

Abstract
Down syndrome (DS) is a condition where a complete or segmental chromosome 21 trisomy causes variable intellectual disability, and progressive memory loss and neurodegeneration with age. Many research groups have examined development of the brain in DS individuals, but studies on age-related changes should also be considered, with the increased lifespan observed in DS. DS leads to pathological hallmarks of Alzheimer's disease (AD) by 40 or 50 years of age. Progressive age-related memory deficits occurring in both AD and in DS have been connected to degeneration of several neuronal populations, but mechanisms are not fully elucidated. Inflammation and oxidative stress are early events in DS pathology, and focusing on these pathways may lead to development of successful intervention strategies for AD associated with DS. Here we discuss recent findings and potential treatment avenues regarding development of AD neuropathology and memory loss in DS.
1. Introduction
The most common cause of dementia is Alzheimer's disease (AD), with rates of prevalence increasing steadily from 60 years of age to reach almost 40% by the age of 85 [1]. AD is defined as the presence of neuritic plaques, which are composed of extracellular deposits of amyloid beta, and neurofibrillary tangles [2]. Neurodegeneration in the later stages of AD is widespread, with massive synapse loss and an overall decline in grey matter resulting from neuronal loss in cortical and hippocampal regions. Cortical neuronal loss is preceded by degeneration of certain subcortical neuronal populations, including basal forebrain cholinergic neurons (BFCNs) [3] and noradrenergic neurons of the locus coeruleus (LC-NE) [4, 5].
While the majority of AD cases are considered sporadic, mutations in amyloid precursor protein (APP) and presenilins 1 and 2 (PS-1 and PS-2) genes are responsible for most of the cases of AD considered “familial” [6]. These mutations lead to alterations in APP metabolism that result in an overabundance of amyloid plaques. Similarly, APP processing is also affected in Down syndrome (DS), a population who exhibit histopathology consistent with AD by the 4th and 5th decades of life with near uniformity, as well as increased risk for dementia [7, 8]. Located on chromosome 21, APP is triplicated in DS, and amyloid-beta deposition is frequently profound in these individuals [9–11]. Recently, cases of familial AD resulting from duplication of only the APP locus have been discovered [12], further defining a role for APP in AD dementia. However, few studies have been able to correlate plaque load with dementia severity. Rather, cognitive function correlates most strongly with the degeneration of cholinergic neurons in the basal forebrain. Reversal of cholinergic hypofunction in AD with choline acetyl transferase inhibitors has been shown to facilitate memory function, albeit to a moderate degree [13]. However, it is still not known what causes the cholinergic degeneration, or if other parallel factors also contribute to the disease. Some potential mechanisms include neuroinflammation, oxidative stress, amyloid toxicity, and abnormal phosphorylation of proteins including the microfilament-associated protein tau; etiological causes include genetic mutations, diet, sedentary lifestyle, and environmental toxins [14]. While familial causes of AD are rare and idiopathic AD is difficult to model, DS presents a large and relatively homogenous population with relevant animal models that can serve to illuminate possible etiologies or treatment paradigms in AD.
In the current paper, we will discuss current theories regarding biological mechanisms and potential treatment paradigms for DS individuals with AD-like dementia (DSD). We include data from animal models, as well as from humans with DSD, and propose potential early prevention models for this difficult and progressive condition.
2. Down Syndrome: A Genetic Insight into AD
The uniformity with which individuals with DS acquire AD neuropathology makes this population important to study, not only to gain a better understanding of AD, but also because there are currently no effective treatment paradigms for DSD [8, 15]. Because they have physiological alterations in cardiac and metabolic systems, cholinesterase inhibitors may be contraindicated in some DSD patients [15, 16]. DS is the most common aneuploidy, occurring as frequently as approximately 1 in every 700 live births in the US [17]. DS results in variable levels of intellectual disability, along with congenital defects, and increased risk of certain cancers, such as leukemias [18]. As maternal age continues to increase and medical interventions have increased the lifespan of DS individuals, the prevalence of DSD continues to grow. The diverse and heterogeneous neurodegeneration in AD and in normal aging are accelerated in DS, and lessons learned from DSD patients may uncover therapeutic targets with widespread implications. In fact, DS can be considered a form of segmental progeroid syndrome, or accelerated aging [19, 20].
Studies assessing the effects of age on cognition in DS demonstrate a greater incidence of short-term memory impairment in DS individuals over 35 years of age, as well as increasing rates of dementia, aphasia, and agnosia [23] while detriments in executive function are evident already in adolescence [24]. As in idiopathic AD, DSD patients display dysfunction of language and motor skills, seizure onset, and behavioral abnormalities [25], in addition to AD-like pathology, including amyloid-beta deposits, neurofibrillary tangles, loss of BFCNs, and pathological alterations in mitochondria and endosomes [26–29]. While trisomy 21 constitutes the triplication of over 300 genes [30, 31], recent animal studies have sought to elucidate which genes may contribute to the observed neurodegenerative pathology. Based on genetic studies in mouse models of DS, several specific genes contained within the triplicated region of murine chromosome 16 (which corresponds to an equivalent section on human Chr. 21; see Figure 1) have been implicated in the DSD neuropathology. One of the most important genes associated with DS is the amyloid precursor protein (APP) gene—increased APP production may partially contribute to DSD-related oxidative stress as well as inflammation. Accumulation of amyloid-beta monomers can directly impair mitochondrial function resulting in energy depletion [32], and it is also well known that accumulation of amyloid—either in tissue culture or in vivo—leads to activation of inflammatory cascades [33, 34], most likely via both microvascular dysfunction and activation of resident glial cells in brain parenchyma. Furthermore, cortical DS neurons exhibit impaired mitochondrial function that results in reduced energy production and elevations in reactive oxygen species (ROS) [35]. Studies using the Ts1Cje mouse model for DS, which does not include triplication of the SOD or APP genes [36], suggest that other triplicated genes may be involved in mitochondrial abnormalities observed in DS. In addition, while APP and SOD-1 each may contribute to the disease, neither gene is solely responsible for the degenerative changes that occur in DS [37]. Other genes located on the critical region include Ets-2 and DSCR1 (Figure 1), which have both been linked to neurodegeneration [35, 38]. In this paper, we will provide evidence, from our recent work and others, suggesting that inflammation and oxidative stress are early dysregulations which may be responsible for age-related dementia and associated pathology in DSD.
Figure 1.
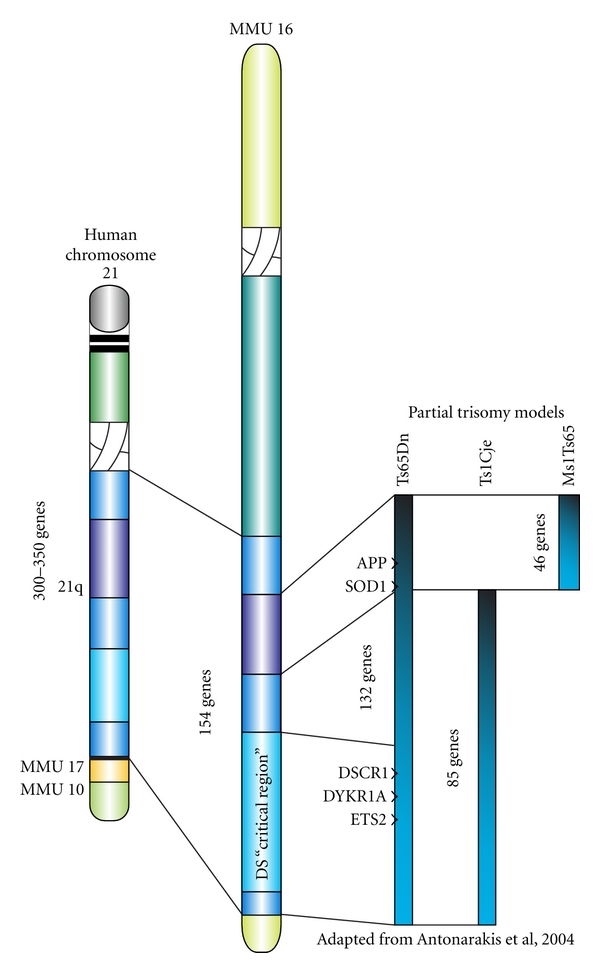
Mouse models for DS. Schematic of the gene segments involved in the so-called “Down syndrome critical region” (DSCR) in human chromosome 21, as well as in different mouse models of the condition. Note that the Ts65Dn mouse contains all genes included in the DSCR, as well as a set of 132 other genes including SOD and APP. Modified from Antonarakis et al. 2004 [21].
