

Abstract
Yersinia pestis, which causes bubonic plague, forms biofilms in fleas, its insect vectors, as a means to enhance transmission. Biofilm development is positively regulated by hmsT, encoding a diguanylate cyclase that synthesizes the bacterial second messenger cyclic-di-GMP. Biofilm development is negatively regulated by the Rcs phosphorelay signal transduction system. In this study, we show that Rcs-negative regulation is accomplished by repressing transcription of hmsT.
INTRODUCTION
Yersinia pestis, the agent of bubonic plague, infects fleas, its vectors, by producing bacterial biofilms that can colonize the insect foregut (19). Growth of the biofilm interferes with blood feeding by the infected fleas and potentiates regurgitative transmission. Complete blockage of the foregut by the bacterial biofilm can occur, and blocked fleas bite mammals repeatedly in futile feeding attempts, further enhancing transmission. Y. pestis biofilms, in which bacteria are surrounded by a self-synthesized polysaccharide-rich matrix, appear to be made only when the bacteria colonize fleas. Several proteins required for Y. pestis biofilms are proteolytically degraded at mammalian body temperatures (25), in vitro biofilms are greatly diminished at 37°C, and a Y. pestis strain defective for biofilm genes was nevertheless highly virulent in a mouse infection (3, 22, 32).
Y. pestis biofilms are positively regulated by cyclic-di-GMP (c-di-GMP), which is synthesized by diguanylate cyclase (DGC) enzymes. The Y. pestis genome encodes several putative DGCs, but only two of them, HmsT and Y3730, are related to biofilm formation (3, 8, 22, 32). Yersinia pseudotuberculosis, a bacterium closely related to Y. pestis, also makes biofilms that are regulated by hmsT (10). The biofilm-promoting activity of c-di-GMP has not been determined, but in other systems, c-di-GMP has been shown to be an allosteric activator of glycosyltransferases (27, 28).
Y. pestis and Y. pseudotuberculosis biofilms are negatively regulated by the Rcs phosphorelay system (33). Rcs consists of two membrane-bound proteins, RcsC and RcsD, a DNA-binding response regulator, RcsB, and an accessory protein, RcsA. In phosphorelays of this type, outputs can be dependent on RcsB alone, acting in homodimers, or on heterodimers of RcsB and RcsA (23). Genetic investigation showed that in Y. pestis, rcsA is a nonfunctional pseudogene, while in Y. pseudotuberculosis, rcsA is functional and represses biofilms (33). Although rcsD has a frameshift in Y.pestis, it is still functional and may dephosphorylate RcsB to derepress biofilms (33).
The factors determining hmsT transcription have not been examined. In the present study, we show that in Y. pestis and Y. pseudotuberculosis, Rcs represses hmsT transcription.
MATERIALS AND METHODS
Bacterial strains and plasmids.
The strains and plasmids used are shown in Table 1. CDY497 was made by a method to insert PCR products into the chromosome using the Red recombination system (11, 33). For strains containing ΔlacZ::Cm, the same method was used, after which reporter constructs containing hmsP::lacZ and hmsH::lacZ fusions were inserted by a modification of the Red method (17). In strains that are ΔlacZ but without chloramphenicol resistance (CDY622, CDY635, CDY636, CDY637, CDY638, and CDY639), the chloramphenicol resistance gene was cured (11). The RcsAB box mutation strains CDY983 and CDY984 were made by two-step allelic replacement (9, 12). A 1.5-kb PCR product containing the RcsAB box of hmsT was amplified from Y. pestis KIM6+ and cloned into pUC18. Using the resulting plasmid as the template, the CC dinucleotide was mutated to AG using a mutagenic PCR primer, and the product was substituted into the Y. pestis chromosome by allelic replacement (12). The oligonucleotide primers used are shown in Table S1 in the supplemental material. All strains were verified by PCR, DNA sequencing, or plasmid complementation, as appropriate.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype and/or description | Reference or source |
|---|---|---|
| Strains | ||
| Y. pestis | ||
| KIM6+ | Wild type (pCD1-) | 14 |
| CDY319 | ΔrcsD::kan | 33 |
| CDY325 | ΔrcsB rcsD::kan | 33 |
| CDY326 | ΔrcsB::kan | 33 |
| CDY330 | ΔrcsA::rcsAYPSTB | 33 |
| CDY377 | ΔrcsD::kan (pCBD190) | 33 |
| CDY421 | ΔrcsB::kan ΔrcsA::rcsAYPSTB | 33 |
| CDY475 | KIM6+ (pCBD209) | This study |
| CDY497 | ΔhmsT::cat | This study |
| CDY567 | ΔlacZ::cat hmsH::lacZ | This study |
| CDY568 | ΔlacZ::cat hmsH::lacZ ΔrcsA::rcsAYPSTB | This study |
| CDY614 | ΔlacZ::cat hmsP::lacZ | This study |
| CDY615 | ΔlacZ::cat hmsP::lacZ ΔrcsA::rcsAYPSTB | This study |
| CDY622 | ΔlacZ | This study |
| CDY635 | ΔlacZ hmsT::lacZ | This study |
| CDY636 | ΔlacZ hmsT::lacZ (pCBD209) | This study |
| CDY637 | ΔlacZ hmsT::lacZ (pBAD/Myc-His) | This study |
| CDY638 | ΔlacZ hmsT::lacZ (pCBD178) | This study |
| CDY639 | ΔlacZ hmsT::lacZ (pCBD179) | This study |
| CDY641 | KIM6+ (pCBD253) | This study |
| CDY642 | KIM6+ (pCBD254) | This study |
| CDY981 | ΔhmsT::cat (pCBD26) | 32 |
| CDY982 | ΔrcsA::rcsAYPSTB (pCBD26) | This study |
| CDY983 | RcsAB* | This study |
| CDY984 | ΔrcsA::rcsAYPSTB RcsAB* | This study |
| CDY1071 | ΔrcsA::rcsAYPSTB ΔlacZ hmsT::lacZ | This study |
| SY743 | KIM6+ (pYC219) | This study |
| SY744 | KIM6+ (pYC220) | This study |
| Y. pseudotuberculosis | ||
| IP32953 | Wild type; serogroup O1 | 7 |
| CDY564 | ΔrcsA::rcsAYPE | 33 |
| CDY985 | IP32953(pCBD26) | This study |
| Plasmids | ||
| pBAD/Myc-His | Expression vector | Invitrogen |
| pCBD26 | hmsT in pCR2.1-TOPO | 31 |
| pCBD178 | rcsAYPE in pUC18 | 33 |
| pCBD179 | rcsAYPSTB in pUC18 | 33 |
| pCBD190 | rcsDYPSTB in PET32a | 33 |
| pCBD209 | rcsB in pBAD/Myc-His | This study |
| pCBD253 | rcsAYPSTB in pMal-4x | This study |
| pCBD254 | rcsAYPE in pMal-4x | This study |
| pCR2.1-TOPO | Cloning vector for PCR products | Invitrogen |
| pUC18 | Cloning vector | 35 |
| pMal-c4x | Expression vector | NEB |
| pYC219 | rcsAYPE-His6 in pUC18 | This study |
| pYC220 | rcsAYPSTB-His6 in pUC18 | This study |
In vitro biofilms.
