

Abstract
Polyglutamine tract-binding protein-1 (PQBP-1) is a 265-residue nuclear protein that is involved in transcriptional regulation. In addition to its role in the molecular pathology of the polyglutamine expansion diseases, mutations of the protein are associated with X-linked mental retardation. PQBP-1 binds specifically to glutamine repeat sequences and proline-rich regions, and interacts with RNA polymerase II and the spliceosomal protein U5-15kD. In this work, we obtained a biophysical characterization of this protein by employing complementary structural methods. PQBP-1 is shown to be a moderately compact but largely disordered molecule with an elongated shape, having a Stokes radius of 3.7 nm and a maximum molecular dimension of 13 nm. The protein is monomeric in solution, has residual β-structure, and is in a premolten globule state that is unaffected by natural osmolytes. Using small-angle x-ray scattering data, we were able to generate a low-resolution, three-dimensional model of PQBP-1.
Introduction
Polyglutamine tract-binding protein-1 (PQBP-1) is a nuclear protein that is expressed predominantly in the central nervous system. It was initially isolated as a binding partner of Brn-2, a brain-specific transcription factor that harbors a glutamine repeat (polyQ) tract (1–3). Although the exact cellular function of PQBP-1 has yet to be elucidated, its temporal expression pattern suggests a role in neuron proliferation and development (3). Studies of the PQBP-1 homologs in mice, nematode, and fly revealed its involvement in lipid storage, learning, and long-term memory processes (4). At the molecular level, PQBP-1 has been shown to be involved in transcription activity (2,5) and pre-mRNA splicing (6).
PQBP-1 consists of the WW domain, the polar amino-acid-rich domain (PRD), and the C-terminal domain (CTD; Fig. 1 a). The WW domain (residues 48–81) plays a role in transcription regulation, owing to its interaction with RNA polymerase II and proline-rich motifs (6–8). The PRD features five hepta-polar amino-acid-rich repeats spanning residues 104–138, followed by an imperfect run of repeating pairs of oppositely charged residues, aspartate-arginine or glutamate-arginine, (DR/ER repeat) up to residue 163. This region of high charge density mediates binding to polyQ tracts (2). The CTD (residues 190–265) is highly conserved among PQBP-1 homologs and interacts with the spliceosomal protein U5-15kD (9), linking PQBP-1 to the RNA processing machinery of the cell.
Figure 1.
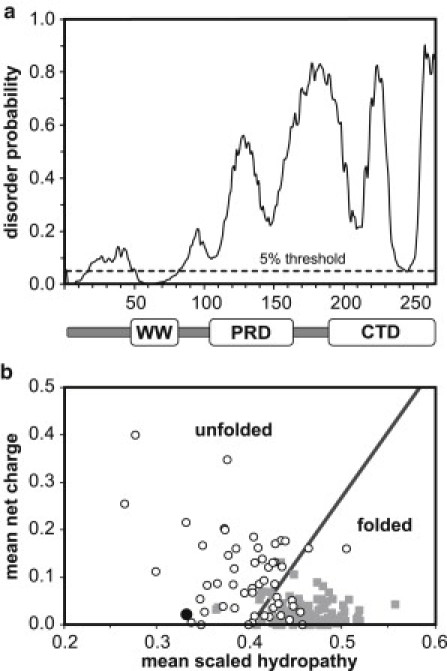
Primary sequence analysis of PQBP-1. (a) DISOPRED2 prediction of local disorder as a function of residue number. The dotted line indicates a 5% false-positive threshold. The domain structure of PQBP-1 is shown at the bottom. (b) Plot of net charge versus hydropathy as a predictor of disorder, compared with a database of folded (gray squares) and unfolded (open circles) proteins. The position of PQBP-1 is shown as a black circle. The boundary condition is shown as a sloping line having the equation , where is the absolute mean net charge and is the mean scaled hydropathy (20).
The binding of PQBP-1 to polyQ tracts led researchers to investigate the potential roles the protein plays in the polyglutamine expansion diseases. These inherited neurodegenerative disorders are characterized by the abnormal lengthening of an otherwise harmless glutamine repeat in the respective disease proteins. The role of PQBP-1 has been specifically addressed in the case of spinocerebellar ataxia type-1. Like the disease protein Ataxin-1, PQBP-1 is expressed at high levels in affected regions of the brain, and both proteins localize to the nucleus and deposit as inclusions (10). The ternary complex of PQBP-1, Ataxin-1 with an expanded polyQ tract and RNA polymerase II, was shown to cause cell death by synergistically repressing transcription (10).
In recent years, investigators have identified 11 mutations in the PQBP-1 gene, all of which result in X-linked mental retardation (XLMR) (11–17). In syndromic types, typical sufferers present mental retardation, microcephaly, leanness, and mild short stature (17), collectively known as Renpenning syndrome. Carrier females do not show remarkable phenotypic affects (12), but the details are not yet known. Five XLMR mutations have been located in the PRD of PQBP-1 (12), the most frequently reported one being p.Arg153SerfsX41 in the DR/ER repeat as a result of mutation in a particularly unstable AG-rich region of exon 4 (13,14,16,17); five other mutations are C-terminal to the PRD. Eight of these 10 mutations are frame shifts that result in PQBP-1 proteins with mutated and truncated C-termini, thus affecting its interaction with the spliceosomal protein U5-15kD. A missense mutation, Y65C, within the WW domain of PQBP-1 causes Golabi-Ito-Hall syndrome (11).
Given the particular medical significance of PQBP-1, we sought to investigate its molecular structure. In a previous study, PQBP-1 was established to be intrinsically unstructured, with a small folded WW domain (18). Spectroscopic evidence suggests that the Y65C mutation disrupts the fold of the isolated WW domain and reduces the domain's affinity for proline-rich ligands (8). Here we employed small-angle x-ray scattering (SAXS) in combination with other biophysical techniques to obtain a structural model of full-length PQBP-1 in solution.
Materials and Methods
Primary sequence analysis
The primary sequence of PQBP-1 was analyzed with the use of DISOPRED2 (19) to identify regions of disorder. The mean net charge and hydropathy of PQBP-1 were calculated and plotted (20) with the use of PONDR (21). The frequency of order- and disorder-promoting residues was calculated based on the classification proposed by Dunker et al. (22).