3. Modeling DS Pathology: The Ts65Dn Mouse
As discussed elsewhere in this issue, a spontaneous translocation of a portion of murine chromosome 16 onto chromosome 17 led to the formation of a DS model, the Ts65Dn mouse [39]. The translocated segment of chromosome 16, syntenic to a significant portion of human chromosome 21 (Figure 1), thus provided a genetic triplication which can be passed on to offspring [39]. Nearly 140 known genes are triplicated in Ts65Dn mice, of which 60% are also located on human chromosome 21 [40]. More importantly, these mice exhibit normal lifespans, allowing for the analysis of progressive neurodegenerative alterations. While Ts65Dn mice fail to develop amyloid plaques, they do exhibit elevated levels of APP and associated peptides in the hippocampus [41–43] and increased phosphorylation of tau protein [44, 45]. Ts65Dn mice also show increased inflammatory morphology with aging [22, 46] (see also Figure 2) synaptic dysfunction [47, 48], and a failure of neurotrophic signaling, particularly involving the retrograde transport of nerve growth factor (NGF) to the basal forebrain [42, 46, 49, 50], and downregulation of brain-derived neurotrophic growth factor (BDNF) levels [51, 52]. In addition, they exhibit age-related degeneration of LC-NE and BFCN neurons [22, 53–55]. Memory deficits are progressive in these mice and onset coincides with BFCN atrophy [43, 46, 56]. Interestingly, a study by Belichenko et al. [57] suggested that 33 genes, included in the so-called “DS critical region” (DSCR) of genes in humans, and triplicated in a novel mouse model (Ts1Rhr), might be responsible for many of the physiological and behavioral detriments observed in the Ts65Dn mice, narrowing the search for the set of genes involved in DSD neuropathology [57]. However, other studies have shown that although this “critical region” is necessary for cognitive impairment and pathology to develop [58], overexpression of these particular genes is not sufficient to generate DSD, at least not in mouse models, demonstrating the complex nature of DS-related dementia and neuropathology with aging.
Figure 2.

Gliosis in hippocampus of Ts65Dn mice. Brain tissue sections from Ts65Dn mice and age-matched normosomic littermates, showing typical hippocampal morphology of astrocytes, using the glial marker GFAP (a) and (b). The sections were from 12-month-old normosomic (NS, a) or Ts65Dn (TS, b) mice. Note increased number of astrocytes in TS mice, as well as elevated expression of GFAP and an activated morphology, with more branching and thicker branches in the TS compared to NS mouse. (c) Astrocyte branching measurements (GFAP labeling) in the hippocampus reveal increased branching in TS mice compared to NS age-matched controls, a sign of activation following inflammatory or other pathological processes. Astrocytosis is increased with aging in the TS mice to a greater extent than in NS mice. (d) Density of a marker for microglial cells, Cd45, is also increased with age in Ts65Dn (TS) but not in age-matched normosomic (NS) mice, indicating ongoing microglial activation in this brain region. Inset in (b) represent 100 microns. Data were not published previously.
While degeneration of basal forebrain cholinergic neurons (BFCNs) occurs during normal aging, DSD and AD are defined by rapidly accelerated loss of these projection neurons, and cholinergic dysfunction correlates strongly with the progression of cognitive decline in both diseases [59, 60]. Ts65Dn mice show consistent learning and memory deficits on spatial reference and working memory tasks [56, 61–67]. Most of these deficits become apparent between 4 and 12 months of age [56], suggesting, indeed, that the behavioral dysfunction developing in the Ts65Dn mouse mimics the segmental progeria syndrome observed in terms of brain function in humans with DS. Ts65Dn mice exhibit deficits in novel object tasks, which are reversed by the partial N-Methyl-D-aspartic acid (NMDA) glutamate receptor blocker Memantine (Namenda) [68–70]. These findings suggest that glutamate and GABA transmitter systems are affected by the genetic alterations in Ts65Dn, directly or indirectly, in Ts65Dn mice, something that has been suggested by work from other research groups as well [71, 72]. In a manuscript by Rueda et al. [71], they found that treatment with memantine in aged Ts65Dn mice improved spatial learning but did not affect the number of dentate granule cells, suggesting that the effects of memantine may be pharmacological, rather than neuroprotective. These data were further supported by our findings, that memantine increased working memory performance, particularly in a novel object task, but did not rescue hippocampal, cholinergic, or locus coeruleus neurons from progressive neurodegeneration [70]. The cognitive impairment observed over time in Ts65Dn mice parallels cognitive impairment in adult DS individuals with early or moderate AD, tested on the WISC-R behavioral battery, showing progressive deterioration in executive function, comprehension, picture completion, vocabulary, and digit span [73]. The memory deficits indicate hippocampal and frontal cortex dysfunction and together with septohippocampal degeneration indicate that the Ts65Dn mouse is a unique model to understand the progression of neuropathology and memory loss in DSD.
4. Locus Coeruleus Degeneration in DSD
LC-NE degeneration, while less studied than BFCN loss, is another hallmark of AD [74]. NE neurotransmission exerts effects on neurons, glia, and blood vessels throughout the neuraxis. LC-NE lesions, using the selective NE neurotoxin DSP-4, give rise to aggravated amyloid accumulation, oxidative stress, and memory loss in transgenic AD models [75–77]. Findings suggest that LC-NE effects are mediated both directly, via neurotransmission changes in the limbic system, and indirectly, via aggravation of amyloid accumulation, inflammation, and oxidative stress pathways. NE-mediated neuroprotection of oxidative stress on BCFNs in vitro is independent of adrenergic receptor activation or intracellular accumulation, [78] suggesting a role for NE in the neutralization of hydroxyl radicals. The antioxidant activity of NE provides a pharmacological link between LC-NE and cholinergic survival. NE circuitry also exhibits a direct influence on memory formation. BFCNs activity is modulated by NE via adrenergic receptor activity [79], and pharmacological stimulation of NE receptors leads to improved cognitive performance both in rodent models and in humans [80]. While NE is an essential modulator of memory through its ability to regulate synaptic mechanisms, NE depletion is not sufficient to significantly alter memory function in intact animals [22]. Yet, NE depletion in the presence of cholinergic dysfunction exacerbates memory impairments [22] and may therefore aggravate deficits in memory systems dependent on the basal forebrain cholinergic neurons. In a recent study, Ts65Dn and NS mice were lesioned using the NE neurotoxin DSP-4 at 4 months of age and were then studied at 8–10 months of age in terms of behavior and neurochemistry. As can be seen in Figure 3 and in [22], the NE lesion gave rise to a significant aggravation of both memory loss and neuropathology in Ts65Dn but not in NS mice, including degeneration of hippocampal and BFCNs as well as increased inflammatory markers. These findings suggest that NE neurotransmission, albeit important for normal function of the brain, plays a particularly important role for curbing age-related pathology in the form of inflammation and neuronal loss. This notion has been supported by other investigators, showing enhanced effects of DSP-4 lesions in APP transgenic mice [76, 81, 82]. These investigators also found that administration of the NE precursor L-threo-DOPS restored microglial functions in NE-depleted mice [76], suggesting a reciprocal system where the amyloid cascade, inflammatory markers, and NE innervation systems affect each other. Interestingly, others have also shown that LC neurons spontaneously degenerate in AD mouse models [83], again suggesting a specific link between accelerated amyloid accumulation and degeneration of LC neurons.
Figure 3.

Effects of the NE neurotoxin DSP-4 on Ts65Dn and normosomic mice. Note significant aggravation of performance in a memory task (a) coupled with aggravated activation of microglial cells (b–d) in the hippocampal formation, as evidenced by Cd45 immunohistochemistry. (a) Average number of errors in a water radial arm maze. The NE lesion exhibited more pronounced effects on errors in the maze in TS than in NS mice, and TS mice performed more errors than NS mice, regardless of NE lesions (DSP) or not (Sal). (b–d) Cd45 staining of microglial cells in the hippocampus in a normosomic mouse (NS) treated with saline (b), a Ts65Dn mice on saline (c), and a Ts65Dn mouse that received DSP-4 lesions of the LC-NE neurons (d). Note significant activation of individual microglial cells as a result of the NE lesion in TS mice compared to controls. Quantitation of inflammatory processes is available in Lockrow et al., 2011 [22].
