

Abstract
The population of group B streptococci (GBS) associated with invasive infections in nonpregnant adults from 2001 to 2008 was analyzed in isolates submitted from 24 hospital laboratories in Portugal (n = 225). The isolates were characterized by antimicrobial susceptibility, pulsed-field gel electrophoresis (PFGE), multilocus sequence typing (MLST), and surface protein gene profiling. GBS invasive cases were found more frequently among men in all age groups. In addition, serotype Ia was the most frequent in our collection, whereas serotype V is dominant elsewhere. Serotype Ia was represented mainly by a single PFGE cluster defined by sequence type 23 (ST23) and surface protein gene eps and by ST24 and bca, similarly to neonatal invasive infections in Portugal, indicating that the same genetic lineages can be responsible for both vaginal colonization and invasive disease in all age groups. In contrast, the hypervirulent serotype III/ST17 neonatal lineage was responsible for a minority of infections. Serotype V isolates were distributed into two genetic lineages, one defined by ST1 and surface protein gene alp3 and macrolide resistant, and another presenting with ST2 and eps and fully susceptible to all antimicrobials tested. The erm(TR) gene was the most frequently found among erythromycin-resistant isolates, while the bovine-associated tet(O) gene was found in a minority of tetracycline-resistant isolates. Our data emphasize the importance of local identification of the genetic lineages responsible for GBS invasive infections in nonpregnant adults. The dominance of serotype Ia in invasive disease in Portugal highlights the importance of this serotype in GBS pathogenesis.
INTRODUCTION
Streptococcus agalactiae (group B streptococcus [GBS]) is well established as a colonizing agent in pregnant women and as an important cause of neonatal sepsis and meningitis (55). Nevertheless, in the past decade, GBS has been increasingly associated with invasive disease in nonpregnant adults (23, 57). Such infections are considered to be responsible for substantial morbidity and mortality, particularly in individuals with chronic underlying conditions, such as malignancies, diabetes mellitus, cirrhosis, and HIV infection, but several other risk factors have been identified in recent years (29). The spectrum of GBS disease in adults is broad, including most frequently bacteremia without a focus and skin and soft tissue, osteoarticular, and urinary tract infections (23). Less frequent clinical presentations include meningitis and endocarditis, which are, however, associated with significantly higher morbidity and mortality (21, 40, 53). Although GBS infections are most frequently community acquired, nosocomial disease is also of concern (29).
With the introduction of antenatal screening guidelines and effective prophylaxis, a significant decrease in neonatal invasive infections was observed worldwide in the last decade where these measures were implemented. On the other hand, the incidence of invasive infections unrelated to pregnancy in adults seems to be increasing worldwide (47, 54), justifying their increased study. Since the 1990s, serotype V emerged in the United States as the most frequent GBS serotype causing invasive disease in nonpregnant adults (33, 57). Later, other serotypes, such as Ia and III, have also been recognized worldwide as significant causes of invasive disease (23, 62), with the exception of Japan, where serotype Ib is reported as the leading cause of invasive disease in adults (44). In some European countries, serotypes Ia, Ib, and III have also been pointed to as important causes of invasive disease in nonpregnant adults (4, 35, 45).
Most GBS isolates remain susceptible to penicillin, the first drug of choice for the prophylaxis and treatment of GBS disease. However, increased levels of erythromycin and clindamycin resistance are being described worldwide, raising concerns as to their use as alternative second-line agents (5, 12, 27). Intrapartum antibiotic prophylaxis is proposed to contribute to the emergence and dissemination of antibiotic-resistant clones (13), as would also be expected from the high antibiotic usage among the older age groups. Therefore, the need for new preventive strategies for GBS disease is now focusing on the development of vaccines. While the first studies exclusively targeted the polysaccharide capsule, its poor immunogenicity and variable prevalence in different countries led to the suggestion to explore vaccination approaches that could overcome serotype specificity. Polysaccharide-protein conjugate vaccines, showing more favorable immune responses than polysaccharide-only vaccines, have undergone clinical trials with promising, although still preliminary, results (2, 22). Currently, purely protein vaccines, based on components of the recently described GBS pilus-like structures, are being discussed in the literature (10, 38).
DNA-based typing methods, such as pulsed-field gel electrophoresis (PFGE) and multilocus sequence typing (MLST), are now extensively used in molecular typing of GBS isolates, alongside classical serotyping and antimicrobial susceptibility testing. These methods have been helpful in discriminating GBS populations into specific genetic lineages, with some recognized for their enhanced virulence potential (6, 28, 36, 42). Nevertheless, the diversity of genotype-phenotype combinations found among GBS isolates suggests ongoing diversification of GBS genetic lineages, to which capsular transformation is likely contributing, although perhaps not as frequently as initially thought (41). Therefore, continuous surveillance and epidemiological studies are needed to ensure an accurate knowledge of how GBS serotypes and proteins are distributed among clones, across countries, and over time, as this will impact on the vaccine strategies under development.
The aim of our study was to characterize the isolates recovered from invasive disease in nonpregnant adults in Portugal; to evaluate how serotype, antimicrobial resistance, and surface protein genes are distributed among PFGE- and MLST-based genetic lineages; and to compare them to those responsible for neonatal invasive infections.
MATERIALS AND METHODS
Bacterial isolates.
A collection of 225 GBS isolates recovered from 2001 to 2008 in nonpregnant adults (≥18 years old) in Portugal was analyzed. This was a laboratory-based surveillance program in which the microbiology laboratories of 24 Portuguese hospitals were asked to isolate, identify, and submit to a central laboratory all nonduplicate GBS isolates recovered in cases of GBS invasive disease. A case of invasive disease was defined as the recovery of an S. agalactiae isolate from a normally sterile body site. Whenever GBS isolates were available from more than one sample from the same patient, only the first isolate was included in the study. The submitted isolates included those recovered from blood (n = 183), ascitic fluid (n = 15), synovial fluid (n = 14), cerebrospinal fluid (CSF) (n = 7), and pleural effusion (n = 6). Confirmation of the isolates' identification to the species level was done by Gram stain, colony morphology, catalase test, and a commercial latex agglutination technique (Slidex Strepto B; bioMérieux, Marcy L'Étoile, France).
Serotyping.
Capsular serotyping of all isolates was carried out by a latex agglutination assay with a GBS serotyping kit (Essum, Umeå, Sweden) according to the manufacturer's instructions.
Antimicrobial susceptibility testing and macrolide resistance phenotype.
Susceptibility testing for penicillin G, erythromycin, clindamycin, tetracycline, chloramphenicol, and levofloxacin was performed by disk diffusion. Clinical and Laboratory Standards Institute (CLSI) methods and interpretation criteria were used for all antimicrobial agents (14). The macrolide resistance phenotype was determined according to a double-disk test with erythromycin and clindamycin, as previously described (43). The M phenotype corresponds to resistance to erythromycin only, while the MLSB phenotype corresponds to resistance to both antibiotics.
Macrolide resistance genotypes and tetracycline resistance determinants.
