

Abstract
Aggregation of α-synuclein is involved in the pathogenesis of Parkinson's disease (PD). Studies of in vitro aggregation of α-synuclein are rendered complex because of the formation of a heterogeneous population of oligomers. With the use of confocal single-molecule fluorescence techniques, we demonstrate that small aggregates (oligomers) of α-synuclein formed from unbound monomeric species in the presence of organic solvent (DMSO) and iron (Fe3+) ions have a high affinity to bind to model membranes, regardless of the lipid-composition or membrane curvature. This binding mode contrasts with the well-established membrane binding of α-synuclein monomers, which is accompanied with α-helix formation and requires membranes with high curvature, defects in the lipid packing, and/or negatively charged lipids. Additionally, we demonstrate that membrane-bound α-synuclein monomers are protected from aggregation. Finally, we identified compounds that potently dissolved vesicle-bound α-synuclein oligomers into monomers, leaving the lipid vesicles intact. As it is commonly believed that formation of oligomers is related PD progression, such compounds may provide a promising strategy for the design of novel therapeutic drugs in Parkinson's disease.
Introduction
Aggregation and amyloid formation of disease-specific proteins are key events in a number of neurodegenerative disorders such as Alzheimer's disease and Parkinson's disease (PD) (1,2). A common feature of the aggregation cascade is the formation of oligomeric species that eventually mature to form larger insoluble deposits. Several findings suggest that oligomeric intermediates, rather than mature amyloid fibrils, represent the principal neurotoxic aggregate species (2–8). A characteristic feature of PD is the intracellular deposition of Lewy bodies in which α-synuclein (αS) represents the major protein component (9). This 140-amino-acid protein is widely distributed throughout the brain (10). A remarkable property of αS is its structural flexibility. In dilute aqueous solutions, the protein is essentially unstructured, whereas it adopts α-helical structure upon binding to lipids (11). In contrast, misfolding leads to formation of ordered fibrils with a rigid β-sheet-like structure both in vitro (12,13) and in vivo in Lewy bodies (14). Aggregation of αS also involves the formation of cytotoxic oligomeric assemblies (14–18). The biophysical and functional properties of these disease-associated oligomer species are unclear (6). One mechanism of oligomer toxicity that has been proposed is the formation of pores that lead to permeabilization of cellular membranes (5,7,19–23). Metal ions trigger the generation of oligomeric species and accelerate the formation of amyloid αS fibrils (4,13,24,25). Several lines of evidence indicate that particularly iron plays an important role in PD pathogenesis. In vitro, association of αS monomers with iron ions has also been reported (13,26,27). Iron exposure has been shown to increase PD risk in epidemiological studies (28,29), increased iron levels correlate with the severity of neuropathological changes in PD patients (30), and high levels of Fe3+ have been found in Lewy bodies (31).
When monomeric αS binds to a lipid-water interface, the N-terminus acquires a helical structure whereas the C-terminus remains unstructured (5,11,32). This lipid-binding mode has been analyzed in vitro by various biophysical techniques (33–38). The affinity of αS to membranes depends on curvature and lipid composition, i.e., αS binds with high affinity to membranes with negative charge and high curvature (5,32,39–41). αS also binds to small unilamellar vesicles (SUVs) of zwitterionic lipids in the gel-phase (DPPC, below chain-melting temperature, Tm) (5,33,34,42). On the other hand, αS has a low affinity to bind to SUVs of zwitterionic lipids in the liquid-crystalline phase (such as POPC) (43,44). Furthermore, binding of αS depends on defect structures present in ordered membranes of high curvature. In stressed membranes, αS is capable of annealing defects, hence preventing membranes from fusion (33,34,45). The membrane affinity and concomitant structural transition of αS led to the assumption that binding of αS monomers to synaptic vesicles is related to its physiological role at synaptic nerve terminals (5,14,46–48). Association of αS to mitochondria has also been described (45,49,50).
With regard to structural studies on the formation of oligomeric assemblies, the most common techniques include immunoblotting, electron microscopy, nuclear magnetic resonance, electron spin resonance, atomic force microscopy, and size-exclusion chromatography. In addition, confocal single-particle fluorescence spectroscopy techniques have been developed to analyze aggregation of proteins (51–56). Using these techniques, we could characterize two different αS oligomer species that were both on-pathway to amyloid fibrils. Organic solvents (ethanol or dimethyl sulfoxide (DMSO)) triggered aggregation resulting in the formation of small (i.e., intermediate-I) oligomers (4). Subsequent addition of Fe3+ ions close to physiological serum levels (5–20 μM) induced formation of larger oligomer species (i.e., intermediate-II). The latter species are SDS-resistant, form ion-permeable pores in a planar lipid bilayer system (4), and are toxic in neuronal cell cultures (57). These findings go along with several lines of evidence implicating the involvement of Fe3+ iron in disease progression in PD, as mentioned above (13,24,26,58,59).
The aim of this study was to improve the understanding of the molecular pathophysiology of the αS aggregation processes in PD. In light of the well-reported binding mode of monomeric αS to lipid membranes, we evaluated the iron-induced formation of αS oligomers in the presence of model lipid vesicles using fluorescence intensity distribution analyses (FIDA) for qualitative and quantitative evaluation of the heterogeneous population of oligomers that is usually formed in in vitro aggregation assays. By labeling αS and vesicles with a different color, the interaction between αS and vesicles was measured. In particular, we were interested to investigate 1), whether the direct interaction of monomeric αS with lipid vesicles of varying phospholipid composition would influence iron-induced oligomer formation, 2), whether αS aggregation results in oligomers with different properties in regard to membrane binding compared to αS monomers, and 3), whether this approach can be used as a model system for characterization of compounds blocking αS aggregation in regard to their effects on physiological and pathological interaction of αS with lipid membranes.
Materials and Methods
Chemicals
Phospholipids (1-palmityl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and dipalmitoyl-sn-glycero-3-phosphocholine (DPPC)) were purchased from Avanti Polar Lipids (Alabaster, AL). BODIPY-FL-DHPE (BODIPY-PE) and Alexa Fluor-647-O-succinimidylester were from Invitrogen (Carlsbad, CA).
Purification and labeling of recombinant wild type αS
Expression and purification was performed as described previously (34). Protein labeling was performed with the amino-reactive fluorescent dye Alexa Fluor-647-O-succinimidylester (“red”) as described previously (4).