Importantly, individuals with DSD exhibit early and progressive degeneration of LC-NE neurons [84]. Recently, a study by Salehi et al. [55] demonstrated successful recovery from memory loss in Ts65Dn mice using the NE precursor Droxidopa (L-threo-dihydroxyphenylserine). These results are promising and should be considered in future clinical treatment paradigms for DSD patients. Since LC-NE degeneration is common to both Parkinson's disease (PD) and AD patients [85–87], future pharmaceutical interventions for dementia may include enhancement of NE neurotransmission also for these neurological conditions. Promising clinical pilot studies have already been initiated in terms of the NE reuptake inhibitor Atomoxetine and memory loss in PD [88] and in Alzheimer's disease [89, 90] even though much remains to be done in terms of incorporating NE enhancement treatment for dementia. LC-NE neurons partake in the regulation of blood vessels, microglial cells, as well as neurons, and degeneration of this monoaminergic cell group can be an active player in neuropathological processes in age-related dementia of different etiology.
5. Inflammatory Pathology in AD and DSD
As in AD, individuals with DSD consistently exhibit chronic inflammation in limbic system areas of the brain, with increases in microglial and astrocytic activation coupled with IL-1β and TNF-α cytokine release [91–93]. Microglial activation typically arises in the entorhinal cortex before developing in the hippocampus and surrounding cortex as well as the basal forebrain [26, 27]. BFCNs are highly sensitive to inflammation and oxidative stress [94], but specific biological mechanisms for their selective loss in AD and in DSD have not been revealed. There is also evidence that TNF-α-induced cortical inflammation at cholinergic terminals leads to retrograde degeneration of BFCNs [95]. Recent work suggests that inflammation due to loss of noradrenergic innervation from the LC-NE innervation of BFCNs is a plausible explanation for the selective vulnerability of these neurons in DSD and AD [22]. β-adrenergic receptors are expressed in astrocytes and microglia and modulate the cytokine release [96]. The reduction of noradrenergic neurons in the LC correlates with amyloid plaques and dementia severity in AD [97, 98]. NE treatment of cholinergic cells in vitro reduces expression of IL-1β and TNF-α, as well as proinflammatory proteins such as iNOS [96]. Since Ts65Dn mice exhibit significant degeneration of both BFCNs and LC-NE neurons, it is not surprising that we found accelerated and age-related astrocytosis and microgliosis in the hippocampus of this mouse model of DS (Figures 2 and 3). As mentioned above, depletion of noradrenergic terminals in murine models of AD results in increased inflammatory cytokine production, activated microglial morphology, and amyloid deposition [76, 82, 99]. NE terminal destruction also impeded cholinergic neurotransmission in AD models which otherwise show no cholinergic deficits [81]. Thus, while inflammation may affect many of these neurodegenerative processes, it also can increase in response to early abnormalities in ACh and NE signaling, since there is a reciprocal relationship between neuronal and glial modulation of inflammatory processes, especially during neurodegenerative disease [96]. Based on these studies, it is difficult to determine whether BFCN and LC-NE degeneration activates the inflammatory pathways, or if the cytokine production by astrocytes and microglia, in turn, causes the neuronal degeneration in DSD and AD. Most likely, all of these processes have interactive and escalating effects on each other, leading, in the end, to memory loss and AD pathology.
6. Neurotrophic Factors and DS
The survival and maintenance of BFCNs depend on neurotrophic support from NGF and BDNF [100]. NGF mRNA is expressed at high levels in regions innervated by cholinergic terminals, such as the neocortex, dentate gyrus, and the hippocampal pyramidal layer [3]. Upon release from postsynaptic neurons, NGF binds to its high-affinity receptor, TrkA, on BFCN nerve terminals, initiating receptor oligomerization which leads to signaling cascades through PI3K and ERK activation and endocytosis of the ligand-receptor complex [101]. This complex is retrogradely transported to the soma where it facilitates signal transduction of phenotypic markers such as choline acetyltransferase [101, 102]. Exogenous administration of NGF rescues BFCNs from age- or toxin-related degeneration and reverses cognitive dysfunction in animal models of AD or normal aging [103]. While the production of NGF in the hippocampus and cerebral cortex has been shown to be unaltered or even increased in AD [103], NGF levels in the basal forebrain exhibit significant decline [104]. A compensatory increase in NGF expression in target regions may be due in part to loss of TrkA receptor expression in BFCN neurons, which occurs early in AD and is recapitulated in aged rodents [3, 105]. Murine models for DS show reductions in retrograde NGF trafficking which occurs in part due to enlarged, dysfunctional endosomes [42, 49, 101]. Recent studies have shown that these endosomal changes can be caused by overexpression of APP [42, 106]. Abnormal endosomes are present in both AD and DSD brains [29] and localize to the vulnerable regions such as the basal forebrain and the hippocampus [107] suggesting that endosomal trafficking of NGF linked to TrkA may be a pathological pathway to explore further in DSD brains.
BDNF also promotes BFCN survival and cholinergic signaling [108–110]. BDNF expression is reduced in AD [109], and BDNF levels are reduced in serum from DS individuals [111], and in brain tissue from the Ts65Dn mouse model for DS [52], and has been shown to be linked to memory function, as well as synaptic plasticity and neurogenesis [112]. BDNF expression is increased following exercise and may therefore contribute to the beneficial effects of voluntary exercise observed in AD as well as in normal aging in humans and animal models [113–117]. Interestingly, several studies have shown that LC-NE innervation into cortical regions regulates the expression of BDNF, suggesting a close link between loss of BDNF expression and LC-NE degeneration in DS [118]. In a recent manuscript by Counts and Mufson [119], the authors demonstrated that administration of NE protected cultured neurons from amyloid-beta-mediated toxicity by upregulating both NGF and BDNF expression. Further, the authors found that NE inhibited increased reactive oxygen species (ROS) and caspase activation caused by the neurotoxin, suggesting also a direct link between the neurotrophic factors, NE innervation, and oxidative stress. Treatment with functional blocking agents for NGF and BDNF removed the beneficial effects, indeed suggesting that NE effects were mediated by the trophic factors. This paper therefore linked several pathological processes in DSD and AD, providing direction for future research and treatment options. Our recent study using Ts65Dn mice extended these findings in vivo, by showing that an LC-NE lesion, using the neurotoxin DSP-4, decreased BDNF expression in frontal cortex, a region associated with working memory loss in the Ts65Dn mouse model [22]. We also found a significant correlation between BDNF expression and NE levels, as well as between BDNF expression and working memory errors, suggesting a clear link between BDNF expression and memory function dependent on this region. BDNF and NGF have been associated with neuroprotection against oxidative stress in neurons [119, 120], suggesting that DSD patients may exhibit increased sensitivity to oxidative stress because of reduced expression of these neurotrophic factors.
7. Oxidative Stress and DSD Pathology
Individuals with DS exhibit elevated oxidative stress early in life [121]. Oxidizing free radicals, also known as ROS, are cytotoxic byproducts of normal mitochondrial metabolism and are normally processed by endogenous antioxidants. But when levels of mitochondrial ROS production exceed the intracellular antioxidant defenses, oxidative molecules can disrupt cellular functions, negatively affecting synaptic plasticity and eventually leading to neuronal injury and apoptosis [122]. The hippocampal formation exhibits a high vulnerability to both ischemic and neurotoxic injury associated with oxidative stress [123]. A marker of RNA oxidative damage, 8-hydroxyguanosine (8-OHG), is elevated in neurons of the hippocampus and cortex early in the progression of AD and precedes much of the pathology in these regions, suggesting that oxidative stress may be the earliest event in AD-related disease processes [124]. Postmortem analysis revealed that 8-OHG immunoreactivity increased significantly in cortical neurons of DS individuals in their teens and twenties, while amyloid-beta burden was increased only after 30 years of age [125], strongly suggesting that oxidative stress is an early event also in DS. The central question is why is oxidative stress so rampant in the brain of DS individuals?