Total bacterial DNA was isolated by treatment of the cells with mutanolysin and boiling. The presence of the erm(B), erm(A) [erm(TR) subclass], and mef genes was detected by a multiplex PCR, as described elsewhere (24). Two isolates that were erythromycin resistant and did not amplify the erm(B), erm(A), or mef gene were also tested for the presence of the erm(T) gene (59). To further discriminate mef genes into mef(A) or mef(E), an additional PCR was performed (56). All tetracycline-resistant isolates were screened for the presence of the tet(K), tet(L), tet(M), and tet(O) genes, as previously described (58).
Pulsed-field gel profiling and MLST.
PFGE was performed for all isolates, as previously described (42). Briefly, chromosomal DNA of the strains was prepared and digested with SmaI or, whenever necessary due to incomplete digestion, its isoschizomer, Cfr9I, and separated by PFGE. Bionumerics software (Applied Maths, Sint-Martens-Latem, Belgium) was used to generate a dendrogram for comparison of the PFGE patterns by the unweighted pair group method with arithmetic averages (UPGMA). The Dice similarity coefficient was used, with optimization and position tolerance settings of 1.0 and 1.5%, respectively. PFGE-based clusters were defined as groups of 3 or more isolates with ≥80% relatedness on the dendrogram (42). MLST was performed by sequencing seven housekeeping genes, as described previously (31), and sequence type (ST) assignment was done by using the S. agalactiae MLST database (http://pubmlst.org/sagalactiae) and analyzed using the entire database and goeBURST (26). Analysis of DNA sequences was done by using Bionumerics software (Applied Maths, Sint-Martens-Latem, Belgium). Alleles and sequence types not previously described were deposited in the S. agalactiae MLST database.
Surface protein gene profile.
Total bacterial DNA was isolated by treatment of the cells with mutanolysin and boiling. A multiplex PCR assay was performed for direct identification of GBS alpha-protein-like genes, as described elsewhere (15). The GBS alpha-C protein gene (bca) and the epsilon (eps), rib, alp2/3, and alp4 surface protein genes were identified by direct analysis of the amplicon size. A similar multiplex PCR assay was performed to distinguish the alp2 and alp3 surface protein genes, as previously described (41).
Typing concordance and statistics.
The Wallace coefficient (W) provides a quantitative measure of the clustering concordance between different typing methods (11, 48). In our collection, the Wallace coefficient was calculated to determine the concordance between PFGE-based clustering, serotyping, and surface protein gene profiling. Simpson's index of diversity (SID) was used to evaluate the diversity found among the isolates studied (11). Both these calculations, as well as the 95% confidence intervals (CI95%) were performed with the Web tools available at http://www.comparingpartitions.info. Information about the sex ratios in different age groups in the Portuguese population was obtained from Statistics Portugal (http://www.ine.pt). The Fisher exact test was used to evaluate associations. The odds ratio (OR) with 95% Wald confidence intervals (CI95%) (1) was calculated against all other serotypes or PFGE clusters and used to identify particular serotypes or PFGE clusters associated with certain characteristics, controlling for a false-discovery rate (FDR) less than or equal to 0.05 (3).
RESULTS
Isolates.
The collection of 225 GBS invasive isolates was recovered from patients with ages ranging from 19 to 92 years, with a mean age of 63 years. Two subpopulations were considered in this study: the younger subpopulation, defined as patients ranging in age from 18 to 64 years, and the elderly subpopulation (≥65 years old). Overall, the GBS isolates were more frequently recovered from men (n = 132; 58.7%) than from women (n = 93; 41.3%), but this varied markedly according to the age group (Table 1). To test if there was an overrepresentation of men, we made two simplifying assumptions: (i) there was no sex or age bias in reporting, and (ii) the entire Portuguese population could be considered at risk of developing GBS invasive infection. According to the Portuguese demographic data available at Statistics Portugal (http://www.ine.pt), in the period from 2001 to 2008, the mean sex ratio in Portugal among adults (15 to 64 years old) was 0.97 males/female and 0.72 males/female in the elderly (≥65 years old). In our collection of GBS isolates, the mean sex ratios of invasive infections among the adults and the elderly were 2.1 and 1.1 males/female, respectively. Considering the Statistics Portugal sex ratios, there is a 2-fold increase in invasive disease among younger men compared to the number of expected cases in a 0.97 males/female ratio and a 1.5-fold increase in invasive disease among elderly men compared to the expected number of cases in a 0.72 males/female ratio, suggesting that men are always at increased risk of having GBS invasive infection independent of age.
Table 1.
Distribution of the 225 GBS isolates by sex and age group
| Sexa | No. (%) |
||
|---|---|---|---|
| 18–64 yr | ≥65 yr | Total | |
| M | 64 (28.4) | 68 (30.2) | 132 (58.7) |
| F | 31 (13.8) | 62 (27.6) | 93 (41.3) |
| Total | 95 (42.2) | 130 (57.8) | 225 (100.0) |
M, male; F, female.
Serotype and MLST distribution among PFGE clusters.
The results of serotyping are summarized in Table 2, reflecting considerable overall serotype diversity (SID = 0.794; CI95%, 0.765 to 0.822). To our knowledge, this is the first time a serotype VI isolate has been identified in Portugal. This serotype is known to be one of the most prevalent among colonizing serotypes of pregnant women in Japan and Malaysia (34) but has rarely been found elsewhere.
Table 2.
Serotype distribution among age groups
| Serotype | No. (%) |
||
|---|---|---|---|
| 18–64 yr | ≥65 yr | Total | |
| Ia | 42 (18.6) | 36 (16.0) | 78 (34.7) |
| Ib | 8 (3.6) | 12 (5.3) | 20 (8.9) |
| II | 11 (4.9) | 15 (6.7) | 26 (11.6) |
| III | 14 (6.2) | 20 (8.9) | 34 (15.1) |
| IV | 1 (0.4) | 1 (0.4) | 2 (0.9) |
| V | 14 (6.2) | 30 (13.3) | 44 (19.6) |
| VI | 0 | 1 (0.4) | 1 (0.4) |
| VII | 0 | 1 (0.4) | 1 (0.4) |
| NT | 5 (2.2) | 14 (6.2) | 19 (8.4) |
| Total | 95 (42.2) | 130 (57.8) | 225 (100.0) |
Serotypes were differently distributed according to patient age groups (Table 2). In the younger subpopulation, serotype Ia was isolated three times more frequently than other serotypes, such as II, III, and V, while in the elderly subpopulation, although serotype Ia was still the most prevalent, serotypes such as III and V were almost as frequent. We therefore tested if there was a different distribution of each serotype in the two subpopulations. Although serotype Ia reached significance in the younger subpopulation (Fisher's exact test, P = 0.011), it did not remain significant after FDR correction, indicating that, in spite of the differences described, none of the different distributions of the serotypes between the two age groups considered was in fact statistically significant.
Serotype II was found to be more frequently isolated from synovial fluid than expected from its representation in invasive isolates (Fisher's exact test, P = 0.01). No other significant associations were found between the biological source of the isolate and either patient characteristics (sex or age) or any of the bacterial features studied, such as serotype or antimicrobial resistance.