Construction of expression plasmid
The cDNA encoding PQBP-1 was amplified from the plasmid GST-B83-4/PCR (1,10) by polymerase chain reaction (PCR) with the following primers:
5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCCCTGGAAGTTCTGTTCCAGGGGCCCATGCCGCTGCCCGTTGCG-3′ and
5′-GGGGACCACTTTGTACAAGAAAGCTGGGTTCAATCCTGCTGCTTGGTTCG-3′
The PCR product was subcloned into the Gateway donor vector pDONR221 (Life Technologies, Grand Island, NJ) to generate the entry clone, which was subsequently recombined with the destination vector pHMGWA (23) to create the expression vector, pHMGW-PQBP1. This plasmid encodes for PQBP-1 fused with an N-terminal hexahistidine and maltose-binding protein (His6-MBP) tag, which is cleavable with PreScission protease.
Expression and purification
The fusion protein His6-MBP-PQBP-1 was overexpressed in Escherichia coli strain Rosetta 2 DE3 (Merck, Darmstadt, Germany). The cells were cultured at 37°C to an absorbance at 600 nm of 0.6 when expression was induced by addition of isopropyl-β-d-thiogalactopyranoside to 0.5 mM, followed by growth at 18°C for 16 h. Cells were pelleted and resuspended in binding buffer (20 mM Na2HPO4 pH 7.4, 150 mM NaCl) with 40 mM imidazole. The cleared lysate was loaded onto a HisTrap column (GE Healthcare, Little Chalfont, UK), and purification was performed according to the manufacturer's instructions. The purified sample was dialyzed against PreScission buffer (20 mM Tris-HCl pH 7.0, 150 mM NaCl, 1 mM DTT, 1 mM EDTA), and digested with PreScission protease at 4°C overnight. The digested samples were loaded onto a HisTrap column again, and PQBP-1 was eluted with 150 mM imidazole. Finally, the proteins were loaded onto a Superdex 75 column (GE Healthcare) and eluted in phosphate-buffered saline (PBS, pH 7.4). Their identity was confirmed by electrospray ionization mass spectrometry.
Size exclusion chromatography
We performed analytical gel filtration with a Superdex 75 16/60 column to determine the molecular mass of PQBP-1 by comparing its elution volume (Ve) with that of reference proteins. The void volume (V0) was determined with Dextran blue. We determined the Stokes radius (RS) of PQBP-1 using a linear calibration plot of [–log(Kav)]1/2 against RS of the standards, where the partition coefficient, Kav = (Ve – V0) / (Vt – V0), with Vt being the total volume of the column.
Analytical ultracentrifugation
Sedimentation velocity experiments were performed on a Beckman Coulter model XL-A analytical ultracentrifuge equipped with UV scanning optics. PQBP-1 in PBS (380 μL) at concentrations of 0.83, 0.42, and 0.17 mg/mL was loaded into 12 mm double-sector cells with quartz windows along with a reference buffer (PBS). Experiments were performed with centrifugation at 50,000 rpm at 20°C, with 324 scans monitored at 280 nm collected continuously.
Data from the first 160 scans were fitted to a continuous sedimentation coefficient distribution c(s) via the program SEDFIT (24). This model employs direct boundary modeling with a distribution of Lamm equation solutions to derive a differential s distribution, with diffusion effects (24). The buffer density (ρ = 1.005235 g/mL), buffer viscosity (η = 1.018906 cP), and partial specific volume of the sample ( = 0.711319 mL/g) at 20°C were computed with the program SEDNTERP (25). Regularization of the c(s) distribution was based on the simplex algorithm by maximum entropy method with a confidence interval of P = 0.95, with the frictional coefficient ratio (f/f0) as a parameter. Model fitting was repeated until the lowest root-mean-square deviation between the data and the calculated curves was achieved. The sedimentation coefficients of the peaks were corrected to s20,w, the standard s-values in water at 20°C. The values of s20,w and f/f0 were plotted against sample concentration, and the corresponding values at infinite dilution, and f/f00, were obtained by extrapolation. The molar mass M corresponding to each peak in the c(s) distribution was calculated with the equation M = (sRT) / [D (1– ρ)], where T is the temperature, R is the gas constant, D is the diffusion coefficient of the sedimenting species characterized by the sedimentation coefficient s, and ρ and are defined as above. The Stokes radius is calculated as implemented in the calculator tool of SEDFIT.
NMR
The 1H NMR spectrum of PQBP-1 in native buffer (20 mM Tris-HCl pH 7.0, 150 mM NaCl, 1 mM DTT) was acquired at a concentration of 0.2–0.5 μM (0.06–0.16 mg/mL) on a Varian 600 MHz spectrometer at 25°C. The water signal was suppressed by the WATERGATE pulse sequence (26). The final spectrum was acquired by summing 128 scans.
Circular dichroism
Simultaneous UV and circular dichroism (CD) spectra of PQBP-1 at 0.35 mg/mL in CD buffer (20 mM Tris-HCl pH 7.0, 150 mM NaF) were measured on a Chirascan-Plus spectrometer (Applied Photophysics Ltd., Leatherhead, UK). Spectral data were collected from 260–180 nm in a 0.01 mm rectangular spacer cell at 20°C. Measurements were recorded with a 1 nm step size, a 1 s acquisition time-per-point, and a spectral bandwidth of 1–2 nm. For variable temperature measurements, the CD spectra of 0.17 mg/mL PQBP-1 were recorded in a 0.5 mm pathlength cell. The sample was heated from 6°C to 94°C and then cooled to 20°C at a rate of 1°C per minute with a 2°C step size. A 0.8 s time-per-point acquisition time and 1 nm step-size were employed in the 260–190 nm region with a 2 nm spectral bandwidth.
We calculated the relative frequency of secondary structural elements of PQBP-1 via the DichroWeb server (27) using the CDSSTR algorithm (28) and reference set 3, for data from 185 –240 nm. Deconvolution yielded a best solution and many other valid solutions, as assessed by the normalized standard deviation (NRMSD). The top 20 solutions were analyzed for the general trend in secondary structure composition.