Vesicle preparation
A green fluorescent dye was incorporated into the vesicles by mixing 0.1% (w/w) BODIPY-PE with the lipids (POPC or DPPC) in chloroform. Chloroform was evaporated with nitrogen gas and 1 h vacuum. Buffer was added to the lipid film, which was vortexed and then hydrated for at least 1 h. Large unilamellar vesicles (LUVs) were made by extrusion, and small unilamellar vesicles (SUVs) were made by sonication as described in Kamp et al. (60). The size of the vesicles was checked with dynamic light scattering measurements using a High Performance Particles Sizer (Malvern Instruments, Herrenberg, Germany). SUVs and LUVs usually had a diameter of 25–35 nm and 70–80 nm, respectively. Vesicles were diluted to a final lipid concentration of 100 μM and stored at 4°C.
Aggregation assay
Experiments were started by diluting the stock solution of αS-647, pH 7.0, in a total assay volume of 20 μL. The concentration of αS-647 was adjusted to ∼10 molecules per focal volume, (≈10 nM). Vesicles were added at a final lipid concentration of 10 μM. To induce αS oligomerization we added either 10 μM FeCl3, DMSO (1% v/v), or DMSO (1% v/v) plus 10 μM FeCl3, respectively. All experiments were performed in 96-well-plates (Evotec-Technologies, Hamburg, Germany). At least five measurements were performed and averaged after 2 h of incubation in two-to-four independent wells per experimental group.
Confocal single particle analysis
Fluorescence correlation spectroscopy (FCS) and scanning for intensely fluorescent targets (SIFT) measurements were carried out on an Insight Reader (Evotec-Technologies) with dual color excitation at 488 and 633 nm as described before (4). Measurement time was 10 s for SIFT measurements and 300 s for FCS measurements. For fluorescence intensity distribution analysis (FIDA), fluorescence from the two different fluorophores was recorded simultaneously. Photons were summed over time intervals of constant length (bins) using a bin length of 40 μs (i.e., 5 × 10 s measurement time generates 1,250,000 bins). All measurements were performed at room temperature.
Results
Quantitative analysis of lipid binding of αS with confocal single particle fluorescence techniques
We used SUVs of zwitterionic lipids in the gel-phase (DPPC-SUV) for the first set of binding studies, in light of the reported strong association of αS to these vesicles (34). To evaluate protein aggregation and lipid binding we always used red-labeled αS (αS-647) and green-labeled vesicles. Fig. 1 A shows 2D-FIDA histograms acquired from SIFT measurements of αS alone, DPPC-SUV alone, and a mixture of αS and DPPC-SUV, respectively. FIDA histograms depict the distribution of the amount of photons that were detected within the bin length (40 μs) of the SIFT measurements, not the average brightness of the detected particles. It should be noted that a homogeneous population of vesicles with on average four fluorophores per vesicle leads to a distribution of intensity ranging from 0 to 250 photons/bin (Fig. 1 A, middle panel). The detection of bins with high photon counts in both the red and green channel demonstrates association of αS to DPPC-SUV (Fig. 1 A, right panel). This could be corroborated by quantitative FIDA evaluation of 1D-FIDA histograms (Fig. 1 B), which can be used to calculate the average brightness and concentration of the particles (see Supporting Material and Kask et al. (55)).
Figure 1.
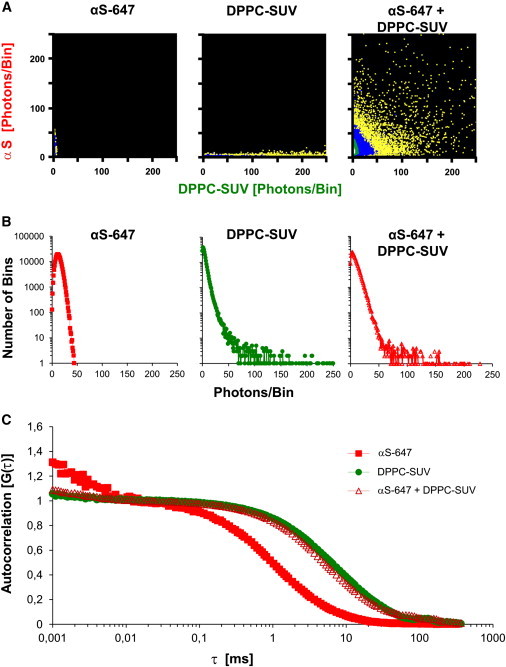
Quantitative evaluation of binding of αS to DPPC-SUV. (A) 2D-FIDA histograms of incubations of red-labeled monomeric αS in solution (αS-647, left panel), green-labeled (BODIPY-PE) DPPC vesicles (DPPC-SUV, middle panel) in solution, and a mixture αS and DPPC-SUV (right panel). X axis, green fluorescence; Y axis, red fluorescence. Excitation at 488 and 633 nm. (B) 1D-FIDA histograms of monomeric αS in solution (left), DPPC-SUV in solution (middle), and vesicle-bound αS (right). (C) Normalized autocorrelation plots derived from FCS measurements of monomeric αS in solution, DPPC-SUV in solution, and αS in the presence of DPPC-SUV. (In panels B and C, red traces represent αS-647, green traces represent BODIPY-PE fluorescence.)
For αS incubated with DPPC-SUV, there was a four- to fivefold increase of red particle brightness, indicating that ∼4–5 αS molecules bound per lipid vesicle (assuming that the quantum yield of Alexa647 did not alter due to binding of αS to the membrane). Furthermore, it could be calculated that ∼74% of the total amount of αS was bound to DPPC-SUV in this assay (Table S1 in the Supporting Material). In a different set of experiments using FCS, we first determined independently the diffusion times for unbound αS (ταS) and DPPC-SUV (τsuv). From the measured τsuv (∼6600 μs) we calculated a hydrodynamic radius of the SUV of 34 nm, which matched the radius determined independently with dynamic light scattering (see Supporting Material). From the measured ταS (∼1000 μs) a hydrodynamic radius for unbound monomeric αS of ∼3 nm was calculated, which also matches the expected value (61). When αS was incubated in the presence of DPPC-SUV, the autocorrelation of αS shifted dramatically toward the curve of DPPC-SUV (Fig. 1 C). Because the diffusion time of DPPC-SUV changes only marginally with a few αS molecules bound to it (45), the fraction of bound αS could be calculated by fitting the autocorrelation curve of αS to ταS and τsuv with a two-component fit (44). We found that 83% of the total amount of αS was bound to DPPC-SUV, which is in close agreement with the fraction bound calculated from the FIDA data (Table S1).