PFGE analysis revealed 33 different profiles defined at ≥80% similarity that grouped into 13 PFGE clusters (having three or more isolates), the major five of which accounted for nearly 70% of the overall collection (Fig. 1). The remaining isolates (n = 26) were included in minor PFGE clusters (containing two or fewer isolates) or had unique profiles (Fig. 1). The SID for the classification of the isolates in PFGE clusters was 0.858 (CI95%, 0.824 to 0.893), indicating that the collection analyzed was quite diverse. The concordance between PFGE-based genotypes and the serotype, as given by WPFGE→serotype = 0.730 (CI95%, 0.638 to 0.821), showed that 73% of any pair of isolates in the same PFGE cluster also share the same serotype.
Fig 1.
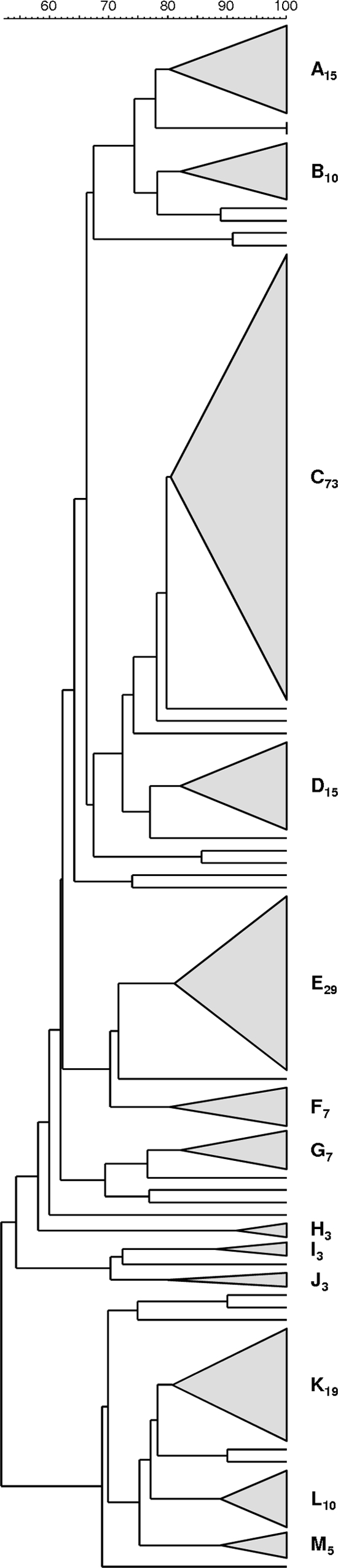
Dendrogram of the PFGE profiles of 225 GBS isolates. The dendrogram was constructed using the UPGMA method. Dice coefficients (percentages) are indicated in the scale above the dendrogram. Each cluster (defined as a group of three or more isolates with a Dice coefficient of ≥80%) is represented by a triangle proportional to the number of isolates included in the cluster. The clusters are designated by capital letters and a subscript number indicating the number of isolates included in the cluster.
Further analysis of the genetic lineages associated with each PFGE cluster included the characterization by MLST of representative isolates of all serotypes (n = 81, corresponding to at least 30% of the isolates of each serotype). Among the isolates studied, we identified five novel alleles [adhP(87), pheS(35), glnA(49), and sdhA(43 and 44)], as well as five novel STs (ST472 to ST474, ST497, and ST498).
The detailed characterization of the clusters depicted in Fig. 1 is summarized in Table 3. While most serotype Ia isolates grouped together in a particular cluster (C73), serotype III isolates distributed mostly into two different clusters (A15 and D15), and serotype V isolates were mainly found in three clusters (K19, L10, and M5). Serotype Ia isolates presented mainly with ST23 and ST24, in agreement with similar observations in neonatal invasive disease and colonization in pregnant women (39, 42). Interestingly, a small number of serotype Ia isolates (n = 7) grouped in a different cluster (E29), together with other isolates exhibiting mainly serotype Ib (n = 15). These serotype Ia isolates presented mostly with ST8 (n = 5), a sequence type that was previously found in Portugal among invasive and colonizing GBS isolates presenting exclusively the type Ib capsular serotype (40).
Table 3.
Distributions of serotypes, MLST-based sequence types, and antimicrobial susceptibility profiles among PFGE clones
| PFGE clustera | Serotype (no.)b | STs in PFGE cluster (no.)d | Genotype (no.) for macrolide resistance phenotype: |
Tetracycline resistance genotype (no.)e | ||
|---|---|---|---|---|---|---|
| cMLSB | iMLSB | M | ||||
| A15 | III (11) | [ST10 (1), ST19 (1), ST286 (1)]h, ST498 (1) | 0 | erm(TR) (3) | 0 | tet(M) (9) |
| II (2) | ST28 (1)h | 0 | 0 | 0 | tet(M) (2) | |
| V (2) | ST19 (2)h | 0 | 0 | 0 | tet(M) (2) | |
| B10 | II (7) | ST28 (1)h | 0 | 0 | 1f | tet(M) (6) |
| V (2) | ST19 (1)h | 0 | 0 | 0 | tet(M) (2) | |
| NTc (1) | ST28 (1) | 0 | 0 | 0 | tet(M) (1) | |
| C73 | Ia (66) | [ST23 (13), ST24 (6), ST144 (1)]h | 0 | 0 | mef(E) (2) | tet(M) (64) |
| NT (3) | None | 0 | 0 | 0 | tet(M) (3) | |
| II (2) | ST28 (1) | 0 | 0 | 0 | tet(M) (2) | |
| Ib (1) | ST23 (1)h | 0 | 0 | 0 | tet(M) (1) | |
| III (1) | ST17 (1) | 0 | 0 | 0 | tet(M) (1) | |
| D15 | III (13) | ST17 (4) | erm(B) (1) | 0 | 0 | tet(M) (10), tet(O) (1) |
| NT (2) | None | 0 | 0 | 0 | tet(M) (2) | |
| E29 | Ia (7) | ST8 (5)h, ST23 (2) | erm(B) (1), erm(TR) (1) | 0 | 0 | tet(M) (7) |
| Ib (15) | [ST8 (2), ST9 (1), ST10 (3), ST12 (1)]h | erm(TR) (1) | erm(TR) (1) | 0 | tet(M) (13) | |
| V (5) | ST10 (1)h | erm(B) (2) | 0 | 0 | tet(M) (4) | |
| II (1) | ST12 (1)h | erm(TR) (1) | 0 | 0 | tet(M) (1) | |
| NT (1) | None | 0 | 0 | 0 | tet(M) (1) | |
| F7 | II (4) | ST474 (1)h | 0 | 0 | 0 | tet(M) (2) |
| Ib (2) | ST10 (1)h | 0 | 0 | 0 | 0 | |
| NT (1) | ST10 (1) | 0 | 0 | 0 | 0 | |
| G7 | NT (3) | ST12 (1)h | 0 | 0 | 0 | tet(O) (3) |
| II (2) | ST12 (1)h | erm(TR) (1) | 0 | 0 | tet(O) (2) | |
| V (2) | ST12 (1)h | 0 | 0 | 0 | tet(O) (2) | |
| H3 | NT (3) | ST130 (1) | 1f | 0 | 0 | 0 |
| I3 | Ia (1) | ST88 (1)h | 0 | 0 | 0 | 0 |
| Ib (1) | None | 0 | 0 | 0 | 0 | |
| II (1) | ST88 (1)h | 0 | 0 | 0 | 0 | |
| J3 | III (2) | ST23 (1)h | 0 | 0 | 0 | tet(M) (1), tet(O) (1) |
| Ia (1) | ST23 (1)h | 0 | 0 | 0 | 0 | |
| K19 | V (13) | [ST1 (4), ST497 (1)]h | erm(B) (1), erm(TR) (1) | erm(TR) (5) | 0 | tet(M) (12) |
| NT (5) | ST1 (1) | 0 | 0 | 0 | tet(M) (5) | |
| VII (1) | ST1 (1)h | 0 | 0 | 0 | tet(M) (1) | |
| L10 | V (10) | ST2 (3) | 0 | 0 | 0 | 0 |
| M5 | V (5) | ST1 (2) | erm(TR) (1) | erm(TR) (2) | 0 | tet(M) (5) |
| Otherg | II (7) | [ST28 (1), ST472 (1)]h | 0 | 0 | 0 | tet(M) (4), tet(O) (1) |
| III (7) | None | erm(B) (2) | 0 | 0 | tet(M) (2) | |
| V (5) | None | 0 | 0 | 0 | tet(M) (2), tet(O) (1) | |
| Ia (3) | None | 0 | 0 | mef(E) (1) | tet(M) (2) | |
| IV (2) | [ST196 (1), ST473 (1)]h | 0 | 0 | 0 | tet(M) (2) | |
| Ib (1) | None | 0 | 0 | 0 | tet(M) (1) | |
| VI (1) | ST10 (1)h | 0 | 0 | 0 | 0 | |
| Total | 225 | 14 | 11 | 4 | 181 | |
PFGE clusters are presented as indicated in Fig. 1. Clusters are designated by capital letters and a subscript number indicating the number of isolates included in the cluster.