Florescence spectroscopy
1-Anilino-8-napthalene-sulfonate (ANS; Sigma, St. Louis, MO) is a fluorescence probe that is commonly used to detect the molten globule state of proteins (29). The fluorescence spectra of 20 μM ANS was monitored at 20°C upon incubation with 20 μM (0.62 mg/mL) PQBP-1 in PBS, PBS with 1 M l-proline, PBS with 1 M trimethylamine N-oxide (TMAO), and PBS with 8 M urea in a FluoroMax-4 spectrofluorometer (Horiba Scientific, Edison, NJ). The fluorescence spectra of ANS in the same buffers without protein were also recorded. An excitation wavelength of 370 nm was used, and the emission spectra were recorded from 400–600 nm. All samples were incubated in the cell compartment at 20°C for >5 min before measurements were taken.
SAXS experiments and data processing
SAXS experiments were performed at the European Molecular Biology Laboratory X33 beamline of the Deutsches Elektronen-Synchrotron (Hamburg, Germany) using a robotic sample changer. The scattering by PQBP-1 in native buffer was measured at five concentrations ranging from 1 to 13 mg/mL on a Pilatus 1M detector (Dectris, Baden, Switzerland). The sample–detector distance was 2.7 m covering the momentum transfer range 0.07 < q < 6.24 nm−1, with q = (4π/λ) sinθ, where 2θ is the scattering angle, and wavelength λ = 1.5 Å. A standard acquisition time of 2 min was used for all samples, and divided into eight 15-s frames to assess and correct for radiation damage.
We processed the data using standard procedures for ATSAS programs (30). We evaluated the forward scattering I(0) and the radius of gyration Rg using the Guinier approximation (31). We also computed these parameters from the entire scattering patterns using the indirect transform package GNOM (32), which also provided the pair distribution function of the particle p(r) and the maximum size Dmax. We evaluated the molecular mass of the sample by comparing the forward scattering I(0) with that from a reference solution of bovine serum albumin.
Structural modeling of the SAXS data
A Kratky plot was employed to qualitatively assess the overall conformational state of PQBP-1 (33). To obtain quantitative estimates of the degree of the dynamics and conformational heterogeneity, we analyzed the SAXS data using an ensemble optimization method (EOM) (34). First, we used the program RANCH to generate a pool of 10,000 random structures, and calculated the scattering curve of each structure with the use of CRYSOL (35). Initial pools were generated with either native-like (more compact) or completely random chains. In each case, two models, one with and one without a structured WW domain (built with MODELLER as described previously (6)), were generated. Each of these four initial pools was subjected to a genetic algorithm (GAJOE) for selection of subsets of ∼20 protein structures such that the discrepancy (the χ2 value) between the average scattering of the ensemble model and the experimental data was minimized. For each pool, 12 independent EOM runs were performed, and the obtained subsets were analyzed to yield the Rg and Dmax.
The low-resolution ab initio shape of PQBP-1 was reconstructed with the use of DAMMIF (36). Compact models of interconnected beads were used to represent the molecule, and simulated annealing was employed to minimize the discrepancy between the experimental and calculated data. Ten DAMMIF models were generated and analyzed using DAMAVER (37), which characterizes the similarity between the individual ab initio models by the normalized spatial discrepancy (NSD) (38). The most representative model (with the lowest average NSD) was visualized in the graphics program QuteMol (39) with the sphere radius set to 2.7 Å.
Availability of materials
The coordinates of the average bead model of PQBP-1 and the color version of all figures can be obtained from the corresponding authors.
Results
PQBP-1 is predicted to be disordered
The primary sequence of PQBP-1 was analyzed to predict disorder in the protein at both local and global scales. Its amino-acid composition shows a significantly higher proportion of disorder-promoting residues (59.6%) and a lower proportion of order-promoting residues (17%) relative to the typical composition for globular proteins (47.4% and 36.2%, respectively). The DISOPRED2 server predicts order at the region corresponding to the WW domain (Fig. 1 a). A stretch of ∼100 residues then follows, encompassing the PRD, which is predicted with confidence to be disordered. The CTD is predicted to be disordered, but with lower confidence. The prediction mirrors a previous disorder prediction of PQBP-1 made with PONDR (18). Overall, PQBP-1 has a low net charge and low overall hydropathy, and falls in the disordered region of the charge/hydropathy plot (Fig. 1 b).
PQBP-1 is monomeric and nonglobular
During size exclusion chromatography (SEC), PQBP-1 eluted from the column in two peaks (a small one followed by a major peak; Fig. 2 a). The two peaks correspond to apparent molecular masses of 89.7 (±2.3) kDa and 63.3 (±1.5) kDa, and Stokes radii of 4.36 (±0.63) nm and 3.50 (±0.55) nm, respectively. The first peak corresponds to an apparent molecular mass exceeding the resolving power of the S75 column (>75 kDa) and contains aggregated proteins. The major peak has an elution volume that corresponds to two times the molecular mass of PQBP-1 (30.6 kDa), suggesting that the protein is dimeric if it adopts a compact, globular conformation. However, it is well established that elongated or unfolded proteins elute at a volume corresponding to a higher apparent molecular mass (40).
Figure 2.
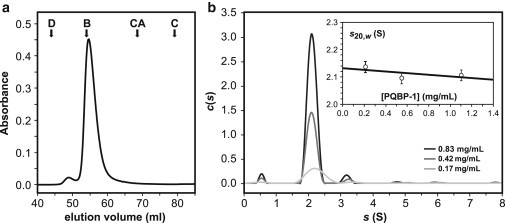
Hydrodynamic properties of PQBP-1. (a) SEC elution profile of PQBP-1 with the corresponding elution volume of molecular mass markers indicated: D, dextran; B, bovine serum albumin (66 kDa, RS = 3.64 nm); CA, carbonic anhydrase (29 kDa, RS = 2.22 nm); C, cytochrome C (12 kDa, RS = 1.69 nm). The small peak at an elution volume of ∼50 ml corresponds to aggregated samples. The apparent molecular mass of the major peak was determined from a calibration plot made with the standard proteins. (b) Velocity sedimentation AUC results analyzed with SEDFIT are presented as the concentration distribution c(s) of sedimentation species at three sample concentrations. The inset shows the sedimentation coefficients corresponding to the major peak in c(s) plotted against their respective sample concentrations to obtain the value at infinite dilution.