Aggregation of αS in the presence of vesicles reveals two separate lipid-depending binding modes
To investigate if there is a specific interaction between oligomeric species and lipid bilayers, we initially chose POPC-SUV to which monomeric αS has a low binding affinity (5,34). When we incubated αS-647 with green-labeled POPC-SUV, high-intensity bins could only be detected along the X axis of the 2D-FIDA histogram (Fig. 2 B). These bins are generated by POPC-SUV devoid of bound αS. Unbound monomeric αS species generated bins close to the origin of the FIDA histogram. Thus, as expected, monomeric αS did not bind to POPC-SUV under control buffer conditions. Also, the addition of Fe3+ ions alone did not cause any changes in the binding and aggregation of αS. In contrast, the presence of 1% DMSO induced generation of intermediate-I oligomers (4), as can be seen by the accumulation of bins along the Y axis with higher red fluorescence intensity compared to αS monomers (Fig. 2 B). However, the absence of bins with both green and red fluorescence indicates that these intermediate-I oligomers were not able to bind to the vesicles.
Figure 2.
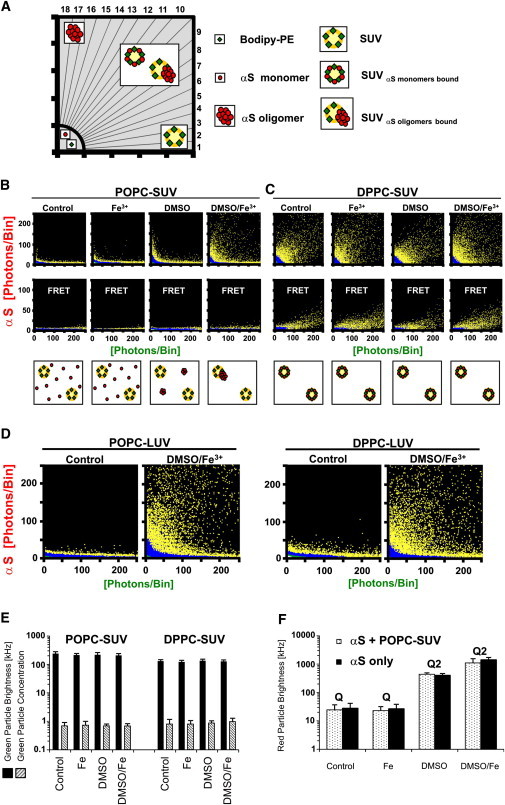
Binding of αS to vesicles of different lipid composition reveals two different binding modes. (A) Scheme of the segments of a 2D-FIDA histogram used for quantitative SIFT analysis. Data points in segments 1 and 2 are generated by vesicles devoid of αS (“purely green bins”); data points in segments 3–16 are caused by monomeric or aggregated αS bound to vesicles (“dual colored bins”); and data points in segments 17 and 18 are caused by unbound αS aggregates (“purely red bins”). Monomeric αS generates data points only in the segment close to the origin. The cartoons illustrate which type of particles are detected: free vesicles with green labels (green diamonds), free αS-monomers (red dots), αS-aggregates, vesicles with bound αS-monomers, or vesicles with bound αS-aggregates. (B) 2D-FIDA histograms of incubations of POPC-SUV and αS-647. (Top panels) Excitation at 488 and 633 nm. (Middle panels) FRET measurements (excitation with 488 nm laser only). (Cartoons, bottom row) The type of particles detected. (Panels from the left to the right) Control experiments (vesicles with αS), incubation with 10 μM FeCl3, 1% DMSO, and 1% DMSO + 10 μM FeCl3, respectively. (C) As panel B for DPPC-SUV. (D) 2D-FIDA histograms of LUV-POPC and LUV-DPPC reveal that αS monomers do not bind to these vesicles, whereas the DMSO/Fe3+-induced oligomers had a high binding propensity. (E) Quantitative FIDA analysis of panels B and C shows that the average green particle brightness and concentration of POPC- and DPPC-SUV is not altered due to additions of iron and/or DMSO. (F) Average red particle brightness of αS with and without POPC-SUV under different aggregation conditions. Q is the fitted brightness of αS monomers in solution. Only with DMSO +/− Fe3+ a second population of αS aggregates with brightness Q2 was found in the data fit.
In contrast, under conditions generating intermediate-II oligomers, i.e., in presence of DMSO and Fe3+ ions, we could detect a significant formation of two-colored bins demonstrating αS aggregates bound to POPC-SUV. Compared to aggregation controls of αS in presence of DMSO and Fe3+ ions in absence of lipid vesicles (see Fig. 3, upper-left panel), that results in large αS oligomers yielding high intensity fluorescent bins along the Y axis (i.e., “purely red bins”), in the presence of POPC-SUV almost no purely red bins with high fluorescent intensity, i.e., free intermediate-II oligomers, could be detected. Thus, the intermediate-II oligomers had all bound to the POPC-SUV. In contrast, a large number of high intensity fluorescent bins along the X axis (i.e., “purely green bins”), representing vesicles devoid of bound αS, were still detected (see also Fig. 3, POPC-SUV only). These results are also confirmed by quantitative analysis of the number of high-intensity bins in sectors 1–18 of the FIDA histograms (Fig. 2 A and Fig. S1).
Figure 3.
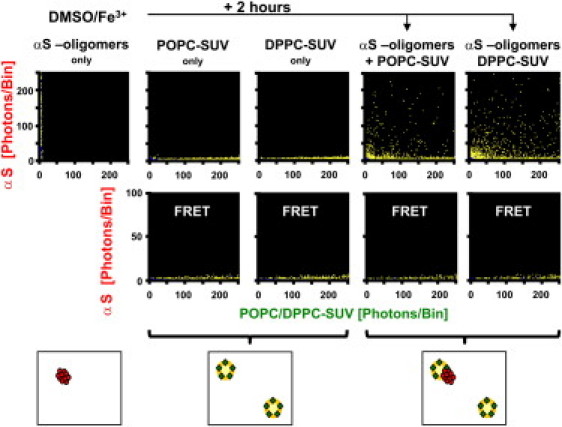
Binding of preformed αS intermediate-II oligomers to vesicles. (Upper lanes) 2D-FIDA histograms of incubations of vesicles and αS. (Leftmost panel) αS in the absence of vesicles after 2 h incubation with 1% DMSO + 10 μM FeCl3 showing the formation of intermediate-II aggregates. (Two middle panels) POPC-SUV and DPPC-SUV in solution, respectively. (Two right panels) To the sample of the leftmost panel (intermediate-II aggregates), an aliquot of POPC-SUV or DPPC-SUV was added, respectively, and the SIFT measurement was repeated. All aggregates had bound to the vesicles. (Middle lanes) FRET measurements of the incubations depicted in the upper lanes. (Cartoons, lower panels) The type of particles detected.