Part of the answer to that question may be the triplication of both APP and SOD-1 genes in DS (Figure 1). The balance between ROS production and the scavenger enzyme pathways is tightly regulated in the cell during normal conditions. We propose that the increase in expression of SOD-1 in DS leads to a reduction in superoxide but an increase in the accumulation of hydrogen peroxide (H2O2) in tissues. This hypothesis is based on a superarray using pooled samples of tissue from the hippocampus of Ts65Dn mice revealing significant elevations in hippocampal SOD-1 expression with only a moderate increase in the other scavenger enzymes, including glutathione reductase and catalase (Figure 4). Elevated rates of conversion from superoxide to H2O2 would lead to lipid peroxidation in neurons and glia, accumulating with time, and leading to the neuropathology observed in Ts65Dn mice with age, as well as in DS individuals. This hypothesis was recently validated by studies from Harris-Cerruti et al. [37], showing that a mouse model consisting of double SOD-1/APP overexpression leads to memory loss and neuropathology, as well as elevated ROS in the brain, while APP overexpression alone was less effective in generating neurodegeneration or ROS accumulation. When the investigators examined hippocampal slices for long-term potentiation (LTP), they found that LTP was impaired in both tg-SOD and tg-APP-SOD mice, but not in tg-APP mice, suggesting that the APP overexpression alone did not affect this cellular component of hippocampal plasticity. SOD-1 overexpression alone also gave rise to ROS accumulation, but not to the extent observed in APP/SOD-1 overexpression mice, suggesting a comodulation of oxidative stress pathways by the APP and SOD-1 genetic overexpression [37].
Figure 4.

(a) Superarray (SABiosciences, Frederick, MD) against oxidative stress and inflammatory markers was used on hippocampal tissue from pooled samples (3 per group) of Ts65Dn and Normosomic mice at 4, 8, 10, and 13 months of age. Note the increased expression in APP and SOD-1 due to increased gene dosage of these genes. However, glutathione peroxidase 1 and 3 (GPX 1 and 3), as well as catalase levels, were not increased to the same extent. Further investigation of the glutathione enzymatic pathway revealed increased GSSG:GSH ratio in Ts65Dn compared to normosomic brain (b), suggesting, a compensatory processing of free radicals, but not sufficient to eliminate peroxidation in neurons. Glutathione exists in two forms: GSH (reduced form) and GSSG (oxidized form). Normally the relationship between these two forms is 1 : 10 in healthy cells. (c) Schematic representation of the ROS scavengers, demonstrating that elevated SOD levels may lead to increased H2O2 levels, leading to enhanced stress on the glutathione and catalase pathways. Data were not published previously.
There is a controversy in the literature regarding beneficial or damaging effects of SOD overexpression. While some investigators show that SOD-1 or SOD-2 overexpression rescues neuropathology in AD transgenic mouse models [126], others demonstrate aggravated pathology when over-expressing SOD-1 [126], suggesting that there is a complicated relationship between SOD-1 and SOD-2 function in the CNS. Gardner and colleagues [127] investigated this question using a minimal mathematical model. The authors concluded that the outcome depended on a balance between processes consuming superoxide without forming H2O2 and those consuming superoxide with high H2O2 yield [127]. Our investigations shed some light on this particular question for DS brains, since Ts65Dn mice exhibited elevated expression of both glutathione and catalase (Figure 4), presumably as a response to elevated H2O2 levels in the brain. However, since most investigators use indirect methods of measuring H2O2, such as measuring lipid peroxidation or associated markers, it has not been shown, at least not to our knowledge, whether neurons or glia from DSD patients or Ts65Dn mice exhibit elevated H2O2 levels, even though studies of postmortem brain tissue have shown that levels of peroxiredoxin, which is an enzyme involved in eliminating H2O2, are elevated in both DSD and AD [128]. The role of oxidative stress in development of pathology in DS individuals is further discussed in other sections of this issue.
Early increases in ROS suggest that antioxidant therapy may benefit DS individuals with AD pathology. While clinical results for vitamin E treatment in AD patients have been mixed to this point [129], there have been minimal studies to determine whether antioxidants could be beneficial in DSD, despite a recent study of vitamin E administration during childhood in DS [130]. We recently reported beneficial effects of long-term vitamin E treatment in Ts65Dn mice [131] and suggest that this may be a viable future option for DSD. Ts65Dn mice were given vitamin E in their diet from 4–10 months of age, and cognitive performance was tested, followed by brain pathology. BFCN and hippocampal cell loss were reduced significantly, and neuroinflammation associated with microglial activation was also significantly reduced, suggesting a strong connection between inflammatory and oxidative stress pathways [131]. Oxidative stress measures correlated with improved cognitive performance, supporting the hypothesis that oxidative stress plays an important role for memory loss associated with DSD. Based on these encouraging findings, and the relatively minor risks associated with vitamin E treatment, we would suggest future development of this treatment paradigm for individuals with DS as a prevention strategy.
8. Overexpression of APP: Disease Modifier
An involvement of the amyloid cascade in the progressive memory loss and neuropathology in DS cannot be denied. It is likely that the overproduction of APP in DS individuals (Figure 1) converges upon both oxidative stress and inflammation pathways in the brain, to cause added harm to the DSD patient with time. Amyloid-beta-induced oxidative stress appears to be mediated through an NMDA receptor-mediated increase in Ca2+ influx [132]. Elevated intracellular Ca2+ disrupts mitochondrial function [133] and may explain the reduced mitochondrial efficiency seen in AD. As previously shown by our laboratory, Ts65Dn mice have deficits in expression of calbindin, a neuronal calcium-binding protein, in the hippocampus [46], suggesting further dysregulation of intracellular Ca2+ pathways. It is also possible that other genetic components of the triplicated gene segment aggravate DS-related AD pathology. The regulator of calcineurin 1 (RCAN1 or DSCR1) is also over-expressed in DS and in Ts65Dn mice (Figure 1). A recent manuscript by Porta et al. [134] demonstrated that RCAN1 knockout neurons (RCAN1−/−) exhibited a reduced response to oxidative stress, and the investigators therefore suggested vulnerability to oxidative stress downstream from the SOD-1-mediated accumulation of H2O2 in DS and in AD. These findings are important for continued efforts in determining the role of different genes in DS to provide additional substrates for neuroprotection strategies.
9. Outstanding Questions
Outstanding questions in this field should focus on prevention and/or treatment options for DSD. As individuals with DS live longer and medical interventions have been able to modify cardiovascular problems or other health issues, the incidence of DSD will go up dramatically in the next couple of decades. Based on recent findings related to vitamin E and antioxidant capacity, we feel that it is important to assess prevention in DS individuals at an early stage using vitamin E and/or other antioxidants. Further, treatment with NE enhancing drugs, such as Atomoxetine (Strattera) [88, 135], has shown promising results in children with ADHD and in PD; it is possible that these pharmaceutical interventions may be beneficial for working memory deficits and early onset problems with executive function in persons with DSD as well. It is important to note that several disease processes, related to inflammation, oxidative stress, cholinergic cell loss, calcium homeostasis, amyloid accumulation, and locus coeruleus degeneration, all converge on the progressive deficits observed in the limbic system of individuals with DS with age. Combination therapy targeting several aspects, or working upstream from the observed pathology, should therefore be developed. Finally, a national registry for DSD and age-matched control brain tissue and associated tissues is long overdue. The development of such a repository will allow centralized and streamlined studies into etiology but also possible treatment paradigms for DSD and finally render this field well-deserved attention, using a nation-wide collaboration for DSD-related studies.
Glossary
Alzheimer's disease (AD): the most common form of dementia.
Down syndrome (DS): whole or segmental triplication of chromosome 21 in humans.
Basal Forebrain cholinergic neurons: small group of neurons in basal forebrain carrying acetylcholine as their transmitter, and innervating large portions of the CNS.
Locus coeruleus noradrenergic neurons (LC-NE): small population consisting of a few thousand neurons in humans, located in the brainstem and innervating most portions of the brain and spinal cord.
Amyloid beta: cleavage form of amyloid precursor protein that accumulates in the brain of people with AD and DSD and has both inflammatory and oxidative stress effects on neurons.
Proinflammatory cytokines: small molecules that are released either in the blood or directly in the brain by inflammatory cells and contribute to inflammatory damage in the brain.
Oxidative stress scavengers: a set of enzymes, including superoxide dismutates, catalase, and glutathione, that reduce free radicals to water via a set of enzymatic reactions.
Long-term potentiation (LTP): a form of cellular potentiation of specific processes often used for studies of cellular learning and memory mechanisms.
