

Abstract
SQ109, a 1,2-diamine related to ethambutol, is currently in clinical trials for the treatment of tuberculosis, but its mode of action remains unclear. Here, we demonstrate that SQ109 disrupts cell wall assembly, as evidenced by macromolecular incorporation assays and ultrastructural analyses. SQ109 interferes with the assembly of mycolic acids into the cell wall core of Mycobacterium tuberculosis, as bacilli exposed to SQ109 show immediate inhibition of trehalose dimycolate (TDM) production and fail to attach mycolates to the cell wall arabinogalactan. These effects were not due to inhibition of mycolate synthesis, since total mycolate levels were unaffected, but instead resulted in the accumulation of trehalose monomycolate (TMM), the precursor of TDM and cell wall mycolates. In vitro assays using purified enzymes showed that this was not due to inhibition of the secreted Ag85 mycolyltransferases. We were unable to achieve spontaneous generation of SQ109-resistant mutants; however, analogs of this compound that resulted in similar shutdown of TDM synthesis with concomitant TMM accumulation were used to spontaneously generate resistant mutants that were also cross-resistant to SQ109. Whole-genome sequencing of these mutants showed that these all had mutations in the essential mmpL3 gene, which encodes a transmembrane transporter. Our results suggest that MmpL3 is the target of SQ109 and that MmpL3 is a transporter of mycobacterial TMM.
INTRODUCTION
Tuberculosis (TB) remains the number-one killer of people infected with a single infectious agent worldwide and it is estimated that a third of the world's population is latently infected with Mycobacterium tuberculosis, the etiologic agent of TB (70). Current short-course chemotherapy requires a minimum of 6 months of treatment consisting of a 2-month intensive phase of treatment with the drugs isoniazid (INH), rifampin, pyrazinamide, and ethambutol (EMB) followed by a 4-month continuation of therapy with INH and rifampin alone (72). This treatment regimen can achieve 95% cure rates in clinical trial settings where optimal patient care is ensured, but global cure rates are much lower. A driving factor for the low cure rates is default of patients on chemotherapy due to the extended treatment duration, where early discontinuation of chemotherapy increases the risk for development of drug resistance. Almost 4% of all TB cases globally are estimated to be multidrug resistant (71), with the number of drug-resistant cases increasing annually. Thus, new drugs are urgently needed to shorten the duration of TB chemotherapy, as well as target drug-resistant TB (4).
Mycobacterial cell wall biosynthesis has historically been a particularly vulnerable target for chemotherapeutic intervention, with many of the drugs in clinical use for TB targeting synthesis of the mycolic acid (INH, ethionamide, and thiacetazone) or arabinan (EMB) components of the cell envelope (3, 6, 34, 39). The mycobacterial cell wall is composed of a covalently linked complex of three heteropolymers, peptidoglycan, arabinogalactan, and mycolic acids, often referred to as the mycolyl-arabinogalactan-peptidoglycan complex (12, 13). This complex assembly forms an asymmetrical, nonfluid monolayer decorated on its surface with noncovalently associated “capsular” lipids, such as trehalose monomycolate (TMM), trehalose dimycolate (TDM), the sulfolipids, phenolic glycolipids, and phthiocerol dimycoserosates (PDIMs), as well as complex polysaccharides and small amounts of protein (Fig. 1) (17, 22, 35). Despite our understanding of the chemical structures involved in the cell wall complex, our knowledge of the mechanistic details of their assembly remains incomplete and our understanding of their precise organization is still evolving (27, 57, 74).
Fig 1.

Diagrammatic representation of the mycobacterial cell wall and proposed pathway involved in mycolic acid processing and export (12, 63) (adapted with permission from reference 16a). The cartoon depicts some components of the cell wall while primarily focusing on trehalose monomycolate (TMM) and trehalose dimycolate (TDM). Hypothetical or proposed steps are indicated by gray arrows, and question marks in parentheses represent hypothetical proteins/enzymes not yet identified. The inset legend represents the shapes used to depict the components shown. Note that the diagram is not to scale and does not include detailed structures or all components of the mycobacterial cell wall.
Mycolic acids are long-chain (C70 to C90) α-alkyl, β-hydroxy fatty acids found anchored directly to terminal arabinose units of the core complex, as well as in abundant esters of the disaccharide trehalose as TMM and TDM (5, 50). These long-chain lipids are the primary mediators of the hydrophobic character of the cell envelope and form a closely packed, impermeable monolayer (12, 44, 45, 51, 67). The front-line agent INH inhibits mycolic acid biosynthesis by the mycobacterial fatty acid synthase II complex via multiple mechanisms (66), resulting in loss of TMM and TDM and of mycolates attached to cell wall arabinan. EMB, on the other hand, inhibits the biosynthesis of the arabinogalactan component of the mycobacterial cell wall (62). The exact mode of action of EMB is unclear, although it is known to target the integral membrane arabinosyltransferases involved in transfer of arabinan to the galactan component of the cell wall, with resistance mutations predominantly mapping to the arabinosyltransferase encoded by embB (68). EMB treatment results in accumulation of both TDM and TMM due to loss of the arabinan acceptor sites for the mycolates in the cell wall (49).
EMB was selected from analogs of the lead molecule N,N′-diisopropyl ethylenediamine which was found to have activity against M. tuberculosis in vitro and in vivo (59). EMB was introduced into standard chemotherapy regimens when it was found to be better tolerated than para-aminosalicylic acid and allowed the reduction of the total duration of chemotherapy from 24 to 18 months in clinical trials (21, 41). Ethambutol has a modest potency against M. tuberculosis in vitro (MIC, 10 μM) and, due to ocular toxicity, can only be administered to humans at doses that give peak serum concentrations less than 2-fold above the MIC, resulting in suboptimal exposure due to its rapid elimination (serum half-life of 4 h) (73). However, the favorable physicochemical properties of ethambutol combined with its low cytotoxicity spurred the search for improved ethylenediamine analogs with improved potency against M. tuberculosis in vitro. Using a combinatorial solid-phase synthetic approach, more than 63,000 analogs were tested and almost 3,000 rescreened for inhibition of M. tuberculosis growth (40). Of the 69 most potent analogs that were resynthesized in milligram scale, two compounds had submicromolar MIC values and one, SQ109, {N′-(2-adamantyl)-N-[(2E)-3,7-dimethylocta-2,6-dienyl]ethane-1,2-diamine}, had low cytotoxicity as well as improved in vivo efficacy against M. tuberculosis (55). SQ109 was shown to be superior to EMB in vitro as well as in vivo (33), has favorable pharmacokinetic properties and, importantly, has been shown to accumulate in the lung, the site of M. tuberculosis infection (32). Accumulation at the site of infection may be a critical parameter that determines drug efficacy (54), further underscoring the potential utility of SQ109 for TB chemotherapy. Replacement of EMB with SQ109 in drug combination regimens leads to improved bacillary clearance from infected animal tissues, which has prompted clinical evaluation of this compound in phase I clinical trials to determine safety (53). SQ109 is now entering trials to evaluate early bactericidal activity in patients with pulmonary TB (Michael Hoelscher, clinicaltrials.gov ID NCT01218217, National Institutes of Health, 2011; http://clinicaltrials.gov/).
Despite this progress of SQ109 in clinical development, the mode of action of SQ109 has remained elusive. Its synthesis was based on the common N,N′-isopropyl ethylenediamine core that led to the discovery of EMB. The initial screen that identified SQ109 was based on the upregulation of the iniBAC operon promoter (40), with the effects of SQ109 on the expression of the proteins encoded by iniBAC being subsequently confirmed by proteomic studies (31). The iniBAC operon encodes a set of genes with unknown function, although they appear to be essential for the activity of an unidentified pump which confers low-level drug tolerance in Mycobacterium bovis and M. tuberculosis (15). These genes are highly upregulated by many inhibitors of cell wall synthesis (1, 11). However, SQ109's mechanism of action was found to be different from those of EMB and N,N′-isopropyl ethylenediamine since it retained full activity against EMB-resistant isolates of M. tuberculosis (55), resulted in M. tuberculosis transcriptional profiles that were distinct from the results for EMB and N,N′-isopropyl ethylenediamine (11), and did not cause the rapid depletion of cell wall-linked arabinose sugars associated with EMB treatment (11).