Based on our expectation that the interaction of monomeric αS with a lipid membrane-water interface can strongly modulate misfolding and aggregation, we repeated the aggregation assay in the presence of DPPC-SUV. In this case two-colored bins representing vesicles with bound αS were already observed without the addition of DMSO and/or Fe3+ ions (Figs. 1 A and 2 C). Only very few purely green bins could be detected, indicating that virtually all vesicles had a few monomeric αS molecules bound. In contrast to the POPC-SUV, formation of free intermediate-I oligomers in the presence of 1% DMSO could not be detected. Even addition of Fe3+ ions and 1% DMSO, i.e., intermediate-II oligomer promoting conditions, did not alter the FIDA histogram (Fig. 2 C and see Fig. S2). Finally, we performed our aggregation assays with LUVs made of POPC and DPPC. Due to the lower curvature of LUVs compared to SUVs, we had previously found that monomeric αS does not bind to DPPC-LUV (34). This was confirmed in these experiments (Fig. 2 D). The DMSO/Fe3+ induced Intermediate-II oligomers, however, bound with similar affinity to both POPC-LUV and DPPC-LUV (Fig. 2 D).
Binding of αS to lipid vesicles revealed by single-particle FRET
To obtain information about the interaction between the fluorophores attached to αS and the fluorophores attached to the lipids of the vesicles, we used a fluorescence resonance energy transfer (FRET) approach within our experimental setup (51,62). The same aggregation assay and confocal setup was used as above, but with excitation only at 488 nm. Virtually no FRET signal was recorded under any condition with POPC-SUV (Fig. 2 B). Even the generation of intermediate-II oligomers by DMSO and Fe3+, although bound to vesicles, did not generate a FRET signal. These findings corroborate, first, that monomeric αS did not bind to POPC-SUV, and second, that the interaction between BODIPY-PE in the lipids and oligomeric αS-647 aggregates, bound to the vesicles, was too weak to allow for FRET. In contrast, we could detect a significant FRET signal for BODIPY-PE-labeled DPPC-SUV and αS-647 monomers under all conditions, suggesting a close distance between the fluorophore of monomeric bound αS and the BODIPY label of the lipids. Addition of DMSO and/or Fe3+ did not alter the FRET signals (Fig. 2 C). This implies that the binding mode of monomeric αS to DPPC-SUV was not altered by the presence of DMSO and Fe3+, i.e., that aggregation of αS was prevented.
Quantitative FIDA analysis of oligomer formation by αS
2D-FIDA graphs show the full raw data of SIFT measurements and allow unbiased detection of different particle species, which differ in regard to brightness and ratio of two fluorescently labeled components. These data can be used for quantitative FIDA analysis, as described in detail in the Supporting Material. Control experiments demonstrated that the quantum yield of the used red and green fluorophores did not change upon additions of iron, DMSO, or SDS (Fig. S3). The average green particle brightness and concentration of the POPC- and DPPC-SUV did not differ in the various assay conditions (Fig. 2 E), showing that the vesicles had not dissolved upon the additions of DMSO and/or iron. Furthermore, binding of αS monomers or oligomers to the membrane did not dissolve the vesicles. The concentration of the green particles also matched the number expected from the incubated lipid concentration (10 μM) assuming ∼4000 lipid molecules per SUV (as expected for vesicles with a diameter of ∼25 nm). The brightness of the green particles corresponded to four BODIPY fluorophores per vesicles, as expected.
Evaluation of the average red particle brightness and concentration by fitting the FIDA data was also informative, despite the heterogeneity of the size of the oligomers that were formed in our aggregation assay. Apart from the monomers, a second population of brighter particles could be detected upon additions of DMSO +/− Fe3+ in the absence of lipid vesicles (control) and in the sample with POPC-SUV (Fig. 2 F). The brightness of the monomers (Q1) was always at ∼27 kHz for αS alone or incubated with Fe3+, whereas in the presence of DMSO a second population with Q2 of ∼400 could be detected for both the control and in the presence of POPC-SUV. Assuming that the quantum yield of Alexa647 was not different in the aggregates, the intermediate-I oligomers contained on average Q2/Q1 = 14 monomers. In the presence of DMSO/Fe3+, Q2 increased to ∼1400 for the control and 1100 in the presence of POPC-SUV. The fraction of aggregated αS in latter case was 13%, showing that despite oligomer formation the majority of αS in the sample had remained monomeric. The intermediate-II oligomers comprised ∼40–50 monomers. A remarkable difference between experiments without vesicles and samples with vesicles was that in the absence of vesicles, after the 2 h incubation with DMSO/Fe3+, the concentration of αS was <10% compared to t = 0, whereas almost no reduction occurred when vesicles had been present. Apparently the vesicles provided a sink for αS oligomers to bind to, preventing them from adsorbing to the wall of the well. When the data fit was performed for the case of DPPC-SUV, the particle brightness for vesicles in the red channel (Q2) did not change upon addition of DMSO and/or Fe3+ indicating that no aggregation occurred when αS monomers were tightly bound to the vesicles.
Binding of preformed oligomers to membranes
In a different set of experiments, we first allowed red-fluorescent αS to aggregate for 2 h after addition of 1% DMSO and 10 μM FeCl3. Upon a SIFT measurement of the sample, formation of intermediate-II oligomers was confirmed along the Y axis of the FIDA histogram (Fig. 3). Subsequently, green fluorescent POPC-SUVs were added to the oligomers and the sample again subjected to SIFT measurements. The FIDA histogram showed no free aggregates along the Y axis anymore (Fig. 3). On the contrary, bins with dual high red and green fluorescence intensities were observed, implying that virtually all oligomers had bound to the vesicles. In addition, a number of green bins remained along the X axis, representing vesicles devoid of bound αS. Similar results were obtained when DPPC-SUV were added to the preformed intermediate-II oligomers, showing that the oligomers—once formed—also had stably bound to these membranes. The absence of FRET signals in all incubation mixtures corroborated that oligomers (and no monomers) had bound to the vesicles. Quantitative analysis revealed that due to the 2 h preincubation of αS before the addition of the vesicles, ∼90% of the original amount of αS was lost from the solution, probably because it had associated with the wall of the well. The remaining oligomers were still detectable by SIFT measurement due to their high fluorescence intensity. The number of dual-color high-intense particles, however, was drastically reduced. Due to the low concentration of monomeric αS, signal of monomers bound to DPPC-SUV was below the detection limit in the SIFT and FRET measurements.