Analytical ultracentrifugation (AUC) was carried out to clarify the oligomeric state of PQBP-1. Sedimentation velocity studies showed that PQBP-1 species were homogeneous in solution, with a single species observed for each preparation (Fig. 2 b). Over the concentration range tested, PQBP-1 showed no sign of self-interaction. A distribution analysis revealed a single peak at = 2.13 (±0.02) S, which accounts for >95% of the sedimenting materials and corresponds to a molar mass of 31.0 (±0.5) kDa, in excellent agreement with the monomeric molecular mass. The frictional coefficient ratio (f/f0) has a value of 1.74 (±0.09), which suggests that PQBP-1 is highly elongated. The Stokes radius of PQBP-1 calculated from these experiments is 3.67 (±0.02) nm.
PQPB-1 is disordered but retains residual structure
We recorded a one-dimensional (1D) 1H NMR spectrum to assess the overall solution state of PQBP-1 (Fig. S1 in the Supporting Material). The chemical shift range covered by the spectrum is narrow, with the highest shifts visible around 8.5 ppm, and the lowest at ∼0.5 ppm. The resonances of the amide and aromatic protons, expected to be at the left of the central water signal (4.7 ppm), are all clustered within ∼2 ppm (6.5–8.5 ppm). This lack of chemical shift dispersion is typical of unfolded and/or highly flexible proteins. However, the presence of a peak at ∼0.2 ppm and of two very faint resonances at 10.4 and 10.9 ppm is in agreement with locally folded regions, such as the WW domain. These resonances are likely to account for at least two of the four tryptophan indole protons. One would expect to see more dispersed peaks in the backbone region due to the folded WW domain. However, the resonances of the more-substantial unfolded regions can dominate and determine the dynamic range of the spectrum. Folded and unfolded regions are also expected to have different degrees of flexibility and exchange regime, thus resulting in resonances with very different linewidths.
The CD spectrum of PQBP-1 (Fig. 3 a) is typical of a protein or peptide without regular conformation: it lacks spectral features or any positive signals in the far-UV range, and has a characteristic negative maximum at 200 nm (41,42). The absence of a positive CD signal below 200 nm indicates that there is very little or no helical structure. The spectrum was deconvoluted satisfactorily, with the back-calculated spectrum closely resembling the experimental data, as reflected in a low NRMSD value of 0.017 (Fig. 3 a). From the top 20 solutions (data not shown), the spectrum is found to consist of a significant amount of β-strand structure (∼20–50%) and turns (∼15–40%), but little helical content (∼5–10%). The remaining regions of the protein (∼25–55%) are irregular structures. The variable temperature CD results are also typical of a disordered protein, with a broad melting profile (Fig. 3 b). The CD spectra of PQBP-1 recorded at high temperatures differ significantly from those recorded at 20°C, indicating that the protein undergoes reversible denaturation, with a melting temperature (Tm) of ∼34°C (Fig. 3 b, inset). Native conformations are restored when the protein is subsequently cooled.
Figure 3.
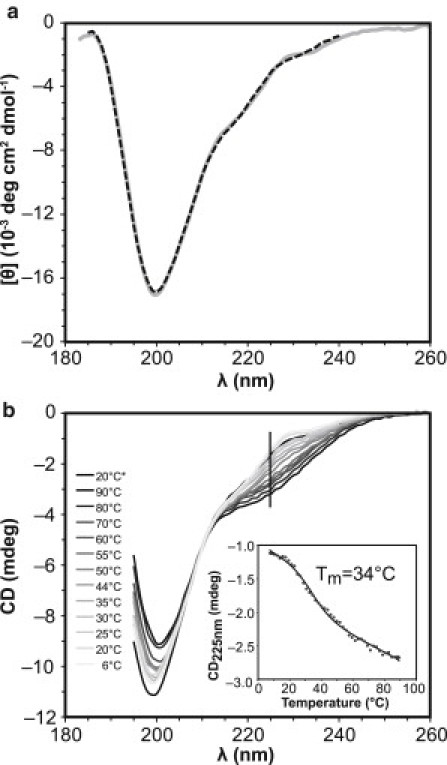
CD of PQBP-1. (a) The far-UV CD spectrum (molar ellipticity) is shown as a gray line. The dashed black line (185–240 nm) is the back-calculated spectrum after deconvolution by the CDSSTR algorithm. (b) Temperature dependence of the CD spectra of PQBP-1. The sample was heated from 6°C (lightest gray) to 90°C (dark gray), and then cooled to 20°C (∗, black). The inset shows the CD at 225 nm as a function of temperature.
PBQB-1 is in the premolten globule state
PQBP-1 is natively disordered yet possesses a substantial amount of secondary structure contents, which fits a description of the molten globule, an intermediate state between the unfolded and folded states of proteins. Incubation of PQBP-1 with ANS in the native condition (PBS) results in a blue shift of the fluorescence spectral peak from 510 nm to 475 nm accompanied by a threefold increase in quantum yield (Fig. 4 a). These effects are characteristic of ANS binding to a species adopting a premolten or molten-globule conformation. The relatively small increase in quantum yield suggested that ANS binding to PQBP-1 is not hydrophobic but instead is electrostatic in nature (43). Next, we tested whether the natural osmolytes TMAO and proline could promote a more folded conformation of PQBP-1, because it is known that these small molecules can induce structure in certain partially folded or denatured proteins (44,45). We found that addition of TMAO or proline did not alter the blue shift or intensity increase effects upon PQBP-1 binding to ANS (Fig. 4, c and d). In 8 M urea, the ANS fluorescence spectrum collected in the presence of PQBP-1 showed a small increase in peak height (at 515–520 nm) but no blue shift, suggesting that PQBP-1 is totally unfolded in denaturing conditions (Fig. 4 b).