Microtiter plate biofilm assays were carried out as previously described (32). Briefly, bacteria were grown in LB broth supplemented with 4 mM CalCl2 and 4 mM MgCl2 overnight at room temperature, diluted to an optical density at 600 nm (OD600) of 0.02, and then aliquoted to 96-well polystyrene plates, which were incubated with shaking at 250 rpm for 24 h at room temperature. Media and planktonic cells were removed; the wells were washed, and the adherent biofilm was stained with crystal violet, which was subsequently solubilized with 80% ethanol–20% acetone before the A600 was measured. Background absorbance for uninoculated control wells was subtracted. Results from three independent experiments with five replicates per experiment were analyzed by one-way analysis of variance (ANOVA) with Dunnett's posttest to compare the wild type to the other strains.
Caenorhabditis elegans biofilms.
Biofilms were assayed for their ability to inhibit nematode growth as described previously (33). Briefly, adult C. elegans adults were allowed to lay eggs on lawns of bacteria for 2 to 4 h. After the adults were removed, the plates were incubated for 2 days at 20°C, and the worm development to the fourth larval (L4) stage was scored. Three independent experiments were carried out with at least 1,200 worms per sample.
β-Galactosidase assays.
Lysates were prepared from cells with chromosomal hmsT::lacZ, hmsH::lacZ, or hmsP::lacZ (Table 1). β-Galactosidase activities were measured spectrophotometrically following cleavage of ONPG (4-nitrophenyl-β-d-galactopyranoside) at 37°C and are expressed in Miller units (24). Results from at least two independent experiments done in triplicate were analyzed by two-tailed Student's t test.
qRT-PCR.
Quantitative real-time reverse transcription-PCR (qRT-PCR) was carried out as previously described (32). Briefly, cells were grown in LB broth overnight, diluted to an OD600 of 0.05, and grown in LB broth at room temperature to OD600 of about 0.8. Total RNA was isolated from cells with the RNeasy minikit (Qiagen). Residual DNA was removed by treatment with ribosomal DNase I (rDNase I) (Ambion) and confirmed by PCR. cDNA was synthesized from the RNA and used for quantitative PCR on an ABI Prism 7900 sequence detection system (TaqMan; Applied Biosystems). The quantity of mRNA was normalized relative to the quantity of the reference gene crr (y1485), whose expression level was not affected by in vivo or in vitro growth conditions (29). The ratio of the normalized quantity of hmsT mRNA in different strains to the normalized quantity in the wild-type samples was calculated. The primers and probe sets used are listed in Table S1 in the supplemental material. Results from three independent experiments done in triplicate were analyzed by one-way ANOVA with Bonferroni's test.
Western blotting.
Procedures for Western blotting were performed essentially as described previously (31). For HmsT detection, proteins were separated on a 12% Bis-Tris polyacrylamide gel (Invitrogen) and blotted onto nitrocellulose. The immunoblots were probed with HmsT rabbit antibody (25). Following incubation of horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (Invitrogen), the immunoreactive proteins were detected with Immobilon Western HRP substrate (Millipore). For RcsA-His6 detection, proteins were treated with 8 M urea and separated on a 15% SDS-PAGE gel and blotted onto polyvinylidene difluoride (PVDF) membrane. The immunoblots were further detected by anti-His antibody (Invitrogen) following the same protocol described above.
Transcription start site of hmsT.
The transcription start site of the hmsT gene was determined by using the FirstChoice RLM-RACE (random amplification of cDNA ends) kit (Ambion) according to the manufacturer's instructions. Primer sequences are listed in Table S1 in the supplemental material.
Protein purification.
The RcsB-His6 expression plasmid pCBD209 was constructed by inserting a PCR-amplified rcsB fragment into pBAD/Myc-His vector (Invitrogen). Expression plasmids for rcsAYPSTB (pCBD253) and rcsAYPE (pCBD254) were made by PCR amplification from Y. pseudotuberculosis and Y. pestis, respectively, and cloning the products into pMal-c4x. RcsB with a C-terminal His6 tag was purified by growing strain CDY475 at 26°C in LB medium with 100 mg/liter ampicillin to an OD600 of ∼0.6. Arabinose (0.2%) was added to the culture, and growth was continued for 4 h before cells were harvested by centrifugation and stored at −80°C. Frozen cells were thawed, resuspended in buffer, and disrupted by sonication. The protein was then purified by using a Ni-nitrilotriacetic acid (NTA) His · Bind purification kit (Novagen), as recommended by the manufacturer. This single step provided RcsB-His6 with greater than 80% purity, as assessed by SDS-polyacrylamide gel electrophoresis with Coomassie blue staining. MalE-RcsAYPSTB fusion and MalE-RcsAYPE fusion proteins were purified from strains carrying the expression plasmid pCBD253 or pCBD254, using the pMal purification system (New England BioLabs) following the manufacturer's protocols.