In the current study, we conducted biochemical analyses to identify the pathway perturbed by SQ109. SQ109 inhibits the incorporation of de novo-synthesized mycolic acids into the M. tuberculosis cell envelope more potently than EMB without affecting total mycolate levels. Analysis of cellular mycolate pools indicated that SQ109 causes depletion of TDM pools with concomitant upregulation of TMM levels, and this effect was shown to be unrelated to the activity of the antigen 85 (Ag85) mycolyltransferases, known to be involved in the conversion of TMM to TDM (Fig. 1) (7) and transfer of mycolates to the cell wall (18, 29, 56). Our hypothesis was that SQ109 inhibited the activity of an efflux system for TMM, and this was further confirmed by the finding that mutations in the essential mmpL3 gene, encoding a transmembrane transporter of unknown function, conferred resistance to SQ109 and related ethylenediamine analogs but not EMB. This finding further supports the notion that the mycolate donor to the arabinogalactan-linked mycolates is TMM.
MATERIALS AND METHODS
Strains, media, and growth conditions.
M. tuberculosis H37Rv was maintained in logarithmic growth phase in liquid culture using Middlebrook 7H9 medium (Becton Dickinson, NJ) containing 0.05% (vol/vol) Tween 80 supplemented with ADC (albumin-dextrose-catalase), and solid cultures were maintained on 7H11 agar (Becton Dickinson, NJ) supplemented with OADC (ADC containing oleic acid). All antibiotics were purchased from Sigma-Aldrich (St. Louis, MO), and all radiochemicals were purchased from PerkinElmer, Inc. ADT (6-azido-6-deoxy-α, α′-trehalose) for Ag85 inhibition assays was kindly provided by K. M. Backus (2). M. tuberculosis was grown at 37°C, and liquid cultures were propagated using roller bottles. Escherichia coli BL21(DE3) was purchased from Invitrogen (Life Technologies), and cultures were grown in LB medium for preparing Ag85 proteins as specified by the TB Vaccine Testing and Research Materials Contract at Colorado State University (TBVTRM-CSU) (http://www.cvmbs.colostate.edu/mip/tb/).
MIC determination and early aerobic kill assays.
MICs were determined using the 2-fold broth dilution method with the MIC reported as the concentration of drug that completely inhibits all visible growth after 1 week of growth (69). Aerobic kill assays were carried out using 10-fold MIC concentrations of the respective compounds. Liquid cultures grown to an optical density at 650 nm (OD650) of ∼0.3 were diluted 100-fold before adding the respective compounds. One hundred-microliter samples were removed on days 0, 1, 2, 3, 4, 6, 8, 11, 14, and 21 and serially diluted in triplicate, and the dilutions were plated in duplicate and incubated for 4 to 6 weeks before assessing M. tuberculosis growth by counting the number of CFU.
Macromolecular incorporation assays.
Five-milliliter M. tuberculosis cultures were grown to an OD650 of ∼0.3 before adding radiolabeled precursors. Ten microcuries per milliliter of [4,5-3H]l-leucine (60 Ci/mmol) and [1-3H]N-acetyl-d-glucosamine (5 Ci/mmol) were used for monitoring protein and peptidoglycan biosynthesis, respectively. Ten microcuries per milliliter of [5,6-3H]uracil (40 Ci/mmol) was used for labeling total nucleic acids (TNA) based on reports describing its efficient incorporation into both RNA and DNA in mycobacteria (61). Specific activities and exposure were obtained from similar studies in M. tuberculosis, where uptake was shown to be linear over this time (38, 47). The cultures were incubated for 1 h, followed by the addition of SQ109 at concentrations ranging from 0 to 10-fold MIC values. After 10 h of SQ109 exposure, cultures were centrifuged and the cell pellets were resuspended in 500 μl of phosphate-buffered saline (PBS) and disrupted by bead beating using a MagNA Lyser Instrument (Roche) and zirconia beads. Macromolecules were precipitated by adding an equal volume of 20% ice-cold trichloroacetic acid (TCA) followed by incubation on ice for 30 min. The precipitate was harvested on Whatman GF/C filters and washed extensively with ice-cold 10% TCA, and incorporated radioactivity was measured by liquid scintillation counting.
Electron microscopy analyses.
Mid-log-phase M. tuberculosis (OD650 = 0.2) was treated with 10 times the MIC of SQ109 (20 μM), EMB (62.5 μM), or INH (2 μM) for 48 h before harvesting the cells and washing twice with PBS containing 0.05% Tween 80 (vol/vol) (PBST). For scanning electron microscopy (SEM), the cell pellets were resuspended in 1/10 volume of PBST and a 50-μl suspension was spotted on Si chips (Ted Pella, Inc.). The cells were allowed to adhere on the chip for 15 to 20 min, followed by two rinses with PBST. The chips were then fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. All subsequent processing was carried out in a Pelco Biowave laboratory microwave system (Ted Pella, Inc.) at 250 W and 20 in. Hg vacuum. The chips were postfixed with 1% osmium tetroxide–0.8% potassium ferricyanide in 0.1 M sodium cacodylate, followed by rinsing with water and dehydration in a graded ethanol series. The specimen was critical point dried in a Bal-Tec CPD 030 drier (Bal-Tec AG) and coated with 80 Å of iridium using an IBS ion beam sputter (South Bay Technology, Inc.). SEM samples were imaged using a Hitachi SU8000 SEM (Hitachi High Technologies). For transmission electron microscopy, the cell pellets were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 6.5). The pellets were postfixed in a microwave with 1% osmium tetroxide–0.8% potassium ferricyanide in 0.1 M sodium cacodylate, followed by 1% tannic acid in distilled water, and stained en bloc with 1% aqueous uranyl acetate. They were then rinsed with distilled water and dehydrated in a graded ethanol series. The pellets were then infiltrated and embedded in Spurr's resin which was polymerized overnight in a 68°C oven. Thin sections (90 nm) were cut using a UC6 ultramicrotome (Leica Microsystems) and stained with 4% aqueous uranyl acetate and Reynold's lead citrate prior to viewing on a Hitachi H-7500 (Hitachi High Technologies) at 80 kV. Digital images were acquired with a Hammamatsu XR-100 digital camera system (AMT).
Lipid labeling, extraction, and analysis.
Radiolabeling of M. tuberculosis lipids with 14C-acetate was carried out by adding the sodium salt of [1,2-14C]acetic acid (PerkinElmer Inc.) at a final concentration of 2 μCi/ml (1 Ci/mmol) of culture with an OD650 of ∼0.3. Radiolabel incorporation was found to be linear up to 18 h under these conditions (results now shown). Five-milliliter culture aliquots were drawn at specified times and centrifuged, and the cell pellet was used directly to analyze total cellular lipids using the methyl esterification protocol for preparing mycolic acid methyl esters (MAMEs) and fatty acid methyl esters (FAMEs) as described earlier (60). To analyze extractable polar lipids, the cell pellet was extracted with 5 ml CHCl3-CH3OH (2:1) at 50°C overnight and the tubes were then recentrifuged. The pellet contained the fraction of lipids covalently associated with the cell wall, whereas the solvent extract contained the extractable noncovalently associated polar lipid fraction (including TMM and TDM). The cell pellet was treated to isolate MAMEs and FAMEs for analysis as described above, whereas the extracted lipids were dried down and redissolved in CHCl3-CH3OH (2:1) for subsequent analysis.
Thin-layer chromatography (TLC) to analyze the extractible lipids was carried out using 20- by 20-cm silica gel 60 plates (EMD Chemicals, Inc.), and different solvent systems were used for separation based on the lipid species and required resolution. For visualizing MAMEs and FAMEs, material containing 10,000 cpm was diluted in an equal volume of acetone and spotted on to TLC plates that were developed three times using petroleum ether-diethyl ether (85:15) (60). For single-dimensional analysis of extractable polar lipids, TLC plates were loaded with samples as described above (except that the extractable lipids were dissolved in CHCl3-CH3OH, 2:1) and the plates were developed in either CHCl3-CH3OH-NH4OH (80:20:2) (7) or CHCl3-CH3OH-H2O (62:25:4) (49). Two-dimensional TLC analysis of extractable polar lipids was carried out using the following solvent system: first dimension, CHCl3-CH3OH-H2O (62:25:4), followed by CHCl3-CH3COOH-CH3OH-H2O (50:60:2.5:3) for separation in the second dimension. The TLC plates were air dried and visualized using a phosphorimager (Typhoon Trio), and densitometry was carried out using the accompanying ImageQuant software (GE Healthcare).