Serotypes are given in decreasing order of frequency in the clusters.
NT, nontypeable.
Brackets indicate STs that were grouped into the same PFGE cluster and belonged to the same clonal complex by goeBURST and that expressed the same serotype.
Number of isolates resistant to tetracycline.
Isolate that failed to amplify the erm(B), erm(TR), mef(A/E), and erm(T) genes.
Other included 26 isolates distributed into 20 different profiles.
STs that were grouped into the same PFGE cluster and belonged to the same clonal complex by goeBURST but expressed different serotypes.
Serotype V was the second most prevalent serotype in our collection. While the isolates in clusters K19 and M5 all presented with ST1, suggesting the existence of two sublineages within this sequence type distinguishable only by PFGE, the isolates found in cluster L10 were ST2. These observations suggest the existence of two main lineages presenting this serotype. Serotype III isolates were predominantly represented in clusters A15 and D15, the former comprising mostly ST19, previously associated with colonization (31, 36), and the latter accounting for the “hypervirulent” ST17 lineage (37, 50). However, this lineage is poorly represented in our collection compared to its overrepresentation in neonatal invasive disease (42).
Surface protein gene profiling.
All except two isolates were positive for the presence of only one surface protein gene that was diversely distributed in the bacterial population (SID = 0.747; CI95%, 0.727 to 0.768). The surface protein gene bca was the most prevalent, followed by the eps, rib, alp3, and alp2 genes, showing variable distributions across serotypes (Table 4). The alp4 gene was not found among the isolates. There was a significant association of most serotypes with a particular surface protein gene, namely, Ia and eps, Ib and bca, II and rib, III and rib, and V and alp3 (all Fisher's exact test, P < 0.0001). Nevertheless, the concordance between PFGE-based genotypes and surface protein genes was low—WPFGE→surface protein = 0.566 (CI95%, 0.491 to 0.641)—as was the concordance between serotype and surface protein genes, Wserotype→surface protein = 0.466 (CI95%, 0.409 to 0.523). These results reflect the fact that some serotypes frequently present with more than one surface protein gene, as seen for serotype Ia isolates, which are mainly associated with the surface protein genes eps and bca, and for serotype V, which presents with alp3 and eps, although only one of these genes reached significance in each serotype (Table 4). Furthermore, serotype Ia isolates are mainly grouped together in a particular PFGE cluster (C73), regardless of presenting with surface protein gene eps or bca, thus making a significant contribution to a lower Wallace coefficient.
Table 4.
Distribution of genes encoding surface proteins across serotypes
| Serotype | No. of isolates with surface protein genea: |
Total no. of isolatesc | ||||
|---|---|---|---|---|---|---|
| bca | eps | rib | alp2 | alp3 | ||
| Ia | 25 | 47 | 2 | 2 | 2 | 78 |
| Ib | 15 | 2 | 1 | 2 | 20 | |
| II | 7 | 1 | 17 | 1 | 26 | |
| III | 3 | 29 | 2 | 34 | ||
| IV | 2 | 2 | ||||
| V | 7 | 15 | 2 | 17 | 41 | |
| VI | 1 | 1 | ||||
| VII | 1 | 1 | ||||
| NTb | 8 | 3 | 4 | 5 | 20 | |
| Total | 66 | 70 | 55 | 7 | 25 | 223 |
Boldface indicates that a significant correlation between the surface protein gene and the serotype was found (see the text).
NT, nontypeable.
The total number of isolates excludes two serotype V isolates that failed to amplify any surface protein gene.
Antimicrobial susceptibility testing.
The frequency of resistance to the antimicrobials tested differed among serotypes and was found unevenly in the PFGE clusters (Table 3). All isolates were susceptible to penicillin and chloramphenicol, and 1.3% (n = 3) were resistant to levofloxacin and 80.4% (n = 181) to tetracycline.
The overall rate of erythromycin resistance (Eryr) was 12.9% (n = 29), higher than that documented previously (10.7%) in a Portuguese study analyzing the GBS isolates responsible for invasive and noninvasive disease, as well as colonization (24). Two isolates (one presenting with the M and the other with the constitutive MLSB [cMLSB] phenotype) failed to amplify any of the genes erm(B), erm(A) [erm(TR) subclass], or mef and were further tested for the presence of the erm(T) gene, frequently found among Streptococcus bovis isolates but already detected in GBS isolates (19), and again, there was no amplification. Among the remaining 27 PCR-positive isolates, the erm(B) gene was present in 25.9% (n = 7), the erm(TR) gene in 63.0% (n = 17), and the mef gene in 11.1% (n = 3). All inducible MLSB (iMLSB) isolates carried the erm(TR) gene, whereas the cMLSB isolates presented with either erm(B) or erm(TR). Such an association between iMLSB and erm(TR) was previously reported in Canada (17). Even though the MLSB phenotype was found to be distributed across serotypes and not particularly clustered, the M phenotype (in the mef-positive isolates) was found exclusively in serotype Ia isolates. Erythromycin resistance was more frequent among serotype V isolates, in agreement with previous observations in other countries (12, 18, 60, 61); however, this association was not significant after FDR correction. In an analysis of individual clusters, erythromycin resistance was found to be overrepresented in cluster K19 (OR = 7.453; CI95% 1.783 to 66.65), which clustered together most isolates of serotype V presenting with ST1, and underrepresented in serotype Ia cluster C73 (OR = 0.207; CI95%, 0.066 to 0.689), both significant after FDR correction (P = 0.021 and P = 0.044, respectively). Also, the high concordance between PFGE-based genotypes and erythromycin resistance, as given by WPFGE→EryR = 0.873 (CI95%, 0.815 to 0.931), indicates that PFGE clustering is a good predictor of erythromycin resistance. For instance, despite the fact that cluster E29 grouped together isolates presenting four different serotypes, all of these serotypes included at least one macrolide-resistant isolate.
