraction under these optimal conditions. We then used these optimized conditions to perform FRAP in TBP, and subsequently applied our mathematical procedure to correct for the residual reversible fraction. We obtained estimates of TBP free fractions that showed ∼5% change before and after correction (35 ± 2.7% vs. 30 ± 3.0%), indicating that the correction procedure had a minimal effect once optimal photobleaching conditions were used.
To test the effects on quantification when optimal conditions were not used for the photobleach, we reexamined TBP FRAPs under conditions 1 and 2, used to mimic previous FRAP studies of TBP. As noted above, our implementation of the correction method cannot estimate free fractions from these FRAPs, so instead we estimated the size of the fast component for conditions 1 and 2 and compared that to the size of the fast component using our optimized condition before and after correction. Here, we found large differences in the size of the fast component between our optimized condition and either conditions 1 or 2 (Table 1), indicating that optimizing the photobleach condition is critical. Again, however, we found a small difference in the size of the corrected fast component once the optimized photobleach condition was selected.
Conclusions
Our principal finding is that the reversible fraction arising in a GFP photobleach is highly variable, ranging up to ∼60% under typical conditions used in a FRAP experiment. This has potentially serious consequences, since most FRAP studies with GFP presume that the reversible fraction is negligible. We found that the reversible fraction grows with weaker photobleaches, contributing substantially to the first seconds of the FRAP recovery. In many cases, the first few seconds of the recovery must be recorded to capture the true fast component of the protein under study. Consequently, some FRAP data may inadvertently incorporate significant reversible fractions that can lead to overestimation of mobility.
We found that the reversible fraction contributed to the fast component in FRAP curves of both H2B-GFP and TBP-GFP. Under typical FRAP conditions, the H2B fast component was as large as 60% even though H2B is known to be tightly bound to chromatin. Therefore we developed a correction procedure for this photoswitching that accounted for the behavior of the reversible molecules during a FRAP. By applying this procedure to H2B-GFP FRAPs performed under different photobleaching conditions, we obtained a consistent fast component of 0.8 ± 0.8%. This suggests that our correction procedure is effective over a large range of reversible fractions, but we recommend first optimizing the photobleach conditions to minimize the reversible effect rather than relying exclusively on the laborious and time consuming correction procedure.
One potential concern with this H2B calibration procedure is that the reversible fraction of GFP likely depends on pH (9), which might vary from one cell type to the next. Nevertheless, we found very similar reversible behaviors for H2B-GFP in HeLa and mouse adenocarcinoma cells (F. Mueller and T. Morisaki, unpublished observations), suggesting that reversible fractions of GFP do not vary widely at least in common tissue culture cells. If there is uncertainty about whether the reversible fraction may change significantly in another cell type, then the correction procedure that we have devised here can be applied.
Our data suggest that the H2B test to minimize reversible behavior will suffice for the vast majority of FRAP experiments. We found that under conditions suitable for FRAP of TBP, we could minimize the reversible fraction to ∼5%. Then our correction procedure produced only small changes in the various quantitative parameters that could be estimated from the TBP FRAP.
Another option for the future to reduce the impact of reversible behaviors may be the identification or creation of other fluorescent proteins more suitable for FRAP. Although none of the fluorescent proteins that we tested were optimal, other fluorescent proteins might be identified or engineered that would have better reversible behaviors, namely very small reversible fractions that recover very quickly. An alternate possibility might be to use organic fluorophores, which may be less subject to reversible behavior than the fluorescent proteins. Such fluorophores can be bound by certain fusion proteins, for example, the SNAP tag (21).
At the moment, given the prevalence of GFP FRAPs, the H2B live-cell calibration procedure should be a useful assay to choose photobleaching conditions that minimize reversible behavior. We used the H2B test to investigate the very different GFP-TBP FRAP curves reported in two published studies (15,20). We were able to closely match these discrepant published FRAP curves using photobleach and imaging conditions based on the published experimental protocols. We showed that the difference in the resultant TBP FRAP curves in our experiments was due partly to the different time intervals between the photobleach and first image of the bleached region and partly to different amounts of photoswitching.
These effects are probably responsible for at least some of the difference reported in the two published studies, but other factors, such as the expression levels of TBP and the details of the TBP-GFP constructs, could also contribute to differences. For example, it appears that Chen et al. (20) and de Graaf et al. used N-terminal GFP fusions while we used a C-terminal fusion. The expression level of TBP is certainly a key variable as both we and de Graaf et al. (15) found that overexpression of TBP could reduce its fast component. One piece of evidence that favors some role for photoswitching in the published difference is that de Graaf et al. (15), who reported the larger TBP fast component also performed FRAP of H2B obtaining a 20% fast component. This might reflect a significant reversible fraction in their experiments, since our correction procedure suggests that the H2B fast component is ∼1%. We should point out however that de Graaf et al. (15) fit their TBP data with a diffusion and binding model that included a procedure to account for photoswitching (A. Houtsmuller, Josephine Nefkens Institute, personal communication, 2011), so they did not ignore this effect in their published quantitative estimates for the binding rates of TBP.
With respect to the previous TBP studies, it is important to point out that our analysis in no way alters the conclusions that have been reported (15) about the role of other cellular factors in mobilizing TBP. By measuring altered FRAP curves after perturbing the levels of TBP interacting factors, BTAF1 and NC2, de Graaf et al. (15) demonstrated that both factors regulate TBP residence times on chromatin, with BTAF1 reducing TBP residence times and NC2 increasing TBP residence times. The conclusions from these and other comparative studies are in general not affected by photoswitching, since as long as the comparisons are done under similar conditions, the reversible fraction will be relatively constant, and so differences in the FRAP curves reflect the underlying biological mechanisms. Reversible fractions may become significant when comparing FRAP curves reported by different laboratories, as we suggest here, or whenever any form of quantitative information is extracted from an early phase of a FRAP curve that is not optimized to minimal reversible behavior. In these circumstances, it is vital to perform the photoswitching test as described above for H2B to minimize the reversible fraction.