References
- 1.Woolley JD, Khan BK, Murthy NK, Miller BL, Rankin KP. The diagnostic challenge of psychiatric symptoms in neurodegenerative disease: rates of and risk factors for prior psychiatric diagnosis in patients with early neurodegenerative disease. Journal of Clinical Psychiatry. 2011;72(2):126–133. doi: 10.4088/JCP.10m06382oli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mancuso M, Orsucci D, LoGerfo A, Calsolaro V, Siciliano G. Clinical features and pathogenesis of Alzheimer’s disease: involvement of mitochondria and mitochondrial DNA. Advances in Experimental Medicine and Biology. 2010;685:34–44. doi: 10.1007/978-1-4419-6448-9_4. [DOI] [PubMed] [Google Scholar]
- 3.Schliebs R, Arendt T. The cholinergic system in aging and neuronal degeneration. Behavioural Brain Research. 2011;221(2):555–563. doi: 10.1016/j.bbr.2010.11.058. [DOI] [PubMed] [Google Scholar]
- 4.Grudzien A, Shaw P, Weintraub S, Bigio E, Mash DC, Mesulam MM. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiology of Aging. 2007;28(3):327–335. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Haglund M, Sjöbeck M, Englund E. Locus ceruleus degeneration is ubiquitous in Alzheimer’s disease: possible implications for diagnosis and treatment. Neuropathology. 2006;26(6):528–532. doi: 10.1111/j.1440-1789.2006.00725.x. [DOI] [PubMed] [Google Scholar]
- 6.Shepherd C, McCann H, Halliday GM. Variations in the neuropathology of familial Alzheimer’s disease. Acta Neuropathologica. 2009;118(1):37–52. doi: 10.1007/s00401-009-0521-4. [DOI] [PubMed] [Google Scholar]
- 7.Moncaster JA, Pineda R, Moir RD, et al. Alzheimer’s disease amyloid-β links lens and brain pathology in down syndrome. PLoS ONE. 2010;5(5) doi: 10.1371/journal.pone.0010659. Article ID e10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lott IT, Dierssen M. Cognitive deficits and associated neurological complications in individuals with Down’s syndrome. The Lancet Neurology. 2010;9(6):623–633. doi: 10.1016/S1474-4422(10)70112-5. [DOI] [PubMed] [Google Scholar]
- 9.Teller JK, Russo C, DeBusk LM, et al. Presence of soluble amyloid β-peptide precedes amyloid plaque formation in Down’s syndrome. Nature Medicine. 1996;2(1):93–95. doi: 10.1038/nm0196-93. [DOI] [PubMed] [Google Scholar]
- 10.Nelson LD, Siddarth P, Kepe V, et al. Positron emission tomography of brain β-amyloid and tau levels in adults with down syndrome. Archives of Neurology. 2011;68(6):768–774. doi: 10.1001/archneurol.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathologica. 2011;121(2):171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- 12.Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AM. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. Journal of Neurology, Neurosurgery and Psychiatry. 2007;78(10):1158–1159. doi: 10.1136/jnnp.2006.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pepeu G, Giovannini MG. Cholinesterase inhibitors and memory. Chemico-Biological Interactions. 2010;187(1–3):403–408. doi: 10.1016/j.cbi.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 14.Lahiri DK, Maloney B, Basha MR, Yuan WG, Zawia NH. How and when environmental agents and dietary factors affect the course of Alzheimer’s disease: the “LEARn” model (Latent Early-Life Associated Regulation) may explain the triggering of AD. Current Alzheimer Research. 2007;4(2):219–228. doi: 10.2174/156720507780362164. [DOI] [PubMed] [Google Scholar]
- 15.Mohan M, Carpenter PK, Bennett C. Donepezil for dementia in people with Down syndrome. Cochrane Database of Systematic Reviews. 2009;(1) doi: 10.1002/14651858.CD007178.pub2. Article ID CD007178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohan M, Bennett C, Carpenter PK. Rivastigmine for dementia in people with Down syndrome. Cochrane Database of Systematic Reviews. 2009;(1) doi: 10.1002/14651858.CD007658. Article ID CD007658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rasmussen SA, Whitehead N, Collier SA, Frías JL. Setting a public health research agenda for Down syndrome: summary of a meeting sponsored by the centers for disease control and prevention and the national down syndrome society. American Journal of Medical Genetics, Part A. 2008;146(23):2998–3010. doi: 10.1002/ajmg.a.32581. [DOI] [PubMed] [Google Scholar]
- 18.Fonatsch C. The role of chromosome 21 in hematology and oncology. Genes Chromosomes and Cancer. 2010;49(6):497–508. doi: 10.1002/gcc.20764. [DOI] [PubMed] [Google Scholar]
- 19.Rao KS. Mechanisms of disease: DNA repair defects and neurological disease. Nature Clinical Practice Neurology. 2007;3(3):162–172. doi: 10.1038/ncpneuro0448. [DOI] [PubMed] [Google Scholar]
- 20.Martin GM. Syndromes of accelerated aging. National Cancer Institute Monograph. 1982;60:241–247. [PubMed] [Google Scholar]
- 21.Antonarakis SE, Lyle R, Dermitzakis ET, Reymond A, Deutsch S. Chromosome 21 and Down syndrome: from genomics to pathophysiology. Nature Reviews Genetics. 2004;5(10):725–738. doi: 10.1038/nrg1448. [DOI] [PubMed] [Google Scholar]
- 22.Lockrow J, Boger H, Gerhardt G, Aston-Jones G, Bachman D, Granholm A-C. A noradrenergic lesion exacerbates neurodegeneration in a down syndrome mouse model. Journal of Alzheimer's Disease. 2011;23(3):471–489. doi: 10.3233/JAD-2010-101218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tyrrell J, Cosgrave M, McCarron M, et al. Dementia in people with Down’s syndrome. International Journal of Geriatric Psychiatry. 2001;16(12):1168–1174. doi: 10.1002/gps.502. [DOI] [PubMed] [Google Scholar]
- 24.Lanfranchi S, Jerman O, Dal Pont E, Alberti A, Vianello R. Executive function in adolescents with Down Syndrome. Journal of Intellectual Disability Research. 2010;54(4):308–319. doi: 10.1111/j.1365-2788.2010.01262.x. [DOI] [PubMed] [Google Scholar]
- 25.Prasher VP. Review of donepezil, rivastigmine, galantamine and memantine for the treatment of dementia in Alzheimer’s disease in adults with Down syndrome: implications for the intellectual disability population. International Journal of Geriatric Psychiatry. 2004;19(6):509–515. doi: 10.1002/gps.1077. [DOI] [PubMed] [Google Scholar]
- 26.Lott IT, Head E. Down syndrome and alzheimer’s disease: a link between development and aging. Mental Retardation and Developmental Disabilities Research Reviews. 2001;7(3):172–178. doi: 10.1002/mrdd.1025. [DOI] [PubMed] [Google Scholar]
- 27.Lott IT, Head E. Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiology of Aging. 2005;26(3):383–389. doi: 10.1016/j.neurobiolaging.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Cataldo AM, Mathews PM, Boiteau AB, et al. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. American Journal of Pathology. 2008;173(2):370–384. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid β deposition in sporadic alzheimer’s disease and down syndrome: differential effects of APOE genotype and presenilin mutations. American Journal of Pathology. 2000;157(1):277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lockstone HE, Harris LW, Swatton JE, Wayland MT, Holland AJ, Bahn S. Gene expression profiling in the adult Down syndrome brain. Genomics. 2007;90(6):647–660. doi: 10.1016/j.ygeno.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 31.Lubec G, Engidawork E. The brain in Down syndrome (Trisomy 21) Journal of Neurology. 2002;249(10):1347–1356. doi: 10.1007/s00415-002-0799-9. [DOI] [PubMed] [Google Scholar]
- 32.Busciglio J, Pelsman A, Wong C, et al. Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron. 2002;33(5):677–688. doi: 10.1016/s0896-6273(02)00604-9. [DOI] [PubMed] [Google Scholar]
- 33.McGeer EG, McGeer PL. Neuroinflammation in Alzheimer’s disease and mild cognitive impairment: a field in its infancy. Journal of Alzheimer’s Disease. 2010;19(1):355–361. doi: 10.3233/JAD-2010-1219. [DOI] [PubMed] [Google Scholar]
- 34.Fuller S, Steele M, Münch G. Activated astroglia during chronic inflammation in Alzheimer’s disease-Do they neglect their neurosupportive roles? Mutation Research. 2010;690(1-2):40–49. doi: 10.1016/j.mrfmmm.2009.08.016. [DOI] [PubMed] [Google Scholar]