Ag85 protein purification and activity assays.
The recombinant Ag85A, -B, and -C proteins were expressed and purified using the pMRLB41 (for Ag85A), pMRLB47 (for Ag85B), and pMRLB16 (for Ag85C) plasmid systems and protocols as specified by TBVTRM-CSU (http://www.cvmbs.colostate.edu/mip/tb/). Native Ag85A, -B, and -C were also obtained from TBVTRM-CSU for activity analysis. Ag85 enzymatic assays were performed using a previously described protocol (36).
Synthesis of SQ109 and its analogs.
SQ109 was synthesized as described previously (40). Detailed methodology and schemes used in the chemical synthesis of SQ109 analogs used in the current study are included in the supplemental material. The compounds DA5 and DA8 were purchased from ChemBridge Corporation (San Diego).
Isolation and genetic characterization of mutants cross resistant to SQ109.
The compounds DA5 (SRI 356596) and DA8 (SRI 391844) were initially identified in a screen for growth inhibition of M. tuberculosis which was run at the Southern Research Institute as part of the MLPCN program and part of the Tuberculosis Antimicrobial Acquisition and Coordinating Facility program contracts N01-AI-95364 and NO1-AI-15449 (24). M. tuberculosis mutants resistant to DA5 and DA8 were isolated by plating 10-fold serial dilutions of liquid cultures grown to an OD600 of ∼1.5 onto plates containing 40 μM DA5 and 21.1 or 42.2 μM DA8, respectively. The plates were incubated for 4 weeks, after which growth was observed. Colonies were picked, grown in liquid medium, and subjected to MIC testing using DA5, DA8, and SQ109. A single DA5-resistant isolate and one colony from each of two DA8-resistant isolates were sent for chromosomal DNA sequencing and single nucleotide polymorphism (SNP) analysis. Whole-genome fragment libraries were prepared using Illumina's paired-end sample preparation kit. DNA samples were hybridized onto the flow cell by using the paired-end cluster generation kit, version 2 (Illumina, San Diego, CA), and transferred to the genome analyzer IIx, and 50 cycles of sequencing were performed using the 36-cycle sequencing kit, version 3 (Illumina, San Diego, CA). Sequencing Control Studio version 2.5 was used with Integrated Real Time Analysis version 1.5.35.0 on the genome analyzer IIx, and Pipeline version 1.5.0 was used for analysis. The resulting 8 to 11 million reads were aligned to the H37Rv genome using bwa, version 0.5.9 (42), and converted to a .sam file and then to a .bam file with samtools version 1.17 (43). The .bam file was sorted, duplicates were removed, and SNPs were called with SAMtools mpileup (extended BAQ calculations were engaged). Only high quality (>200) homozygous SNPs were reported. The online programs TMHMM (http://www.cbs.dtu.dk/services/TMHMM-2.0/) and TMpred (http://www.ch.embnet.org/software/TMPRED_form.html) were used to predict the topology of the putative MmpL3 protein.
RESULTS
In vitro kill kinetics of SQ109.
To determine if SQ109 exhibited bacteriostatic or bactericidal activity against M. tuberculosis, in vitro survival assays were performed. Aerobic batch cultures were exposed to EMB, INH, the SQ109 analog NIH59 (40, 55), and SQ109, and cell survival was followed for 21 days (Fig. 2). SQ109 and its analog NIH59 reduced early-culture cell viability and exhibited activity similar to that of INH, a known M. tuberculosis bactericidal agent (26). In addition, EMB and SQ109 reduced mycobacterial survival by approximately 5 logs by day 21 (Fig. 2).
Fig 2.
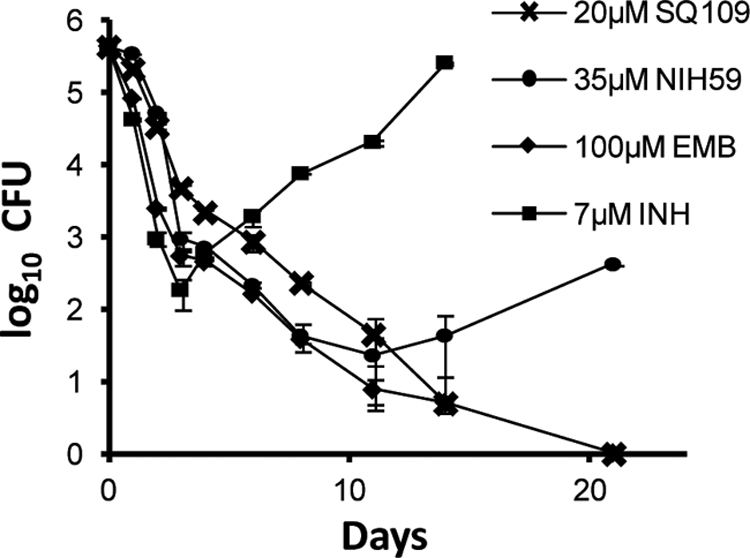
In vitro activity of SQ109. Survival of M. tuberculosis following 10× MIC exposure to EMB, INH, NIH59, and SQ109 under aerobic conditions. Batch cultures were propagated in the presence of the indicated drugs, and samples were withdrawn for determining CFU counts by plating on the days indicated. The increase in CFU counts in the INH treatment is due to the emergence of spontaneous resistance as reported previously (8).
There was an increase in the number of bacilli in the batch culture exposed to INH after 4 days of exposure, which can be attributed to the high rate of emergence of INH resistance observed under in vitro conditions (8). We were also unable to isolate spontaneous SQ109 mutants when M. tuberculosis was plated on agar containing 5- or 10-fold the MIC of SQ109 or by attempts to generate resistant mutants in serial liquid culture at different SQ109 concentrations, whereas INH mutants were isolated at reported frequencies (data not shown). These results suggested that SQ109 inhibited an essential target where mutations in residues critical for SQ109 binding are not tolerated, or it is possible that this compound inhibited multiple targets.
SQ109 affects covalent attachment of lipids to the cell envelope.
Transcriptional profiling studies had suggested that SQ109 has a mechanism of action distinct from that of EMB (11). To confirm that the primary effect of SQ109 was still directed toward the cell envelope, we performed macromolecular incorporation assays to quantify protein, nucleic acid, and cell wall biosynthesis using radiolabeled precursors. These results demonstrated that SQ109 did not inhibit the incorporation of uracil, l-leucine, or N-acetyl-d-glucosamine as markers of nucleic acid, protein, and peptidoglycan synthesis, respectively, at sub-MIC concentrations, although inhibition was evident, especially for peptidoglycan biosynthesis, at concentrations above its MIC (Fig. 3A). In contrast, treatment of cells with SQ109 followed by radiolabeling with 14C-acetate showed that there was a rapid and dramatic reduction of lipids covalently bound to the cell wall upon SQ109 treatment. EMB, in contrast, resulted in a more gradual decline of covalently attached lipids (Fig. 3B). This rapid inhibition of covalently attached lipids was not mirrored in the total free lipids with either SQ109 or EMB (Fig. 3C). Thus, SQ109 appears to specifically affect attachment, not biosynthesis, of lipids to the mycobacterial cell wall during assembly.
Fig 3.

Macromolecular incorporation assays indicate that SQ109 inhibits precursor incorporation during cell wall biosynthesis. (A) Cultures were labeled with [4,5-3H]l-leucine, [1-3H]N-acetyl-d-glucosamine, and [5,6-3H]uracil for 1 h to monitor protein, peptidoglycan, and total nucleic acid biosynthesis, respectively, upon being exposed to different concentrations of SQ109 based on its MIC. (B and C) SQ109 inhibits precursor incorporation into cell wall lipids. Batch cultures were exposed to 10× MIC concentrations of EMB and SQ109 for the indicated times followed by labeling with 14C-acetate for 1 h. Cells were harvested and extracted with CHCl3-CH3OH (2:1), and the solvent extract contained polar extractable lipids, whereas lipids covalently associated with the cell wall remained in the insoluble material. (B) Radioactivity recovered from the solvent-extracted pellet after saponification (representing covalently bound cell wall lipids) showing the rapid decrease in the incorporation of 14C into cell wall-bound lipids on SQ109 treatment. (C) Radioactivity recovered in solvent extract prior to saponification (representing extractable polar lipids).