Among tetracycline-resistant isolates, the tet(K) and tet(L) genes were not detected in any of the isolates tested. The majority of the isolates carrying the tet(O) gene grouped together in the same PFGE cluster (G7), regardless of the different capsular types expressed by the isolates included in the cluster (Table 3).
DISCUSSION
In this study, GBS invasive infections were more frequent in the elderly, reflecting the possible impact of risk factors that increase with age, such as comorbidities and immune senescence. GBS invasive infections were also substantially more frequent in men, regardless of age. Our findings are in agreement with a higher proportion of invasive infections among younger and elderly men reported elsewhere (35, 57), but the reasons behind the higher susceptibility of men remain to be clarified. It was suggested previously that sex could be a surrogate marker for other risk factors, including underlying medical conditions (29); however, our study was not designed to address this issue.
The GBS isolates characterized here revealed substantial diversity of genetic lineages defined by both PFGE and MLST (Fig. 1 and Table 3), similar to that reported among isolates causing neonatal invasive infections in both Portugal (42) and Barcelona, Spain (39). However, the collection characterized here did present higher diversity in terms of serotype (42) and surface protein genes (39) than collections of invasive isolates from neonates, with higher SID values and nonoverlapping CI95% values. In adults, we also found GBS isolates of serotypes VI and VII and isolates carrying the alp2 gene for the first time as agents of infection in Portugal. Taken together, these observations suggest a more diverse genetic structure of the GBS population causing invasive disease in adults than in neonates. A possible explanation for this difference could be the multiplicity of clinical presentations of invasive infections in adults, together with frequent and varied underlying medical conditions presented by these patients. A history of antimicrobial consumption is another factor that may increase the diversity of the GBS population by selecting for resistant isolates.
A very conserved and consistent serotype distribution is observed among the GBS isolates causing neonatal invasive disease in Europe, including Portugal, and the United States, where capsular types Ia and III are preponderant (4, 42, 49). However, in the GBS isolates causing invasive disease in nonpregnant adults in Portugal, we observed that the dominant serotypes contrast with those found in other countries. Our data show that serotype Ia was significantly more prevalent (35%), followed by serotypes V (20%), III (15%), II (12%), and Ib (9%). In other countries, such as Spain (8), Sweden (46), Norway (4), the United States (57), and Australia and New Zealand (62), serotype V is the leading cause of invasive infections in nonpregnant adults, including the elderly, and serotype Ia is much less frequent. It is possible that in Portugal there are specific lineages of serotype Ia that are well adapted to their particular niches and that may be expanding. We have previously described the increased invasive potential of serotype Ia lineages presenting with ST23 and ST24, mainly when causing early-onset disease in neonates (42). Considering the similar clonal structure observed in this study, it is possible that the same lineages are particularly prone to cause invasive infections in adults. Taken together, the dominance of serotype Ia in invasive disease in adults, as well as the previously documented importance of this serotype in neonatal invasive infections and in colonization of pregnant women (39, 42), make it clear that the same genetic lineages can be responsible for both colonization and invasive disease in all age groups. We also found a small number of ST8 serotype Ia isolates grouping in a different cluster (E29), together with other isolates exhibiting mainly serotype Ib, but also other serotypes, and presenting with ST8 and its single-locus variants (SLVs) ST9, ST10, and ST12. Moreover, these STs share only two alleles (in the atr and glcK loci) with ST24 and none with ST23, indicating that this lineage of serotype Ia isolates is unrelated to the dominant ST23 and ST24 lineages and may have resulted from capsular switching. The recipient of this putative capsular-switching event would belong to the lineage defined by PFGE cluster E29, as most of these serotype Ia and Ib isolates are not only grouped into the same PFGE cluster, but also share the same surface protein (encoded by bca) and closely related STs. This particular lineage was not found among GBS serotype Ia isolates in Portugal characterized in previous studies but had already been recovered from invasive and colonizing isolates in adults presenting exclusively the type Ib capsular serotype (40). This observation is consistent with this new serotype-genotype combination having arisen by capsular switching and possibly having an adaptive advantage for causing invasive infections in adults, explaining why the Ia/ST8/bca lineage was not previously associated with vaginal colonization or neonatal infections.
Serotype V was the second most prevalent serotype, distributed into three PFGE clusters and characterized by different sequence types and surface protein genes. The isolates found in cluster L10 were fully susceptible to all antimicrobials tested, including tetracycline, causing serotype V to be the only serotype significantly associated with tetracycline susceptibility.
The prevalence of serotype III was much lower than that found in neonatal invasive disease in Portugal (42). The two main clusters of serotype III isolates, A15 and D15, presented mainly with ST19 and ST17, respectively. Whereas ST19 was previously associated with colonization, ST17 is recognized as a major lineage responsible for neonatal invasive infections (31, 36). The underrepresentation of this lineage in our collection, in agreement with similar observations elsewhere (37), reinforces the differences between neonatal and adult invasive infections and the importance of other serotypes, particularly serotype Ia, in invasive infection.
We found a strong correlation between the serotype and the genes encoding surface proteins (Table 4). However, the correspondence is not absolute, and the surface protein gene proved helpful in discriminating different genetic lineages within serotypes. Similar to what was reported previously (39), serotype Ia was associated with two surface protein genes, bca and eps, although only eps reached significance. Considering the high number of bca-carrying isolates in our collection and the perfect association of this surface protein gene with ST24 (39), this particular sublineage seems equally adept at causing infections in adults and in neonates. Future studies, particularly in other European countries, should help evaluate whether serotype Ia is expanding in the continent or whether, on the contrary, it represents successful clones within the more limited geographical boundaries of the Mediterranean region. Nevertheless, previous reports of the ST24 sublineage as a rare clone in both Europe (52) and the United States (7) may indicate ongoing changes in the clonal composition of GBS causing invasive infections worldwide.
Two distinct surface protein genes, alp3 and eps, were found to be similarly distributed among serotype V isolates, even though only the first was significantly associated with the serotype, mainly because eps is also overrepresented in the ST23/serotype Ia isolates. Considering that most studies describe a strong association of serotype V with the alp3 gene (27, 32, 51), it is possible that our observation of a similar frequency of the genetic lineage serotype V/ST2/eps reflects the expansion of a sublineage not frequent elsewhere.