- 35.Helguera P, Pelsman A, Pigino G, Wolvetang E, Head E, Busciglio J. ets-2 promotes the activation of a mitochondrial death pathway in down’s syndrome neurons. Journal of Neuroscience. 2005;25(9):2295–2303. doi: 10.1523/JNEUROSCI.5107-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shukkur EA, Shimohata A, Akagi T, et al. Mitochondrial dysfunction and tau hyperphosphorylation in Ts1Cje, a mouse model for Down syndrome. Human Molecular Genetics. 2006;15(18):2752–2762. doi: 10.1093/hmg/ddl211. [DOI] [PubMed] [Google Scholar]
- 37.Harris-Cerruti C, Kamsler A, Kaplan B, Lamb B, Segal M, Groner Y. Functional and morphological alterations in compound transgenic mice overexpreszing Cu/Zn superoxide dismutaze and amyloid precursor protein. European Journal of Neuroscience. 2004;19(5):1174–1190. doi: 10.1111/j.1460-9568.2004.03188.x. [DOI] [PubMed] [Google Scholar]
- 38.Lott IT, Head E, Doran E, Busciglio J. Beta-amyloid, oxidative stress and down syndrome. Current Alzheimer Research. 2006;3(5):521–528. doi: 10.2174/156720506779025305. [DOI] [PubMed] [Google Scholar]
- 39.Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Progress in Clinical and Biological Research. 1990;360:263–280. [PubMed] [Google Scholar]
- 40.Gardiner K, Fortna A, Bechtel L, Davisson MT. Mouse models of Down syndrome: how useful can they be? Comparison of the gene content of human chromosome 21 with orthologous mouse genomic regions. Gene. 2003;318(1-2):137–147. doi: 10.1016/s0378-1119(03)00769-8. [DOI] [PubMed] [Google Scholar]
- 41.Choi JHK, Berger JD, Mazzella MJ, et al. Age-dependent dysregulation of brain amyloid precursor protein in the Ts65Dn Down syndrome mouse model. Journal of Neurochemistry. 2009;110(6):1818–1827. doi: 10.1111/j.1471-4159.2009.06277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salehi A, Delcroix JD, Belichenko PV, et al. Increased app expression in a mouse model of Down’s Syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51(1):29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 43.Hunter CL, Bimonte-Nelson HA, Nelson M, Eckman CB, Granholm AC. Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: effects of estrogen. Neurobiology of Aging. 2004;25(7):873–884. doi: 10.1016/j.neurobiolaging.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 44.Kern DS, Maclean KN, Jiang H, Synder EY, Sladek JR, Bjugstad KB. Neural stem cells reduce hippocampal tau and reelin accumulation in aged Ts65Dn down syndrome mice. Cell Transplantation. 2011;20(3):371–379. doi: 10.3727/096368910X528085. [DOI] [PubMed] [Google Scholar]
- 45.Lomoio S, Scherini E, Necchi D. β-Amyloid overload does not directly correlate with SAPK/JNK activation and tau protein phosphorylation in the cerebellar cortex of Ts65Dn mice. Brain Research. 2009;1297:198–206. doi: 10.1016/j.brainres.2009.08.052. [DOI] [PubMed] [Google Scholar]
- 46.Hunter CL, Bachman D, Granholm AC. Minocycline prevents cholinergic loss in a mouse model of Down’s syndrome. Annals of Neurology. 2004;56(5):675–688. doi: 10.1002/ana.20250. [DOI] [PubMed] [Google Scholar]
- 47.Di Filippo M, Tozzi A, Ghiglieri V, et al. Impaired plasticity at specific subset of striatal synapses in the Ts65Dn mouse model of down syndrome. Biological Psychiatry. 2010;67(7):666–671. doi: 10.1016/j.biopsych.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 48.Belichenko PV, Kleschevnikov AM, Masliah E, et al. Excitatory-inhibitory relationship in the fascia dentata in the Ts65Dn mouse model of down syndrome. Journal of Comparative Neurology. 2009;512(4):453–466. doi: 10.1002/cne.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper JD, Salehi A, Delcroix JD, et al. Failed retrograde transport of NGF in a mouse model of Down’s syndrome: reversal of cholinergic neurodegenerative phenotypes following NGF infusion. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(18):10439–10444. doi: 10.1073/pnas.181219298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunter CL, Isacson O, Nelson M, et al. Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down’s syndrome. Neuroscience Research. 2003;45(4):437–445. doi: 10.1016/s0168-0102(03)00005-1. [DOI] [PubMed] [Google Scholar]
- 51.Seo H, Isacson O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Experimental Neurology. 2005;193(2):469–480. doi: 10.1016/j.expneurol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 52.Bimonte-Nelson HA, Hunter CL, Nelson ME, Granholm ACE. Frontal cortex BDNF levels correlate with working memory in an animal model of Down syndrome. Behavioural Brain Research. 2003;139(1-2):47–57. doi: 10.1016/s0166-4328(02)00082-7. [DOI] [PubMed] [Google Scholar]
- 53.Granholm ACE, Sanders LA, Crnic LS. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of down’s syndrome. Experimental Neurology. 2000;161(2):647–663. doi: 10.1006/exnr.1999.7289. [DOI] [PubMed] [Google Scholar]
- 54.Holtzman DM, Santucci D, Kilbridge J, et al. Developmental abnormalities and age-related neurodegeneration in a mouse model of Down syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(23):13333–13338. doi: 10.1073/pnas.93.23.13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salehi A, Faizi M, Colas D, et al. Restoration of norepinephrine-modulated contextual memory in a mouse model of Down syndrome. Science Translational Medicine. 2009;1(7):7–17. doi: 10.1126/scitranslmed.3000258. [DOI] [PubMed] [Google Scholar]
- 56.Hunter CL, Bimonte HA, Granholm ACE. Behavioral comparison of 4 and 6 month-old Ts65Dn mice: age-related impairments in working and reference memory. Behavioural Brain Research. 2003;138(2):121–131. doi: 10.1016/s0166-4328(02)00275-9. [DOI] [PubMed] [Google Scholar]
- 57.Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC. The "Down syndrome critical region" is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. Journal of Neuroscience. 2009;29(18):5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olson LE, Roper RJ, Sengstaken CL, et al. Trisomy for the Down syndrome ’critical region’ is necessary but not sufficient for brain phenotypes of trisomic mice. Human Molecular Genetics. 2007;16(7):774–782. doi: 10.1093/hmg/ddm022. [DOI] [PubMed] [Google Scholar]
- 59.Isacson O, Seo H, Lin L, Albeck D, Granholm AC. Alzheimer’s disease and Down’s syndrome: roles of APP, trophic factors and ACh. Trends in Neurosciences. 2002;25(2):79–84. doi: 10.1016/s0166-2236(02)02037-4. [DOI] [PubMed] [Google Scholar]
- 60.Nardone R, Marth R, Ausserer H, Bratti A, Tezzon F. Reduced short latency afferent inhibition in patients with Down syndrome and Alzheimer-type dementia. Clinical Neurophysiology. 2006;117(10):2204–2210. doi: 10.1016/j.clinph.2006.07.134. [DOI] [PubMed] [Google Scholar]
- 61.Fernandez F, Morishita W, Zuniga E, et al. Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nature Neuroscience. 2007;10(4):411–413. doi: 10.1038/nn1860. [DOI] [PubMed] [Google Scholar]
- 62.Reeves RH, Irving NG, Moran TH, et al. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nature Genetics. 1995;11(2):177–184. doi: 10.1038/ng1095-177. [DOI] [PubMed] [Google Scholar]
- 63.Demas GE, Nelson RJ, Krueger BK, Yarowsky PJ. Impaired spatial working and reference memory in segmental trisomy (Ts65Dn) mice. Behavioural Brain Research. 1998;90(2):199–201. doi: 10.1016/s0166-4328(97)00116-2. [DOI] [PubMed] [Google Scholar]
- 64.Hyde LA, Frisone DF, Crnic LS. Ts65Dn mice, a model for Down syndrome, have deficits in context discrimination learning suggesting impaired hippocampal function. Behavioural Brain Research. 2001;118(1):53–60. doi: 10.1016/s0166-4328(00)00313-2. [DOI] [PubMed] [Google Scholar]
- 65.Galdzicki Z, Siarey R, Pearce R, Stoll J, Rapoport SI. On the cause of mental retardation in Down syndrome: extrapolation from full and segmental trisomy 16 mouse models. Brain Research Reviews. 2001;35(2):115–145. doi: 10.1016/s0926-6410(00)00074-4. [DOI] [PubMed] [Google Scholar]