Effect of SQ109 on cell wall ultrastructure.
We next analyzed the ultrastructural changes that occurred in M. tuberculosis after treatment with SQ109 and other known inhibitors of cell wall assembly by scanning and transmission electron microscopy (Fig. 4). Scanning electron microscopy revealed that M. tuberculosis cells showed significant ruffling of the surface upon SQ109 treatment (Fig. 4Ai). Similar to INH-treated (Fig. 4Aii) and EMB-treated (Fig. 4Aiii) cells, SQ109-treated cells were slightly shorter than untreated cells (Fig. 4Aiv). Quantification of cell length from 150 bacilli for each treatment revealed that all three cell wall inhibitors reduced cell length by about 30% (see Fig. S1 and Table S1 in the supplemental material). Treatment with all three cell wall inhibitors likewise resulted in cells with a larger transverse diameter, with untreated cells having a width of 0.36 ± 0.03 μm (mean ± standard deviation) and SQ109-treated cells having a width of 0.43 ± 0.04 μm (see Table S1). Transmission electron microscopy of sections cut from identically treated fixed cells showed that SQ109 treatment resulted in an accumulation of electron-opaque material in the region separating the plasma membrane from the electron-lucent cell wall skeleton (Fig. 4Bi and ii). This dense zone was more similar to the appearance of INH-treated cells (Fig. 4Biii and iv) but appeared more pronounced. SQ109-treated cells looked very distinct from untreated (Fig. 4Bvii and viii) and EMB-treated (Fig. 4Bv and vi) cells, consistent with a distinct mechanism of cell wall inhibition.
Fig 4.

Ultrastructural changes associated with SQ109 treatment parallel the changes observed with INH and EMB treatment. (A) Scanning electron micrographs of M. tuberculosis treated with 10× MIC of SQ109 (i), EMB (ii), or INH (iii) for 48 h, as opposed to untreated cells (iv), show shortening of cells on drug exposure. Scale bar: 1 μm. (B) Transmission electron micrographs of M. tuberculosis treated with 10× MIC of SQ109 (i, ii), INH (iii, iv), or EMB (v, vi), as opposed to untreated cells (vii, viii), showing longitudinal (i, iii, v, vii) and transverse (ii, iv, vi, viii) sections of representative cells, respectively. Scale bar: 100 nm. Treated cells show cell wall layer thickening with all three agents.
SQ109 inhibits attachment of mycolates to the cell wall and TDM, resulting in TMM accumulation.
The macromolecular incorporation assays and ultrastructural analyses suggested that the target of SQ109 was associated with assembly of the cell wall, consistent with the upregulation of the iniBAC operon and a previously reported increase in cell wall arabinose content on SQ109 exposure (11). To narrow the target down further, we explored the effect of SQ109 on the attachment of mycolic acids to the cell wall skeleton. Following extraction of peripherally associated lipids, saponification of the covalently attached cell wall lipids revealed that all three classes of arabinogalactan-anchored mycolates were rapidly depleted (Fig. 5A). Newly synthesized mycolates attached to the cell wall were undetectable within 75 min of SQ109 treatment. In contrast, EMB resulted in a gradual decline in cell wall-attached mycolates that were still detectable after 5 h of drug treatment (Fig. 5A). Treatment of cells with SQ109 did not affect total fatty acid and mycolate pools (Fig. 6A and B) in the time frame that this drug abolished attachment of mycolates to the cell wall (Fig. 5).
Fig 5.

Mycolate lipid profiles of M. tuberculosis on drug exposure. Bacteria in batch cultures were exposed to 10× MIC concentrations of EMB and SQ109 for 1 h and were then labeled with 14C-acetate. Cells were harvested at indicated time points, and the pellets were extracted with CHCl3-CH3OH (2:1). (A) TLC analysis of MAMEs and FAMEs isolated from pellets after free lipids were removed by CHCl3-CH3OH extraction. Solvent system: petroleum ether-diethyl ether (85:15). The figure demonstrates loss of incorporation of de novo-synthesized mycolic acids into the cell wall on SQ109 exposure, contrary to the results for the EMB control. (B) Extractable lipid profiles (CHCl3-CH3OH extract of pellet) showing accumulation of TDM on EMB exposure, as previously reported (49), and loss of TDM production on SQ109 treatment. Solvent system: CHCl3-CH3OH-NH4OH (80:20:2).
Fig 6.

Total mycolic acid and fatty acid profiles (intracellular and cell wall associated) from M. tuberculosis cells on SQ109 treatment. Cells were processed as described for Fig. 5 without prior chloroform-methanol extraction to remove free lipids, demonstrating that SQ109 does not reduce mycolic acid biosynthesis. (A) TLC analysis of total MAMEs and FAMEs isolated. (B) Radioactivity recovered from cell pellets after esterification to measure total 14C incorporated into mycolic acids and fatty acids.
TDM levels in the extractable lipid fraction gradually increase upon EMB treatment (Fig. 5B), as has been previously reported (49), presumably resulting from the accumulation of mycolates in TDM as a result of the disappearance of terminal hexarabinofuranoside attachment sites for mycolates. In contrast to this, SQ109 treatment resulted in an immediate loss of detectable TDM even at the shortest time points analyzed (Fig. 5B). Despite the rapid loss of mycolic acids in both the cell wall skeleton and TDM, analysis of total fatty acids by saponification of whole cells showed that the net mycolic acid levels were largely unaffected even after 3 h of drug treatment.
The effect of SQ109 on TDM production led us to look at upstream TMM levels, since this molecule is the likely precursor to TDM and cell wall mycolates (65). TLC analysis using a previously described solvent system (49) led to better separation and resolution of these lipids (Fig. 7A). As expected, the spots corresponding to TMM and TDM were absent in the INH-treated culture due to complete inhibition of mycolic acid biosynthesis, whereas TDM accumulation was observed in the EMB-treated sample (Fig. 7A). TDM production was abolished on SQ109 treatment, but there was a corresponding increase in the intensity of the spot corresponding to TMM compared to that of the untreated control (Fig. 7A). Densitometric analysis of the image further confirmed that SQ109-treated M. tuberculosis cells lost the ability to produce TDM, with concomitant accumulation of TMM (Fig. 7B). To rule out any ambiguity due to comigrating lipid species, 2-dimensional TLC was performed using the same samples, which clearly demonstrated the accumulation of TMM in SQ109-treated cultures in comparison to its levels in INH or EMB treatment (Fig. 7C). The inhibition of TDM production occurred in a dose-dependent manner (see Fig. S2 in the supplemental material).
Fig 7.

SQ109 treatment leads to the loss of TDM biosynthesis and the accumulation of TMM. (A) TLC analysis of extractable polar lipids after EMB, INH, and SQ109 treatment (spots corresponding to TDM and TMM are indicated). Solvent system: CHCl3-CH3OH-H2O (62:25:4). (B) Densitometry analysis of spots corresponding to TDM and TMM from panel A on exposure to the respective drugs, showing the relative levels of the two glycolipids. (C) Two-dimensional TLC analysis and resolution of extractable lipids, showing TDM and TMM migration and accumulation of TMM on SQ109 treatment. Solvent system: first dimension (y axis), CHCl3-CH3OH-H2O (62:25:4), and second dimension (x axis), CHCl3-CH3COOH-CH3OH-H2O (50:60:2.5:3). CONT, control.
SQ109 does not inhibit antigen 85-mediated transesterification.