Figure 4.
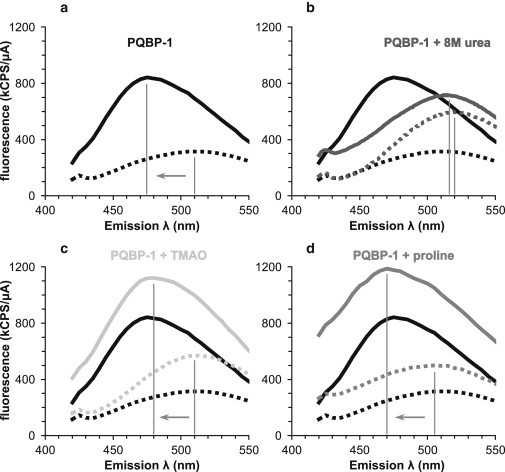
ANS fluorescence spectroscopy of PQBP-1. Each set of measurements consists of two spectra, one with ANS and buffer only (dashed line) and the other with ANS, buffer, and PQBP-1 (solid line). (a and b) Spectra in the (a) native condition (PBS, black lines) and (b) denaturing condition (8 M urea, dark gray lines). (c and d) Spectra with osmolytes in PBS: with (c) 1 M TMAO (light gray lines) and (d) with 1 M proline (gray lines). The peak of each spectrum is identified with a vertical line, and the blue shift caused by PQBP-1 is indicated by an arrow. Spectra in the native condition (black lines) are shown for comparison in all panels.
We then calculated the theoretical Stokes radii of monomeric PQBP-1 in various known conformations (Table 1). There is a linear dependency between log(RS) and the logarithm of molecular mass for proteins in different states (46). The RS of PQBP-1 obtained from gel filtration (3.50 nm) agrees well with that determined from sedimentation velocity experiments (3.67 nm), and that predicted for a natively disordered premolten globule protein (3.70 ± 0.41 nm). It also falls into the broad range (3.53 ± 2.09 nm) covered by the premolten globule state of a folded protein, and marginally into the natively unstructured random coil conformations. The calculated RS for other conformational states do not agree with the experimentally determined value. This analysis strongly supports the notion that PQBP-1 adopts a premolten globule conformation.
Table 1.
Theoretical Stokes radii for monomeric PQBP-1
| Conformation model | RS, nm |
|---|---|
| Globular states | |
| Folded | 2.49 ± 0.18 |
| Molten globule | 2.78 ± 0.85 |
| Premolten globule | 3.53 ± 2.09 |
| Unfolded, urea/GdnHCl | 4.87 ± 2.7 / 5.15 ± 5.4 |
| Natively unstructured states | |
| Premolten globule | 3.70 ± 0.41 |
| Unfolded | 4.57 ± 0.81 |
Values were calculated with a molecular mass of 30,472 Da in different conformation models.
Solution model of PQBP-1
We employed SAXS to study the overall shape and conformational flexibility of PQBP-1 in solution. The processed scattering pattern of PQBP-1 (Fig. 5 a) yields an Rg of 3.8 (±0.2) nm, with a molecular mass of 30 (±3) kDa, suggesting that the protein remains monomeric in solution. The ratio of the Rg to the hydrodynamics radius (determined by AUC and SEC) is an indicator of the compactness of a protein. PQBP-1 has an Rg/RS ratio of 1.0, which is intermediate between the value of a spherical species (0.8) and that of an unstructured random coil (1.5) (47). The Kratky plot (Fig. 5 b, inset) is not indicative of a compact, globular protein (which is typically bell shaped) or a fully unfolded protein, which would show a plateau or upward slope at higher scattering angles. Instead, this plot shows a peak at approximately q = 1 nm−1 followed by a shallow downward slope, suggesting that PQBP-1 is partially unfolded. The p(r) function of PQBP-1 has an asymmetric shape (Fig. 5 c), with its maximum at low r followed by a long tail. This is again consistent with an extended, perhaps partially unfolded molecule with the maximum dimension (Dmax) equal to 13 nm.
Figure 5.
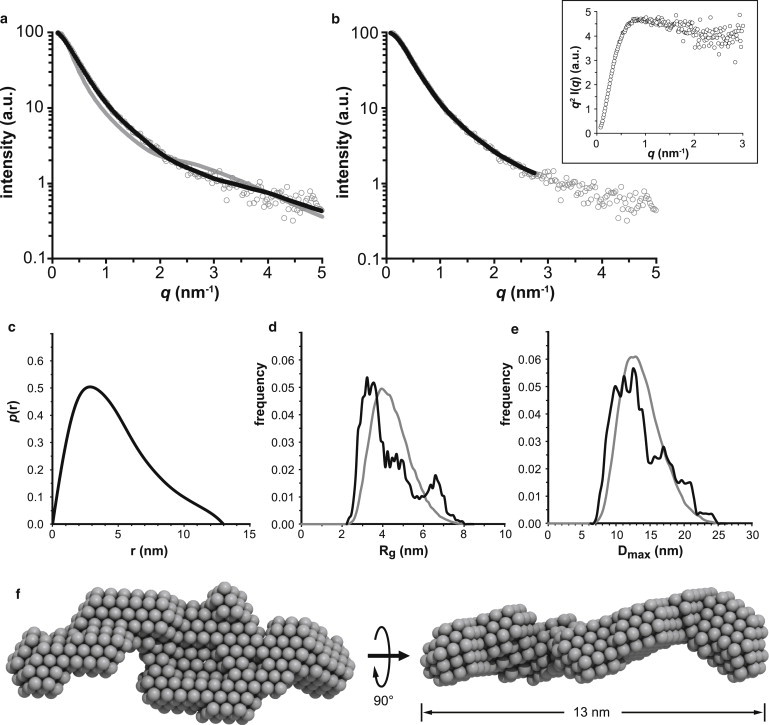
SAXS analysis of PQBP-1. The experimental x-ray scattering data are shown as open circles, along with the calculated scattering curves (black) from (a) EOM analysis and (b) DAMMIF reconstruction. The calculated scattering curve of the initial randomized ensemble before EOM is shown in gray in panel a. The Kratky plot is shown as an inset. (c) The p(r) distribution of PQBP-1. (d and e) Results of the EOM study showing the distribution of the Rg (d) and Dmax (e). The distributions of the random pool are shown in gray, and those of the selected ensemble are in black. (f) Two views of the ab initio bead model of PQBP-1 calculated using DAMMIF. The model shown is the most representative of 10 structures calculated. a.u., arbitrary unit.