When comparing our surface protein gene-profiling results to those found among neonatal invasive isolates (39), a considerable difference was also found in serotype II isolates. Even though the number of serotype II isolates causing invasive disease in neonates was smaller than in adults, the isolates responsible for neonatal infections mainly carried the bca gene, while in this study, the number of isolates carrying the rib gene was more than double that of isolates carrying the bca gene. In addition, we found that the isolates carrying the bca gene presented mostly with ST12 (and its double-locus variant, ST474), whereas the isolates carrying the rib gene presented mainly with ST28 (and its SLV, ST472). Both ST472 and ST474 were newly identified in this study and suggest diversification of two different serotype II lineages carrying distinct surface protein genes. It is possible that the rib-carrying lineage of serotype II isolates shows a higher propensity to cause joint infections (4 out of the 5 serotype II isolates recovered from synovial fluid presented with the surface protein gene rib), being responsible for the overall association of this serotype with synovial fluid.
Erythromycin resistance was found to be similarly distributed among the constitutive and inducible MLSB phenotypes (cMLSB, n = 14; iMLSB, n = 11), while in a previous study in Portugal, the cMLSB phenotype was three times more prevalent than the iMLSB phenotype (24). The number of isolates carrying the erm(TR) gene was more than double the number of isolates carrying the erm(B) gene, also the opposite of what had been previously reported in the country (24). This could be due to the expansion or disappearance of specific clones carrying these genetic elements or to the fact that in the previous study, both invasive and noninvasive isolates were characterized (24). In contrast to the data reported here, several publications indicate that the erm(B) gene is overrepresented in serotype V isolates (9, 25, 27, 30, 61), while a higher prevalence of erm(TR) has been described only in Canada (17). This suggests that there are several distinct erythromycin-resistant genetic lineages expressing serotype V that are differentially distributed in various geographic locations. The fact that macrolide resistance was significantly associated with specific PFGE-based clusters but not with particular serotypes suggests that increasing resistance is due in part to the limited expansion of resistant clones, although horizontal dissemination of genetic elements carrying resistance determinants may also have contributed. Considering the wide consensus on the importance of serotype V in erythromycin resistance, the fact that the serotype is overrepresented in almost all studies of GBS causing invasive infections in nonpregnant adults and the diversity of resistant lineages, a detailed discrimination of macrolide-resistant clones from other regions may help us understand the evolution and dynamics of erythromycin resistance in GBS.
The nearly ubiquitous resistance to tetracycline was mostly mediated by the tet(M) gene, as described elsewhere (52). A small fraction of the isolates carried the tet(O) gene, which has been associated with bovine strains (20). Not all erythromycin-resistant isolates were simultaneously resistant to tetracycline, including those carrying the erm(B) gene, indicating that erythromycin resistance is not necessarily linked to tetracycline resistance and may not be encoded in the same mobile genetic elements, as previously suggested (16). Even though the tet(M) gene is spread throughout all serotypes and major PFGE clusters, the association between the tet(O) gene and a particular genetic lineage, as defined by the same PFGE cluster (G7) and sequence type (ST12), supports the clonal expansion of this resistance determinant.
The main limitation of our study is the fact that this is not a population-based study, although our network does include the majority of hospital-based microbiology laboratories in Portugal. Also, no audits to monitor compliance of the reporting laboratories in submitting all isolates to the central laboratory for characterization were performed. Since the records of the reporting laboratories were not independently reviewed to detect unreported cases, it is possible that not all cases of laboratory-confirmed invasive GBS disease occurring within our surveillance network were effectively reported. Accordingly, we have refrained from calculating the incidence of GBS invasive disease in Portugal, since we would probably underestimate the actual incidence. Another limitation of our study is the absence of clinical data, which prevents us from evaluating the associations between GBS infection and patient history, including underlying medical conditions that were previously shown to be risk factors for GBS invasive infection (29). Despite these limitations, we believe the collection studied is large enough to reflect the bacterial population diversity and to identify the major clones causing invasive disease among adults in Portugal. Also, since it is laboratory-based surveillance, it is unlikely that any sex or age bias exists when reporting GBS invasive cases, indicating that any sex or age imbalance in the collection reflects the actual characteristics of the GBS invasive infection cases.
GBS is a significant agent of invasive disease in adults, and the high morbidity and mortality associated with these infections justifies the continued monitoring of the pathogen. While most serotypes and genetic lineages are capable both of asymptomatic colonization and of causing invasive disease in different age groups, their prevalences are not the same, suggesting that particular lineages may be better adapted to specific lifestyles or age groups. On the other hand, the salient role played by serotype Ia isolates as causes of invasive infections in both adults and neonates in the Iberian Peninsula, together with the high prevalence of ST24, which is rarely found elsewhere, suggests that lineages with enhanced invasiveness may be emerging at a regional level. This dynamic nature of GBS invasive isolates may prove challenging to future strategies for prevention of these infections.
ACKNOWLEDGMENTS
This work was partly supported by a grant from Fundação Calouste Gulbenkian and an unrestricted grant from Glaxo Smithkline Portugal. E.R.M. was supported by a grant from Fundação para a Ciência e a Tecnologia (SFRH/BD/41761/2007).
We thank Andreas Domke for technical support.
Members of the Portuguese Group for the Study of Streptococcal Infections are as follows: Luís Lito and Lurdes Monteiro, Hospital de Santa Maria, Lisbon; Filomena Martins, Maria Ana Pessanha, Elsa Gonçalves, and Teresa Morais, Hospital de São Francisco Xavier, Lisbon; José Diogo, Ana Rodrigues, and Isabel Nascimento, Hospital Garcia de Orta, Almada; Manuela Ribeiro, Fernanda Cotta, and Dolores Pinheiro, Hospital de São João, Porto; Paulo Lopes, Ismália Calheiros, Luísa Felício, and Angelina Lameirão, Centro Hospitalar de Vila Nova de Gaia, Vila Nova de Gaia; Valquíria Alves, Antónia Read, and Margarida Monteiro, Hospital Pedro Hispano, Matosinhos; Ana Paula Castro, Hospital de Vila Real, Vila Real; Ana Florinda Alves and Henrique Oliveira, Centro Hospitalar de Coimbra, Coimbra; Ana Paula Mota and Margarida Tomaz, Hospital Senhora da Oliveira, Guimarães; Elmano Ramalheira and Fernanda Bessa, Hospital Infante Dom Pedro, Aveiro; Graça Ribeiro, Luísa Boaventura, Catarina Chaves, and Teresa Reis, Hospitais da Universidade de Coimbra, Coimbra; Helena Ramos, Ana Paula Castro, Susana Ferreira, and Paulo Pinto, Hospital de Santo António, Porto; Maria Alberta Faustino and Adelaide Alves, Hospital de Braga; Teresa Vaz, Marília Gião, and Rui Ferreira, Centro Hospitalar do Barlavento Algarvio, Portimão; Ana Fonseca, and Adriana Coutinho, Centro Hospitalar de Cascais, Cascais; Fernando Augusto Fonseca, Centro Hospitalar Póvoa do Varzim-Vila do Conde, Póvoa do Varzim; Teresa Silva Afonso and Nuno Canhoto, Hospital Central do Funchal, Funchal; Teresa Pina, M. J. Silvestre, and H. Peres, Hospital Curry Cabral, Lisbon; Ilse Fontes, and Paulo Martinho, Hospital de Santa Luzia, Elvas; Ana Cristina Silva, and Maria Hermínia Costa, Hospital de São Sebastião, Santa Maria da Feira; Margarida F. Pinto and Hermínia Choon, Hospital de Santa Marta, Lisbon; Adriana Coutinho, Hospital Espírito Santo, Évora; Maria Dinah Carvalho, I. Alves, P. Cabral, and Margarida Abecassis, Hospital Pulido Valente, Lisbon; Gina Marrão, Ana Domingos, and José Grossinho, Hospital de Santo André, Leiria; and Maria Luísa Cabral and Olga Neto, Hospital dos SAMS, Lisbon.