The Ag85 proteins are mycolyltransferases capable of catalyzing the transesterification of mycolic acids between a range of acceptors (2). Mycolic acid transfer between two TMM molecules leads to the formation of one TDM and a free trehalose (7). Since SQ109 inhibits TDM production and leads to the accumulation of TMM in M. tuberculosis, the Ag85 proteins were evaluated for their sensitivity to SQ109 inhibition. Native and recombinant Ag85A, -B, and -C proteins were purified and analyzed in mycolyltransferase reactions as previously described, using the known Ag85 inhibitor 6-azido-6-deoxy-α, α′-trehalose (ADT) as a positive control (7). EMB and NIH59 (see Fig. 9A) were also included in this analysis (Fig. 8). When added at 200 μM final concentration, ADT inhibited the mycolyltransferase activity of native Ag85C protein by 69%, as reported previously (7), whereas EMB, NIH59, and SQ109 at similar concentrations did not inhibit the enzyme under these conditions (Fig. 8). Instead, a slight enhancement of enzyme activity was observed, possibly due to improved solubilization of the substrates and products (Table 1). This effect was observed for all three isoforms of Ag85 (Table 1). These results demonstrate that the in vivo loss of mycolate transfer to the cell wall arabinogalactan and accumulation of TMM by SQ109 was not due to direct inhibition of the Ag85 mycolyltransferases.
Fig 9.

Identification of adamantyl diamines and related compounds that retain the ability to inhibit TDM production in M. tuberculosis. (A) Identification of the SQ109 analogs and adamantyl amines/amides that inhibit the growth of M. tuberculosis. Structures of the compounds synthesized in the current study (DBK series) and identified in a screen for growth inhibition of M. tuberculosis (DA series) are shown, and their MICs are indicated in parenthesis. (B) Ability of the compounds to inhibit TDM production in M. tuberculosis.
Fig 8.

SQ109 is not an Ag85 mycolyltransferase inhibitor. Inhibition assays using native Ag85C from M. tuberculosis are shown (36). The known Ag85 inhibitor 6-azido-6-deoxy-α, α′-trehalose (ADT) was used as a positive control, and EMB and NIH59 were used as negative controls. The spots corresponding to TDM and TMM are indicated. Solvent system for TLC analysis: CHCl3-CH3OH-NH4OH (80:20:2).
Table 1.
Effect of SQ109 and ADTa on Ag85A, Ag85B, and Ag85C mycolyltransferase activities
6-Azido-6-deoxy-α, α'-trehalose (ADT) is a known inhibitor of the Ag85 proteins and was used as a positive control.
The respective compounds were used at 200 μM final concentration.
The assay measures 14C-TDM production (36). TDM production in a control reaction for each enzyme treated with dimethyl sulfoxide only was taken to be 100%, respectively. Negative values indicate increased levels of TDM production and lack of inhibition.
Native enzymes purified from M. tuberculosis culture supernatants and supplied by TBVTRM-CSU were used in the analysis.
Mutations in MmpL3 confer resistance to SQ109 and analogs.
Despite repeated attempts on both solid and liquid media, we were unable to select for spontaneous mutants that were SQ109 resistant. We therefore examined related compounds in the hope that they would at least partially share the same binding site and perhaps allow for the generation of resistance. NIH59 produced transcriptional profiles in treated M. tuberculosis cells that were very similar to those of SQ109-treated cells, suggesting that this compound affected a similar target (11). To verify that other SQ109 analogs have a similar mechanism of action to SQ109, we monitored the characteristic disappearance of TDM and accumulation of TMM in cells upon exposure to the compounds. MIC testing showed that complete reduction of the geranyl group of SQ109 (DBK7) or replacement of this with another adamantyl group (DBK13) led to only a slight reduction in activity compared to that of SQ109 (Fig. 9A). The replacement of the adamantyl and geranyl groups with other bulky groups also diminished activity only slightly, as seen in the case of NIH59. Treatment of cells at 10-fold their respective MIC concentrations followed by 14C-acetate labeling and lipid analysis showed that all of these analogs of SQ109 (DBK7, DBK13, and NIH59) similarly inhibited TDM synthesis concomitant with TMM accumulation (Fig. 9). In addition, we analyzed two related compounds (DA5 and DA8) that had been identified in a high-throughput chemical screen for M. tuberculosis growth inhibition and which also potently upregulated the iniBAC operon (D. A. Alland, unpublished results). These compounds also contained the characteristic amino-adamantyl (or amido-adamantyl) group of SQ109 and similarly inhibited TDM synthesis concomitant with TMM accumulation (Fig. 9).
DA5 and DA8 had significantly higher MICs than SQ109, suggesting that they had reduced affinity for the target and, therefore, might more easily allow mutation of the target which could lead to resistance. Plating M. tuberculosis cells on medium containing either DA5 or DA8 resulted in the appearance of resistant mutants at a moderate frequency of approximately 1 in 107 cells. Two DA5-resistant (5_1 and 5_2) and three DA8-resistant (8_1, 8_2, and 8_3) mutants were isolated, and all five mutants were found to be cross-resistant to SQ109 (Table 2). To identify the mutation(s) associated with DA5, DA8, and SQ109 resistance, the genomes of three mutants, 5_1, 8_2, and 8_3, respectively, were sequenced. All three mutants had single nucleotide transition mutations leading to replacement polymorphisms in the gene encoding the membrane transport protein MmpL3 (Rv0206c), which is conserved across all mycobacteria for which genome sequences are available. The 5_1 mutant had a G2098A transition leading to an A700T amino acid change, whereas the 8_2 and 8_3 mutants had an A119G transition leading to a Q40R change in the predicted amino acid sequence of MmpL3. The 5_1 mutant was sequenced independently a second time, producing exactly the same result. In addition to the mmpL3 mutation, the 8_2 and 8_3 mutants had a T → C transition mutation at position 2055375 in the M. tuberculosis H37Rv genome, which is in the intergenic region between the Rv1812c and Rv1813c open reading frames, encoding a putative dehydrogenase and a small conserved hypothetical protein, respectively, both of which are predicted to be secreted and nonessential in M. tuberculosis (37, 58). Further sequencing of mmpL3 of a DA5- and DA8-resistant mutant further confirmed that single nucleotide polymorphisms in this gene were associated with resistance, with one of these (8_1) carrying a novel mutation leading to an L567P amino acid change (Table 2). These results strongly suggest that the target of SQ109 and related analogs is the essential transmembrane transporter encoded by mmpL3. Treatment of the resistant mutants with SQ109 or analogs showed that TDM production was inhibited at concentrations above their respective MIC concentrations (see Fig. S3 and S4 in the supplemental material). The role of MmpL3 in TMM transport to the cell wall would suggest that accumulated TMM would be consumed by the extracellular mycolyltransferases after removal of SQ109. To verify this, we treated cells with 10-fold the MIC of SQ109 followed by radiolabeling of lipids. Cells were subsequently washed to remove radiolabel and drug, followed by outgrowth. Subsequent TLC analysis of extractable lipids demonstrated that the accumulated TMM disappeared, with radiolabel appearing in a time-dependent manner in TDM (see Fig. S2 in the supplemental material), with most radiolabel expected to convert to arabinan-linked mycolates.
Table 2.
Resistance and single nucleotide polymorphism profiles of SQ109 cross-resistant mutants
| M. tuberculosis H37Rv isolatea | Fold resistanceb |
Gene mutationc | |||||
|---|---|---|---|---|---|---|---|
| DA5 | DA8 | SQ109 | INH | Rif | EMB | ||
| WT | 1 | 1 | 1 | 1 | 1 | 1 | Not applicable |
| 5_1 | >4 | ND | 4 | 1 | 1 | 1 | mmpL3 G2098A/A700Td |
| 5_2 | 4 | ND | 2 | ND | ND | ND | mmpL3 G2098A/A700Td |
| 8_1 | 4 | 8–10 | 2–8 | 1 | 1 | 1 | mmpL3 T1700C/L567Pd |
| 8_2 | ND | 8–10 | 2–4 | ND | ND | ND | mmpL3 A119G/Q40Rd and T2055375Ce |
| 8_3 | 4 | 8–10 | 2–4 | ND | ND | ND | mmpL3 A119G/Q40Rd and T2055375Ce |
The wild-type strain (WT) was used to isolate spontaneous mutants resistant to DA5 and DA8, which were used to check for cross-resistance to SQ109.
Fold increase in MIC compared to that of the WT strain. ND, not done.
Mutations for 5_1, 8_1, and 8_3 were identified by whole-genome sequencing. The gene and the specific amino acid change are indicated. Mutations for 5_2 and 8_2 were confirmed by sequencing of mmpL3.
Nucleotide number, based on the sequence of the mmpL3 open reading frame where the first nucleotide of the start codon was assigned as nucleotide number 1, and amino acid number, based on the sequence of mmpL3.