- 66.Faizi M, Bader PL, Tun C, et al. Comprehensive behavioral phenotyping of Ts65Dn mouse model of Down Syndrome: activation of β1-adrenergic receptor by xamoterol as a potential cognitive enhancer. Neurobiology of Disease. 2011;43(2):397–413. doi: 10.1016/j.nbd.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chang Q, Gold PE. Age-related changes in memory and in acetylcholine functions in the hippocampus in the Ts65Dn mouse, a model of Down syndrome. Neurobiology of Learning and Memory. 2008;89(2):167–177. doi: 10.1016/j.nlm.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Costa ACS. On the promise of pharmacotherapies targeted at cognitive and neurodegenerative components of down syndrome. Developmental Neuroscience. 2011;33(5):414–427. doi: 10.1159/000330861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Costa ACS, Scott-McKean JJ, Stasko MR. Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology. 2008;33(7):1624–1632. doi: 10.1038/sj.npp.1301535. [DOI] [PubMed] [Google Scholar]
- 70.Lockrow J, Boger H, Bimonte-Nelson H, Granholm A-C. Effects of long-term memantine on memory and neuropathology in Ts65Dn mice, a model for Down syndrome. Behavioural Brain Research. 2011;221(2):610–622. doi: 10.1016/j.bbr.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rueda N, Llorens-Martín M, Flórez J, et al. Memantine normalizes several phenotypic features in the Ts65Dn mouse model of down syndrome. Journal of Alzheimer’s Disease. 2010;21(1):277–290. doi: 10.3233/JAD-2010-100240. [DOI] [PubMed] [Google Scholar]
- 72.Siarey RJ, Kline-Burgess A, Cho M, et al. Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. Journal of Neurochemistry. 2006;98(4):1266–1277. doi: 10.1111/j.1471-4159.2006.03971.x. [DOI] [PubMed] [Google Scholar]
- 73.Devenny DA, Krinsky-McHale SJ, Sersen G, Silverman WP. Sequence of cognitive decline in dementia in adults with Down’s syndrome. Journal of Intellectual Disability Research. 2000;44(6):654–665. doi: 10.1046/j.1365-2788.2000.00305.x. [DOI] [PubMed] [Google Scholar]
- 74.Weinshenker D. Functional consequences of locus coeruleus degeneration in Alzheimer’s disease. Current Alzheimer Research. 2008;5(3):342–345. doi: 10.2174/156720508784533286. [DOI] [PubMed] [Google Scholar]
- 75.Heneka MT, Galea E, Gavriluyk V, et al. Noradrenergic depletion potentiates β-amyloid-induced cortical inflammation: implications for Alzheimer’s disease. Journal of Neuroscience. 2002;22(7):2434–2442. doi: 10.1523/JNEUROSCI.22-07-02434.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heneka MT, Nadrigny F, Regen T, et al. Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(13):6058–6063. doi: 10.1073/pnas.0909586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jardanhazi-Kurutz D, Kummer MP, Terwel D, et al. Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochemistry International. 2010;57(4):375–382. doi: 10.1016/j.neuint.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 78.Traver S, Salthun-Lassalle B, Marien M, Hirsch EC, Colpaert F, Michel PP. The neurotransmitter noradrenaline rescues septal cholinergic neurons in culture from degeneration caused by low-level oxidative stress. Molecular Pharmacology. 2005;67(6):1882–1891. doi: 10.1124/mol.104.007864. [DOI] [PubMed] [Google Scholar]
- 79.Acquas E, Wilson C, Fibiger HC. Pharmacology of sensory stimulation-evoked increases in frontal cortical acetylcholine release. Neuroscience. 1998;85(1):73–83. doi: 10.1016/s0306-4522(97)00546-0. [DOI] [PubMed] [Google Scholar]
- 80.Harley CW. Norepinephrine and the dentate gyrus. Progress in Brain Research. 2007;163:299–318. doi: 10.1016/S0079-6123(07)63018-0. [DOI] [PubMed] [Google Scholar]
- 81.Heneka MT, Ramanathan M, Jacobs AH, et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. Journal of Neuroscience. 2006;26(5):1343–1354. doi: 10.1523/JNEUROSCI.4236-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalinin S, Gavrilyuk V, Polak PE, et al. Noradrenaline deficiency in brain increases β-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiology of Aging. 2007;28(8):1206–1214. doi: 10.1016/j.neurobiolaging.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 83.Gekman DC, Nelson O, Liang F, Liang CL, Games D. The PDAPP mouse model of Alzheimer’s disease: Locus coeruleus neuronal shrinkage. Journal of Comparative Neurology. 2005;492(4):469–476. doi: 10.1002/cne.20744. [DOI] [PubMed] [Google Scholar]
- 84.German DC, Manaye KF, White CL, et al. Disease-specific patterns of locus coeruleus cell loss. Annals of Neurology. 1992;32(5):667–676. doi: 10.1002/ana.410320510. [DOI] [PubMed] [Google Scholar]
- 85.Mann DMA, Yates PO, Hawkes J. The pathology of the human locus ceruleus. Clinical Neuropathology. 1983;2(1):1–7. [PubMed] [Google Scholar]
- 86.Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Archives of Neurology. 2003;60(3):337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- 87.Brunnström H, Friberg N, Lindberg E, Englund E. Differential degeneration of the locus coeruleus in dementia subtypes. Clinical Neuropathology. 2011;30(3):104–110. doi: 10.5414/npp30104. [DOI] [PubMed] [Google Scholar]
- 88.Marsh L, Biglan K, Gerstenhaber M, Williams JR. Atomoxetine for the treatment of executive dysfunction in Parkinson’s disease: a pilot open-label study. Movement Disorders. 2009;24(2):277–282. doi: 10.1002/mds.22307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mohs RC, Shiovitz TM, Tariot PN, Porsteinsson AP, Baker KD, Feldman PD. Atomoxetine augmentation of cholinesterase inhibitor therapy in patients with alzheimer disease: 6-month, randomized, double-blind, placebo-controlled, parallel-trial study. American Journal of Geriatric Psychiatry. 2009;17(9):752–759. doi: 10.1097/JGP.0b013e3181aad585. [DOI] [PubMed] [Google Scholar]
- 90.Dolder CR, Davis LN, McKinsey J. Use of psychostimulants in patients with dementia. Annals of Pharmacotherapy. 2010;44(10):1624–1632. doi: 10.1345/aph.1P341. [DOI] [PubMed] [Google Scholar]
- 91.Griffin WST. Inflammation and neurodegenerative diseases. American Journal of Clinical Nutrition. 2006;83(2):470S–474S. doi: 10.1093/ajcn/83.2.470S. [DOI] [PubMed] [Google Scholar]
- 92.Mrak RE, Griffin WST. Glia and their cytokines in progression of neurodegeneration. Neurobiology of Aging. 2005;26(3):349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 93.Kálmán J, Juhász A, Laird G, et al. Serum interleukin-6 levels correlate with the severity of dementia in down syndrome and in Alzheimer's disease. Acta Neurologica Scandinavica. 1997;96(4):236–240. doi: 10.1111/j.1600-0404.1997.tb00275.x. [DOI] [PubMed] [Google Scholar]
- 94.McKinney M. Brain cholinergic vulnerability: relevance to behavior and disease. Biochemical Pharmacology. 2005;70(8):1115–1124. doi: 10.1016/j.bcp.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 95.Zassler B, Weis C, Humpel C. Tumor necrosis factor-α triggers cell death of sensitized potassium chloride-stimulated cholinergic neurons. Molecular Brain Research. 2003;113(1-2):78–85. doi: 10.1016/s0169-328x(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 96.Carnevale D, De Simone R, Minghetti L. Microglia-neuron interaction in inflammatory and degenerative diseases: role of cholinergic and noradrenergic systems. CNS and Neurological Disorders. 2007;6(6):388–397. doi: 10.2174/187152707783399193. [DOI] [PubMed] [Google Scholar]
- 97.Bondareff W, Mountjoy CQ. Number of neurons in nucleus locus ceruleus in demented and non-demented patients: rapid estimation and correlated parameters. Neurobiology of Aging. 1986;7(4):297–300. doi: 10.1016/0197-4580(86)90010-2. [DOI] [PubMed] [Google Scholar]
- 98.Zweig RM, Ross CA, Hedreen JC, et al. Neuropathology of aminergic nuclei in Alzheimer’s disease. Progress in Clinical and Biological Research. 1989;317:353–365. [PubMed] [Google Scholar]
- 99.Feinstein DL, Heneka MT, Gavrilyuk V, Dello Russo C, Weinberg G, Galea E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochemistry International. 2002;41(5):357–365. doi: 10.1016/s0197-0186(02)00049-9. [DOI] [PubMed] [Google Scholar]
- 100.Schindowski K, Belarbi K, Buée L. Neurotrophic factors in Alzheimer’s disease: role of axonal transport. Genes, Brain and Behavior. 2008;7(1):43–56. doi: 10.1111/j.1601-183X.2007.00378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Delcroix JD, Valletta J, Wu C, et al. Trafficking the NGF signal: implications for normal and degenerating neurons. Progress in Brain Research. 2004;146:3–23. doi: 10.1016/s0079-6123(03)46001-9. [DOI] [PubMed] [Google Scholar]