Detection of detergent-resistant intermediate-II oligomers
Toxic αS aggregates in PD patients, transgenic mouse and cell culture models of PD have been reported to be resistant to dissolution by detergents such as SDS (17,63,64). In fact, 0.2% SDS efficiently blocked de novo formation of intermediate-I and intermediate-II oligomers in our in vitro aggregation assay. However, once formed, intermediate-II oligomers were resistant to dissolution by SDS whereas intermediate-I oligomers completely dissolved (4). To evaluate these features in the presence of vesicles, we added 0.2% SDS after 2 h of incubation time to wells containing αS, vesicles, and DMSO/Fe3+ and repeated the SIFT measurements. In the case of POPC-SUV, SDS dissolved the lipid vesicles completely as expected. What remained were POPC/SDS mixed micelles generating bins close to the origin of the FIDA histogram, i.e., particles with much less BODIPY-PE-labels per particle compared to the POPC-SUV (Fig. 4 A). In addition, free SDS-resistant intermediate-II oligomers with high fluorescence intensity could still be detected along the Y axis. In the case of DPPC-SUV, SDS also solubilized the vesicles (Fig. 4 B). However, compared to the POPC-SUV, only signals at low photons/bin ratios were detected along the Y axis, thus representing αS monomers most likely bound to SDS/DPPC micelles. In contrast to the case of POPC-SUV, no SDS-resistant oligomers were detectable upon dissolution of the DPPC-SUV. These findings further support our earlier conclusion that binding of αS-monomers to DPPC membranes had prevented formation of DMSO/Fe3+-induced aggregates.
Figure 4.
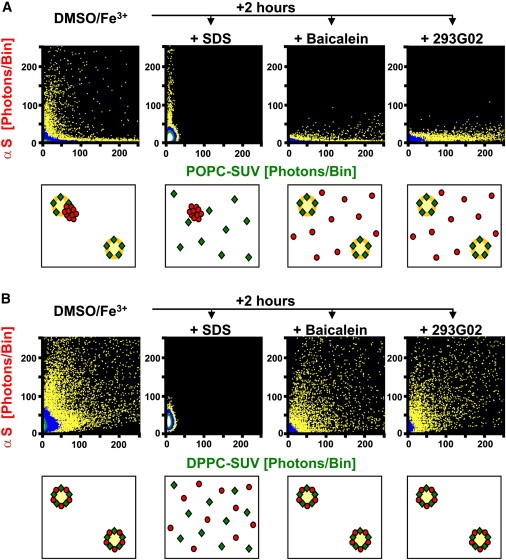
Solubilization of vesicles and αS oligomers. 2D-FIDA histograms of incubations of vesicles and αS aggregates. (A) POPC-SUV; (B) DPPC-SUV. (Panels from the left to the right) Vesicles with αS after 2 h incubation with 1% DMSO + 10 μM FeCl3, followed by solubilization with 0.2% SDS, 10 μM Baicalein, or 10 μM 293GO2, respectively. SDS dissolved the vesicles; Baicalein and 293GO2 dissolved the αS aggregates. (Cartoons, lower panels) The type of particles detected.
Aggregation inhibitors can dissolve membrane bound intermediate-II oligomers
Compounds inhibiting aggregation of αS-monomers have been identified in high throughput assays (4,17,65–73). We added two potent inhibitors (Baicalein and 293G02) after 2 h of incubation time to wells containing αS, vesicles, and DMSO/Fe3+ and repeated the SIFT measurements. In the case of POPC-SUV, vesicle-bound intermediate-II oligomers dissolved upon addition of the inhibitors, whereas intact vesicles remained detectable along the X axis (Fig. 4 A). The released αS monomers did not bind to the POPC-SUV, as expected. In the case of DPPC-SUV, upon addition of the inhibitors, the FIDA analysis displayed αS monomers bound to vesicles, similar to the case before the addition of the inhibitors (Fig. 4 B).
Discussion
The working hypothesis in many studies is that oligomeric aggregation intermediates rather than mature amyloid fibrils represent the principal toxic species in various neurodegenerative diseases. However, many issues remain unclear: 1), Numerous different oligomer species, formed in vivo as well as in vitro, have been described and it is not always clear how they are formed (6,74). 2), In vivo probably a heterogeneous population of αS oligomers coexists with monomers and fibrils, and it is not clear which subfraction of oligomer species is toxic. 3), The underlying molecular events causing their neurotoxicity have not been completely elucidated. Oligomers might bind to specific cytosolic proteins, organelles, or membrane receptors, change their function, or affect signaling pathways (6,75–77). Alternatively, annular oligomers might insert into membranes and make them leaky to ions (4,7,74,78,79). Whether the latter occurs by a pore-forming mechanism (i.e., insertion of protein aggregates into the membrane) or by some other kind of membrane-damaging mechanism remains to be clarified (4,74,80). In this study, association of the oligomers to vesicles was strong and might have permeabilized the vesicles but did not dissolve the vesicular structure. The latter was only found when detergent was added (Figs. 2 E and 4 A).
With regard to αS, studies on its aggregation are further complicated by its structural flexibility. Whether lipid binding impedes or enhances its aggregation is controversial. Using different techniques and various lipids, opposing conclusions were drawn (5,43,44,74,81–84). A high concentration of αS at the membrane surface and the hydrophobic interface provided by the membrane might enhance αS clustering and aggregation (12,85–87). Conversely, the α-helical structure of membrane-bound αS might impede misfolding to β-sheet-like conformers on pathway to fibrillar αS (88). This article provides evidence that the latter is the case. In this study, strong binding of αS to DPPC-SUV was quantitatively evaluated (Fig. 1, Supporting Material) and was consistent with other studies (33,34). The established structural switch of monomeric αS when it binds to lipid surfaces seems to impede its aggregation (Fig. 2). In contrast, when monomeric αS did not bind to lipids, i.e., in the presence of POPC-SUV, at the vesicle concentration used, aggregation of αS was not impeded (Fig. 2). The oligomers formed subsequently had a high propensity to bind to the vesicles.