EMSA.
For the electrophoretic mobility shift assay (EMSA), a 117-bp PCR product containing the RcsAB box or mutated RcsAB box of the hmsT promoter region was amplified using the primers shown in Table S1 in the supplemental material. Purified recombinant protein was added to DNA binding reaction mixtures containing 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, 10 mM dithiothreitol (DTT), 500 μg/ml bovine serum albumin (BSA), and 35 ng PCR product. The binding assays were performed in a volume of 16 μl at room temperature for 30 min. After incubation, 4 μl of loading buffer (40% sucrose, 0.01% bromophenol blue, in binding buffer) was added, and the samples were electrophoresed at 70 V for 1 h in 6% DNA retardation gels (Invitrogen). The gels were stained with ethidium bromide.
RESULTS
The Y. pestis rcsAYPSTB biofilm defect is rescued by hmsT overexpression.
In a previous study, we reported evidence that in Y. pestis, rcsA is a nonfunctional pseudogene, which we designated rcsAYPE (33). We showed that replacing rcsAYPE with the functional Y. pseudotuberculosis allele (rcsAYPSTB) resulted in strong loss of biofilms. In the reciprocal experiment, replacing the Y. pseudotuberculosis gene with the pseudogene allele, Y. pseudotuberculosis biofilm development was derepressed.
A hypothesis consistent with these results is that RcsAYPSTB represses transcription of the diguanylate cyclase gene hmsT, a known positive regulator of biofilms (4, 20, 32). If the hypothesis is correct, overexpressing hmsT in the Y. pestis ΔrcsAYPE::rcsAYPSTB (rcsAYPSTB) background could be expected to restore biofilm production. We tested this with both in vitro and in vivo biofilm assays. In the in vitro assay, bacteria in growth medium are incubated in polystyrene dishes for approximately 24 h. After washing to remove planktonic bacteria, the adherent biofilm is stained with crystal violet, which is then solubilized and quantified. No biofilm was detectable in the Y. pestis rcsAYPSTB background (Fig. 1A). When the rcsAYPSTB strain was transformed with a high-copy hmsT plasmid, biofilms were restored, and in fact exceeded the wild-type level by about 4-fold. Essentially the same overproduction was observed when a biofilm-defective hmsT mutant was transformed with the hmsT overexpression plasmid.
Fig 1.

HmsT overexpression suppresses the biofilm defect of RcsAYPSTB expression. (A) Y. pestis biofilms produced in polystyrene culture dishes and quantified by crystal violet staining (Materials and Methods). The mean and standard deviation (SD) are indicated; all of the strains except for the rcsAYPSTB RcsAB* and RcsAB* strains differed significantly from the wild type (P < 0.05). (B) Biofilms adhering to the head of C. elegans were assayed for the ability to inhibit nematode growth by blocking feeding. Nematode eggs were deposited on Y. pestis lawns, and the fraction (mean ± SD) of animals developing to the fourth larval (L4) stage was scored (Materials and Methods).
Deleting rcsB in the rcsAYPSTB background also caused biofilm overproduction (Fig. 1A). This indicates that RcsAYPSTB-mediated repression requires RcsB, consistent with the known functioning of Rcs phosphorelays (23) and our previous results (33). It also indicates that although rcsA is a pseudogene in Y. pestis, other genes encoding proteins of the Rcs phosphorelay are functional. In particular, the response regulator RcsB is capable of RcsA-independent repression.
In the in vivo assay, biofilms form on the nematode C. elegans and block the animal's feeding and development. Beginning with eggs at time zero, C. elegans normally develops to the fourth larval stage (L4) in 2 days at 20°C. Wild-type Y. pestis makes biofilms that prevent almost all worms from becoming L4s. An hmsT mutant had a strong, although somewhat variable, defect; plasmid overexpression restored growth inhibition to the wild-type level (Fig. 1B). In the same experiments, a strain with functional rcsAYPSTB substituted for the rcsAYPE pseudogene had a complete biofilm defect (i.e., all nematodes developed normally). Plasmid overexpression of hmsT fully rescued this defect. (If biofilm overproduction occurred it would not have been detected with this in vivo assay because wild-type Y. pestis completely inhibits nematode growth.)
The in vitro and in vivo experiments together suggest that hmsT is downstream of the Rcs phosphorelay and repressed by functional RcsA in an RcsB-dependent manner.
Rcs negatively regulates Y. pestis hmsT transcription.
To further examine Rcs control of hmsT, we first deleted an endogenous Y. pestis lacZ gene (5) and then constructed transcriptional fusions using E. coli lacZ as the reporter. All fusions were integrated into the chromosome at their native locus.
We first compared two isogenic strains: one with the wild-type rcsAYPE pseudogene and the other with the rcsAYPSTB functional allele. Cells were grown in broth to mid-exponential, early stationary, and late stationary phases and were also grown overnight on agar and assayed for β-galactosidase activity. Under each growth condition, hmsT::lacZ transcription was strongly reduced in the rcsAYPSTB background (Table 2).
Table 2.