Nucleotide number, based on the complete genome sequence of M. tuberculosis H37Rv; the mutation lies in the intergenic region between Rv1812c and Rv1813c.
DISCUSSION
Although SQ109 was developed as an analog of EMB, a known inhibitor of cell wall arabinogalactan biosynthesis, biochemical and transcriptional data have previously suggested that the target of SQ109 is different from that of EMB. Here, we showed by using macromolecular incorporation assays to probe for effects on protein, nucleic acid, peptidoglycan, and lipid biosynthesis that SQ109 rapidly inhibits the attachment of lipids to the cell wall. This confirms our earlier supposition (11, 40) that upregulation of the iniBAC operon is indicative of inhibition of cell wall biosynthesis. Recent work suggests that this operon encodes an uncharacterized pump conferring multidrug tolerance (15). Ultrastructural analysis of cells treated with SQ109 confirmed that it resulted in cell shortening and widening similar to that seen with EMB and INH, which affect the assembly of the arabinan and mycolate components of the mycolyl-arabinogalactan-peptidoglycan complex of the cell wall, respectively (Fig. 4).
Current models of the cell wall core are consistent with an inner leaflet composed of a monolayer of parallel mycolic acid chains linked to arabinan, interdigitated by an outer leaflet composed of TDM and TMM (and other amphiphiles), resulting in a tightly packed bilayer. There are several theoretical models proposed to explain the unexpectedly thin and symmetrical nature of the core (27, 74). Inhibition of mycolate attachment either directly by inhibition of its synthesis by INH or by inhibiting the formation of cell wall arabinan to which mycolic acids are attached by EMB could compromise the permeability of the cell wall core to the staining reagents or increase the association of this layer with material with increased staining characteristics (48). SQ109 results in a rapid decrease in the attachment of mycolic acids to the cell wall arabinogalactan complex, as well as to TDM (Fig. 5), and is associated with a drop in cellular viability (Fig. 2). The loss of cell wall mycolates was not due to inhibition of their biosynthesis as total mycolate pools were unaffected (Fig. 6). Concurrent with the loss of mycolate transfer to the cell wall or TDM, TMM accumulated in SQ109-treated cells (Fig. 7). This series of metabolic effects is distinct from the effects of any known inhibitors of M. tuberculosis cell wall assembly, including EMB and INH.
The biosynthesis of mycolic acids by the Fas-II complex and the acyl-coenzyme A carboxylase (AccA3/AccD4)/acyl-AMP ligase (FadD32)/polyketide synthase 13 (Pks13) complex has been described in detail (reviewed in references 5 and 50), but the steps relating to their transport across the cell membrane and attachment to the cell wall are not clear (63). The current model suggests that the reductase encoded by the Rv2509 gene catalyzes the final reaction during mycolic acid biosynthesis, converting the β-keto group remaining from condensation of the α-branch with the meromycolate precursor to a β-hydroxy moiety (10), most probably while they are still attached to Pks13. This is followed by sequential steps catalyzed by the hypothetical mycolyltransferases I and II, which mediate the transfer of the mycolic acids from Pks13 to a mannose-P-heptaprenol carrier (9) and trehalose, respectively, eventually leading to the formation of TMM within the cell (Fig. 1) (63). TMM is then exported out of the cell by an unidentified transporter, predicted to be an ATP binding cassette (ABC) family transporter (63). Once outside the cell, the antigen 85 proteins (Ag85A, -B, and -C) mediate mycolate transfer from one TMM molecule to another to give TDM and, also, from TMM to the cell wall arabinogalactan (Fig. 1) (7). It has also been demonstrated that the purified Ag85 mycolyltransferases can convert TMM to TDM in vitro (7) and that deletion of the Ag85C gene significantly reduces the mycolic acid content of the M. tuberculosis cell wall (29). The events between reduction of the β-keto group of the mycolates and their attachment to the cell wall or deposition into TDM are unknown (Fig. 1).
The effect of SQ109 on TDM and TMM levels mimics the phenotype of mycolyltransferase mutants of Corynebacterium. Individual disruption of any of its five Ag85-like genes reduces transfer of mycolates to the cell wall, leading to TDM depletion and TMM accumulation (18, 56). The demonstration that the inhibition of transfer of mycolates to form TDM and cell wall arabinogalactan is not due to inhibition of the secreted Ag85 mycolyltransferases (Fig. 8 and Table 1), the only known proteins capable of performing the two described processes (7), suggests that SQ109 and the other active compounds identified in Fig. 9 must inhibit a step prior to the one catalyzed by the Ag85 proteins, thereby preventing access of the mycolyltransferases to TMM. According to the currently accepted model (Fig. 1), there are three key hypothetical proteins that yet remain to be identified. These include the two additional mycolyltransferases (I and II) involved in TMM formation and the membrane transporter that pumps TMM out of the cell (63). Since TMM is still produced in the presence of SQ109, the hypothetical mycolyltransferases I and II are presumably unaffected by this compound, leaving the TMM transporter as the prime candidate for inhibition by SQ109.
We were unable to generate spontaneous SQ109-resistant mutants to confirm this hypothesis, suggesting that the compound inhibited an essential target where the amino acid residues critical for binding of SQ109 are equally important for the function of the target or that the binding interactions were sufficiently strong that single mutations were unable to confer resistance. Given the relatively large and highly hydrophobic nature of SQ109, a large binding surface is not unlikely, making the development of resistance through single amino acid changes difficult. Nonetheless, the possibility cannot be excluded that this compound affects other secondary targets in the cells. Using analogs of SQ109, which we biochemically demonstrated have the same mechanism of action but showing weaker activity, we were able to generate resistant mutants that showed 2- to 8-fold cross-resistance to SQ109 (Table 2). Most of these analogs shared the ethylenediamine core structure and resulted in loss of TDM formation with concomitant TMM accumulation (Fig. 9). The fact that these compounds allowed the generation of spontaneously resistant mutants suggests that the amino acid residues critical for binding of these compounds were not the same residues that drive the binding of SQ109. The modest cross-resistance of these mutants to SQ109 (Table 2) supports this notion. Whole-genome resequencing of three of these mutants identified mutations in the essential mmpL3 gene (19). Mutations in mmpL3 were also identified in a further two resistant mutants (Table 2). MmpL3 was a very attractive candidate as a TMM pump since this is the only protein of this family with an essential function.
The MmpL proteins belong to the resistance, nodulation, and cell division (RND) family of membrane proteins that are involved in the transport of diverse ligands out of the cell, driven by energy from the proton motive force (52). These proteins generally contain 12 transmembrane helices with large loops between transmembrane segments 1 and 2 and 7 and 8 that are extracytoplasmic and are involved in substrate recognition (23). Using topology prediction algorithms, the mutation in the 5_1 strain (A700T) was mapped to the base of the last transmembrane helix (number 12) and is predicted to be located on the outer face of the cell membrane. The Q40R mutations in the 8_2 and 8_3 strains mapped to the first extracytoplasmic loop between transmembrane helices 1 and 2, which is known to be important for substrate recognition in other related proteins. Since both mutations changed amino acids predicted to reside outside the transmembrane helices, it is possible that these mutations decrease/alter the ability of SQ109 and other related compounds to bind to and inhibit the function of the mutant MmpL3 proteins, leading to resistance. In a study that interrogated the role of all 13 of the M. tuberculosis mmpL genes, it was demonstrated that the mmpL3 gene was the only essential member of this family since attempts to delete this gene were unsuccessful (19). Therefore, our results suggest that the MmpL3 protein is the most probable cellular target of SQ109 and related compounds (Fig. 9A) and that MmpL3 is thus the likely TMM transporter. Other MmpL proteins also encode transporters of cytoplasmically synthesized lipids, and their nonessentiality is rationalized by the fact that none of these lipids are essential for in vitro growth. For example, the MmpL7 protein has been shown to be involved in the transport of the surface-exposed lipid phthiocerol dimycocerosate (PDIM) across the cell membrane (14). In addition, the MmpL8 protein has been shown to be involved in the transport of the precursor of the sulfolipid SL-1 across the cell membrane (16, 20). Interestingly, the final stages of biosynthesis and transport are coupled for both MmpL7 and MmpL8. In both systems, the terminal polyketide synthase enzyme is attached to the lipid substrate to be transported, and in the case of MmpL7, it has been shown to physically interact with PpsE, the last polyketide synthase involved in PDIM biosynthesis (30). In the case of MmpL8, the esterification of hydroxyphthioceranic acids to a diacyl-trehalose-sulfate is directly associated with the export of sulfolipid-1 (16, 20). Comparison of the known PDIM and SL-1 export mechanisms with the final stages of mycolic acid biosynthesis suggests additional similarities between the three processes. The precursors of the mycolic acids remain linked to the acyl carrier domain of Pks13 to a late, as-yet-undefined stage (63). Therefore, it is possible that Pks13 and MmpL3 function in a manner similar to that of the PksE-MmpL7 and the Pks2-MmpL8 proteins that are involved in PDIM and SL-1 export, respectively. The transferase that esterifies the mycolic acid to trehalose is not dependent on the function of MmpL3 since TMM accumulation occurs when the function of MmpL3 is inhibited, unlike MmpL8, whose deletion abrogates the transfer of hydroxyphthioceranic acids (16, 20).