Traces of organic solvents induce generation of a partially folded αS intermediates and accelerate amyloid formation (12,74,89). We used this approach as a model system to study aggregation pathways of αS in vitro at concentrations in the nanomolar range, allowing efficient oligomer characterization by confocal single particle fluorescence techniques. Additionally, we reasoned that oligomeric αS aggregation intermediates might be kinetically trapped, as amyloid formation is highly inefficient at low αS concentrations. Indeed, quantitative FIDA analyses of our aggregation studies showed that, in the case of DMSO/Fe3+, 87% of αS remained as monomers. Addition of traces of organic solvent lowers the dielectric constant of the buffer. The latter favors hydrophobic interactions. It should be noted that agitation, often used to accelerate aggregation, provides hydrophobic-hydrophilic interfaces created by air bubbles (85). Hence the driving force to accelerate aggregation by agitation might be the same as by adding traces of organic solvent. In light of the involvement of iron ions in the pathogenesis of PD we investigated the enhancement of αS aggregation in the presence of Fe3+ ions (4,13). Because of the very low solubility of Fe3+ and its association with αS, Fe3+ might seed αS aggregation (26). In our assay, Fe3+ alone did not enhance aggregation (Fig. 2, B and F). This is at variance with other reports where much higher αS and iron concentrations were used (13,25).
Conclusion
Association of monomeric αS to lipids and concomitant α-helical folding is a well-established binding mode. This article establishes a second binding mode of specific αS-oligomers formed in solution in the presence of Fe3+. Whereas binding of monomeric αS requires lipid surfaces with negative charges and/or packing defects, binding affinity of oligomers to membranes is high, regardless of their lipid composition or curvature. The two different binding modes could also be distinguished by FRET analysis. Bound monomers had a detectable FRET signal between the Alexa-647-labels of αS and the BODIPY-PE incorporated into the membranes, despite the small spectral overlap of the emission spectra of BODIPY-PE and the excitation-spectra of Alexa-647. On the other hand, bound oligomers displayed no detectable FRET signal. This is probably due to the larger distance between the two labels when oligomers are bound to the membranes and the much slower lateral diffusion of the oligomers compared to the monomers. Thus, despite the heterogeneity of the oligomers, the different single-molecule fluorescence techniques used in this study provided a powerful platform to distinguish between the two lipid binding modes of αS. Interestingly, membrane-bound oligomers readily dissolved in the presence of aggregation inhibitors (Fig. 4). The compounds neither dissolved the vesicles nor did they alter the binding of monomeric αS to membranes. This makes them an attractive tool for drug development in PD.
Acknowledgments
This work was supported by Collaborative Research Center SFB 596 (B13) of the Deutsche Forschungsgemeinschaft and the Lüneburg Foundation.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Koo E.H., Lansbury P.T., Jr., Kelly J.W. Amyloid diseases: abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Kuperstein I., Broersen K., De Strooper B. Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 2010;29:3408–3420. doi: 10.1038/emboj.2010.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kostka M., Högen T., Giese A. Single particle characterization of iron-induced pore-forming α-synuclein oligomers. J. Biol. Chem. 2008;283:10992–11003. doi: 10.1074/jbc.M709634200. [DOI] [PubMed] [Google Scholar]
- 5.Beyer K. Mechanistic aspects of Parkinson's disease: α-synuclein and the biomembrane. Cell Biochem. Biophys. 2007;47:285–299. doi: 10.1007/s12013-007-0014-9. [DOI] [PubMed] [Google Scholar]
- 6.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 7.Kayed R., Sokolov Y., Glabe C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- 8.Bucciantini M., Giannoni E., Stefani M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- 9.Spillantini M.G., Schmidt M.L., Goedert M. α-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 10.Iwai A., Masliah E., Saitoh T. The precursor protein of non-Aβ component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 11.Bartels T., Ahlstrom L.S., Beyer K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010;99:2116–2124. doi: 10.1016/j.bpj.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Munishkina L.A., Phelan C., Fink A.L. Conformational behavior and aggregation of α-synuclein in organic solvents: modeling the effects of membranes. Biochemistry. 2003;42:2720–2730. doi: 10.1021/bi027166s. [DOI] [PubMed] [Google Scholar]
- 13.Santner A., Uversky V.N. Metalloproteomics and metal toxicology of α-synuclein. Metallomics. 2010;2:378–392. doi: 10.1039/b926659c. [DOI] [PubMed] [Google Scholar]
- 14.Uversky V.N. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J. Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 15.Karpinar D.P., Balija M.B., Zweckstetter M. Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson's disease models. EMBO J. 2009;28:3256–3268. doi: 10.1038/emboj.2009.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dev K.K., Hofele K., van der Putten H. Part II: α-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology. 2003;45:14–44. doi: 10.1016/s0028-3908(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 17.Nonaka T., Watanabe S.T., Hasegawa M. Seeded aggregation and toxicity of α-synuclein and tau: cellular models of neurodegenerative diseases. J. Biol. Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glabe C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glabe C.G., Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006;66:S74–S78. doi: 10.1212/01.wnl.0000192103.24796.42. [DOI] [PubMed] [Google Scholar]
- 20.Volles M.J., Lansbury P.T., Jr. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 2002;41:4595–4602. doi: 10.1021/bi0121353. [DOI] [PubMed] [Google Scholar]
- 21.Caughey B., Lansbury P.T. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- 22.van Rooijen B.D., Claessens M.M., Subramaniam V. Membrane binding of oligomeric α-synuclein depends on bilayer charge and packing. FEBS Lett. 2008;582:3788–3792. doi: 10.1016/j.febslet.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 23.van Rooijen B.D., Claessens M.M., Subramaniam V. Lipid bilayer disruption by oligomeric α-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim. Biophys. Acta. 2009;1788:1271–1278. doi: 10.1016/j.bbamem.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 24.Uversky V.N., Li J., Fink A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure. J. Biol. Chem. 2001;276:44284–44296. doi: 10.1074/jbc.M105343200. [DOI] [PubMed] [Google Scholar]