RcsB and RcsA-PSTB repression of hmsT transcription in Y. pestis
| Y. pestis strain | Descriptiona | β-Galactosidase activity (Miller units) under growth conditionb: |
|||
|---|---|---|---|---|---|
| EP | ES | LS | Agar | ||
| CDY632 | hmsT::lacZ | 5.3 ± 1.4 | 5.9 ± 0.9 | 8.3 ± 1.6 | 9.5 ± 1.6 |
| CDY1071 | rcsAYPSTB hmsT::lacZ | 1.3± 0.2c | 1.4± 0.2c | 1.7± 0.3c | 2.2± 0.4c |
| CDY636 | hmsT::lacZ (empty vector) | 6.0 ± 1.4 | 7.1 ± 1.2 | 9.6 ± 1.9 | 9.8 ± 1.1 |
| CDY637 | hmsT::lacZ (p-rcsB) | 4.2 ± 1.1 | 4.1± 1.2d | 4.0± 1.3d | 4.1± 1.1d |
| CDY640 | hmsT::lacZ (empty vector) | 5.4 ± 1.4 | 7.0 ± 1.4 | 6.1 ± 1.1 | 9.0 ± 1.6 |
| CDY639 | hmsT::lacZ (p-rcsAYPSTB) | 1.3± 0.3e | 1.4± 0.2e | 0.9± 0.2e | 1.4± 0.1e |
| CDY638 | hmsT::lacZ (p-rcsAYPE) | 6.7 ± 1.1 | 5.6 ± 1.1 | 6.8 ± 1.1 | 8.8 ± 1.1 |
| CDY567 | hmsH::lacZ | 10.5 ± 1.6 | 11.0 ± 1.5 | 9.8 ± 2.5 | 11.1 ± 1.8 |
| CDY568 | rcsAYPSTB hmsH::lacZ | 8.6 ± 1.4 | 8.8 ± 2.3 | 9.4 ± 1.6 | 9.5 ± 1.7 |
| CDY614 | hmsP::lacZ | 1.8 ± 0.2 | 1.6 ± 0.1 | 1.8 ± 0.1 | 1.8 ± 0.1 |
| CDY615 | rcsAYPSTB hmsP::lacZ | 1.7 ± 0.1 | 1.7 ± 0.6 | 1.7 ± 0.4 | 1.8 ± 0.1 |
| CDY622 | ΔlacZ | 0.1 ± 0.1 | 0.2 ± 0.1 | 0.3 ± 0.1 | 0.2 ± 0.0 |
All strains had deletion of the chromosomal lacZ. See Table 1 for further description of the plasmids.
EP, exponential phase; ES, early stationary phase; LS, late stationary phase, 26°C, LB cultures; agar, colonies from LB agar plate incubated at 26°C for 24 h. Statistically significant differences are highlighted in boldface.
P < 0.01 compared to CDY632.
P < 0.05 compared to CDY636.
P < 0.01 compared to CDY640.
We next used the hmsT::lacZ reporter to determine the effects of overexpression of RcsB, RcsAYPSTB, and RcsAYPE. Overexpression of RcsB produced moderate decreases in hmsT transcription under all conditions tested (Table 2); this is consistent with our previous study showing that RcsB overexpression reduces in vitro biofilms (33). Overexpression of functional RcsAYPSTB greatly decreased hmsT transcription, while the pseudogene-encoded RcsAYPE resulted in weak effects in both decreasing and increasing directions, suggestive of experimental noise.
To confirm the results of lacZ reporter assays, we used quantitative reverse transcription-PCR (qRT-PCR). Transcription of hmsT was strongly decreased when functional rcsAYPSTB was substituted for pseudogene rcsAYPE (Fig. 2). Deletion of rcsB in a strain with the pseudogene rcsAYPE produced a substantial increase in hmsT transcription over the wild-type level, suggesting that RcsB represses hmsT to some extent in the absence of functional RcsA. Deletion of rcsD repressed hmsT transcription, while double deletion of rcsB and rcsD produced an increase in transcription of hmsT (Fig. 2). This indicates that rcsD is functional and derepresses hmsT transcription through rcsB, consistent with the biofilm assays (Fig. 1A) (33).
Fig 2.
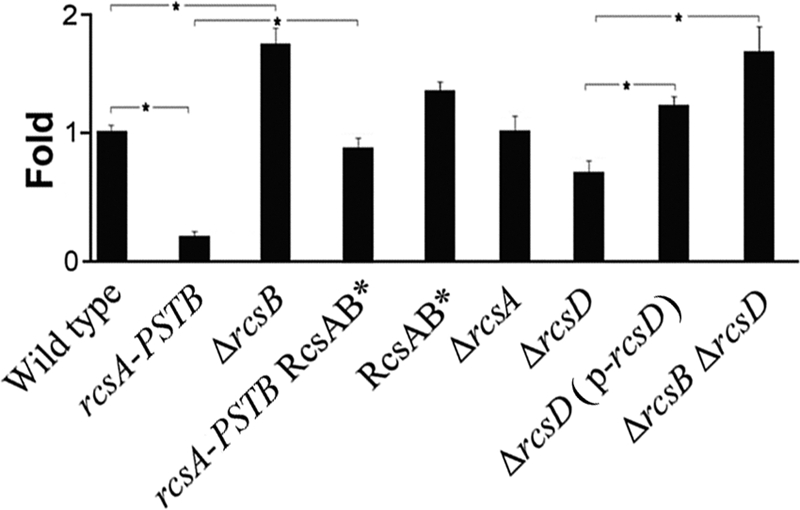
Effect of Rcs on hmsT transcription in Y. pestis. hmsT mRNA levels were determined by quantitative real-time PCR (Materials and Methods) and normalized to wild type. The means and standard deviations from three independent experiments with three replicates are indicated. *, P < 0.05.
We also examined HmsT expression by Western blotting. In Y. pestis with functional rcsAYPSTB, HmsT was reduced to a nearly undetectable level (Fig. 3). When rcsB was deleted in the rcsAYPSTB background, expression was restored, further confirming that RcsA function requires RcsB. Consistent with the result that hmsT transcription was increased in an rcsB deletion strain (Fig. 2), HmsT expression also appeared to be slightly greater in the rcsB deletion strain (Fig. 3).
Fig 3.

HmsT expression in Y. pestis. Western blots of total protein-matched lysates prepared from stationary-phase room temperature LB cultures and probed with polyclonal anti-HmsT antibody. The strain designations (Table 1) are as follows: ΔhmsT, CDY497; wild type, KIM6+; ΔrcsB, CDY326; rcsA-PSTB (rcsAYPSTB), CDY330; and ΔrcsB rcsA-PSTB (rcsAYPSTB), CDY421.