- 102.Counts SE, Mufson EJ. The role of nerve growth factor receptors in cholinergic basal forebrain degeneration in prodromal Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2005;64(4):263–272. doi: 10.1093/jnen/64.4.263. [DOI] [PubMed] [Google Scholar]
- 103.Williams BJ, Eriksdotter-Jonhagen M, Granholm AC. Nerve growth factor in treatment and pathogenesis of Alzheimer’s disease. Progress in Neurobiology. 2006;80(3):114–128. doi: 10.1016/j.pneurobio.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 104.Mufson EJ, Conner JM, Kordower JH. Nerve growth factor in Alzheimer’s disease: defective retrograde transport to nucleus basalis. NeuroReport. 1995;6(7):1063–1066. doi: 10.1097/00001756-199505090-00028. [DOI] [PubMed] [Google Scholar]
- 105.Williams B, Granholm AC, Sambamurti K. Age-dependent loss of NGF signaling in the rat basal forebrain is due to disrupted MAPK activation. Neuroscience Letters. 2007;413(2):110–114. doi: 10.1016/j.neulet.2006.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang Y, Mullaney KA, Peterhoff CM, et al. Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1630–1635. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ginsberg SD, Alldred MJ, Counts SE, et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biological Psychiatry. 2010;68(10):885–893. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Koshimizu H, Kiyosue K, Hara T, et al. Multiple functions of precursor BDNF to CNS neurons: negative regulation of neurite growth, spine formation and cell survival. Molecular Brain. 2009;2(1, article 27) doi: 10.1186/1756-6606-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fahnestock M, Garzon D, Holsinger RMD, Michalski B. Neurotrophic factors and Alzheimer's disease: are we focusing on the wrong molecule? Journal of Neural Transmission. 2002;(62, supplement):241–252. doi: 10.1007/978-3-7091-6139-5_22. [DOI] [PubMed] [Google Scholar]
- 110.Navakkode S, Korte M. Cooperation between cholinergic and glutamatergic receptors are essential to induce BDNF-dependent long-lasting memory storage. Hippocampus. 2012;22(22):335–346. doi: 10.1002/hipo.20902. [DOI] [PubMed] [Google Scholar]
- 111.Dogliotti G, Galliera E, Licastro F, Corsi MM. Age-related changes in plasma levels of BDNF in Down syndrome patients. Immunity and Ageing. 2010;7, article 2 doi: 10.1186/1742-4933-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.von Bohlen O, Halbach Involvement of BDNF in age-dependent alterations in the hippocampus. Frontiers in Aging Neuroscience. 2010;2, article 36 doi: 10.3389/fnagi.2010.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Berchtold NC, Castello N, Cotman CW. Exercise and time-dependent benefits to learning and memory. Neuroscience. 2010;167(3):588–597. doi: 10.1016/j.neuroscience.2010.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Berchtold NC, Chinn G, Chou M, Kesslak JP, Cotman CW. Exercise primes a molecular memory for brain-derived neurotrophic factor protein induction in the rat hippocampus. Neuroscience. 2005;133(3):853–861. doi: 10.1016/j.neuroscience.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 115.Liu H-L, Zhao G, Cai K, Zhao H-H, Shi L-D. Treadmill exercise prevents decline in spatial learning and memory in APP/PS1 transgenic mice through improvement of hippocampal long-term potentiation. Behavioural Brain Research. 2011;218(2):308–314. doi: 10.1016/j.bbr.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 116.Liu YF, Chen HI, Yu L, et al. Upregulation of hippocampal TrkB and synaptotagmin is involved in treadmill exercise-enhanced aversive memory in mice. Neurobiology of Learning and Memory. 2008;90(1):81–89. doi: 10.1016/j.nlm.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 117.Nichol K, Deeny SP, Seif J, Camaclang K, Cotman CW. Exercise improves cognition and hippocampal plasticity in APOE ε{lunate}4 mice. Alzheimer's and Dementia. 2009;5(4):287–294. doi: 10.1016/j.jalz.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Willis L, Quintero EM, Nelson M, Granholm AC. Regulation of trophic factor expression by innervating target regions in intraocular double transplants. Cell Transplantation. 2005;14(1):21–29. doi: 10.3727/000000005783983313. [DOI] [PubMed] [Google Scholar]
- 119.Counts SE, Mufson EJ. Noradrenaline activation of neurotrophic pathways protects against neuronal amyloid toxicity. Journal of Neurochemistry. 2010;113(3):649–660. doi: 10.1111/j.1471-4159.2010.06622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jellinger KA. Recent advances in our understanding of neurodegeneration. Journal of Neural Transmission. 2009;116(9):1111–1162. doi: 10.1007/s00702-009-0240-y. [DOI] [PubMed] [Google Scholar]
- 121.Strydom A, Dickinson MJ, Shende S, Pratico D, Walker Z. Oxidative stress and cognitive ability in adults with Down syndrome. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(1):76–80. doi: 10.1016/j.pnpbp.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 122.Nikonenko AG, Radenovic L, Andjus PR, Skibo GG. Structural features of ischemic damage in the hippocampus. Anatomical Record. 2009;292(12):1914–1921. doi: 10.1002/ar.20969. [DOI] [PubMed] [Google Scholar]
- 123.Cruz-Sánchez FF, Gironès X, Ortega A, Alameda F, Lafuente JV. Oxidative stress in Alzheimer’s disease hippocampus: a topographical study. Journal of the Neurological Sciences. 2010;299(1-2):163–167. doi: 10.1016/j.jns.2010.08.029. [DOI] [PubMed] [Google Scholar]
- 124.Nunomura A, Perry G, Aliev G, et al. Oxidative damage is the earliest event in Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2001;60(8):759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 125.Nunomura A, Perry G, Pappolla MA, et al. Neuronal oxidative stress precedes amyloid-β deposition in down syndrome. Journal of Neuropathology and Experimental Neurology. 2000;59(11):1011–1017. doi: 10.1093/jnen/59.11.1011. [DOI] [PubMed] [Google Scholar]
- 126.Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(32):13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gardner R, Salvador A, Moradas-Ferreira P. Why does SOD overexpression sometimes enhance, sometimes decrease, hydrogen peroxide production? A minimalist explanation. Free Radical Biology and Medicine. 2002;32(12):1351–1357. doi: 10.1016/s0891-5849(02)00861-4. [DOI] [PubMed] [Google Scholar]
- 128.Kim SH, Fountoulakis M, Cairns N, Lubec G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer's disease and Down Syndrome. Journal of Neural Transmission. 2001;(61, supplement):223–235. doi: 10.1007/978-3-7091-6262-0_18. [DOI] [PubMed] [Google Scholar]
- 129.Praticò D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: lights and shadows. Annals of the New York Academy of Sciences. 2008;1147:70–78. doi: 10.1196/annals.1427.010. [DOI] [PubMed] [Google Scholar]
- 130.Ellis JM, Hooi KT, Gilbert RE, et al. Supplementation with antioxidants and folinic acid for children with Down’s syndrome: randomised controlled trial. British Medical Journal. 2008;336(7644):594–597. doi: 10.1136/bmj.39465.544028.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lockrow J, Prakasam A, Huang P, Bimonte-Nelson H, Sambamurti K, Granholm AC. Cholinergic degeneration and memory loss delayed by vitamin E in a Down syndrome mouse model. Experimental Neurology. 2009;216(2):278–289. doi: 10.1016/j.expneurol.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.De Felice FG, Velasco PT, Lambert MP, et al. Aβ oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. Journal of Biological Chemistry. 2007;282(15):11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 133.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407(6805):802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 134.Porta S, Serra SA, Huch M, et al. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: a potential pathogenic process in neurodegeneration. Human Molecular Genetics. 2007;16(9):1039–1050. doi: 10.1093/hmg/ddm049. [DOI] [PubMed] [Google Scholar]
- 135.Faraone SV, Biederman J, Spencer T, et al. Atomoxetine and stroop task performance in adult attention-deficit/ hyperactivity disorder. Journal of Child and Adolescent Psychopharmacology. 2005;15(4):664–670. doi: 10.1089/cap.2005.15.664. [DOI] [PubMed] [Google Scholar]