- 25.Binolfi A., Rasia R.M., Fernández C.O. Interaction of α-synuclein with divalent metal ions reveals key differences: a link between structure, binding specificity and fibrillation enhancement. J. Am. Chem. Soc. 2006;128:9893–9901. doi: 10.1021/ja0618649. [DOI] [PubMed] [Google Scholar]
- 26.Peng Y., Wang C., Zhou F. Binding of α-synuclein with Fe(III) and with Fe(II) and biological implications of the resultant complexes. J. Inorg. Biochem. 2010;104:365–370. doi: 10.1016/j.jinorgbio.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies P., Moualla D., Brown D.R. α-synuclein is a cellular ferrireductase. PLoS ONE. 2011;6:e15814. doi: 10.1371/journal.pone.0015814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zayed J., Ducic S., Roy M. Environmental factors in the etiology of Parkinson's disease. Can. J. Neurol. Sci. 1990;17:286–291. [PubMed] [Google Scholar]
- 29.Logroscino G., Gao X., Ascherio A. Dietary iron intake and risk of Parkinson's disease. Am. J. Epidemiol. 2008;168:1381–1388. doi: 10.1093/aje/kwn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Götz M.E., Double K., Riederer P. The relevance of iron in the pathogenesis of Parkinson's disease. Ann. N. Y. Acad. Sci. 2004;1012:193–208. doi: 10.1196/annals.1306.017. [DOI] [PubMed] [Google Scholar]
- 31.Gaeta A., Hider R.C. The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy. Br. J. Pharmacol. 2005;146:1041–1059. doi: 10.1038/sj.bjp.0706416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davidson W.S., Jonas A., George J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- 33.Kamp F., Beyer K. Binding of α-synuclein affects the lipid packing in bilayers of small vesicles. J. Biol. Chem. 2006;281:9251–9259. doi: 10.1074/jbc.M512292200. [DOI] [PubMed] [Google Scholar]
- 34.Nuscher B., Kamp F., Beyer K. α-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J. Biol. Chem. 2004;279:21966–21975. doi: 10.1074/jbc.M401076200. [DOI] [PubMed] [Google Scholar]
- 35.Drescher M., Godschalk F., Huber M. Spin-label EPR on α-synuclein reveals differences in the membrane binding affinity of the two antiparallel helices. ChemBioChem. 2008;9:2411–2416. doi: 10.1002/cbic.200800238. [DOI] [PubMed] [Google Scholar]
- 36.Drescher M., Veldhuis G., Huber M. Antiparallel arrangement of the helices of vesicle-bound α-synuclein. J. Am. Chem. Soc. 2008;130:7796–7797. doi: 10.1021/ja801594s. [DOI] [PubMed] [Google Scholar]
- 37.Ulmer T.S., Bax A., Nussbaum R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005;280:9595–9603. doi: 10.1074/jbc.M411805200. [DOI] [PubMed] [Google Scholar]
- 38.Ferreon A.C., Gambin Y., Deniz A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kubo S., Nemani V.M., Fortin D.L. A combinatorial code for the interaction of α-synuclein with membranes. J. Biol. Chem. 2005;280:31664–31672. doi: 10.1074/jbc.M504894200. [DOI] [PubMed] [Google Scholar]
- 40.Middleton E.R., Rhoades E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010;99:2279–2288. doi: 10.1016/j.bpj.2010.07.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shvadchak V.V., Falomir-Lockhart L.J., Jovin T.M. Specificity and kinetics of α-synuclein binding to model membranes determined with fluorescent excited state intramolecular proton transfer (ESIPT) probe. J. Biol. Chem. 2011;286:13023–13032. doi: 10.1074/jbc.M110.204776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bartels T., Lankalapalli R.S., Brown M.F. Raftlike mixtures of sphingomyelin and cholesterol investigated by solid-state 2H NMR spectroscopy. J. Am. Chem. Soc. 2008;130:14521–14532. doi: 10.1021/ja801789t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narayanan V., Scarlata S. Membrane binding and self-association of α-synucleins. Biochemistry. 2001;40:9927–9934. doi: 10.1021/bi002952n. [DOI] [PubMed] [Google Scholar]
- 44.Rhoades E., Ramlall T.F., Eliezer D. Quantification of α-synuclein binding to lipid vesicles using fluorescence correlation spectroscopy. Biophys. J. 2006;90:4692–4700. doi: 10.1529/biophysj.105.079251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamp F., Exner N., Haass C. Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010;29:3571–3589. doi: 10.1038/emboj.2010.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nemani V.M., Lu W., Edwards R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scott D.A., Tabarean I., Roy S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J. Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burré J., Sharma M., Südhof T.C. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura K., Nemani V.M., Edwards R.H. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 2008;28:12305–12317. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parihar M.S., Parihar A., Ghafourifar P. Mitochondrial association of α-synuclein causes oxidative stress. Cell. Mol. Life Sci. 2008;65:1272–1284. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Behrends C., Langer C.A., Hartl F.U. Chaperonin TRiC promotes the assembly of polyQ expansion proteins into nontoxic oligomers. Mol. Cell. 2006;23:887–897. doi: 10.1016/j.molcel.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 52.Bieschke J., Giese A., Kretzschmar H. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc. Natl. Acad. Sci. USA. 2000;97:5468–5473. doi: 10.1073/pnas.97.10.5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Giese A., Levin J., Kretzschmar H. Effect of metal ions on de novo aggregation of full-length prion protein. Biochem. Biophys. Res. Commun. 2004;320:1240–1246. doi: 10.1016/j.bbrc.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 54.Levin J., Bertsch U., Giese A. Single particle analysis of manganese-induced prion protein aggregates. Biochem. Biophys. Res. Commun. 2005;329:1200–1207. doi: 10.1016/j.bbrc.2005.02.094. [DOI] [PubMed] [Google Scholar]
- 55.Kask P., Palo K., Gall K. Fluorescence-intensity distribution analysis and its application in biomolecular detection technology. Proc. Natl. Acad. Sci. USA. 1999;96:13756–13761. doi: 10.1073/pnas.96.24.13756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Giese A., Bader B., Kretzschmar H. Single particle detection and characterization of synuclein co-aggregation. Biochem. Biophys. Res. Commun. 2005;333:1202–1210. doi: 10.1016/j.bbrc.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 57.Hüls S., Högen T., Herms J. AMPA-receptor-mediated excitatory synaptic transmission is enhanced by iron-induced α-synuclein oligomers. J. Neurochem. 2011;117:868–878. doi: 10.1111/j.1471-4159.2011.07254.x. [DOI] [PubMed] [Google Scholar]
- 58.Berg D., Gerlach M., Becker G. Brain iron pathways and their relevance to Parkinson's disease. J. Neurochem. 2001;79:225–236. doi: 10.1046/j.1471-4159.2001.00608.x. [DOI] [PubMed] [Google Scholar]