We used lacZ reporters to examine two other possible targets of RcsAYPSTB regulation. The operon hmsHFRS directs exopolysaccharide synthesis and is essential for Y. pestis and Y. pseudotuberculosis biofilms (10, 14, 15, 19). We assayed hmsH::lacZ transcriptional fusions to determine whether RcsA negatively regulates hmsHFRS (Table 2). Under all conditions tested, β-galactosidase was somewhat reduced in the rcsAYPSTB background, but this difference was not statistically significant, suggesting that Rcs has little direct effect on hmsHFRS expression. We also examined transcription of hmsP, encoding a c-di-GMP phosphodiesterase that negatively regulates biofilms. Transcription of an hmsP::lacZ fusion was unchanged in the rcsAYPSTB background, and plasmid overexpression of rcsB also did not affect hmsP transcription (Table 2). We conclude that hmsP is not an Rcs regulatory target.
RcsA represses hmsT in Y. pseudotuberculosis.
We showed previously that Y. pseudotuberculosis strain IP32953 does not make biofilms in the C. elegans model, but will do so if its functional rcsAYPSTB is replaced by the Y. pestis pseudogene rcsAYPE (33). This suggested that, in Y. pseudotuberculosis as in Y. pestis, Rcs regulates hmsT transcription. We tested this with qPCR assays and Western blotting of the wild-type IP32953 strain and an isogenic ΔrcsAYPSTB::rcsAYPE (rcsAYPE) substitution strain. Transcription of hmsT was increased 5-fold when the wild-type rcsAYPE allele was replaced by pseudogene rcsAYPE (Fig. 4A). In Western blotting, HmsT was undetectable in the wild type but was detected with the rcsAYPE substitution (Fig. 4B).
Fig 4.
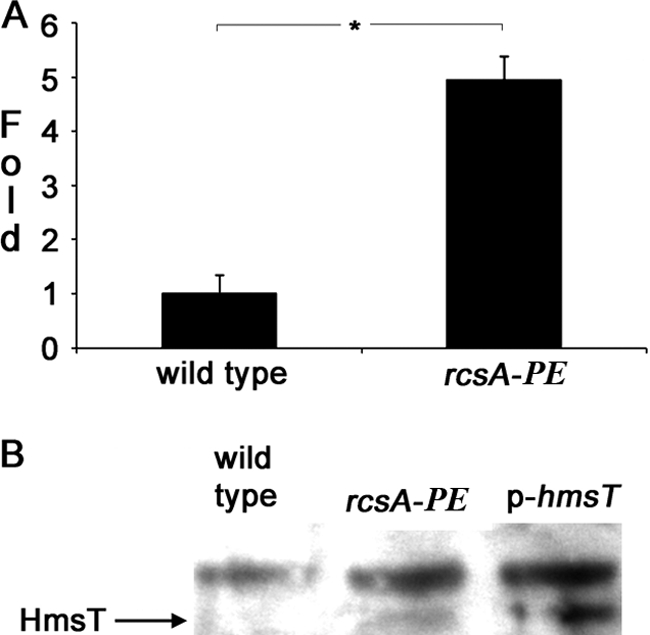
Effect of pseudogene rcsAYPE on hmsT expression in Y. pseudotuberculosis. Shown are relative amounts of hmsT mRNA (A) and HmsT protein (B) made by the wild type (IP32953) with a functional rcsA allele (rcsAYPSTB) and the isogenic strain (CDY564) with the pseudogene (rcsAYPE) replacement. The transcript level was determined by qRT-PCR as described in the legend to Fig. 2, and the protein level was determined by Western blotting as described in the legend to Fig. 3. The p-hmsT strain (CDY985) was included in panel B to allow the HmsT band to be distinguished from an unidentified cross-reacting protein.
An RcsAB box in the Y. pestis hmsT promoter mediates downregulation.
Using a PCR-based method (Materials and Methods), we determined the hmsT transcriptional start site to be 128 bp upstream of the initial ATG. The hmsT transcriptional start site is located in a palindromic RcsAB box that matches the consensus sequence at 12 of 14 nucleotides, including all 6 of the most conserved (34). The box is found at approximately the same position upstream of hmsT in all sequenced Yersinia spp. (see Table S2 in the supplemental material).
To determine whether Rcs transcriptional regulation acts on the RcsAB box, we mutated the box on the chromosome of wild-type and rcsAYPSTB Y. pestis strains by changing the CC dinucleotide to AG (designated RcsAB*). The mutation rescued the in vitro biofilm defect of the strain carrying functional rcsAYPSTB (Fig. 1A), and it restored hmsT transcription to the wild-type level (Fig. 2). These findings suggest that the mutation reduced or abolished binding by RcsB-RcsAYPSTB.
hmsT promoter binding by RcsB alone and by RcsB with functional RcsA.
We directly assayed RcsB and RcsA binding to the RcsAB box using an electrophoretic mobility shift assay (EMSA). The DNA probe was a 117-bp promoter sequence comprising the RcsAB box and 38 bp 5′ and 65 bp 3′ of the box. RcsB was purified with a C-terminal His Tag, and RcsA was purified with an N-terminal MalE fusion. The promoter DNA was electrophoresed alone or after incubation with purified proteins (see Materials and Methods).
Purified RcsB retarded migration of the hmsT promoter in a concentration-dependent manner (Fig. 5A), indicating that RcsB binds the DNA without an accessory protein. This is consistent with genetic results (Fig. 2) suggesting that RcsB downregulates hmsT transcription.
Fig 5.