Because the Kratky plot, p(r) function, and other biophysical analyses of PQBP-1 all indicated that the protein is partially unfolded, we employed an ensemble method (EOM) to quantitatively assess its flexibility. The best EOM solution was obtained from the most compact initial pool that employs the fixed WW domain and the native-like chains, with a χ2 value of 0.86 (Fig. 5 a). A value of <1 indicates a good agreement between the calculated scattering of the ensemble model and the experimental scattering (35). In contrast, the data generated from the average over the random conformations (i.e., the initial pool for the EOM ensemble) display significant systematic deviations from the data (Fig. 5 a).
The EOM-selected ensemble shows a major fraction of the population (Fig. 5 d) displays Rg values of ∼3.5 nm, which is much smaller than the average Rg of the random population of 4.2 nm. This suggests that the optimized ensemble was even more compact and more enriched with the structures than the random pool (Fig. 5, d and e). There is also a peak with Rg values well over 6 nm. Together, these findings indicate that the protein is compact but explores a conformation space that includes rather extended dimensions.
Because the results from the CD, NMR, and EOM analyses provided evidence that PQBP-1 contains some structure and is not fully unfolded, we performed ab initio modeling to visualize its average solution conformation. Ten independent models were generated by DAMMIF, and all fit the experimental data well with χ2-values of ∼0.9 (Fig. 5 b). The models align with an average NSD value of 0.78, indicating a stable shape reconstruction (48). For comparison, we also computed the scattering curves from the best-fit ellipsoid and a sphere model, and the results show that the data cannot be reconciled with a simple shape model (Fig. S2). The most representative DAMMIF model, presented in Fig. 5 f, has an extended conformation, with both ends protruding from a bulkier core. The longest molecular dimension of the model is 13 nm, in agreement with the Dmax obtained from the p(r) function.
Discussion
From this combined biophysical study, a molecular view of PQBP-1 is emerging. The protein is monomeric but its shape deviates greatly from globularity. The hydrodynamic behavior of PQBP-1 is intermediate between that of a fully folded protein and that of a completely random coil. It contains some local structure and is moderately compact but lacks a folded core. The ab initio molecular shape reconstructed from the SAXS data results in an elongated molecule.
Our results on the residual structure agree well with previous findings (18). Although the NMR spectrum is too broad to allow appreciation of details, due to the molecular mass of the protein, it does reveal some chemical shift dispersion, indicating the presence of some structure. The far-UV CD spectrum also agrees with that described in the earlier study (18) and is typical of a mostly unstructured peptide. Here, we extended that study significantly by including CD data beyond 200 nm, which is important in the deconvolution of the spectrum. We interpreted the CD results with caution given that current methods of CD spectral analysis are not ideally adaptable to proteins with a large degree of disorder, because the reference databases consist mainly of globular folded proteins (27). Nevertheless, the top 20 deconvolution solutions show a clear consensus regarding the range of secondary structures present, reflecting the fact that PQBP-1 adopts dynamic and transient conformations in the solution state. The variable-temperature CD experiment shows that PQBP-1 follows a noncooperative melting between an extended and an unfolded state. With a Tm of ∼34°C, any order is likely to be relatively transient at physiological temperatures.
The experiments reported here demonstrate conclusively that PQBP-1 is best described as being in the premolten globule state, which was originally investigated in the context of protein folding, i.e., the intermediate state en route to becoming a globular protein (49). The characteristics of the molten globule are that it has similar secondary structural elements with the native folded state, but the tertiary interactions are absent; the protein remains globular but loosely packed. The premolten globule is a state in which only ∼50% of native secondary structures are present and the overall conformation is extended and nonglobular (33). The molten globule and premolten globule states can be detected when natively globular proteins are subjected to various denaturing conditions. The propensity for proteins to be unstructured under physiological conditions was later appreciated by Wright and Dyson (50). In particular, this has been observed in the eukaryotic proteome: it was calculated that whereas 2–4% of prokaryotic proteins contain disordered regions, 33% of eukaryotic proteins contain sizable regions (>30 residues long) of native disorder (19).
The persistence of intrinsically disordered states in the proteomes of organisms demonstrates the crucial role that conformations play in protein function. Proteins containing disorder are overrepresented in certain functional classes (19), including transcriptional regulation and development processes in which PQBP-1 is involved. Furthermore, these proteins are disproportionately found in the nucleus, including in transcription factor complexes (19), as is PQBP-1 (10). A protein that adopts a flexible, adaptable conformation can bind to multiple different targets. The interactions tend to be highly specific but have a low affinity (51). Of interest, PQBP-1 was observed to bind to nickel resin during preparative purification of the protein, presumably via its highly charged disordered PRD (Fig. 1 c). This is the region that binds to polyQ tracts, which is also disordered, as revealed in a number of experiments (52–54). It is conceivable that its conformational variability offers PQBP-1 the advantage of adaptive binding to disordered targets, and links to its specific role in the pathogenesis of polyQ diseases. Although some disordered regions of proteins can form structure on binding to targets, many proteins do not exhibit this behavior (55). The CTD of PQBP-1 has been shown to remain disordered upon binding to U5-15kD (56), and here we have shown that PQBP-1 cannot be promoted into a state of higher folding by natural osmolytes. However, we cannot rule out the possibility that other binding partners can induce structure formation.
In this work, we acquired knowledge about the unique molecular properties of PQBP-1. Our hydrodynamic and biophysical characterizations of this protein, along with the low-resolution solution model, will pave the way for studying the molecular interactions of PQBP-1, which have many medical implications.