Footnotes
Published ahead of print 4 January 2012
REFERENCES
- 1. Altman DG. 1999. Practical statistics for medical research. Chapman & Hall, Boca Raton, FL [Google Scholar]
- 2. Baker CJ, Edwards MS. 2003. Group B streptococcal conjugate vaccines. Arch. Dis. Child. 88:375–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate—a practical and powerful approach to multiple testing. J. R. Stat Soc. Ser. B Stat. Methodol. 57:289–300 [Google Scholar]
- 4. Bergseng H, Rygg M, Bevanger L, Bergh K. 2008. Invasive group B streptococcus (GBS) disease in Norway 1996–2006. Eur. J. Clin. Microbiol. Infect. Dis. 27:1193–1199 [DOI] [PubMed] [Google Scholar]
- 5. Betriu C, et al. 2003. Erythromycin and clindamycin resistance and telithromycin susceptibility in Streptococcus agalactiae. Antimicrob. Agents Chemother. 47:1112–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blumberg HM, et al. 1996. Invasive group B streptococcal disease: the emergence of serotype V. J. Infect. Dis. 173:365–373 [DOI] [PubMed] [Google Scholar]
- 7. Bohnsack JF, et al. 2008. Population structure of invasive and colonizing strains of Streptococcus agalactiae from neonates of six U.S. Academic Centers from 1995 to 1999. J. Clin. Microbiol. 46:1285–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bolaños M, Hernández A, Santana O, Molina J, Martín-Sánchez AA. 2005. Distribution of Streptococcus agalactiae serotypes in samples from non-pregnant adults. Clin. Microbiol. Newsl. 27:151–153 [Google Scholar]
- 9. Brzychczy-Wloch M, et al. 2010. Genetic characterization and diversity of Streptococcus agalactiae isolates with macrolide resistance. J. Med. Microbiol. 59:780–786 [DOI] [PubMed] [Google Scholar]
- 10. Buccato S, et al. 2006. Use of Lactococcus lactis expressing pili from group B Streptococcus as a broad-coverage vaccine against streptococcal disease. J. Infect. Dis. 194:331–340 [DOI] [PubMed] [Google Scholar]
- 11. Carrico JA, et al. 2006. Illustration of a common framework for relating multiple typing methods by application to macrolide-resistant Streptococcus pyogenes. J. Clin. Microbiol. 44:2524–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castor ML, et al. 2008. Antibiotic resistance patterns in invasive group B streptococcal isolates. Infect. Dis. Obstet. Gynecol. 2008:727505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Centers for Disease Control and Prevention 2002. Prevention of perinatal group B streptococcal disease. JAMA 51:1–22 [Google Scholar]
- 14. Clinical and Laboratory Standards Institute 2009. Performance standards for antimicrobial susceptibility testing—nineteenth informational supplement, vol, no. 3 (M100-S19). Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 15. Creti R, Fabretti F, Orefici G, von Hunolstein C. 2004. Multiplex PCR assay for direct identification of group B streptococcal alpha-protein-like protein genes. J. Clin. Microbiol. 42:1326–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Culebras E, Rodriguez-Avial I, Betriu C, Redondo M, Picazo JJ. 2002. Macrolide and tetracycline resistance and molecular relationships of clinical strains of Streptococcus agalactiae. Antimicrob. Agents Chemother. 46:1574–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DE Azavedo JC, McGavin M, Duncan C, Low DE, McGeer A. 2001. Prevalence and mechanisms of macrolide resistance in invasive and noninvasive group B streptococcus isolates from Ontario, Canada. Antimicrob. Agents Chemother. 45:3504–3508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Diekema DJ, et al. 2003. Molecular epidemiology of macrolide resistance in neonatal bloodstream isolates of group B streptococci. J. Clin. Microbiol. 41:2659–2661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dipersio LP, Dipersio JR. 2007. Identification of an erm(T) gene in strains of inducibly clindamycin-resistant group B Streptococcus. Diagn. Microbiol. Infect. Dis. 57:189–193 [DOI] [PubMed] [Google Scholar]
- 20. Dogan B, Schukken YH, Santisteban C, Boor KJ. 2005. Distribution of serotypes and antimicrobial resistance genes among Streptococcus agalactiae isolates from bovine and human hosts. J. Clin. Microbiol. 43:5899–5906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domingo P, et al. 1997. Group B streptococcal meningitis in adults: report of twelve cases and review. Clin. Infect. Dis. 25:1180–1187 [DOI] [PubMed] [Google Scholar]
- 22. Edwards MS. 2008. Group B streptococcal conjugate vaccine: a timely concept for which the time has come. Hum. Vaccin. 4:444–448 [DOI] [PubMed] [Google Scholar]
- 23. Farley MM. 2001. Group B streptococcal disease in nonpregnant adults. Clin. Infect. Dis. 33:556–561 [DOI] [PubMed] [Google Scholar]
- 24. Figueira-Coelho J, Ramirez M, Salgado MJ, Melo-Cristino J. 2004. Streptococcus agalactiae in a large Portuguese teaching hospital: antimicrobial susceptibility, serotype distribution, and clonal analysis of macrolide-resistant isolates. Microb. Drug Resist. 10:31–36 [DOI] [PubMed] [Google Scholar]
- 25. Fluegge K, Supper S, Siedler A, Berner R. 2004. Antibiotic susceptibility in neonatal invasive isolates of Streptococcus agalactiae in a 2-year nationwide surveillance study in Germany. Antimicrob. Agents Chemother. 48:4444–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Francisco AP, Bugalho M, Ramirez M, Carriço JA. 2009. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gherardi G, et al. 2007. Molecular epidemiology and distribution of serotypes, surface proteins, and antibiotic resistance among group B streptococci in Italy. J. Clin. Microbiol. 45:2909–2916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ip M, et al. 2006. Identification of a Streptococcus agalactiae serotype III subtype 4 clone in association with adult invasive disease in Hong Kong. J. Clin. Microbiol. 44:4252–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jackson LA, et al. 1995. Risk factors for group B streptococcal disease in adults. Ann. Intern. Med. 123:415–420 [DOI] [PubMed] [Google Scholar]
- 30. Janapatla RP, Ho YR, Yan JJ, Wu HM, Wu JJ. 2008. The prevalence of erythromycin resistance in group B streptococcal isolates at a University Hospital in Taiwan. Microb. Drug Resist. 14:293–297 [DOI] [PubMed] [Google Scholar]
- 31. Jones N, et al. 2003. Multilocus sequence typing system for group B Streptococcus. J. Clin. Microbiol. 41:2530–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kong F, Gowan S, Martin D, James G, Gilbert GL. 2002. Serotype identification of group B streptococci by PCR and sequencing. J. Clin. Microbiol. 40:216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kothari NJ, et al. 2009. Invasive group B streptococcal disease in the elderly, Minnesota, USA, 2003–2007. Emerg. Infect. Dis. 15:1279–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lachenauer CS, et al. 1999. Serotypes VI and VIII predominate among group B streptococci isolated from pregnant Japanese women. J. Infect. Dis. 179:1030–1033 [DOI] [PubMed] [Google Scholar]