RcsB and RcsB-RcsAYPSTB bind the hmsT promoter. (A) Electrophoretic mobility shift assays (EMSAs) of hmsT promoter DNA incubated with increasing concentrations of RcsB. Lanes 1 and 12, hmsT probe alone; lanes 2 to 11, hmsT probe with 100, 200, 400, 600, 800, 1,000, 1,500, 2,000, 3,000, or 5,000 ng of RcsB in the 16-μl reaction mixture. (B) Supershift of hmsT promoter by RcsAYPSTB but not RcsAYPE. Lane 1, hmsT probe alone; lanes 2 to 12, hmsT probe with 250 ng RcsB and with 250, 500, 1,000, 1,500, or 2,000 ng of RcsAYPSTB (lanes 3 to 7) or RcsAYPE (lanes 8 to 12).(C) RcsAB box mutation alters protein binding to the hmsT promoter. hmsT promoters with wild-type RcsAB box (top) or with the mutated RcsAB* box (bottom) were tested with identical protein combinations. Lanes 1 and 2, free hmsT probe in the absence or presence of bovine serum albumin (BSA); lanes 3 to 12 also contained BSA. Lanes 3 to 6 and 9 to 12 contained RcsB at 250, 500, 800, or 1,200 ng per reaction mixture; lanes 7 to 12 contained RcsAYPSTB at 2,000 ng (lane 7) or 3,000 ng (lanes 8 to 12) per reaction mixture.
RcsAYPSTB and RcsAYPE were used in the EMSA together with a fixed concentration of RcsB that produced a mobility shift. RcsAYPSTB with RcsB produced a concentration-dependent supershift of the promoter (Fig. 5B). RcsAYPE with RcsB did not supershift even at the maximum concentration tested (2,000 ng per 16-μl reaction). In fact, at high RcsAYPE concentrations the RcsB shift was almost undetectable (Fig. 5B, lanes 10 to 12). This suggests that the aberrant RcsAYPE accessory protein binds RcsB in a manner that prevents RcsB promoter binding.
In the absence of RcsB, RcsAYPSTB had no effect on mobility even at very high concentrations (Fig. 5C, lanes 7 and 8). This is consistent with results for E. coli RcsA, which does not bind DNA in the absence of E. coli RcsB (23).
To examine binding specifically to the RcsAB box, we performed EMSA on both the wild-type and mutated boxes. With the wild-type box, 250 ng of RcsB was sufficient to produce a strong mobility shift, and at 500 ng, the free probe was almost undetectable (Fig. 5C, lanes 3 and 4). In contrast, with the mutated box, it required 800 ng of RcsB to produce a strong shift, and 1,200 ng for essentially complete shifting (Fig. 5C, lanes 5 and 6). The mutations also affected RcsB-RcsAYPSTB binding, which was assayed using a fixed amount (2,000 ng) of RcsAYPSTB and increasing concentrations of RcsB. With the wild-type RcsAB box, 800 ng of RcsB was sufficient to render the free probe undetectable (Fig. 5C, lane 11); with the mutated box, the free probe was still detectable even with 1,200 ng of RcsB (Fig. 5C, lane 12).
rcsA is not a biofilm-related gene in Y. pestis.
The above EMSA results showed RcsAYPE prevents RcsB binding to hmsT promoter (Fig. 5B). However, deletion or overexpression of rcsAYPE did not affect biofilm formation or hmsT transcription in Y. pestis (Fig. 1A, 2, and 6A and Table 2), indicating that rcsAYPE is a nonfunctional pseudogene. One explanation for the apparent discrepancy is that RcsAYPE is not expressed in vivo. To test this and due to the absence of an RcsA antibody, plasmids expressing RcsAYPSTB-His6 and RcsAYPE-His6 were constructed. Addition of a His6 tag on the C terminus of RcsA did not affect the role of RcsAYPSTB or RcsAYPE (data not shown). Expression of RcsAYPE and RcsAYPSTB was further analyzed by immunoblotting. As shown in Fig. 6B, both RcsAYPE and RcsAYPSTB proteins were detected. Taken together, these results suggested RcsAYPE can be expressed in vivo, but it does not function to downregulate hmsT.
Fig 6.

RcsAYPE does not have a biofilm-related regulatory function in Y. pestis. (A) Relative amounts of adherent biofilm made by Y. pestis KIM6+ derivatives. The mean and standard deviation from at least three independent experiments with three replicates are indicated. (B) Western blots of total protein-matched lysates prepared from stationary-phase room temperature LB cultures and probed with polyclonal anti-His antibody. As predicted from genome sequence data, RcsAYPE is 1 kDa bigger than RcsAYPSTB because of a 10-amino-acid internal duplication. Strain designations (Table 1) are as follows: ΔrcsA, CDY327; rcsA-PE (rcsAYPE) SY743; and rcsA-PSTB (rcsAPSTB), SY744.
DISCUSSION
Y. pestis and Y. pseudotuberculosis diverged from a common ancestor within the past 20,000 years (1, 2). Despite many genetic and genomic similarities, the two have markedly different life cycles (6, 21, 26, 30). Y. pestis exists enzootically, shuttling between rodents and their fleas, and because of multiple auxotrophies is apparently an obligate parasite. Y. pseudotuberculosis is a prototroph capable of life outside a host, and is often found in association with warm-blooded animals. As a pathogen, Y. pseudotuberculosis is infectious orally; it has no known invertebrate vector and was unable to block fleas in extensive tests (13).
We previously obtained evidence suggesting that, during the evolution of Y. pestis, Y. pestis rcsA mutated from a functional allele (rcsAYPSTB) to a pseudogene (rcsAYPE) was the product of natural selection and not genetic drift (33). The mutation apparently was required for Y. pestis to colonize its flea vector with a biofilm, because Y. pestis in which rcsAYPSTB was substituted for rcsAYPE were strongly defective for flea blockage (33).
In the present study, we show that a target of Rcs regulation is hmsT, encoding a DGC that regulates biofilms. Several lines of evidence support this conclusion. First, in both in vivo and in vitro assays, hmsT overexpression restores biofilms to Y. pestis expressing rcsAYPSTB (Fig. 1). Second, hmsT transcription is reduced when rcsAYPSTB is present in the cells (Table 2 and Fig. 2), and HmsT protein levels are also reduced (Fig. 3). Consistent with the Y. pestis results, when pseudogene rcsAYPE was placed in Y. pseudotuberculosis, hmsT transcription was markedly increased, and HmsT protein became detectable (Fig. 4); the same substitution conferred biofilm competence in the C. elegans assay to a Y. pseudotuberculosis strain that did not otherwise form detectable biofilms in that system (33). Third, hmsT transcription is reduced in an rcsD deletion strain, while hmsT transcription is increased in an rcsB and rcsD double deletion strain (Fig. 2). Finally, we showed that RcsB alone, or RcsB combined with RcsAYPSTB, binds the hmsT promoter, while RcsAYPSTB alone does not (Fig. 5).