Acknowledgments
We thank Rohanah Hussain and Giuliano Siligardi at beamline B23 (Diamond Light Source, Oxfordshire, UK) for assistance in data collection, processing, and interpretation; Eikki Lanckriet of Expedeon for supplying samples of Nvoy polymer for testing; and Anthony Keeble and Andrew Beavil for providing access to the fluorescence spectroscopy equipment.
M.R. was supported by an Engineering and Physical Sciences Research Council studentship. D.S. received support from EU e-Infrastructure grant WeNMR 261572. Measurements obtained at the European Molecular Biology Laboratory beamline X33 were supported by the EU's Seventh Framework Programme (FP7/2007-2013) under grant agreement 226716.
Contributor Information
Dmitri Svergun, Email: svergun@embl-hamburg.de.
Yu Wai Chen, Email: yu-wai.chen@kcl.ac.uk.
Supporting Material
References
- 1.Imafuku I., Waragai M., Okazawa H. Polar amino acid-rich sequences bind to polyglutamine tracts. Biochem. Biophys. Res. Commun. 1998;253:16–20. doi: 10.1006/bbrc.1998.9725. [DOI] [PubMed] [Google Scholar]
- 2.Waragai M., Lammers C.H., Okazawa H. PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum. Mol. Genet. 1999;8:977–987. doi: 10.1093/hmg/8.6.977. [DOI] [PubMed] [Google Scholar]
- 3.Qi Y., Hoshino M., Okazawa H. PQBP-1 is expressed predominantly in the central nervous system during development. Eur. J. Neurosci. 2005;22:1277–1286. doi: 10.1111/j.1460-9568.2005.04339.x. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura N., Horiuchi D., Okazawa H. Expression of human PQBP-1 in Drosophila impairs long-term memory and induces abnormal courtship. FEBS Lett. 2006;580:2335–2340. doi: 10.1016/j.febslet.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 5.Enokido Y., Maruoka H., Okazawa H. PQBP-1 increases vulnerability to low potassium stress and represses transcription in primary cerebellar neurons. Biochem. Biophys. Res. Commun. 2002;294:268–271. doi: 10.1016/S0006-291X(02)00477-1. [DOI] [PubMed] [Google Scholar]
- 6.Tapia V.E., Nicolaescu E., Sudol M. Y65C missense mutation in the WW domain of the Golabi-Ito-Hall syndrome protein PQBP1 affects its binding activity and deregulates pre-mRNA splicing. J. Biol. Chem. 2010;285:19391–19401. doi: 10.1074/jbc.M109.084525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sudol M., Sliwa K., Russo T. Functions of WW domains in the nucleus. FEBS Lett. 2001;490:190–195. doi: 10.1016/s0014-5793(01)02122-6. [DOI] [PubMed] [Google Scholar]
- 8.Komuro A., Saeki M., Kato S. Association of two nuclear proteins, Npw38 and NpwBP, via the interaction between the WW domain and a novel proline-rich motif containing glycine and arginine. J. Biol. Chem. 1999;274:36513–36519. doi: 10.1074/jbc.274.51.36513. [DOI] [PubMed] [Google Scholar]
- 9.Waragai M., Junn E., Okazawa H. PQBP-1/Npw38, a nuclear protein binding to the polyglutamine tract, interacts with U5-15kD/dim1p via the carboxyl-terminal domain. Biochem. Biophys. Res. Commun. 2000;273:592–595. doi: 10.1006/bbrc.2000.2992. [DOI] [PubMed] [Google Scholar]
- 10.Okazawa H., Rich T., Kanazawa I. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron. 2002;34:701–713. doi: 10.1016/s0896-6273(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 11.Lubs H., Abidi F.E., Schwartz C.E. Golabi-Ito-Hall syndrome results from a missense mutation in the WW domain of the PQBP1 gene. J. Med. Genet. 2006;43:e30. doi: 10.1136/jmg.2005.037556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalscheuer V.M., Freude K., Ropers H.H. Mutations in the polyglutamine binding protein 1 gene cause X-linked mental retardation. Nat. Genet. 2003;35:313–315. doi: 10.1038/ng1264. [DOI] [PubMed] [Google Scholar]
- 13.Martínez-Garay I., Tomás M., Martínez F. A two base pair deletion in the PQBP1 gene is associated with microphthalmia, microcephaly, and mental retardation. Eur. J. Hum. Genet. 2007;15:29–34. doi: 10.1038/sj.ejhg.5201717. [DOI] [PubMed] [Google Scholar]
- 14.Stevenson R.E., Bennett C.W., Schwartz C.E. Renpenning syndrome comes into focus. Am. J. Med. Genet. A. 2005;134:415–421. doi: 10.1002/ajmg.a.30664. [DOI] [PubMed] [Google Scholar]
- 15.Lenski C., Abidi F., Schwartz C.E. Novel truncating mutations in the polyglutamine tract binding protein 1 gene (PQBP1) cause Renpenning syndrome and X-linked mental retardation in another family with microcephaly. Am. J. Hum. Genet. 2004;74:777–780. doi: 10.1086/383205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cossée M., Demeer B., Mandel J.L. Exonic microdeletions in the X-linked PQBP1 gene in mentally retarded patients: a pathogenic mutation and in-frame deletions of uncertain effect. Eur. J. Hum. Genet. 2006;14:418–425. doi: 10.1038/sj.ejhg.5201593. [DOI] [PubMed] [Google Scholar]
- 17.Germanaud D., Rossi M., des Portes V. The Renpenning syndrome spectrum: new clinical insights supported by 13 new PQBP1-mutated males. Clin. Genet. 2011;79:225–235. doi: 10.1111/j.1399-0004.2010.01551.x. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi M., Mizuguchi M., Kawano K. Polyglutamine tract binding protein-1 is an intrinsically unstructured protein. Biochim. Biophys. Acta. 2009;1794:936–943. doi: 10.1016/j.bbapap.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Ward J.J., Sodhi J.S., Jones D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004;337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 21.Li X., Romero P., Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform. Ser. Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 22.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 23.Busso D., Delagoutte-Busso B., Moras D. Construction of a set Gateway-based destination vectors for high-throughput cloning and expression screening in Escherichia coli. Anal. Biochem. 2005;343:313–321. doi: 10.1016/j.ab.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 24.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philo, J. 2006. Trial ware data analysis programs SVEDBERG and DCDT+. http://www.jphilo.mailway.com. Accessed 11 October 2011.