- 59.Riederer P., Sofic E., Youdim M.B. Transition metals, ferritin, glutathione, and ascorbic acid in Parkinsonian brains. J. Neurochem. 1989;52:515–520. doi: 10.1111/j.1471-4159.1989.tb09150.x. [DOI] [PubMed] [Google Scholar]
- 60.Kamp F., Hamilton J.A., Hamilton J.A. Movement of fatty acids, fatty acid analogues, and bile acids across phospholipid bilayers. Biochemistry. 1993;32:11074–11086. doi: 10.1021/bi00092a017. [DOI] [PubMed] [Google Scholar]
- 61.Uversky V.N., Lee H.J., Lee S.J. Stabilization of partially folded conformation during α-synuclein oligomerization in both purified and cytosolic preparations. J. Biol. Chem. 2001;276:43495–43498. doi: 10.1074/jbc.C100551200. [DOI] [PubMed] [Google Scholar]
- 62.Jares-Erijman E.A., Jovin T.M. FRET imaging. Nat. Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 63.Kahle P.J., Neumann M., Haass C. Selective insolubility of α-synuclein in human Lewy body diseases is recapitulated in a transgenic mouse model. Am. J. Pathol. 2001;159:2215–2225. doi: 10.1016/s0002-9440(10)63072-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharon R., Bar-Joseph I., Selkoe D.J. The formation of highly soluble oligomers of α-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 65.Schiffer N.W., Broadley S.A., Schmid B. Identification of anti-prion compounds as efficient inhibitors of polyglutamine protein aggregation in a zebrafish model. J. Biol. Chem. 2007;282:9195–9203. doi: 10.1074/jbc.M607865200. [DOI] [PubMed] [Google Scholar]
- 66.Bieschke J., Russ J., Wanker E.E. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA. 2010;107:7710–7715. doi: 10.1073/pnas.0910723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Masuda M., Suzuki N., Hasegawa M. Small molecule inhibitors of α-synuclein filament assembly. Biochemistry. 2006;45:6085–6094. doi: 10.1021/bi0600749. [DOI] [PubMed] [Google Scholar]
- 68.Bertsch U., Winklhofer K.F., Giese A. Systematic identification of antiprion drugs by high-throughput screening based on scanning for intensely fluorescent targets. J. Virol. 2005;79:7785–7791. doi: 10.1128/JVI.79.12.7785-7791.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu M., Rajamani S., Fink A.L. The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J. Biol. Chem. 2004;279:26846–26857. doi: 10.1074/jbc.M403129200. [DOI] [PubMed] [Google Scholar]
- 70.Caruana M., Högen T., Vassallo N. Inhibition and disaggregation of α-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 2011;585:1113–1120. doi: 10.1016/j.febslet.2011.03.046. [DOI] [PubMed] [Google Scholar]
- 71.Hillmer A.S., Putcha P., Giese A. Converse modulation of toxic α-synuclein oligomers in living cells by N′-benzylidene-benzohydrazide derivates and ferric iron. Biochem. Biophys. Res. Commun. 2010;391:461–466. doi: 10.1016/j.bbrc.2009.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Necula M., Kayed R., Glabe C.G. Small molecule inhibitors of aggregation indicate that amyloid β oligomerization and fibrilization pathways are independent and distinct. J. Biol. Chem. 2007;282:10311–10324. doi: 10.1074/jbc.M608207200. [DOI] [PubMed] [Google Scholar]
- 73.Wang J., Ho L., Pasinetti G.M. Grape-derived polyphenolics prevent Aβ oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer's disease. J. Neurosci. 2008;28:6388–6392. doi: 10.1523/JNEUROSCI.0364-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kayed R., Pensalfini A., Glabe C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 2009;284:4230–4237. doi: 10.1074/jbc.M808591200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cisse M., Halabisky B., Mucke L. Reversing EphB2 depletion rescues cognitive functions in Alzheimer model. Nature. 2011;469:47–52. doi: 10.1038/nature09635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Resenberger U.K., Harmeier A., Tatzelt J. The cellular prion protein mediates neurotoxic signaling of β-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li S., Jin M., Selkoe D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 2011;31:6627–6638. doi: 10.1523/JNEUROSCI.0203-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kagan B.L., Hirakura Y., Lin M.C. The channel hypothesis of Alzheimer's disease: current status. Peptides. 2002;23:1311–1315. doi: 10.1016/s0196-9781(02)00067-0. [DOI] [PubMed] [Google Scholar]
- 79.Kim H.Y., Cho M.K., Zweckstetter M. Structural properties of pore-forming oligomers of α-synuclein. J. Am. Chem. Soc. 2009;131:17482–17489. doi: 10.1021/ja9077599. [DOI] [PubMed] [Google Scholar]
- 80.van Rooijen B.D., Claessens M.M., Subramaniam V. Membrane permeabilization by oligomeric α-synuclein: in search of the mechanism. PLoS ONE. 2010;5:e14292. doi: 10.1371/journal.pone.0014292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith D.P., Tew D.J., Cappai R. Formation of a high affinity lipid-binding intermediate during the early aggregation phase of α-synuclein. Biochemistry. 2008;47:1425–1434. doi: 10.1021/bi701522m. [DOI] [PubMed] [Google Scholar]
- 82.Bosco D.A., Fowler D.M., Kelly J.W. Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization. Nat. Chem. Biol. 2006;2:249–253. doi: 10.1038/nchembio782. [DOI] [PubMed] [Google Scholar]
- 83.Necula M., Chirita C.N., Kuret J. Rapid anionic micelle-mediated α-synuclein fibrilization in vitro. J. Biol. Chem. 2003;278:46674–46680. doi: 10.1074/jbc.M308231200. [DOI] [PubMed] [Google Scholar]
- 84.Perrin R.J., Woods W.S., George J.M. Exposure to long chain polyunsaturated fatty acids triggers rapid multimerization of synucleins. J. Biol. Chem. 2001;276:41958–41962. doi: 10.1074/jbc.M105022200. [DOI] [PubMed] [Google Scholar]
- 85.Pronchik J., He X., Talaga D.S. In vitro formation of amyloid from α-synuclein is dominated by reactions at hydrophobic interfaces. J. Am. Chem. Soc. 2010;132:9797–9803. doi: 10.1021/ja102896h. [DOI] [PubMed] [Google Scholar]
- 86.Pandey A.P., Haque F., Hovis J.S. Clustering of α-synuclein on supported lipid bilayers: role of anionic lipid, protein, and divalent ion concentration. Biophys. J. 2009;96:540–551. doi: 10.1016/j.bpj.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drescher M., van Rooijen B.D., Huber M. A stable lipid-induced aggregate of α-synuclein. J. Am. Chem. Soc. 2010;132:4080–4082. doi: 10.1021/ja909247j. [DOI] [PubMed] [Google Scholar]
- 88.Bodner C.R., Maltsev A.S., Bax A. Differential phospholipid binding of α-synuclein variants implicated in Parkinson's disease revealed by solution NMR spectroscopy. Biochemistry. 2010;49:862–871. doi: 10.1021/bi901723p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Munishkina L.A., Fink A.L., Uversky V.N. Conformational prerequisites for formation of amyloid fibrils from histones. J. Mol. Biol. 2004;342:1305–1324. doi: 10.1016/j.jmb.2004.06.094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.