RcsAYPE prevents RcsB binding to hmsT promoter in vitro (Fig. 5B) and can be expressed in Y. pestis (Fig. 6B), but it did not have a biofilm-related function under our tested conditions (Fig. 1A, 2, and 6B and Table 2), indicating rcsAYPE is a pseudogene rather than a functional mutant allele. However, we cannot preclude the possibility that RcsAYPE plays a role under a specific environmental condition or in other Rcs-regulated phenotypes.
It has been reported that Rcs system negatively regulates flhDC transcription in Escherichia coli (16). An RcsAB box is present at 4 bp downstream of the transcription start site of the flhDC gene, and RcsAB heterodimer binding to the box might prevent the RNA polymerase from binding to the promoter (16). In Yersinia spp., the hmsT transcription start site is located in the RcsAB box; thus, the Rcs system might use a similar mechanism to repress hmsT transcription in Y. pestis.
A previous report on Y. pseudotuberculosis Rcs by Hinchliffe et al. produced evidence that Rcs positively regulates biofilms in vitro (18). This appears to contradict our findings, both in the present study (Fig. 1) and in previous results (33), that Rcs negatively regulates biofilms. The reasons for this discrepancy are uncertain, but it is significant that the study by Hinchliffe et al. reported robust biofilm formation at 37°C (18). In both Y. pestis (20) and Y. pseudotuberculosis (C. Darby, unpublished data), biofilms that require the hmsHFRS exopolysaccharide operon and hmsT biofilms have been shown to be downregulated at 37°C. The study by Hinchliffe et al. did not report whether the biofilms observed were dependent on either hmsT or hmsHFRS. It is possible then, that the observed Rcs-dependent biofilms are not hmsT or hmsHFRS dependent, i.e., that Y. pseudotuberculosis has multiple means of making biofilms under different environmental conditions.
Y. pestis hmsT mutation and Y. pestis rcsAYPSTB strains have similar biofilm phenotypes in vitro but different biofilm phenotypes in vivo. Y. pestis rcsAYPSTB forms almost no biofilm on the head of C. elegans or in the digestive tract of fleas, whereas the Y. pestis hmsT mutant forms intermediate levels of biofilm on the head of C. elegans and in the digestive tract of the flea (Fig. 1B) (32). Thus, repressing hmsT is not the only mechanism by which Rcs negatively regulates biofilms. Rcs may regulate other target genes to repress Yersinia biofilm formation.
Our data support a model of multilevel control of hmsT and thus of biofilms. In the presence of RcsB and functional RcsA, as is found in Y. pseudotuberculosis, hmsT transcription is tightly repressed. Presumably Y. pseudotuberculosis has mechanisms to relieve this repression under appropriate conditions, but the place of biofilms in the Y. pseudotuberculosis life cycle is not known. This tight repression apparently was partially relieved during Y. pestis evolution by the conversion of rcsA to a pseudogene (33). There remains a residual repression mediated by Rcs, as indicated by the fact that Y. pestis rcsB deletion strains overproduce biofilms and the rcsD deletion strain produces decreased biofilms. The reason for the apparent continuing need for Rcs is uncertain. It is possible that the biofilm overproduction that occurs when rcsB is deleted reaches an extent that the insects would no longer perform as well as vectors, implying selection to maintain rcsB. Furthermore, deletion of either rcsC or rcsD in Y. pseudotuberculosis resulted in defects in adhesion to, and invasion of, a human epithelial cell line (18), suggesting that the Rcs phosphorelay also functions during mammalian infection. If also true in Y. pestis, this would preclude mutation of rcsB as an adaptive step in Y. pestis evolution. In summary, then, it appears that Y. pestis evolved to occupy the flea niche in part by derepressing hmsT, thereby derepressing biofilms.
Supplementary Material
ACKNOWLEDGMENTS
We thank Alexandra Koumoutsi for assistance with strain construction.
This work was supported by NIH grant AI057512 (to C.D.); the Division of Intramural Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), and by the Institute of Pathogen Biology, Chinese Academy of Medical Sciences.