- 26.Furihata K., Shimotakahara S., Tashiro M. An efficient use of the WATERGATE W5 sequence for observing a ligand binding with a protein receptor. Magn. Reson. Chem. 2008;46:799–802. doi: 10.1002/mrc.2264. [DOI] [PubMed] [Google Scholar]
- 27.Whitmore L., Wallace B.A. Protein secondary structure analyses from circular dichroism spectroscopy: methods and reference databases. Biopolymers. 2008;89:392–400. doi: 10.1002/bip.20853. [DOI] [PubMed] [Google Scholar]
- 28.Johnson W.C. Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins. 1999;35:307–312. [PubMed] [Google Scholar]
- 29.Semisotnov G.V., Rodionova N.A., Gilmanshin R.I. Study of the “molten globule” intermediate state in protein folding by a hydrophobic fluorescent probe. Biopolymers. 1991;31:119–128. doi: 10.1002/bip.360310111. [DOI] [PubMed] [Google Scholar]
- 30.Petoukhov M.V., Konarev P.V., Svergun D.I. Towards automated and web-supported small-angle scattering data analysis. J. Appl. Cryst. 2007;40:s223–s228. [Google Scholar]
- 31.Guinier A. La diffraction des rayons X aux très petits angles: application à l′étude de phénomènes ultramicroscopiques. Ann. Phys. 1939;12:161–237. [Google Scholar]
- 32.Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Cryst. 1992;25:495–503. [Google Scholar]
- 33.Uversky V.N. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernadó P., Mylonas E., Svergun D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007;129:5656–5664. doi: 10.1021/ja069124n. [DOI] [PubMed] [Google Scholar]
- 35.Svergun D., Barberato C., Koch M.H.J. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Cryst. 1995;28:768–773. [Google Scholar]
- 36.Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volkov V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Cryst. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozin M., Svergun D.I. Automated matching of high- and low-resolution structural models. J. Appl. Cryst. 2001;34:33–41. [Google Scholar]
- 39.Tarini M., Cignoni P., Montani C. Ambient occlusion and edge cueing to enhance real time molecular visualization. IEEE Trans. Vis. Comput. Graph. 2006;12:1237–1244. doi: 10.1109/TVCG.2006.115. [DOI] [PubMed] [Google Scholar]
- 40.Andrews P. The gel-filtration behaviour of proteins related to their molecular weights over a wide range. Biochem. J. 1965;96:595–606. doi: 10.1042/bj0960595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siligardi G., Drake A.F. The importance of extended conformations and, in particular, the PII conformation for the molecular recognition of peptides. Biopolymers. 1995;37:281–292. doi: 10.1002/bip.360370406. [DOI] [PubMed] [Google Scholar]
- 42.Drake A.F., Siligardi G., Gibbons W.A. Reassessment of the electronic circular dichroism criteria for random coil conformations of poly(L-lysine) and the implications for protein folding and denaturation studies. Biophys. Chem. 1988;31:143–146. doi: 10.1016/0301-4622(88)80019-x. [DOI] [PubMed] [Google Scholar]
- 43.Burgardt N.I., Ferreyra R.G., Ceolín M. Biophysical characterisation and urea-induced unfolding of recombinant Yarrowia lipolytica sterol carrier protein-2. Biochim. Biophys. Acta. 2009;1794:1115–1122. doi: 10.1016/j.bbapap.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 44.Chang Y.C., Oas T.G. Osmolyte-induced folding of an intrinsically disordered protein: folding mechanism in the absence of ligand. Biochemistry. 2010;49:5086–5096. doi: 10.1021/bi100222h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uversky V.N., Li J., Fink A.L. Trimethylamine-N-oxide-induced folding of α-synuclein. FEBS Lett. 2001;509:31–35. doi: 10.1016/s0014-5793(01)03121-0. [DOI] [PubMed] [Google Scholar]
- 46.Uversky V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- 47.Damaschun G., Damaschun H., Zirwer D. Acid denatured apo-cytochrome c is a random coil: evidence from small-angle X-ray scattering and dynamic light scattering. Biochim. Biophys. Acta. 1991;1078:289–295. doi: 10.1016/0167-4838(91)90571-g. [DOI] [PubMed] [Google Scholar]
- 48.Svergun D.I., Koch M.H. Advances in structure analysis using small-angle scattering in solution. Curr. Opin. Struct. Biol. 2002;12:654–660. doi: 10.1016/s0959-440x(02)00363-9. [DOI] [PubMed] [Google Scholar]
- 49.Kuwajima K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins. 1989;6:87–103. doi: 10.1002/prot.340060202. [DOI] [PubMed] [Google Scholar]
- 50.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 51.Dunker A.K., Brown C.J., Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 52.Chen Y.W., Stott K., Perutz M.F. Crystal structure of a dimeric chymotrypsin inhibitor 2 mutant containing an inserted glutamine repeat. Proc. Natl. Acad. Sci. USA. 1999;96:1257–1261. doi: 10.1073/pnas.96.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masino L., Kelly G., Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–272. doi: 10.1016/s0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 54.Masino L., Musi V., Pastore A. Domain architecture of the polyglutamine protein ataxin-3: a globular domain followed by a flexible tail. FEBS Lett. 2003;549:21–25. doi: 10.1016/s0014-5793(03)00748-8. [DOI] [PubMed] [Google Scholar]
- 55.Marsh J.A., Dancheck B., Peti W. Structural diversity in free and bound states of intrinsically disordered protein phosphatase 1 regulators. Structure. 2010;18:1094–1103. doi: 10.1016/j.str.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi M., Mizuguchi M., Kawano K. Polyglutamine tract-binding protein-1 binds to U5-15kD via a continuous 23-residue segment of the C-terminal domain. Biochim. Biophys. Acta. 2010;1804:1500–1507. doi: 10.1016/j.bbapap.2010.03.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.