- 35. Lambertsen L, Ekelund K, Skovsted IC, Liboriussen A, Slotved HC. 2010. Characterisation of invasive group B streptococci from adults in Denmark 1999 to 2004. Eur. J. Clin. Microbiol. Infect. Dis. 29:1071–1077 [DOI] [PubMed] [Google Scholar]
- 36. Lin FY, et al. 2006. Phylogenetic lineages of invasive and colonizing strains of serotype III group B Streptococci from neonates: a multicenter prospective study. J. Clin. Microbiol. 44:1257–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Luan SL, et al. 2005. Multilocus sequence typing of Swedish invasive group B Streptococcus isolates indicates a neonatally associated genetic lineage and capsule switching. J. Clin. Microbiol. 43:3727–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Margarit I, et al. 2009. Preventing bacterial infections with pilus-based vaccines: the group B streptococcus paradigm. J. Infect. Dis. 199:108–115 [DOI] [PubMed] [Google Scholar]
- 39. Martins ER, et al. 2011. Group B streptococci causing neonatal infections in Barcelona are a stable clonal population: 18-year surveillance. J. Clin. Microbiol. 49:2911–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martins ER, et al. 2007. Streptococcus agalactiae serotype Ib as an agent of meningitis in two adult nonpregnant women. J. Clin. Microbiol. 45:3850–3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martins ER, Melo-Cristino J, Ramirez M. 2010. Evidence for rare capsular switching in Streptococcus agalactiae. J. Bacteriol. 192:1361–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martins ER, Pessanha MA, Ramirez M, Melo-Cristino J, and the Portuguese Group for the Study of Streptococcal Infections 2007. Analysis of group B streptococcal isolates from infants and pregnant women in Portugal revealing two lineages with enhanced invasiveness. J. Clin. Microbiol. 45:3224–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Melo-Cristino J, Fernandes ML. 1999. Streptococcus pyogenes isolated in Portugal: macrolide resistance phenotypes and correlation with T types. Portuguese Surveillance Group for the Study of Respiratory Pathogens. Microb. Drug Resist. 5:219–225 [DOI] [PubMed] [Google Scholar]
- 44. Murayama SY, et al. 2009. Capsular type and antibiotic resistance in Streptococcus agalactiae isolates from patients, ranging from newborns to the elderly, with invasive infections. Antimicrob. Agents Chemother. 53:2650–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Persson E, et al. 2008. Characterisation of invasive group B streptococci based on investigation of surface proteins and genes encoding surface proteins. Clin. Microbiol. Infect. 14:66–73 [DOI] [PubMed] [Google Scholar]
- 46. Persson E, et al. 2004. Serotypes and clinical manifestations of invasive group B streptococcal infections in western Sweden 1998–2001. Clin. Microbiol. Infect. 10:791–796 [DOI] [PubMed] [Google Scholar]
- 47. Phares CR, et al. 2008. Epidemiology of invasive group B streptococcal disease in the United States, 1999–2005. JAMA 299:2056–2065 [DOI] [PubMed] [Google Scholar]
- 48. Pinto FR, Melo-Cristino J, Ramirez M. 2008. A confidence interval for the Wallace coefficient of concordance and its application to microbial typing methods. PLoS One 3:e3696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Poyart C, et al. 2008. Invasive group B streptococcal infections in infants, France. Emerg. Infect. Dis. 14:1647–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Quentin R, et al. 1995. Characterization of Streptococcus agalactiae strains by multilocus enzyme genotype and serotype: identification of multiple virulent clone families that cause invasive neonatal disease. J. Clin. Microbiol. 33:2576–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Radtke A, et al. 2009. Identification of surface proteins of group B streptococci: serotyping versus genotyping. J. Microbiol. Methods 78:363–365 [DOI] [PubMed] [Google Scholar]
- 52. Sadowy E, Matynia B, Hryniewicz W. 2010. Population structure, virulence factors and resistance determinants of invasive, non-invasive and colonizing Streptococcus agalactiae in Poland. J. Antimicrob. Chemother. 65:1907–1914 [DOI] [PubMed] [Google Scholar]
- 53. Sambola A, et al. 2002. Streptococcus agalactiae infective endocarditis: analysis of 30 cases and review of the literature, 1962–1998. Clin. Infect. Dis. 34:1576–1584 [DOI] [PubMed] [Google Scholar]
- 54. Schrag SJ, et al. 2000. Group B streptococcal disease in the era of intrapartum antibiotic prophylaxis. N. Engl. J. Med. 342:15–20 [DOI] [PubMed] [Google Scholar]
- 55. Schuchat A. 1998. Epidemiology of group B streptococcal disease in the United States: shifting paradigms. Clin. Microbiol. Rev. 11:497–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Silva-Costa C, Pinto FR, Ramirez M, Melo-Cristino J. 2008. Decrease in macrolide resistance and clonal instability among Streptococcus pyogenes in Portugal. Clin. Microbiol. Infect. 14:1152–1159 [DOI] [PubMed] [Google Scholar]
- 57. Skoff TH, et al. 2009. Increasing burden of invasive group B streptococcal disease in nonpregnant adults, 1990–2007. Clin. Infect. Dis. 49:85–92 [DOI] [PubMed] [Google Scholar]
- 58. Trzcinski K, Cooper BS, Hryniewicz W, Dowson CG. 2000. Expression of resistance to tetracyclines in strains of methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 45:763–770 [DOI] [PubMed] [Google Scholar]
- 59. Tsai JC, et al. 2005. The erm(T) gene is flanked by IS1216V in inducible erythromycin-resistant Streptococcus gallolyticus subsp. pasteurianus. Antimicrob. Agents Chemother. 49:4347–4350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Uh Y, et al. 2004. Serotypes and genotypes of erythromycin-resistant group B streptococci in Korea. J. Clin. Microbiol. 42:3306–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. von Both U, et al. 2003. A serotype V clone is predominant among erythromycin-resistant Streptococcus agalactiae isolates in a southwestern region of Germany. J. Clin. Microbiol. 41:2166–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhao Z, et al. 2008. Distribution of genotypes and antibiotic resistance genes among invasive Streptococcus agalactiae (group B streptococcus) isolates from Australasian patients belonging to different age groups. Clin. Microbiol. Infect. 14:260–267 [DOI] [PubMed] [Google Scholar]