Footnotes
Published ahead of print 10 February 2012
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1. Achtman M, et al. 2004. Microevolution and history of the plague bacillus, Yersinia pestis. Proc. Natl. Acad. Sci. U. S. A. 101: 17837– 17842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Achtman M, et al. 1999. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 96: 14043– 14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bobrov AG, et al. 2011. Systematic analysis of cyclic di-GMP signalling enzymes and their role in biofilm formation and virulence in Yersinia pestis. Mol. Microbiol. 79: 533– 551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bobrov AG, Kirillina O, Forman S, Mack D, Perry RD. 2008. Insights into Yersinia pestis biofilm development: topology and co-interaction of Hms inner membrane proteins involved in exopolysaccharide production. Environ. Microbiol. 10: 1419– 1432 [DOI] [PubMed] [Google Scholar]
- 5. Bobrov AG, Perry RD. 2006. Yersinia pestis lacZ expresses a beta-galactosidase with low enzymatic activity. FEMS Microbiol. Lett. 255: 43– 51 [DOI] [PubMed] [Google Scholar]
- 6. Brubaker RR. 1991. Factors promoting acute and chronic diseases caused by yersiniae. Clin. Microbiol. Rev. 4: 309– 324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chain PS, et al. 2004. Insights into the evolution of Yersinia pestis through whole-genome comparison with Yersinia pseudotuberculosis. Proc. Natl. Acad. Sci. U. S. A. 101: 13826– 13831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Darby C. 2008. Uniquely insidious: Yersinia pestis biofilms. Trends Microbiol. 16: 158– 164 [DOI] [PubMed] [Google Scholar]
- 9. Darby C, Ananth SL, Tan L, Hinnebusch BJ. 2005. Identification of gmhA, a Yersinia pestis gene required for flea blockage, by using a Caenorhabditis elegans biofilm system. Infect. Immun. 73: 7236– 7242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Darby C, Hsu JW, Ghori N, Falkow S. 2002. Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature 417: 243– 244 [DOI] [PubMed] [Google Scholar]
- 11. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97: 6640– 6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Donnenberg MS, Kaper JB. 1991. Construction of an eae deletion mutant of enteropathogenic Escherichia coli by using a positive-selection suicide vector. Infect. Immun. 59: 4310– 4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Erickson DL, Jarrett CO, Wren BW, Hinnebusch BJ. 2006. Serotype differences and lack of biofilm formation characterize Yersinia pseudotuberculosis infection of the Xenopsylla cheopis flea vector of Yersinia pestis. J. Bacteriol. 188: 1113– 1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fetherston JD, Schuetze P, Perry RD. 1992. Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol. Microbiol. 6: 2693– 2704 [DOI] [PubMed] [Google Scholar]
- 15. Forman S, et al. 2006. Identification of critical amino acid residues in the plague biofilm Hms proteins. Microbiology 152: 3399– 3410 [DOI] [PubMed] [Google Scholar]
- 16. Francez-Charlot A, et al. 2003. RcsCDB His-Asp phosphorelay system negatively regulates the flhDC operon in Escherichia coli. Mol. Microbiol. 49: 823– 832 [DOI] [PubMed] [Google Scholar]
- 17. Gerlach RG, Holzer SU, Jackel D, Hensel M. 2007. Rapid engineering of bacterial reporter gene fusions by using Red recombination. Appl. Environ. Microbiol. 73: 4234– 4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hinchliffe SJ, Howard SL, Huang YH, Clarke DJ, Wren BW. 2008. The importance of the Rcs phosphorelay in the survival and pathogenesis of the enteropathogenic yersiniae. Microbiology 154: 1117– 1131 [DOI] [PubMed] [Google Scholar]
- 19. Jarrett CO, et al. 2004. Transmission of Yersinia pestis from an infectious biofilm in the flea vector. J. Infect. Dis. 190: 783– 792 [DOI] [PubMed] [Google Scholar]
- 20. Kirillina O, Fetherston JD, Bobrov AG, Abney J, Perry RD. 2004. HmsP, a putative phosphodiesterase, and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol. Microbiol. 54: 75– 88 [DOI] [PubMed] [Google Scholar]
- 21. Koornhof HJ, Smego RA, Jr, Nicol M. 1999. Yersiniosis. II. The pathogenesis of Yersinia infections. Eur. J. Clin. Microbiol. Infect. Dis. 18: 87– 112 [DOI] [PubMed] [Google Scholar]
- 22. Lillard JW, Jr, Bearden SW, Fetherston JD, Perry RD. 1999. The haemin storage (Hms+) phenotype of Yersinia pestis is not essential for the pathogenesis of bubonic plague in mammals. Microbiology 145: 197– 209 [DOI] [PubMed] [Google Scholar]
- 23. Majdalani N, Gottesman S. 2005. The Rcs phosphorelay: a complex signal transduction system. Annu. Rev. Microbiol. 59: 379– 405 [DOI] [PubMed] [Google Scholar]
- 24. Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 25. Perry RD, et al. 2004. Temperature regulation of the hemin storage (Hms+) phenotype of Yersinia pestis is posttranscriptional. J. Bacteriol. 186: 1638– 1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prentice MB, Rahalison L. 2007. Plague. Lancet 369: 1196– 1207 [DOI] [PubMed] [Google Scholar]
- 27. Ross P, et al. 1990. The cyclic diguanylic acid regulatory system of cellulose synthesis in Acetobacter xylinum.. Chemical synthesis and biological activity of cyclic nucleotide dimer, trimer, and phosphothioate derivatives. J. Biol. Chem. 265: 18933– 18943 [PubMed] [Google Scholar]
- 28. Ross P, et al. 1987. Regulation of cellulose synthesis in Acetobacter xylinum by cyclic diguanylic acid. Nature 324: 279– 281 [DOI] [PubMed] [Google Scholar]
- 29. Sebbane F, Jarrett CO, Gardner D, Long D, Hinnebusch BJ. 2006. Role of the Yersinia pestis plasminogen activator in the incidence of distinct septicemic and bubonic forms of flea-borne plague. Proc. Natl. Acad. Sci. U. S. A. 103: 5526– 5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smego RA, Frean J, Koornhof HJ. 1999. Yersiniosis. I. Microbiological and clinicoepidemiological aspects of plague and non-plague Yersinia infections. Eur. J. Clin. Microbiol. Infect. Dis. 18: 1– 15 [DOI] [PubMed] [Google Scholar]
- 31. Sun YC, Koumoutsi A, Darby C. 2009. The response regulator PhoP negatively regulates Yersinia pseudotuberculosis and Yersinia pestis biofilms. FEMS Microbiol. Lett. 290: 85– 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sun YC, et al. 2011. Differential control of Yersinia pestis biofilm formation in vitro and in the flea vector by two c-di-GMP diguanylate cyclases. PLoS One 6: e19267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sun YC, Hinnebusch BJ, Darby C. 2008. Experimental evidence for negative selection in the evolution of a Yersinia pestis pseudogene. Proc. Natl. Acad. Sci. U. S. A. 105: 8097– 8101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wehland M, Bernhard F. 2000. The RcsAB box. Characterization of a new operator essential for the regulation of exopolysaccharide biosynthesis in enteric bacteria. J. Biol. Chem. 275: 7013– 7020 [DOI] [PubMed] [Google Scholar]
- 35. Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33: 103– 119 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.