In conclusion, we have shown that photoswitching can be a serious problem in GFP FRAP, but it should now be easy to identify, minimize, and, if necessary, correct using the procedures we describe.
Acknowledgments
We thank Tom Misteli for plasmids and Claudio Fenizia for cDNA. We are grateful to Carolyn Smith for her comments on the manuscript. We thank Tim Stasevich for suggesting the intuitive approach used to modify the FRAP equations in the presence of photoswitching.
This research was supported in part by the intramural program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
Florian Mueller's present address is Institut Pasteur, Imaging and Modeling Group CNRS, Paris, France
Supporting Material
References
- 1.Houtsmuller A.B. Fluorescence recovery after photobleaching: application to nuclear proteins. Adv. Biochem. Eng. Biotechnol. 2005;95:177–199. doi: 10.1007/b102214. [DOI] [PubMed] [Google Scholar]
- 2.Sprague B.L., McNally J.G. FRAP analysis of binding: proper and fitting. Trends Cell Biol. 2005;15:84–91. doi: 10.1016/j.tcb.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 3.Mueller F., Mazza D., McNally J.G. FRAP and kinetic modeling in the analysis of nuclear protein dynamics: what do we really know? Curr. Opin. Cell Biol. 2010;22:403–411. doi: 10.1016/j.ceb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reits E.A.J., Neefjes J.J. From fixed to FRAP: measuring protein mobility and activity in living cells. Nat. Cell Biol. 2001;3:E145–E147. doi: 10.1038/35078615. [DOI] [PubMed] [Google Scholar]
- 5.Dayel M.J., Hom E.F.Y., Verkman A.S. Diffusion of green fluorescent protein in the aqueous-phase lumen of endoplasmic reticulum. Biophys. J. 1999;76:2843–2851. doi: 10.1016/S0006-3495(99)77438-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dickson R.M., Cubitt A.B., Moerner W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature. 1997;388:355–358. doi: 10.1038/41048. [DOI] [PubMed] [Google Scholar]
- 7.Henderson J.N., Ai H.W., Remington S.J. Structural basis for reversible photobleaching of a green fluorescent protein homologue. Proc. Natl. Acad. Sci. USA. 2007;104:6672–6677. doi: 10.1073/pnas.0700059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lemmer P., Gunkel M., Cremer C. Using conventional fluorescent markers for far-field fluorescence localization nanoscopy allows resolution in the 10-nm range. J. Microsc. 2009;235:163–171. doi: 10.1111/j.1365-2818.2009.03196.x. [DOI] [PubMed] [Google Scholar]
- 9.Sinnecker D., Voigt P., Schaefer M. Reversible photobleaching of enhanced green fluorescent proteins. Biochemistry. 2005;44:7085–7094. doi: 10.1021/bi047881x. [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Parajo M.F., Segers-Nolten G.M.J., van Hulst N.F. Real-time light-driven dynamics of the fluorescence emission in single green fluorescent protein molecules. Proc. Natl. Acad. Sci. USA. 2000;97:7237–7242. doi: 10.1073/pnas.97.13.7237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seward H.E., Bagshaw C.R. The photochemistry of fluorescent proteins: implications for their biological applications. Chem. Soc. Rev. 2009;38:2842–2851. doi: 10.1039/b901355p. [DOI] [PubMed] [Google Scholar]
- 12.Phair R.D., Scaffidi P., Misteli T. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 2004;24:6393–6402. doi: 10.1128/MCB.24.14.6393-6402.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sprague B.L., Pego R.L., McNally J.G. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys. J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mueller F., Wach P., McNally J.G. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys. J. 2008;94:3323–3339. doi: 10.1529/biophysj.107.123182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Graaf P., Mousson F., Timmers H.T. Chromatin interaction of TATA-binding protein is dynamically regulated in human cells. J. Cell Sci. 2010;123:2663–2671. doi: 10.1242/jcs.064097. [DOI] [PubMed] [Google Scholar]
- 16.Mazza D., Cella F., Diaspro A. Role of three-dimensional bleach distribution in confocal and two-photon fluorescence recovery after photobleaching experiments. Appl. Opt. 2007;46:7401–7411. doi: 10.1364/ao.46.007401. [DOI] [PubMed] [Google Scholar]
- 17.Wolffe A.P., Hayes J.J. Chromatin disruption and modification. Nucleic Acids Res. 1999;27:711–720. doi: 10.1093/nar/27.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Workman J.L., Kingston R.E. Alteration of nucleosome structure as a mechanism of transcriptional regulation. Annu. Rev. Biochem. 1998;67:545–579. doi: 10.1146/annurev.biochem.67.1.545. [DOI] [PubMed] [Google Scholar]
- 19.Beaudouin J.L., Mora-Bermúdez F., Ellenberg J. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 2006;90:1878–1894. doi: 10.1529/biophysj.105.071241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen D., Hinkley C.S., Huang S. TBP dynamics in living human cells: constitutive association of TBP with mitotic chromosomes. Mol. Biol. Cell. 2002;13:276–284. doi: 10.1091/mbc.01-10-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein T., Löschberger A., Sauer M. Live-cell dSTORM with SNAP-tag fusion proteins. Nat. Methods. 2011;8:7–9. doi: 10.1038/nmeth0111-7b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.