In conclusion, our results suggest that MmpL3 is the transporter of TMM and that TDM formation and anchoring of mycolic acids to the cell envelope require TMM export. This finding ties in well with the currently proposed model for mycolic acid processing and export since the involvement of the MmpL proteins in lipid transport seems to be a common theme in mycobacteria. The initial increase in peptidoglycan biosynthesis observed in the present study, followed by its inhibition, and the previously reported increase in the arabinose content in M. tuberculosis cell walls treated with SQ109 (11) agrees well with this model since proper assembly of the mycolyl-arabinogalactan-peptidoglycan complex requires the coordinated biosynthesis of all three components (25). Therefore, inhibiting the biosynthesis of one of its components could lead to short-term compensatory effects and increased production of the other components, prior to complete cessation and cell death. The inhibition of mycolic acid biosynthesis by drugs such as INH (64), peptidoglycan biosynthesis by beta-lactams (28), branching of cell wall arabinan synthesis by EMB (49, 62), and inhibition of the epimerization of ribose residues required for arabinan synthesis by the benzothiazinones (46) highlight the extreme vulnerability of the assembly mechanism of the cell wall mycolyl-arabinogalactan-peptidoglycan complex in M. tuberculosis and suggest that there may be other, as-yet-untapped targets for TB treatment.
Supplementary Material
ACKNOWLEDGMENTS
This work utilized the NIH Molecular Libraries Production Centers Network (MLPCN; www.mli.nih.gov).
This study was funded (in part) by the Intramural Research Program of NIAID, NIH (to C.E.B.), and in part by a UNCF/Merck Postdoctoral Science Research Fellowship (to R.W.).
We gratefully acknowledge W. R. Jacobs, Jr. (Albert Einstein College of Medicine), for assistance with sequencing efforts.
Footnotes
Published ahead of print 17 January 2012
Supplemental material for this article may be found at http://aac.asm.org/.
REFERENCES
- 1. Alland D, Steyn AJ, Weisbrod T, Aldrich K, Jacobs WR., Jr 2000. Characterization of the Mycobacterium tuberculosis iniBAC promoter, a promoter that responds to cell wall biosynthesis inhibition. J. Bacteriol. 182:1802–1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Backus KM, et al. 2011. Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis. Nat. Chem. Biol. 7:228–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barry CE. 2011. Lessons from seven decades of antituberculosis drug discovery. Curr. Top. Med. Chem. 11:1216–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barry CE, III, Blanchard JS. 2010. The chemical biology of new drugs in the development for tuberculosis. Curr. Opin. Chem. Biol. 14:456–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barry CE, III, et al. 1998. Mycolic acids: structure, biosynthesis and physiological functions. Prog. Lipid Res. 37:143–179 [DOI] [PubMed] [Google Scholar]
- 6. Barry CE, Crick DC, McNeil MR. 2007. Targeting the formation of the cell wall core of M. tuberculosis. Infect. Disord. Drug Targets 7:182–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Belisle JT, et al. 1997. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276:1420–1422 [DOI] [PubMed] [Google Scholar]
- 8. Bergval IL, Schuitema AR, Klatser PR, Anthony RM. 2009. Resistant mutants of Mycobacterium tuberculosis selected in vitro do not reflect the in vivo mechanism of isoniazid resistance. J. Antimicrob. Chemother. 64:515–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Besra GS, et al. 1994. Identification of the apparent carrier in mycolic acid synthesis. Proc. Natl. Acad. Sci. U. S. A. 91:12735–12739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bhatt A, Brown AK, Singh A, Minnikin DE, Besra GS. 2008. Loss of a mycobacterial gene encoding a reductase leads to an altered cell wall containing beta-oxo-mycolic acid analogs and accumulation of ketones. Chem. Biol. 15:930–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boshoff HI, et al. 2004. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J. Biol. Chem. 279:40174–40184 [DOI] [PubMed] [Google Scholar]
- 12. Brennan PJ. 2003. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb.) 83:91–97 [DOI] [PubMed] [Google Scholar]
- 13. Brennan PJ, Nikaido H. 1995. The envelope of mycobacteria. Annu. Rev. Biochem. 64:29–63 [DOI] [PubMed] [Google Scholar]
- 14. Camacho LR, et al. 2001. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J. Biol. Chem. 276:19845–19854 [DOI] [PubMed] [Google Scholar]
- 15. Colangeli R, et al. 2005. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol. Microbiol. 55:1829–1840 [DOI] [PubMed] [Google Scholar]
- 16. Converse SE, et al. 2003. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc. Natl. Acad. Sci. U. S. A. 100:6121–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16a. Crick DC, Mahapatra S, Brennan PJ. 2001. Biosynthesis of the arabinogalactan-peptidoglycan complex of Mycobacterium tuberculosis. Glycobiology 11:107R–118R [DOI] [PubMed] [Google Scholar]
- 17. Daffe M, Draper P. 1998. The envelope layers of mycobacteria with reference to their pathogenicity. Adv. Microb. Physiol. 39:131–203 [DOI] [PubMed] [Google Scholar]
- 18. De Sousa-D'Auria C, et al. 2003. New insights into the biogenesis of the cell envelope of corynebacteria: identification and functional characterization of five new mycoloyltransferase genes in Corynebacterium glutamicum. FEMS Microbiol. Lett. 224:35–44 [DOI] [PubMed] [Google Scholar]
- 19. Domenech P, Reed MB, Barry CE., III 2005. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect. Immun. 73:3492–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Domenech P, et al. 2004. The role of MmpL8 in sulfatide biogenesis and virulence of Mycobacterium tuberculosis. J. Biol. Chem. 279:21257–21265 [DOI] [PubMed] [Google Scholar]
- 21. Doster B, Murray FJ, Newman R, Woolpert SF. 1973. Ethambutol in the initial treatment of pulmonary tuberculosis. U.S. Public Health Service tuberculosis therapy trials. Am. Rev. Respir. Dis. 107:177–190 [DOI] [PubMed] [Google Scholar]
- 22. Draper P. 1998. The outer parts of the mycobacterial envelope as permeability barriers. Front. Biosci. 3:D1253–D1261 [DOI] [PubMed] [Google Scholar]
- 23. Elkins CA, Nikaido H. 2002. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominantly by two large periplasmic loops. J. Bacteriol. 184:6490–6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goldman RC, et al. 2007. Programs to facilitate tuberculosis drug discovery: the tuberculosis antimicrobial acquisition and coordinating facility. Infect. Disord. Drug Targets. 7:92–104 [DOI] [PubMed] [Google Scholar]
- 25. Hancock IC, Carman S, Besra GS, Brennan PJ, Waite E. 2002. Ligation of arabinogalactan to peptidoglycan in the cell wall of Mycobacterium smegmatis requires concomitant synthesis of the two wall polymers. Microbiology 148:3059–3067 [DOI] [PubMed] [Google Scholar]
- 26. Heifets LB, Lindholm-Levy PJ, Flory M. 1991. Comparison of bacteriostatic and bactericidal activity of isoniazid and ethionamide against Mycobacterium avium and Mycobacterium tuberculosis. Am. Rev. Respir. Dis. 143:268–270 [DOI] [PubMed] [Google Scholar]
- 27. Hoffmann C, Leis A, Niederweis M, Plitzko JM, Engelhardt H. 2008. Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. U. S. A. 105:3963–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, III, Blanchard JS. 2009. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323:1215–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackson M, et al. 1999. Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol. Microbiol. 31:1573–1587 [DOI] [PubMed] [Google Scholar]
- 30. Jain M, Cox JS. 2005. Interaction between polyketide synthase and transporter suggests coupled synthesis and export of virulence lipid in M. tuberculosis. PLoS Pathog. 1:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jia L, Coward L, Gorman GS, Noker PE, Tomaszewski JE. 2005. Pharmacoproteomic effects of isoniazid, ethambutol, and N-geranyl-N′-(2-adamantyl)ethane-1,2-diamine (SQ109) on Mycobacterium tuberculosis H37Rv. J. Pharmacol. Exp. Ther. 315:905–911 [DOI] [PubMed] [Google Scholar]
- 32. Jia L, et al. 2006. Interspecies pharmacokinetics and in vitro metabolism of SQ109. Br. J. Pharmacol. 147:476–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jia L, et al. 2005. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 144:80–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kastrinsky DB, McBride NS, Backus KM, LeBlanc JJ, Barry CE., III 2010. Mycolic acid/cyclopropane fatty acid/fatty acid biosynthesis and health relations, p 65–145 In Mander L, Liu H-W. (ed.), Comprehensive natural products II, vol I Elsevier Science, Amsterdam, Netherlands [Google Scholar]
- 35. Kaur D, Guerin ME, Skovierova H, Brennan PJ, Jackson M. 2009. Chapter 2: biogenesis of the cell wall and other glycoconjugates of Mycobacterium tuberculosis. Adv. Appl. Microbiol. 69:23–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kremer L, Maughan WN, Wilson RA, Dover LG, Besra GS. 2002. The M. tuberculosis antigen 85 complex and mycolyltransferase activity. Lett. Appl. Microbiol. 34:233–237 [DOI] [PubMed] [Google Scholar]
- 37. Lamichhane G, et al. 2003. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 100:7213–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Laneelle MA, Daffe M. 2009. Transport assays and permeability in pathogenic mycobacteria. Methods Mol. Biol. 465:143–151 [DOI] [PubMed] [Google Scholar]
- 39. Lange RP, Locher HH, Wyss PC, Then RL. 2007. The targets of currently used antibacterial agents: lessons for drug discovery. Curr. Pharm. Des. 13:3140–3154 [DOI] [PubMed] [Google Scholar]
- 40. Lee RE, et al. 2003. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 5:172–187 [DOI] [PubMed] [Google Scholar]
- 41. Lees AW, Tyrrell WF, Smith J, Allan GW. 1970. Ethambutol in the retreatment of chronic pulmonary tuberculosis. Br. J. Dis. Chest 64:85–89 [DOI] [PubMed] [Google Scholar]
- 42. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H, et al. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liu J, Barry CE, III, Besra GS, Nikaido H. 1996. Mycolic acid structure determines the fluidity of the mycobacterial cell wall. J. Biol. Chem. 271:29545–29551 [DOI] [PubMed] [Google Scholar]
- 45. Liu J, Rosenberg EY, Nikaido H. 1995. Fluidity of the lipid domain of cell wall from Mycobacterium chelonae. Proc. Natl. Acad. Sci. U. S. A. 92:11254–11258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Makarov V, et al. 2009. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 324:801–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McClatchy JK. 1971. Mechanism of action of isoniazid on Mycobacterium bovis strain BCG. Infect. Immun. 3:530–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mdluli K, Swanson J, Fischer E, Lee RE, Barry CE., III 1998. Mechanisms involved in the intrinsic isoniazid resistance of Mycobacterium avium. Mol. Microbiol. 27:1223–1233 [DOI] [PubMed] [Google Scholar]
- 49. Mikusova K, Slayden RA, Besra GS, Brennan PJ. 1995. Biogenesis of the mycobacterial cell wall and the site of action of ethambutol. Antimicrob. Agents Chemother. 39:2484–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Minnikin DE, Kremer L, Dover LG, Besra GS. 2002. The methyl-branched fortifications of Mycobacterium tuberculosis. Chem. Biol. 9:545–553 [DOI] [PubMed] [Google Scholar]
- 51. Nikaido H, Kim SH, Rosenberg EY. 1993. Physical organization of lipids in the cell wall of Mycobacterium chelonae. Mol. Microbiol. 8:1025–1030 [DOI] [PubMed] [Google Scholar]
- 52. Nikaido H, Takatsuka Y. 2009. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 1794:769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Nikonenko BV, Protopopova M, Samala R, Einck L, Nacy CA. 2007. Drug therapy of experimental tuberculosis (TB): improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob. Agents Chemother. 51:1563–1565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Prideaux B, et al. 2011. High-sensitivity MALDI-MRM-MS imaging of moxifloxacin distribution in tuberculosis-infected rabbit lungs and granulomatous lesions. Anal. Chem. 83:2112–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Protopopova M, et al. 2005. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 56:968–974 [DOI] [PubMed] [Google Scholar]
- 56. Puech V, Bayan N, Salim K, Leblon G, Daffe M. 2000. Characterization of the in vivo acceptors of the mycoloyl residues transferred by the corynebacterial PS1 and the related mycobacterial antigens 85. Mol. Microbiol. 35:1026–1041 [DOI] [PubMed] [Google Scholar]
- 57. Sani M, et al. 2010. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog. 6:e1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 59. Shepherd RG, et al. 1966. Structure-activity studies leading to ethambutol, a new type of antituberculous compound. Ann. N. Y. Acad. Sci. 135:686–710 [DOI] [PubMed] [Google Scholar]
- 60. Slayden RA, Barry CE., III 2001. Analysis of the lipids of Mycobacterium tuberculosis. Methods Mol. Med. 54:229–245 [DOI] [PubMed] [Google Scholar]
- 61. Somogyi PA, Foldes I. 1983. Incorporation of thymine, thymidine, adenine and uracil into nucleic acids of Mycobacterium phlei and its phage. Ann. Microbiol. (Paris) 134A:19–28 [DOI] [PubMed] [Google Scholar]
- 62. Takayama K, Kilburn JO. 1989. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 33:1493–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Takayama K, Wang C, Besra GS. 2005. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 18:81–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Takayama K, Wang L, David HL. 1972. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2:29–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tropis M, et al. 2005. The crucial role of trehalose and structurally related oligosaccharides in the biosynthesis and transfer of mycolic acids in Corynebacterineae. J. Biol. Chem. 280:26573–26585 [DOI] [PubMed] [Google Scholar]
- 66. Vilcheze C, Jacobs WR., Jr 2007. The mechanism of isoniazid killing: clarity through the scope of genetics. Annu. Rev. Microbiol. 61:35–50 [DOI] [PubMed] [Google Scholar]
- 67. Wang L, Slayden RA, Barry CE, III, Liu J. 2000. Cell wall structure of a mutant of Mycobacterium smegmatis defective in the biosynthesis of mycolic acids. J. Biol. Chem. 275:7224–7229 [DOI] [PubMed] [Google Scholar]
- 68. Wolucka BA. 2008. Biosynthesis of D-arabinose in mycobacteria—a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 275:2691–2711 [DOI] [PubMed] [Google Scholar]
- 69. Wong SY, et al. 2011. Mutations in gidB confer low-level streptomycin resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 55:2515–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. World Health Organization 2010. Global tuberculosis control 2010. [Google Scholar]
- 71. World Health Organization 2010. Multidrug and extensively drug-resistant TB (M/XDR-TB): 2010 global report on surveillance and response. World Health Organization, Geneva, Switzerland [Google Scholar]
- 72. World Health Organization 2010. Treatment of tuberculosis: guidelines for national programmes, 4th ed World Health Organization, Geneva, Switzerland [Google Scholar]
- 73. Zhu M, et al. 2004. Pharmacokinetics of ethambutol in children and adults with tuberculosis. Int. J. Tuberc. Lung Dis. 8:1360–1367 [PubMed] [Google Scholar]
- 74. Zuber B, et al. 2008. Direct visualization of the outer membrane of mycobacteria and corynebacteria in their native state. J. Bacteriol. 190:5672–5680 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.